

Abstract
Cyclization of linear dipeptidyl precursors derived from nonribosomal peptide synthetases (NRPSs) into 2,5-diketopiperazines (DKPs) is a crucial step in the biosynthesis of a large number of bioactive natural products. However, the mechanism of DKP formation in fungi has remained unclear, despite extensive studies of their biosyntheses. Here we show that DKP formation en route to the fungal virulence factor gliotoxin requires a seemingly extraneous couplet of condensation (C) and thiolation (T) domains in the NRPS GliP. In vivo truncation of GliP to remove the CT couplet or just the T domain abrogated production of gliotoxin and all other gli pathway metabolites. Point mutation of conserved active sites in the C and T domains diminished cyclization activity of GliP in vitro and abolished gliotoxin biosynthesis in vivo. Verified NRPSs of other fungal DKPs terminate with similar CT domain couplets, suggesting a conserved strategy for DKP biosynthesis by fungal NRPSs.
Keywords: gliotoxin, natural products, biosynthesis, cyclization, nonribosomal peptide synthetase
Graphical Abstract
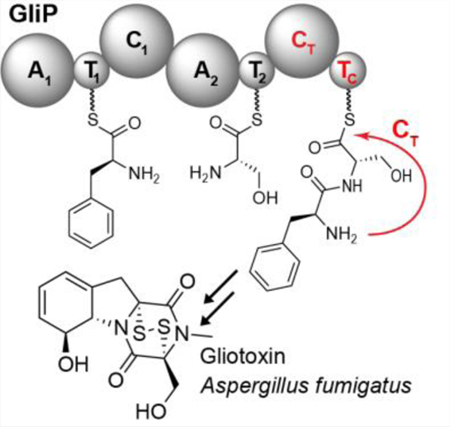
Seemingly extraneous: Diketopiperazines derived from non-ribosomal peptide synthetases, an important family of fungal virulence factors, do not form spontaneously, as presumed; instead cyclization relies on previously unannotated domains of the synthetase.
NRPS-derived DKPs form a large class of natural products with diverse biological activities.[1–5] Gliotoxin (1) is the best-known member of the epipolythiodiketopiperazines (ETPs), a family of toxic DKPs produced by a variety of filamentous fungi.[1–5] The biosynthesis of 1 has been extensively studied, due to its significant contribution to the virulence of the devastating human pathogen, A. fumigatus[6–12] and as a model system for fungal NRPS pathways. Production of 1 is accomplished via the gli biosynthetic gene cluster (BGC) in A. fumigatus and related fungi (Figure 1a). The 13-gene gli BGC encodes the transcriptional regulator, GliZ, one transporter (GliA), several backbone tailoring enzymes, and the core NRPS, GliP (Figure 1a).[13] Whereas, the majority of gli-cluster tailoring enzymes have previously been characterized extensively in vitro and in vivo (blue arrows, Figure 1a), functional analysis of the core enzyme, GliP, is incomplete (Figure 1b).[14]
Figure 1.

Gliotoxin biosynthesis in A. fumigatus. (a) gli-cluster gene annotations, including characterized tailoring enzymes (blue), tailoring enzymes with inferred functions (grey), and genes without predicted function (black). gliZ: transcription factor; gliI: pyridoxal phosphate dependent desulfurase; gliC, gliF: cytochrome P450 oxidases; gliM: O-methyltransferase; gliG: glutathione-S-transferase; gliK: glutamate cyclase; gliA: transporter; gliN: N-methyltransferase; gliT: oxidase; (b) Putative function of GliP and abbreviated biosynthesis of 1 showing the most abundant intermediates or shunt metabolites 2 and 3, as well as the detoxification product, 4.[6,9,16–19]
Homology-based annotation of GliP uncovers two adenylation (A), two condensation (C), and three thiolation (T) domains, referred to as A1-T1-C1-A2-T2-C2-T3.[14] Previous work showed that recombinant GliP converts L-Phe and L-Ser into the DKP cyclo(L-Phe-L-Ser) (2), which was speculated to result from spontaneous cyclization of a GliP-T2-tethered-L-Phe-L-Ser intermediate (Figure 1b).[7,14] Although 2 could plausibly be derived from non-enzymatic cyclization of T2-tethered L-Phe-L-Ser, the presence of the C2 and T3 domains, which in this model have no function, may suggest an alternative mechanism. In 2012, Gao et al. reported that macrocyclization of linear peptidyl precursors produced by a variety of fungal multi-module NRPSs is catalyzed by conserved terminal condensation-like (CT) domains,[15] and that CT domain activity was dependent on a conserved histidine within the amino acid sequence SHXXXDXXS/T. We noted that this CT-conserved sequence exists in the GliP-C2 domain (Figure S1), suggesting that the C2 domain in GliP may be involved in cyclization of a tethered L-Phe-L-Ser dipeptide (Figure 1b). We further hypothesized that the seemingly extraneous T3 domain also plays a role in DKP formation.
To investigate the function of the putative GliP-CT and the GliP-T3 domains in vivo, we constructed two truncation mutants in A. fumigatus, GliP-ΔT3 and GliP-ΔCTT3, and assessed the impact of these truncations on the biosynthesis of 1 and gli-pathway metabolites (1–4, Figure 2a–d). Comparison of whole-metabolome extracts from WT (Af293), GliP-ΔT3, and GliP-ΔCTT3 revealed a complete loss of 1–4 in the GliP-ΔCTT3 strain (Figure 2a–d). In extracts from the GliP-ΔT3 strain we were unable to detect 1 and the most abundant gli-pathway shunt metabolites 2 and 3, however, we observed trace quantities of 4 (99 % less than Af293), the detoxification product of 1 (Figure 2a–d).[20] These data suggested that normal biosynthesis of 1 requires the T3 domain of GliP.
Figure 2.

LC-HRMS analysis of whole-metabolome extracts from WT and mutant A. fumigatus strains. (a-d) Representative overlaid extracted-ion-chromatograms (EICs) for 1–4 in Af293 (black), GliP-ΔT3 (green), and GliP-ΔCTT3 (purple). Darker lines representative overlaid EICs for 1–4 in the indicated strains with added exogenous 1. “10 x” indicates scaling of weak signals applied for comparison. “*” = unrelated peaks.
Gliotoxin (1) serves as a positive feedback loop for its own production through regulation of gli-cluster expression.[21] Accordingly, the inability of the GliP-ΔCTT3 mutant strain to produce 1 resulted in almost complete loss of gli-cluster expression (Figure S2), which could have affected our results. Therefore, we repeated the experiment by growing cultures supplemented with exogenous 1, which largely rescued gli-cluster expression (Figure S3).[21] LC-HRMS comparison of extracts from gliotoxin-supplemented cultures showed that rescue of cluster gene expression did not recover production of gli-pathway metabolites. As in the case without supplementation of 1, the GliP-ΔCTT3 mutant did not produce any 1–3 (Figure 2a–d), whereas the GliP-ΔT3 strain produced small quantities of 2 (96 % less than Af293) and the tailored metabolite, 3 (96 % less than Af293) (Figure 2a–d). These results indicate that normal biosynthesis of 1 requires the T3 domain of GliP, whereas in the absence of T3, only small amounts of 1 and other gli-pathway metabolites are produced, possibly via cyclization of a T2-tethered intermediate.
We further considered the possibility that the GliP-ΔCTT3 and GliP-ΔT3 variants may have reduced adenylation activity and do not load L-Phe and L-Ser as efficiently as the WT GliP. To address this concern, we heterologously produced and purified GliP-WT, GliP-ΔCTT3 and GliP-ΔT3 proteins from E. coli (Figure S4) and conducted an ATP-[32P]pyrophosphate exchange assay. We found that adenylation activity of the A1- and A2 –domains is not reduced in the truncated proteins, GliP-ΔCTT3 and GliP-ΔT3, relative to GliP-WT (Figure S5).[14,22] Notably, the heterologously expressed GliP truncations remained largely functional. Production of cyclo-Phe-Ser (2) was only slightly reduced with the GliP-ΔT3 mutant and still about one third of WT with the GliP-ΔCTT3 mutant (Figure S6). Residual production of 2 by these mutants likely results from spontaneous or CT-catalyzed cyclization of the T2-tethered L-Phe-L-Ser (Figure 1b), which does not appear to occur in vivo in the corresponding A. fumigatus mutants (compare Figures 2a and S6).
Taken together, these results indicate that the GliP-T3 domain is required for DKP formation in vivo, since in the absence of T3, only small amounts of 1 and other gli-pathway metabolites are produced, possibly via cyclization of a T2-tethered intermediate. Therefore, we hypothesized that in full length GliP the L-Phe-L-Ser dipeptide is transferred from the T2 domain to T3 prior to cyclization via CT.
To assess whether the T3 domain is functional, we tested for appropriate post-translational decoration of the predicted active site residue, serine 2095, with a phosphopantetheinyl-(ppant-) moiety. LC-HRMS/MS analysis of tryptic digests of GliP-WT showed diagnostic ppant-fragmentation of the peptide containing S2095, confirming ppant attachment to the active site serine in T3 (Figure S7).[24]
To further probe the roles of the T3 and CT domains for DKP formation, we constructed A. fumigatus strains carrying point mutations at the T3 and CT active sites. For this purpose, we first created a new ΔGliP strain, then reintroduced either GliP(WT), a point mutant of the T3 active site, GliP-S2095A, or a point mutant lacking the putative catalytic histidine of the CT domain, GliP-H1754A (see Figure S2 and Supporting Methods for details). The ΔGliP+GliP(WT) strain produced a distribution of pathway metabolites (Figure 3a) very similar to that of WT A. fumigatus, proving that the reintroduced GliP is fully functional. In contrast, production of gli pathway metabolites was almost completely abolished in the S2095A and H1754A mutant strains (Figure 3b,c). To rescue potentially suppressed expression of gli-cluster genes in the absence of gliotoxin (1), we repeated the experiment with exogenously added 1, which resulted in a production of a small amount (<2% of WT) of cyclo-Phe-Ser in the S2095A mutant while cyclo-Phe-Ser remained undetectable in the H1754A mutant (Figure 3b,c).
Figure 3.

LC-HRMS analysis of whole-metabolome extracts from mutant A. fumigatus strains. Representative EICs for compounds 1, 2 and 4 in (a) A. fumigatus ΔGliP +GliP-WT, (b) A. fumigatus ΔGliP +GliP-S2095A, and (c) A. fumigatus ΔGliP +GliP-S2095A. Dashed chromatograms representative EICs for 1, 2, and 4 in the indicated strains with added exogenous gliotoxin, 1. Note that the extent of conversion of gliotoxin (1) to its detoxification product 4 is highly variable between experiments, e.g. in the example chromatogram shown in (a) no 1 remains following addition of 1.
To confirm the essential role of the GliP-T3 domain, we heterologously expressed a GliP mutant protein in which the first and second T-domains were inactivated by substitution of serines 555 and 1582 for alanine (GliP-ΔT1T2). We then used synthetic L-Phe-L-Ser-N-acetylcysteamine (L-Phe-L-Ser-SNAC, 5) to effect attachment of L-Phe-L-Ser to T3 via transthiolation and monitored production of 2 (Figures 4a and S8a). Since thioester 5 could cyclize non-enzymatically or via catalysis by the CT domain without first getting attached to T3 (Figure S8b), we included control assays without GliP, without Sfp, or without coenzyme A (= no ppant functionalization on T3), and using a GliP mutant protein in which all three T-domains were inactivated (GliP-ΔT1T2T3). Incubation of 5 under these conditions (Figure 4a) demonstrated that the presence of a functional T3 domain significantly increases the cyclization activity of GliP.
Figure 4.

(a) In vitro production of cyclo-Phe-Ser (2) using different GliP variants. Relative yields of 2 were measured via integration of the corresponding peak in LC-MS ion-chromatograms (n = 4). **p < 0.01, ***p < 0.001. (b) Model for CTTC-catalyzed DKP formation in gliotoxin (1) biosynthesis.
Our results demonstrate that cyclization of tethered Phe-Ser dipeptide en route to 1 is not spontaneous, as proposed,[7–11] and rather requires two additional GliP domains, the second condensation-like domain (CT domain) and the enigmatic terminal thiolation domain T3 (“TC” domain) to which the nascent dipeptide appears to be transferred prior to cyclization via CT (Figure 4b). Significantly, mutation of the catalytic histidine in the CT domain or the ppant attachment site within the T3 domain is sufficient to almost completely abrogate gliotoxin biosynthesis. These observations highlight the importance of dedicated cyclization domains in fungal NRPSs.[25,26] Gliotoxin (1) is a representative member of a large class of NRPS-derived DKPs that seem likely to be produced via similar mechanisms. Analysis of available fungal genomes revealed 56 putative NRPSs that feature terminal domains homologous to the CTT3 tandem in GliP (see Supporting Methods, Table S1, and Figure S9), indicating a conserved biosynthetic strategy for DKP formation.[13,27]
Whereas the GliP CT domain is homologous to the recently described CT domains that catalyze cyclization of fungal tripeptides;[15] the requirement of an additional TC domain for DKP formation is perhaps unexpected, given that dipeptide thioesters often cyclize non-enzymatically, as we showed in our in vitro studies. We note that TC domains in DKP-producing NRPSs could serve as a tether for linear dipeptides during tailoring by other cluster enzymes. Although there is substantial evidence that, in the case of gliotoxin, many of the later steps of its biosynthesis proceed via untethered, cyclic intermediates, the structures of shunt metabolites in related DKP biosynthesis pathways may suggest tailoring of tethered dipeptides. For example, in the case of hexadehydroastechrome, which is derived from cyclo-Trp-Ala derivatives, abundant production of prenylated tryptophan in mutants defective in late-stage tailoring enzymes could be due to recycling of a tethered prenylated Trp-Ala dipeptide (Figure S10).[28]
In conclusion, our study suggests a general framework for fungal DKP biosynthesis, wherein the TC domain serves as a tether for dipeptide cyclization by an adjacent CT domain and potentially for tailoring by other cluster enzymes. Furthermore, our characterization of the biosynthetic roles of the CT and TC domains in DKP formation extends the functional repertoire of NRPS domains.
Supplementary Material
Acknowledgements
This research was funded by an NIH Chemical Biology Interface (CBI) Training Grant (5T32GM008500) to J.A.B., an NIH Predoctoral Training Program in Genetics Grant (5T32GM007133–40) to B.T.P., and NIH R01GM112739–01 to N.P.K. and F.C.S. We thank Prof. Robert Cramer for the kind gift of the GliP plasmid.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supporting information for this article is given via a link at the end of the document.
References
- [1].Scharf DH, Brakhage AA, Mukherjee PK, Environ. Microbiol 2016, 18, 1096. [DOI] [PubMed] [Google Scholar]
- [2].Wang Y, Li ZL, Bai J, Zhang LM, Wu X, Zhang L, Pei YH, Jing YK, Hua HM, Chem. Biodiversity 2012, 9, 385. [DOI] [PubMed] [Google Scholar]
- [3].Cui CB, Kakeya H, Okada G, Onose R, Osada H, J. Antibiot 1996, 49, 527. [DOI] [PubMed] [Google Scholar]
- [4].Nilov DK, Yashina KI, Gushchina IV, Zakharenko AL, Sukhanova MV, Lavrik OI, Švedas VK, Biochemistry (Mosc) 2018, 83, 152. [DOI] [PubMed] [Google Scholar]
- [5].Wang X, Li Y, Zhang X, Lai D, Zhou L, Molecules 2017, 22, 2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Forseth R, Fox E, Chung D, Howlett BJ, Keller NP, Schroeder FC, J. Am. Chem. Soc 2011, 133, 9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Scharf DH, Heinekamp T, Remme N, Hortschansky P, Brakhage AA, Hertweck C, Appl. Microbio.l Biotechnol 2011, 93, 467. [DOI] [PubMed] [Google Scholar]
- [8].Scharf DH, Remme N, Habel A, Chankhamjon P, Scherlach K, Heinekamp T, Hortschansky P, Brakhage AA, Hertweck C, J. Am. Chem. Soc 2011, 133, 12322. [DOI] [PubMed] [Google Scholar]
- [9].Scharf DH, Chankhamjon P, Scherlach K, Heinekamp T, Willing K, Brakhage AA, Hertweck C, Angew. Chem. Int. Ed. Engl 2013, 52, 11092. [DOI] [PubMed] [Google Scholar]
- [10].Dolan SK, O’Keeffe G, Jones GW, Doyle S, Trends Microbiol 2015, 23, 419. [DOI] [PubMed] [Google Scholar]
- [11].Chang SL, Chiang YM, Yeh HH, Wu TK, Wang CCC, Bioorg. Med. Chem. Lett 2013, 23, 2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Scharf DH, Dworschak JD, Chankhamjon P, Scherlach K, Heinekamp T, Brakhage AA, Hertweck C, ACS Chem. Biol 2018, 13, 2508. [DOI] [PubMed] [Google Scholar]
- [13].Gardiner DM, Howlett BJ, FEMS Microbiol. Lett 2005, 248, 241. [DOI] [PubMed] [Google Scholar]
- [14].Balibar CJ, Walsh CT, Biochemistry 2006, 45, 15029–15038. [DOI] [PubMed] [Google Scholar]
- [15].Gao X, Haynes SW, Ames BD, Wang P, Vien LP, Walsh CT, Tang Y, Nat. Chem. Biol 2012, 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Schrettl M, Carberry S, Kavanagh K, Haas H, Jones GW, O’Brien J, Nolan A, Stephens J, Fenelon O, Doyle S, PLoS Pathog 2010, 6, e1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Marion A, Groll M, Scharf DH, Scherlach K, Glaser M, Sievers H, Schuster M, Hertweck C, Brakhage AA, Antes I, et al. , ACS Chem. Biol 2017, 12, 1874. [DOI] [PubMed] [Google Scholar]
- [18].Scharf DH, Habel A, Heinekamp T, Brakhage AA, Hertweck C, J. Am. Chem. Soc 2014, 136, 11674. [DOI] [PubMed] [Google Scholar]
- [19].Scharf DH, Chankhamjon P, Scherlach K, Heinekamp T, Roth M, Brakhage AA, Hertweck C, Angew. Chem. Int. Ed. Engl 2012, 51, 10064. [DOI] [PubMed] [Google Scholar]
- [20].Dolan SK, Owens RA, O’Keeffe G, Hammel S, Fitzpatrick DA, Jones GW, Doyle S, Chem. Biol 2014, 21, 999. [DOI] [PubMed] [Google Scholar]
- [21].Cramer RA, Gamcsik MP, Brooking RM, Najvar LK, Kirkpatrick WR, Patterson TF, Balibar CJ, Graybill JR, Perfect JR, Abraham SN, et al. , Eukaryotic Cell 2006, 5, 972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Forseth RR, Amaike S, Schwenk D, Affeldt KJ, Hoffmeister D, Schroeder FC, Keller NP, Angew. Chem. Int. Ed. Engl 2012, 52, 1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yin J, Lin AJ, Golan DE, Walsh CT, Nat. Protoc 2006, 1, 280. [DOI] [PubMed] [Google Scholar]
- [24].Dorrestein PC, Bumpus SB, Calderone CT, Garneau-Tsodikova S, Aron ZD, Straight PD, Kolter R, Walsh CT, Kelleher NL, Biochemistry 2006, 45, 12756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Clevenger KD, Ye R, Bok JW, Thomas PM, Islam MN, Miley GP, Robey MT, Chen C, Yang K, Swyers M, et al. , ACS Chem. Biol 2018, 57, 3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Robey MT, Ye R, Bok JW, Clevenger KD, Islam MN, Chen C, Gupta R, Swyers M, Wu E, Gao P, et al. , Biochemistry 2018, 13, 1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL, Nucleic Acids Res 2008, 36, W5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yin W-B, Baccile JA, Bok JW, Chen Y, Keller NP, Schroeder FC, J. Am. Chem. Soc 2013, 135, 2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.