

ABSTRACT
The dramatic growth that occurs during Drosophila larval development requires rapid conversion of nutrients into biomass. Many larval tissues respond to these biosynthetic demands by increasing carbohydrate metabolism and lactate dehydrogenase (LDH) activity. The resulting metabolic program is ideally suited for synthesis of macromolecules and mimics the manner by which cancer cells rely on aerobic glycolysis. To explore the potential role of Drosophila LDH in promoting biosynthesis, we examined how Ldh mutations influence larval development. Our studies unexpectedly found that Ldh mutants grow at a normal rate, indicating that LDH is dispensable for larval biomass production. However, subsequent metabolomic analyses suggested that Ldh mutants compensate for the inability to produce lactate by generating excess glycerol-3-phosphate (G3P), the production of which also influences larval redox balance. Consistent with this possibility, larvae lacking both LDH and G3P dehydrogenase (GPDH1) exhibit growth defects, synthetic lethality and decreased glycolytic flux. Considering that human cells also generate G3P upon inhibition of lactate dehydrogenase A (LDHA), our findings hint at a conserved mechanism in which the coordinate regulation of lactate and G3P synthesis imparts metabolic robustness to growing animal tissues.
KEY WORDS: Drosophila, Redox balance, Aerobic glycolysis, Lactate dehydrogenase, Glycerol-3-phosphate dehydrogenase
Highlighted Article: The rapid growth of Drosophila larvae is dependent on the coordinated activity of lactate dehydrogenase and glycerol-3-phosphate dehydrogenase 1, a metabolic relationship that is also observed in human cancer cells.
INTRODUCTION
Nearly a century ago, Otto Warburg observed that tumors exhibit high levels of glucose consumption coupled to oxygen-independent lactate production (Warburg, 1956; Warburg et al., 1924). This metabolic program, which is commonly referred to as the Warburg effect or aerobic glycolysis, has become a focal point of cancer metabolism research (Vander Heiden et al., 2009). The manner by which tumors consume glucose and generate lactate, however, is not unique to either cancer cells or diseased tissues. In fact, the hallmark characteristics of aerobic glycolysis are activated under a variety of normal developmental conditions, such as during maturation of human T cells (Cooper et al., 1963; Pearce et al., 2013; Wang et al., 1976), formation of vertebrate somites (Bulusu et al., 2017; Oginuma et al., 2017), development of muscle tissue (Tixier et al., 2013), activation of hair follicle stem cells (Flores et al., 2017) and Drosophila larval growth (Tennessen et al., 2011). Moreover, studies of the mitochondrial pyruvate carrier (MPC1) reveal that forcibly shifting intestinal stem cells towards a more glycolytic state induces overproliferation in both mice and flies (Bricker et al., 2012; Schell et al., 2017). Overall, these examples illustrate how the coordinate regulation of glycolytic flux and lactate metabolism plays a central role in biomass production, cell fate decisions and developmental growth (Miyazawa and Aulehla, 2018).
Although the exact reason why cells activate aerobic glycolysis in vivo remains debatable, one likely explanation revolves around the redox challenges imposed upon highly glycolytic cells (Vander Heiden et al., 2009). Under conditions of elevated glucose catabolism, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) transfers electrons to NAD+, resulting in the formation of NADH (for a review of the relationship between redox balance and glycolysis, see Lunt and Vander Heiden, 2011). These reducing equivalents must be efficiently removed from NADH because the resulting decrease in NAD+ availability can dampen glycolytic flux and restrict growth. LDH relieves this redox burden by coupling NADH oxidation to lactate formation, thus ensuring that NAD+ is regenerated at an adequate rate. Therefore, highly glycolytic cells, whether in diseased or normal tissues, become reliant on lactate dehydrogenase (LDH) to maintain redox balance. This hypothesis has long been attractive to the cancer metabolism field because LDH inhibitors could hypothetically interfere with tumor growth while having lesser impact on normal tissues (Avi-Dor and Mager, 1956). As a result, much of our understanding regarding how LDH influences biosynthesis, growth and cell proliferation is derived from cancer cell studies.
The goal of using LDH inhibitors to disrupt tumor growth has a rich history rooted in the observation that pyruvate analogs, such as oxamate, inhibit the growth of HeLa cells in glucose-rich media (Goldberg and Colowick, 1965; Goldberg et al., 1965). More recent analyses support these early studies, demonstrating that both RNAi knockdown of LDHA transcripts and LDHA inhibitors disrupt cell proliferation in culture and interfere with tumor growth in mouse xenograft experiments (Billiard et al., 2013; Boudreau et al., 2016; Daniele et al., 2015; Fantin et al., 2006; Qing et al., 2010). Moreover, a conditional Ldha mutation prevents the formation of KRAS- and EGFR-induced non-small cell lung cancer in mice, thereby providing in vivo evidence that some tumors require LDHA (Xie et al., 2014).
Despite the ability of LDHA inhibitors to disrupt the growth and tumorigenicity of certain cancer cells, a growing body of evidence suggests that animal cells can compensate for the loss of LDHA activity. Pancreatic cancer cell lines can become resistant to the LDHA inhibitor GNE-140 by increasing oxidative phosphorylation (Boudreau et al., 2016). Similarly, human colon adenocarcinoma and murine melanoma cell lines that lack both LDHA and LDHB increase oxidative phosphorylation and are capable of forming tumors in xenograft experiments (Zdralevic et al., 2018). However, the most significant evidence that cellular metabolism readily adapts to the loss of LDH activity is not based on cancer studies, but instead stems from a rare inborn error of metabolism known as glycogen storage disease type XI (GSD-XI), which results from loss-of-function mutations in the human LDHA gene (Maekawa et al., 1990). Other than reports of skin lesions and symptoms associated with exercise intolerance (Kanno et al., 1980; Yoshikuni et al., 1986), GSD-XI patients develop and grow normally (Kanno et al., 1988) which is surprising given the role of LDHA in several developmental and physiological processes. The mild symptoms experienced by GSD-XI patients not only raise the possibility that LDH inhibitors might be ineffective in a clinical setting, but also suggest that studies of animal development can identify the metabolic mechanisms that function redundantly with LDH. Toward this goal, we examined the metabolic consequences of mutating Ldh (FBgn0001258; also known as dLdh) in the fruit fly Drosophila melanogaster.
Similar to cancer cells, Drosophila larvae increase glycolytic metabolism and LDH activity as a means of supporting the ∼200-fold increase in body mass that occurs during this developmental stage (Graveley et al., 2011; Li et al., 2013; Rechsteiner, 1970; Tennessen et al., 2011, 2014b). Moreover, proper maintenance of redox balance is crucial for normal larval growth. For example, larvae raised on a high sugar diet rely on glucose-6-phosphate dehydrogenase to maintain NADP+/NADPH and glutathione redox balance (Teesalu et al., 2017). When this high sugar response is disrupted in salt-inducible kinase 3 mutants, larvae exhibit growth defects, larval lethality and disruption of sugar metabolism (Teesalu et al., 2017). To determine whether a larval increase in LDH activity is necessary to maintain redox balance and promote growth, we examined how Ldh mutations influence larval development. We found that although Ldh mutants exhibit a decreased NAD+/NADH ratio, this metabolic insult had no noticeable effect on either growth rate or biomass accumulation. Instead, metabolomic analysis revealed that Ldh mutants upregulate glycerol-3-phosphate (G3P) production, which also couples the reduction of a glycolytic intermediate (dihydroxyacetone phosphate) with NAD+ regeneration and potentially supports larval growth despite loss of LDH activity. We observed a similar result in Gpdh1 (FBgn0001128) mutants, which develop normally despite a decreased NAD+/NADH ratio. Larvae that lack both LDH and GPDH1, however, exhibit severe growth defects, synthetic lethality and dramatic changes in metabolism, including decreased ATP levels and aberrant steady state levels of sugars and amino acids, thus demonstrating that these two enzymes cooperatively support larval growth. Considering that both cancer cells lines and human GSD-XI patients also increase G3P production in response to the loss of LDHA (Billiard et al., 2013; Boudreau et al., 2016; Miyajima et al., 1995), our findings suggest that fundamental aspects of this metabolic relationship are similar in both Drosophila and humans.
RESULTS
LDH maintains larval NAD+ redox balance
To understand how lactate synthesis influences Drosophila larval metabolism, we used liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) to measure the NAD+/NADH ratio in larvae harboring a trans-heterozygous combination of the previously described Ldh loss-of-function alleles, Ldh16 and Ldh17, as well as a precise-excision control strain, Ldhprec (Li et al., 2017). Consistent with a model in which LDH regulates larval redox balance, our analysis revealed that Ldh16/17 mutant larvae exhibit a decreased NAD+/NADH ratio (Fig. 1A,B; Table S1). In contrast, the ratios of NADP+/NADPH, reduced glutathione/oxidized glutathione (GSH/GSSG) and ADP/ATP were similar in both mutant and control larvae (Fig. 1A,B; Table S1). The abundance of AMP relative to ATP, however, was slightly elevated in Ldh mutants (Fig. 1B; Table S1). Overall, our results demonstrate that loss of LDH activity interferes with the NAD+/NADH balance of larvae raised under standard culture conditions but has minimal effects on other aspects of redox metabolism and energy production.
Fig. 1.
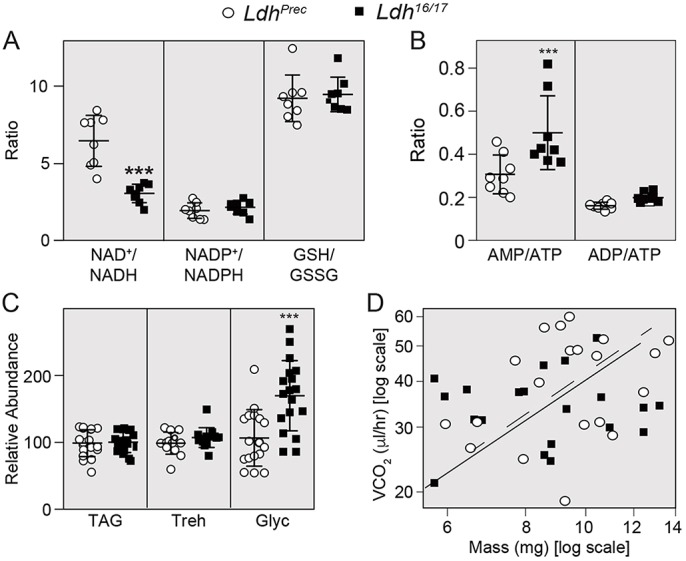
LDH maintains the NAD+/NADH redox balance during larval development. Targeted LC-MS/MS analysis was used to measure metabolites associated with redox balance in Ldhprec controls and Ldh16/17 mutants. (A,B) The ratios of NAD+/NADH, NADP+/NADPH, GSH/GSSG, AMP/ATP and ADP/ATP were determined in control and mutant larvae; n=8 biological replicates were collected from independent populations with 100 mid-L2 larvae per sample. Experiments were repeated twice (see Table S1). (C) Ldhprec controls and Ldh16/17 mutants were collected as mid-L2 larvae and the concentration of triglycerides (TAG), trehalose (Treh) and glycogen (Glyc) were measured in whole animal homogenates. All assays were repeated a minimum of three times; n>10 samples collected from independent populations with 25 mid-L2 larvae per sample. Absolute values for all samples illustrated in C are available in Table S2. (D) The rate of CO2 production was measured in Ldh16/17 mutants and precise excision controls. Diagonal lines represent the slope relating log(mass) and log(metabolic rate) for Ldhprec (broken line) or Ldh16/17 (unbroken line). Error bars represent s.d.; ***P<0.001.
Despite the fact that redox balance is significantly altered in Ldh mutants, the phenotypic consequences of this metabolic disruption are mild. We previously demonstrated that Ldh mutant larvae raised under ideal culture conditions can grow at a normal rate for much of larval development (Li et al., 2017). Furthermore, Ldh mutants that survive the larval mid-third instar (mid-L3) lethal phase develop into adults (Li et al., 2017). To determine whether any other biosynthetic processes are disrupted in Ldh mutants, we quantified the major larval pools of stored energy. Our analysis revealed that loss of LDH activity had no effect on either triglyceride or trehalose levels (Fig. 1C; Table S2). Meanwhile, glycogen levels exhibited a modest, but significant increase in Ldh mutants compared with control larvae (Fig. 1C; Table S2). This latter observation was notable because the epidermis of human GSD-XI patients also appears to accumulate excess glycogen (Yoshikuni et al., 1986), indicating that Ldh mutants phenocopy the subtle metabolic defects observed in humans lacking LDHA.
G3P levels are elevated in Ldh mutants
The ability of Ldh mutants to grow at a normal rate suggests that Drosophila development adapts to the loss of LDH activity. In this regard, human cell culture studies suggest that LDHA inhibition can increase flux from glycolysis into the tricarboxylic acid cycle (TCA cycle) (Billiard et al., 2013; Xie et al., 2014). We found no evidence, however, that this metabolic shift occurs in flies, as Ldh mutant and control larvae produced CO2 at similar rates (Fig. 1D). Furthermore, mitochondrial DNA content was unchanged in Ldh mutants (Fig. S1A), suggesting that loss of LDH activity does not induce excess mitochondrial biogenesis. We also tested the possibility that the rate of citrate synthesis was increased in Ldh mutants by feeding 13C6-glucose to second instar (L2) larvae. Consistent with both the metabolomic analysis and respirometry experiments, the rate of m+2 citrate was similar in both precise excision controls and Ldh mutants (Fig. S1B), indicating that loss of LDH activity does not increase the rate of pyruvate oxidation in the mitochondria.
Because our initial metabolic characterization failed to provide an adequate explanation for how larvae compensate for the loss of LDH, we turned to an untargeted metabolomics approach to poll a larger pool of analytes. Four independent studies based on gas chromatography coupled with mass spectrometry (GC-MS) revealed that Ldh16/17 mutants exhibit reproducible changes in only four metabolites: lactate, pyruvate, 2-hydroxyglutarate (2HG) and G3P (Fig. 2A,B; Tables S3-S7). As previous studies had already examined the relationship between LDH and the metabolites lactate, pyruvate and 2HG (Li et al., 2017), we focused our efforts on understanding why the G3P pool size was increased in Ldh mutants. To confirm that Ldh mutants accumulate excess G3P as a result of decreased LDH activity, we demonstrated that expression of an Ldh transgene (p{Ldh}) in Ldh mutant larvae can restore G3P levels to those observed in Ldhprec control larvae (Fig. 2C). Similarly, ubiquitous expression of UAS-Ldh-RNAi (Ldhi) transgene induced elevated G3P levels (Fig. 2D), thus confirming that Drosophila larvae accumulate excess G3P in response to the loss of LDH activity. Considering that G3P levels are elevated in both GSD-XI patients and pancreatic cancer cells exposed to an LDH inhibitor (Billiard et al., 2013; Boudreau et al., 2016; Miyajima et al., 1995), our findings suggest that both flies and humans accumulate G3P to compensate for the loss of LDH activity.
Fig. 2.
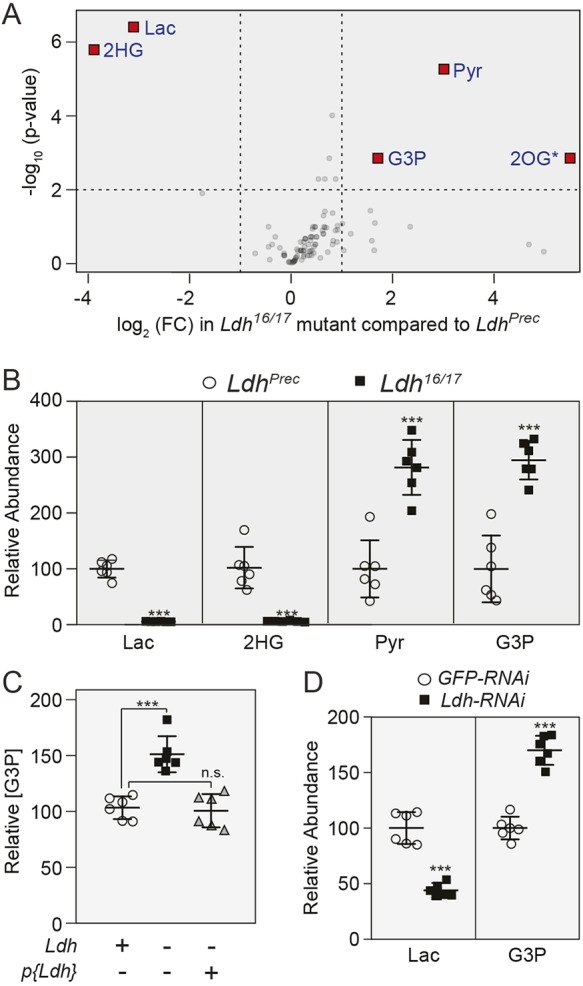
Metabolomic analysis of Ldh mutants. Data from GC-MS metabolomic analysis (Table S4) comparing Ldhprec controls and Ldh16/17 mutants were analyzed using Metaboanalyst. (A) Volcano plot highlighting metabolites that exhibited a >1.5-fold change and a P-value of <0.01. Note that changes in 2OG levels were not reproducible in subsequent experiments. (B) Relative abundance of metabolites that exhibited significant changes in all four GC-MS experiments (P<0.01; see Table S3). (C) Relative abundance of G3P was measured in Ldhprec controls, Ldh16/17 mutants and p{Ldh}; Ldh16/17 rescued animals during the L2 larval stage. (D) Lactate and G3P levels were measured in L2 larvae that ubiquitously expressed either a UAS-GFP-RNAi construct or a UAS-Ldh-RNAi construct under the control of da-GAL4. For all panels, n=6 biological replicates were collected from independent populations with 25 mid-L2 larvae per sample. For C and D, experiments were repeated twice; ***P<0.001. 2HG, 2-hydroxyglutarate; 2OG, 2-oxoglutarate; G3P, glycerol-3-phosphate; Lac, lactate; n.s., not significant; Pyr, pyruvate.
The inverse correlation between lactate and G3P metabolism raises the question of how these metabolic reactions are coordinately regulated at a cellular level. In this regard, neither Gpdh1 mRNA nor GPDH1 protein levels were elevated in Ldh mutants. qRT-PCR revealed that Gpdh1 mRNA levels were similar in both control and mutant larvae (Fig. S2). Similarly, mosaic analysis in the larval brain – a tissue that exhibits both LDH and GPDH1 activity (Rechsteiner, 1970) – revealed that GPDH1 was present at similar levels in both Ldh16 mutant clones and wild-type cells (Fig. S3A,B). Considering that Ldh mutants exhibited elevated G3P levels in every experiment conducted during the course of this study, our findings suggest that Drosophila Ldh mutants can accumulate excess G3P independent of changes in Gpdh1 gene transcription or GPDH1 enzyme abundance.
GPDH1 regulates larval NAD+/NADH redox balance and ATP levels
Because GPDH1 regenerates NAD+ by converting the glycolytic intermediate dihydroxyacetone phosphate (DHAP) into G3P, increased G3P production could provide Ldh mutants with an alternative means of maintaining NAD+ levels (Fig. 3A). Moreover, GPDH1 is a highly abundant protein in Drosophila larvae and could itself represent a key regulator of larval redox balance. To test these possibilities, we generated two Gpdh1 loss-of-function alleles, Gpdh1A10 and Gpdh1B18, both of which represent frameshift mutations that either delete or truncate the C-terminal catalytic domain, which is required for GPDH1 enzyme activity (Fig. S4A,B). Larvae that harbor a trans-heterozygous combination of these alleles, Gpdh1A10/B18, exhibited a significant decrease in both GPDH1 protein expression and G3P levels compared with controls (Fig. 3B; Fig. S5). Furthermore, ubiquitous expression of a UAS-Gpdh1 transgene restored normal G3P levels in mutant larvae (Fig. 3B), confirming that the loss of zygotic GPDH1 reduces G3P synthesis.
Fig. 3.

GPDH1 controls NAD+/NADH redox balance during larval development. (A) Diagram illustrating how GPDH1 and LDH redundantly influence NAD+ levels. (B) GC-MS was used to measure relative G3P abundance in mid-L2 larvae for the following five genotypes (as presented left to right in the figure): Gpdh1B18/+, Gpdh1A10/B18, Gpdh1A10/B18; da-GAL4/+, GpdhA10/B18; UAS-Gpdh1 and Gpdh1A10/B18; da-GAL4 UAS-Gpdh1. (C) NAD+/NADH ratio in mid-L2 larvae of genotypes Gpdh1B18/+, Ldh16/17 and Gpdh1A10/B18. (D) Mid-L2 larvae were fed D-glucose-13C6 for 2 h and the rates of 13C isotope incorporation into lactate and G3P were determined based on m+3 isotopologue abundance. (E) ATP levels were significantly decreased in Gpdh1A10/B18 compared with Gpdh1B18/+ controls. (F) The body mass of Gpdh1A10/B18 larvae was significantly lower than that of Gpdh1B18/+ controls 0-4 h after the L2-L3 molt. In B,C,E,F, n=6 biological replicates per genotype; in D, n=5 biological replicates per genotype. Error bars represent s.d. For B and C, the P-value was adjusted for multiple comparisons using the Bonferroni–Dunn method; ***P<0.001. All experiments were repeated a minimum of two times. G3P, glycerol-3-phosphate; DHAP, dihydroxyacetone phosphate; Lac, lactate.
To determine whether G3P production influences larval redox balance, we measured both NAD+ and NADH levels in Gpdh1 mutants. Similar to Ldh mutants, we observed that the NAD+/NADH ratio in mid-L2 larvae was significantly lower in Gpdh1 mutants compared with a Gpdh1B18/+ heterozygous control (Fig. 3C). We also examined the extent to which GPDH1 influences NAD+ levels by feeding D-glucose-13C6 to mid-L2 larvae and measuring the rate of lactate and G3P synthesis. Because both LDH and GPDH1 must oxidize one molecule of NADH in order to form one molecule of either lactate or G3P, respectively (Fig. 3A), we can indirectly infer the rate at which each enzyme regenerates NAD+ based on synthesis of these metabolites. Our analysis revealed that mid-L2 larvae synthesize m+3 lactate and m+3 G3P at similar rates (Fig. 3D), indicating that GPDH1 and LDH regenerate roughly equivalent amounts of NAD+ during larval development.
Our observation that Drosophila Gpdh1 mutants exhibit a decreased NAD+/NADH ratio balance also implicates this enzyme in coordinating redox balance with larval energy production. G3P synthesized in the cytoplasm can be oxidized on the inner mitochondrial membrane by the FAD-dependent enzyme glycerophosphate oxidase 1 (Gpo-1; FBgn0022160) to generate ATP via oxidative phosphorylation (O'Brien and MacIntyre, 1972). Therefore, any decrease in G3P synthesis could also reduce ATP production. Consistent with this function, ATP levels were significantly decreased in Gpdh1 mutants compared with controls (Fig. 3E). Yet, despite the role for GPDH1 in these crucial metabolic processes, animals lacking zygotic GPDH1 activity exhibit only mild developmental defects. In agreement with previous reports (Bewley and Lucchesi, 1977), we found that Gpdh1 mutants are viable through larval development and are ∼15% smaller at the L2-L3 molt (Fig. 3F; Fig. S4C), a result that again demonstrates how larval development is robust and can compensate for significant metabolic insults.
Gpdh1; Ldh double mutants exhibit severe growth defects
Because LDH and GPDH1 individually regulate larval redox balance and shunt metabolites away from glycolysis, we tested the possibility that simultaneous removal of both enzymes would induce a synthetic growth phenotype. Indeed, Gpdh1; Ldh double-mutant L2 larvae collected 68-72 h after egg-laying were 85% smaller than either the Gpdh1 or Ldh single mutant (Fig. 4A-C; Fig. S6A-D). This growth defect was also apparent at 92-96 h after egg-laying, when single mutants had completed the L2-L3 molt but double-mutant larvae were still L2 larvae (Fig. S6E-H). Moreover, unlike either of the single mutants, Gpdh1; Ldh double mutants die throughout L1 and L2 development, with ∼30% of double-mutant larvae dying prior to the L1-L2 molt and nearly all animals failing to complete the L2-L3 molt (n>100; Fig. 4C); however, during the course of this study, we found that double mutants occasionally escaped this lethal phase and progressed into L3 development.
Fig. 4.

Gpdh1; Ldh double mutants exhibit severe growth phenotypes. (A) Representative images of L2 larvae (68-72 h after egg laying) from synchronized populations of w1118, Ldh16/17, Gpdh1A10/B18 or Gpdh1A10/B18; Ldh16/17 double mutants. (B) The body mass of w1118 and Gpdh1A10/B18; Ldh16/17 double-mutant larvae measured 68-72 h after egg-laying. (C) The viability of the four genotypes listed in A were measured from 0-4 h post-hatching until after completion of the L1-L2 molt (L1-L2), 0-4 h after the L1-L2 molt until after completion of the L2-L3 molt (L2-L3), and from 0-4 h after the L2-L3 molt until the mid-L3 stage (eL3-mL3). In B and C, error bars represent s.d.; n=6 biological replicates per genotype. (D-H) Maximum projections of dorsal half of L2 larval brains stained for Dpn (green) and DAPI (blue) from w1118 controls (D), Ldh16/17 mutants (E), Gpdh1A10/B18 mutants (F), age-matched Gpdh1A10/B18; Ldh16/17 double mutants (G) and size-matched Gpdh1A10/B18; Ldh16/17 double mutants (H). The scale bar in D also applies to E-H. Note that D′-H′ display the Dpn channel alone in grayscale. ***P<0.001.
To characterize further the growth defects of double-mutant larvae, we examined the larval brain, which is reported to display high levels of both LDH and GPDH1 activity (Rechsteiner, 1970). The w1118 controls, Gpdh1 and Ldh single mutants, and Gpdh1; Ldh double mutants were collected 60 h after egg-laying. Brains were fixed and stained with DAPI to visualize overall tissue size and for Deadpan (Dpn) to visualize neuroblasts (Fig. 4D-H). Although the brains of single mutant larvae exhibited no growth defects, the brains of Gpdh1; Ldh double mutants were significantly smaller than controls (Fig. 4D-G). However, if Gpdh1; Ldh double mutants were allowed to develop for an additional 24 h, the brain grew to a similar size as the w1118 control (Fig. 4D,H). To examine these growth phenotypes more closely at a cellular level, we dissected brains from size-matched larvae and incubated them in Grace's Insect Medium with EdU for 2 h. Compared with w1118 controls, Ldh single mutants and Gpdh1 single mutants, the number of cells that stained with EdU was significantly decreased in Gpdh1; Ldh mutant brains (Fig. 5A-E). This finding demonstrates that fewer cells entered S-phase during the 2h incubation period and future studies should determine whether this phenotype results from delays in the cell cycle, increased apoptosis or defects in stem cell maintenance. We observed a similar phenomenon in the larval intestine, where the posterior midgut of age-matched Gpdh1; Ldh mutant larvae was shorter than in either of the single mutants and exhibited a growth delay of approximately 24 h compared with single-mutant controls (Fig. S7A-E). Overall, these results demonstrate that, although larval development can compensate for the loss of either LDH or GPDH1, removal of both enzymes severely restricts tissue growth.
Fig. 5.

EdU labeling of Gpdh1; Ldh double mutants. (A-D) Maximum projections of the dorsal half of size-matched L2 larval brains stained with EdU (red) and DAPI (blue) from w1118 controls (A), Ldh16/17 mutants (B), Gpdh1A10/B18 mutants (C) and Gpdh1A10/B18; Ldh16/17 double mutants (D). The scale bar in A applies to A-D. Note that A′-D′ display EdU staining alone in grayscale. (E) Histogram of the number of EdU-positive cells per dorsal brain lobe per genotype. Error bars represent s.d. P-value adjusted for multiple comparisons using the Bonferroni–Dunn method; ***P<0.001.
Simultaneous loss of LDH and GPDH1 disrupts ATP homeostasis and glycolytic flux
Because our metabolic studies suggested that LDH and GPDH1 cooperatively regulate larval development, we next examined the possibility that the Gpdh1; Ldh double-mutant growth phenotypes stem from a severe metabolic disruption. Indeed, compared with GpdhB18/+; Ldh16/+ heterozygous controls, ATP levels were dramatically decreased in GpdhA10/B18; Ldh16/17 double mutants (Fig. 6A). However, our metabolic characterization also unexpectedly revealed that the ratio of NAD+ to NADH was similar in control and double-mutant larvae (Fig. 6B). One explanation for this counterintuitive result is that glycolytic flux is decreased in GpdhA10/B18; Ldh16/17 double mutants, resulting in a lower rate of NADH formation. Indeed, GpdhA10/B18; Ldh16/17 L2 larvae fed D-glucose-13C6 for a 2h interval synthesized ∼50% less m+3 labeled pyruvate compared with GpdhB18/+; Ldh16/+ heterozygous controls (Fig. 6C). In contrast, the relative abundance of 13C-labeled pyruvate was similar in Gpdh1A10/B18 mutants and Gpdh1B18/+ controls at the end of the 2h feeding interval (Fig. S8A). Moreover, Ldh16/17 mutants exhibited a modest but not significant increase in the abundance of 13C-labeled pyruvate compared with controls (Fig. S8B), a result that is consistent with our observations that these mutants accumulate excess pyruvate, are unable to synthesize lactate and do not increase the rate at which pyruvate enters the TCA cycle (Fig. 2; Fig. S1B).
Fig. 6.
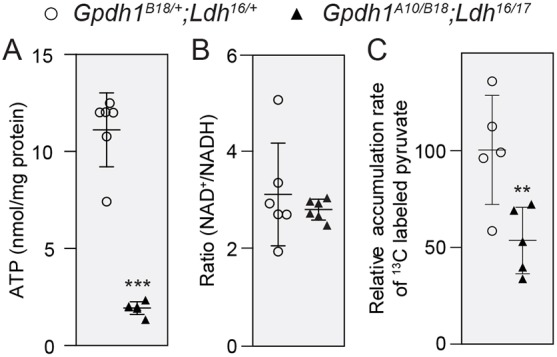
ATP homeostasis and glycolytic flux are disrupted in Gpdh1; Ldh double mutants. (A-C) Size-matched Gpdh1A10/B18; Ldh16/17 double mutants and Gpdh1B18/+; Ldh16/+ control larvae were collected at the mid-L2 stage and evaluated for changes in ATP (A), NAD+/NADH ratio (B) and the relative metabolic flux rates from 13C6-glucose into pyruvate (C). For all panels, error bars represent s.d.; n=6 biological replicates in A and B; n=5 biological replicates in C. Each experiment was repeated twice. **P<0.01. ***P<0.001.
Our stable-isotope tracer analysis indicates that simultaneous loss of both LDH and GPDH1 activity impairs larval glycolytic flux. To further characterize this metabolic defect, we used a targeted GC-MS-based approach to analyze central carbon metabolism of both Gpdh1 single mutants and Gpdh1; Ldh double mutants (Fig. 7A-C; Tables S8 and S9). In the case of the Gpdh1 mutant larvae, metabolomic analysis revealed that steady-state levels of glucose, trehalose and glycolytic intermediates were normal, but amino acid metabolism was significantly disrupted. Not only were levels of aspartate and several essential amino acids decreased, but we also observed elevated levels of urea and the urea cycle intermediate ornithine (Fig. 7A; Table S8). Notably, our analysis uncovered elevated glutamate and proline levels, which, considering that insects can synthesize proline in an NADH-dependent manner (Mccabe and Bursell, 1975), hints at a mechanism by which loss of G3P production induces elevated proline synthesis in response to aberrant redox balance. Although we are uncertain as to the significance of the elevated glutamate levels, we note that both glutamate and proline are intermediates in the proline–alanine cycle, which is used by some insects to move reducing equivalents between tissues (Arrese and Soulages, 2010). If the proline–alanine cycle is active in Drosophila, elevated levels of these two amino acids could represent aberrant regulation of this redox shuttle – a possibility that should be evaluated in future studies. Intriguingly, Gpdh1 mutants do not exhibit an increase in either lactate or 2HG levels (Fig. 7A,B), a finding that could indicate that LDH activity is saturated in developing larvae. Furthermore, xanthine and urate levels, which are produced by purine catabolism, were also increased in Gpdh1 mutants (Fig. 7A; Table S8).
Fig. 7.

Amino acid and glucose metabolism are disrupted in Gpdh1; Ldh double mutants. (A) Heat map summarizing changes in metabolite abundance in Ldh16/17 mutants relative to Ldhprec controls, Gpdh1A10/B18 mutants relative to Gpdh1B18/+ controls, and Gpdh1A10/B18; Ldh16/17 double mutants relative to size-matched Gpdh1B18/+; Ldh16/+ controls. (B,C) Abundance of select metabolites for either Gpdh1A10/B18 mutants relative to Gpdh1B18/+ controls (B) or size-matched Gpdh1A10/B18; Ldh16/17 double mutants relative to Gpdh1B18/+; Ldh16/+ (C). Asp, aspartate; G3P, glycerol-3-phosphate; Glc, glucose; Glu, glutamate; 2HG, 2-hydroxyglutarate; Lac, lactate; Pro, proline; Treh, trehalose. For all panels, n=6 biological replicates per genotype. Each experiment was repeated twice. Error bars represent s.d. P-values adjusted for multiple comparisons using the Bonferroni–Dunn method; *P<0.05, **P<0.01, ***P<0.001.
Nearly all of the metabolic changes observed in the Gpdh1 single mutant were enhanced in the Gpdh1; Ldh double mutants (Fig. 7A,C; Table S9). For example, Gpdh1; Ldh mutant larvae exhibited a 500-fold increase in xanthine levels compared with the heterozygous controls (Fig. 7A; Table S9), suggesting severe disruption of purine metabolism. Overall, the metabolic changes observed in the double mutant represented the combined metabolic disruptions seen in the single mutants (Fig. 7A), but with two major exceptions – the relative abundance of both trehalose and glucose were only significantly elevated in Gpdh1; Ldh mutant larvae (Fig. 7A,C). This result is important as it supports our model in which loss of both enzymes leads to decreased glycolytic flux. Overall, our metabolomic approach not only demonstrates that the growth defects caused by loss of both LDH and GPDH1 are associated with severe disruption of central carbon metabolism but also highlights the plasticity of animal metabolism.
DISCUSSION
Our findings demonstrate that LDH and GPDH1 promote larval growth by cooperatively regulating carbohydrate metabolism. This relationship probably serves multiple purposes, as the production of lactate and G3P metabolism not only influence larval NAD+/NADH redox balance but also control the pool size of glycolytic intermediates and dictate the manner by which cells generate ATP. Considering that human cells also upregulate GPDH1 activity in response to decreased lactate synthesis, our findings indicate that this metabolic relationship is conserved across animal phyla and hints at a mechanism by which GPDH1 activity could render tumors resistant to LDH inhibitors.
The roles of LDH and GPDH1 in cancer and animal development
The possibility of using LDH inhibitors to disrupt tumor growth was first proposed over 60 years ago, shortly after the discovery that the pyruvate analog oxamate disrupts aerobic glycolysis and slows the growth of HeLa cells (Goldberg and Colowick, 1965; Papaconstantinou and Colowick, 1961). During the last decade, the goal of using LDH inhibitors as chemotherapeutic agents has been revisited, with several studies demonstrating that this approach can disrupt cancer cell growth (Billiard et al., 2013; Boudreau et al., 2016; Daniele et al., 2015; Fantin et al., 2006; Qing et al., 2010). Yet, despite the promise of such compounds, studies of human and mouse LDHA mutants raise concerns about the potential effectiveness of inhibiting LDH. First, GSD-XI patients grow and develop normally (Kanno et al., 1988, 1980; Miyajima et al., 1995), suggesting that human developmental metabolism can compensate for loss of this enzyme. Second, although LDHA inhibition induces elevated TCA cycle flux in cell culture, this reliance on the TCA cycle is not observed in tumors derived from a conditional LDHA mutant or in ex vivo tumor slices treated with an LDH inhibitor (Xie et al., 2014). Such observations are important because they suggest that the metabolic plasticity of cells in culture differs significantly from tissues in vivo.
Our studies in the fly support the in vivo mammalian observations that Ldh mutants grow normally and do not increase CO2 production, indicating that flux through the TCA cycle is unchanged. Instead, we observed that Ldh mutants specifically upregulate G3P synthesis as a means of maintaining developmental growth. This finding is consistent with decades of observation in tumors, insects and healthy human tissues, which, on the whole, repeatedly pointed to an inverse correlation between lactate and G3P production (Boxer and Shonk, 1960; Miyajima et al., 1995; Rechsteiner, 1970; Zebe and McShan, 1957). Moreover, recent cell culture studies have also demonstrated that LDH inhibitors induce G3P synthesis, thus demonstrating that this metabolic relationship is present in cultured cells (see supplemental data in Billiard et al., 2013; Boudreau et al., 2016). Overall, our observations in the fly suggest a common metabolic relationship that allows animal cells to adapt to redox stress.
The link between larval redox balance and the role of G3P in ATP production could also explain a contradiction in the literature on Drosophila metabolism. Mutations that disrupt either glycolysis (e.g. ERR, Pfk) or the electron transport chain (ETC) result in severe developmental defects (Mandal et al., 2005; Meiklejohn et al., 2013; Pletcher et al., 2019; Tennessen et al., 2011). In contrast, larvae that harbor mutations in either the mitochondrial pyruvate carrier or malate dehydrogenase 2 are able to complete larval development with relatively mild phenotypes (Bricker et al., 2012; Wang et al., 2010). Similarly, larvae that lack zygotic isocitrate dehydrogenase 3b exhibit developmental delays but are able to survive until metamorphosis (Duncan et al., 2017). These observations suggest that, although glycolysis and oxidative phosphorylation are necessary for development, larvae do not require a fully functional TCA cycle. This arrangement makes sense in that larval metabolism is largely dedicated to shuttling metabolic intermediates into biosynthetic pathways. By activating GPDH1, the production of G3P helps regenerate cytosolic NAD+ without increasing CO2 production while also allowing cells to transfer reducing equivalents to the ETC and generate ATP. In this regard, future studies should examine links between the disruption of larval glycolysis and metabolic sensors such as AMPK, Tor and ChREBP that are known to coordinate ATP homeostasis and central carbon metabolism with developmental growth (Havula and Hietakangas, 2018; Mandal et al., 2005; Tennessen and Thummel, 2011).
Drosophila as a model for studying metabolic plasticity
Our study highlights the remarkable metabolic plasticity that underlies animal development and physiology. Intermediary metabolism adapts to a surprisingly broad range of natural genetic variation, dietary stress and metabolic insults. For example, mutations in the Drosophila mitochondrial pyruvate carrier MPC1, which render cells unable to transport pyruvate into the mitochondria, elicit no obvious phenotypes when mutant larvae are raised under standard growth conditions (Bricker et al., 2012). Moreover, natural populations of Drosophila can buffer larval development against significant variations in mitochondrial oxidative capacity and the scaling relationship between mass and metabolic rate (Matoo et al., 2019). A similar phenomenon is also observed in Caenorhabditis elegans, where entire metabolic pathways are rewired in response to dietary stress or genetic mutations (MacNeil et al., 2013; Watson et al., 2014, 2016). These examples not only highlight the adaptability of animal metabolism, but also emphasize how little we understand about this topic. The molecular mechanisms that control adaptive metabolic rewiring, however, are often difficult to study in a laboratory setting where animals are raised on high nutrient diets and buffered against environmental stress. In this regard, recent advances in metabolomics provide a powerful approach for understanding how metabolism adapts to environmental and genetic insults. By analyzing changes in gene expression within the context of metabolomic data, compensatory changes in metabolic flux can be quickly identified and analyzed using standard model organism genetics. The power of this approach is demonstrated by our studies of Drosophila Ldh. Even without prior knowledge of the link between LDH and GPDH1 activity, our study pinpointed increased G3P synthesis as the adaptive response within Ldh mutants, thus demonstrating how metabolomics holds the potential to illuminate the complex metabolic network that supports animal development.
MATERIALS AND METHODS
Drosophila melanogaster husbandry and genetic analysis
Fly stocks were maintained at 25°C on Bloomington Drosophila Stock Center (BDSC) food. Larvae were raised and collected as previously described (Li and Tennessen, 2017). Briefly, 50 adult virgin females and 25 males were placed into a mating bottle and embryos collected for 4 h on a 35 mm molasses agar plate with a smear of yeast on the surface. Collection plates were stored inside an empty 60 mm plastic plate and placed in a 25°C incubator for 60 h. All mutations and transgenes were studied in a w1118 background. Gpdh1 and Ldh mutations were maintained in trans to the balancer chromosomes CyO, p{GAL4-twi.G}, p{UAS-2xEGFP} (BDSC Stock 6662) and TM3, p{Dfd-GMR-nvYFP}, Sb1 (BDSC Stock 23231), respectively. Unless noted, Ldh mutant larvae harbored a trans-heterozygous combination of Ldh16 and Ldh17 (Ldh16/17) and an Ldh precise excision control strain (Ldhprec) was used in all experiments (for a description of these alleles, see Li et al., 2017). Ldh mutant phenotypes were rescued using the previously describe transgene {pLdh} (Li et al., 2017). RNAi experiments were conducted using transgenes that targeted either Ldh (BDSC stock 33640) or GFP (BDSC stock 41556).
Generation of Gpdh1 mutations
Gpdh1 mutations were generated using a CRISPR/Cas9 approach (Gratz et al., 2013; Sebo et al., 2014). Two oligos encoding guide RNA sequences that targeted either exon 3 (5′-GGCTTCGACAAGGCCGAGGG-3′) or exon 4 (5′-GATCTGATCACGACGTGTTA-3′) were inserted into the BbsI site of pU6-BbSI-gRNA (Addgene). Each gRNA construct was independently injected into BDSC Stock 52669 (y1 M{vas-Cas9.S}ZH-2A w1118) by Rainbow Transgenic Flies (Camarillo). The mutations GpdhA10 (19 bp deletion within exon 3; 5′-TCGACAAGGCCGAGGGCGG-3′) and GpdhB18 (7 bp deletion with exon 4; 5′-ACGTGTT-3′) were isolated using a PCR-based sequencing approach. All experiments described herein used a trans-heterozygous combination of these two alleles (GpdhA10/B18).
Generation of the UAS-Gpdh1 transgene
The UAS-Gpdh1 transgenic strain was generated by PCR amplification of the Gpdh1 cDNA from Drosophila Genomics Resource Center (DGRC) cDNA clone FI03663 using the oligos 5′-AGAATTCATGGCGGATAAAGTAAAT-3′ and 5′-AGCGGCCGCTTAAAGTTTTGGCGACGG-3′. The Gpdh1 PCR product was inserted into the EcoRI and NotI sites of pUAST-attB (DGRC) and the resulting plasmid injected into BDSC Stock 24867 (M{vas-int.Dm}ZH-2A, PBac{y[+]-attP-3B}VK00031) by Rainbow Transgenic Flies (Camarillo, CA, USA).
GC-MS analysis
Ldh mutants and precise excision controls were analyzed using four independent targeted metabolomic analyses based on GC-MS. Samples were collected, processed and analyzed at either the University of Utah metabolomics core facility or Indiana University Mass Spectrometry Facility as previously described (Cox et al., 2017; Li and Tennessen, 2018). Each sample contained 25 mid-L2 larvae. For all experiments, six biological replicates were analyzed per genotype. GC-MS data were normalized based on sample mass and internal succinic-d4 acid standard. Each experiment was statistically analyzed using Metaboanalyst (metaboanalyst.ca) version 4.0 with Pareto scaling (Chong et al., 2018).
LC-MS/MS analysis
For both Ldh mutants and precise excision controls, 100 mid-L2 larvae were collected in a 1.5 ml microfuge tube. Each sample was immediately washed three times using ice-cold PBS. All wash solution was removed and the sample tube drop-frozen in liquid nitrogen. Metabolite extraction and LC-MS analysis was performed by the University of Utah Metabolomics core facility as previously described (Bricker et al., 2012; Cox et al., 2017). Data were analyzed using a two-tailed Student's t-test.
Colorimetric metabolite assays
Glycogen, triglycerides, trehalose and soluble protein were measured in mid-L2 larvae using previously described methods (Tennessen et al., 2014a). Briefly, 25 mid-L2 larvae were collected from the surface of a molasses egg-laying cap that contained ∼1.5 g of yeast paste and then placed in a 1.5 ml microfuge tube. For each assay, at least six biological replicates were collected from independent mating bottles. Samples were washed three times using ice-cold PBS (pH 7.4). All PBS was removed from the samples and the larvae homogenized in appropriate assay buffer. Larval homogenate (10 µl) was removed for measuring soluble protein using a Bradford assay and the remaining homogenate immediately heat treated at 70°C for 5 min. Heat-treated samples were frozen at −80°C until analyzed using the appropriate assay.
NAD+ and NADH were measured using the Amplite fluorimetric NAD+/NADH ratio assay kit (AAT Bioquest; 15263) according to instructions. Ten mid-L2 larvae were washed with cold PBS and homogenized with 100 µl of lysis buffer. The lysates were centrifuged for 10 min at 3000 g and the supernatants collected for analysis. Fluorescence was monitored with a Cytation 3 plate reader (BioTek) at Ex/Em=540/590 nm. The concentrations of NAD+ and NADH were normalized to the soluble protein concentrations.
All metabolite measurements were repeated a minimum of three times with six independent samples analyzed per genotype. Data were analyzed using a two-tailed Student's t-test.
Quantification of mitochondrial genome copy number
Quantitative PCR was used to measure the relative ratio of mitochondrial DNA to genomic DNA in precise excision controls and Ldh mutants based on a previously described strategy (Oliveira and Kaguni, 2011). Total DNA was isolated from 25 mid-L2 larvae using the Qiagen Core Gene 2 extraction kit. For mitochondrial DNA measurements, DNA samples were diluted 1:100 and the Drosophila mitochondrial gene mt:CoI was amplified using the oligos 5′-TGCTCCTGATATAGCATTCCCACGA-3′ and 5′-TCCACCATGAGCAATTCCAGCGG-3′. The relative abundance of genomic DNA in the samples was measured by amplifying the Rpl32 genomic locus using oligos 5′-AGGCCCAAGATCGTGAAGAA-3′ and 5′-TGTGCACCAGGAACTTCTTGAA-3′.
Larval respiration studies
We quantified routine metabolic rate in precise excision controls and Ldh mutants as CO2 production using established flow-through respirometry protocols for larval D. melanogaster (Hoekstra and Montooth, 2013; Hoekstra et al., 2013). We measured metabolic rate for 20 biological replicates per genotype. Each biological replicate consisted of ten mid-L2 larvae that were placed in a small cap containing 0.5 ml of fly food inside a glass respirometry chamber. The amount of CO2 produced by the group of larvae was measured by flowing CO2-free air through the chambers at a rate of 100 ml/min and measuring the CO2 produced as a result of metabolism using an infrared CO2 analyzer (Li-Cor 7000 CO2/H2O Analyzer; LI-COR). Each run of the respirometer used a multiplexed system (Sable Systems International) to cycle through four chambers that contained larvae and a fifth baseline chamber; all chambers were all housed in a thermal cabinet maintained at 26°C (Tritech Research). Genotypes were randomly assigned to chambers within each run. Within each run, two technical replicate measurements were performed for each group of larvae. Technical replicate measures were strongly correlated (r=0.935). We calculated the average rate of CO2 produced across the 10 min time interval for the first replicate measure in each run for each biological sample after correcting for any minimal drift in the baseline signal. Each group of larvae was massed using a Cubis microbalance (Sartorius AG) before being placed in the respirometer. This allowed us to statistically account for the relationship between mass and metabolic rate when testing for differences between genotypes using a Type II Model regression implemented with smatR (Warton et al., 2006) in the R statistical package (R Core Team, 2017). There was no significant difference between genotypes in the slope relating log(mass) and log(metabolic rate) (i.e. the mass-scaling exponent) (P=0.099). We then tested whether genotypes differed in metabolic rate across masses (i.e. for a difference in the y intercept of the relationship between mass and metabolic rate).
qRT-PCR
Total RNA was isolated from Ldh16/17 and LdhPrec mid-2L larvae using Tripure Reagent (Roche). cDNA synthesis was conducted using the Thermo Maxima H Minus First Strand cDNA Synthesis Kit with dsDNase (K1681, ThermoScientific). cDNAs and the appropriate oligonucleotides (see below) were added; FastStart Essential DNA Green Master (Roche) and a Roche LightCycler 96 were used to quantify the relative abundance of Gpdh1 and rp49 was used as an internal reference. The following primer sets were used to measure the relative abundance of Gpdh1 mRNA: rp49 forward, 5′-AAGTGTGCGGCTCGTATTTCG-3′; rp49 reverse, 5′-TCATCTTGAAGCAGGTTGGGC-3′; Gpdh1 forward, 5′-ATCACGACGTGTTACGGTGG-3′; Gpdh1 reverse, 5′-GGCCGTTGAGCATTTCCTTC-3′.
GPDH1 immunofluorescence
The anti-GPDH1 was generously provided by Dr David Maughn on behalf of the late Dr David T. Sullivan (Skuse and Sullivan, 1985). Mid-2L larvae were dissected and fixed at room temperature for 30 min with 4% formaldehyde in PBS as previously described. Fixed samples were washed three times, incubated in normal serum blocking buffer [5% normal donkey serum, 1× PBS with 0.1% Triton X-100 (PBT)] for 30 min at room temperature and stained overnight at 4°C with anti-GPDH1 (1:1000). Samples were washed three times using 1× PBT and stained with 1:200 Alexa Fluor 488 goat anti-rabbit (#R37116; Thermo Fisher) and 1:50 Alexa Fluor 568 phalloidin (#A12380; Thermo Fisher). Stained tissues were cleared and then mounted with ProLong Gold Antifade mountant (#P36941; Thermo Fisher).
Mosaic analysis
Virgin females of the genotype P{ry[+t7.2]=hsFLP}22, y[1] w[*]; P{w[+mC]=arm-lacZ.V}70C P{ry[+t7.2]=neoFRT}80B (Bloomington Stock 6341) were crossed to either w1118; FRT80B males (control) or w1118; Ldh16 FRT80B males. F1 progeny from these crosses were collected as L1 larvae, heat-treated in a 37°C water bath for 1 h and subsequently reared at 25°C. The central nervous system (CNS) from L3 larvae was dissected and processed for immunohistochemistry as described above. Briefly, the larval CNS was dissected in ice cold 1× PBS and fixed in 4% paraformaldehyde for 45 min. CNS samples were then washed in 1× PBT and incubated in blocking buffer containing 0.5% normal goat serum, bovine serum albumin and 1× PBT for 1 h. Samples were incubated overnight at 4°C in 1× PBT containing chicken anti-β-galactosidase (1:1000, Abcam; ab9361) and rabbit anti-GPDH1 (1:1000). Following this incubation, CNS samples were washed three times in 1× PBT and incubated in 1× PBT containing anti-rabbit Alexa Fluor 488 (1:1000, Jackson Laboratories; 711-545-152), anti-chicken Alexa Fluor 568 (1:1000, Jackson Laboratories; 703-545-155) and DAPI (0.5 µg/ml) for 1 h at room temperature. Samples were mounted using Vectashield Antifade Mounting Media (Vector Laboratories, H-1000) and imaged using a Leica SP8 confocal microscope. Ldh mutant clones do not express β-galactosidase, whereas the twin-spot clones display visibly higher levels of β-galactosidase expression compared with neighboring heterozygous cells.
13C-based metabolic turnover rate analysis
Measurements of metabolic turnover rates from 13C-labeled glucose to pyruvate and citrate were conducted as previously described (Li et al., 2018). Briefly, mid-L2 larvae were fed with semidefined medium (Backhaus et al., 1984) containing 50% D-glucose-13C6 (Cambridge Isotopes; CLM-1396-1) for 2 h. Metabolites were detected using an Agilent 6890N gas chromatograph with a 5973 Inert Mass Selective Detector and Gerstel MPS2 autosampler. The isotopologue distributions were corrected based on the natural abundance of elements. The metabolic turnover rate fx was estimated based on the formula XL/XT=p[1−exp(−fx*t/XT)], where XL is the amount of 13C-labeled metabolite (m+3), XT is the amount of total metabolite pool and p is the percentage of glucose-13C6.
Graphical representation of metabolite data
All figures were generated using Graphpad Prism 7.0 (version 7.0c). Metabolomic data are presented as scatter plots, with the error bars representing the standard deviation and the line in the middle representing the mean value.
Larval CNS and gastrointestinal tract staining
CNS and intestine dissection and analysis were performed as previously described (Luhur et al., 2017, 2014). Briefly, size- and age-synchronized larval CNSs and larval intestines were dissected in ice cold 1× PBS and fixed at room temperature for 45 min in 4% paraformaldehyde (Electron Microscopy Services) in 1× PBS with and without 0.3% Triton X-100, respectively. For Dpn staining, CNSs were subsequently washed with 0.3% PBT and blocked (1× PBS, 0.5% BSA) for at least 1 h at room temperature. Primary antibody staining was performed with guinea pig anti-Dpn (provided by James Skeath, Washington University, St. Louis, MO, USA; 1:500) for 5 h at room temperature. Secondary antibody staining was performed overnight with Alexa Fluor 488-conjugated goat anti-guinea pig antibodies (Life Technologies, 1:1000; A-11073) and DAPI (0.5 µg/ml) at 4°C. Larval intestines were washed with 0.1% PBT and incubated with DAPI (0.5 µg/ml) for 15 min. Following these secondary washes, CNSs and intestines were mounted in Vectashield mounting medium (Vector Laboratories, H-1000).
For EdU staining, dissected larval CNSs were immediately incubated in Grace's insect medium supplemented with 1 mM EdU (Thermo Fisher, C10338). Subsequent manipulations were performed as described in the product manual.
For quantifications, multiple z-steps of individual brain lobes or posterior midguts were acquired using the Leica SP5 confocal microscope in the Light Microscopy Imaging Center at Indiana University, Bloomington. The maximum projection of each z-stack was generated using FIJI. Dpn- and EdU-positive cell numbers were manually counted using Adobe Photoshop. The area of larval enterocyte nuclei was measured using Fiji.
Supplementary Material
Acknowledgements
We thank the Bloomington Drosophila Stock Center (NIH P40OD018537) for providing fly stocks, the Drosophila Genomics Resource Center (NIH 2P40OD010949) for genomic reagents, Flybase (NIH 5U41HG000739) and the Indiana University Light Microscopy Imaging Center. Targeted GC-MS analysis was conducted using instruments housed in the Indiana University Mass Spectrometry Facility. Some of the metabolomic analyses described herein were performed at the Metabolomics Core Facility at the University of Utah, which is supported by 1S10OD016232-01, 1S10OD021505-01 and 1U54DK110858-01. Special thanks to Alexander Fitt for technical assistance and Dr David Maughn for generously providing us with an aliquot of the anti-GPDH1 antibody on behalf of the late Dr David T. Sullivan.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: H.L., M.R., K.B., K.L.M., N.S.S., J.M.T.; Methodology: H.L., C.R.J., K.L.M., J.M.T.; Formal analysis: H.L., M.R., A.L., N.H.M., G.C., J.M.T.; Investigation: H.L., M.R., K.B., M.C.S., A.L., N.H.M., C.R.J., R.C.P., G.C., C.J.G., A.K.B., K.L.M., J.M.T.; Resources: H.L., M.C.S., A.L., R.C.P., G.C., C.J.G., A.K.B., J.A.K., K.L.M., N.S.S., J.M.T.; Data curation: H.L., J.M.T.; Writing - original draft: J.M.T.; Writing - review & editing: H.L., M.R., M.C.S., K.L.M., N.S.S., J.M.T.; Visualization: H.L., M.R., K.B., A.L., C.R.J., K.L.M., J.M.T.; Supervision: J.A.K., K.L.M., N.S.S., J.M.T.; Project administration: J.M.T.; Funding acquisition: J.M.T.
Funding
K.B. and N.S.S. were supported by the National Institutes of Health (R01GM124220). C.R.J. was supported by a National Science Foundation (NSF) CAREER award to K.L.M. (IOS-1149178, IOS-1505247). J.M.T. and J.A.K were supported by a NSF MRI Award (1726633). J.M.T. is supported by the National Institute of General Medical Sciences under a R35 Maximizing Investigators' Research Award (MIRA; 1R35GM119557). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.175315.supplemental
References
- Arrese E. L. and Soulages J. L. (2010). Insect fat body: energy, metabolism, and regulation. Annu. Rev. Entomol. 55, 207-225. 10.1146/annurev-ento-112408-085356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avi-Dor Y. and Mager J. (1956). The effect of fluoropyruvate on the respiration of animal-tissue preparations. Biochem. J. 63, 613-618. 10.1042/bj0630613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backhaus B., Sulkowski E. and Schlote F. (1984). A semi-synthetic, general-purpose medium for Drosophila melanogaster. Dros. Inf. Serv. 60, 210-212. [Google Scholar]
- Bewley G. C. and Lucchesi J. C. (1977). Origin of alpha-glycerophosphate dehydrogenase isozymes in Drosophila melanogaster and their functional relationship in the alpha-glycerophosphate cycle. Biochem. Genet. 15, 235-251. 10.1007/BF00484456 [DOI] [PubMed] [Google Scholar]
- Billiard J., Dennison J. B., Briand J., Annan R. S., Chai D., Colón M., Dodson C. S., Gilbert S. A., Greshock J., Jing J. et al. (2013). Quinoline 3-sulfonamides inhibit lactate dehydrogenase A and reverse aerobic glycolysis in cancer cells. Cancer Metab. 1, 19 10.1186/2049-3002-1-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau A., Purkey H. E., Hitz A., Robarge K., Peterson D., Labadie S., Kwong M., Hong R., Gao M., Del Nagro C. et al. (2016). Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat. Chem. Biol. 12, 779-786. 10.1038/nchembio.2143 [DOI] [PubMed] [Google Scholar]
- Boxer G. E. and Shonk C. E. (1960). Low levels of soluble DPN-linked alpha-glycerophosphate dehydrogenase in tumors. Cancer Res. 20, 85-91. [PubMed] [Google Scholar]
- Bricker D. K., Taylor E. B., Schell J. C., Orsak T., Boutron A., Chen Y.-C., Cox J. E., Cardon C. M., Van Vranken J. G., Dephoure N. et al. (2012). A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 337, 96-100. 10.1126/science.1218099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulusu V., Prior N., Snaebjornsson M. T., Kuehne A., Sonnen K. F., Kress J., Stein F., Schultz C., Sauer U. and Aulehla A. (2017). Spatiotemporal analysis of a glycolytic activity gradient linked to mouse embryo mesoderm development. Dev. Cell 40, 331-341.e334. 10.1016/j.devcel.2017.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J., Soufan O., Li C., Caraus I., Li S., Bourque G., Wishart D. S. and Xia J. (2018). MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 46, W486-W494. 10.1093/nar/gky310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper E. H., Barkhan P. and Hale A. J. (1963). Observations on the proliferation of human leucocytes cultured with phytohaemagglutinin. Br. J. Haematol. 9, 101-111. 10.1111/j.1365-2141.1963.tb05446.x [DOI] [PubMed] [Google Scholar]
- Cox J. E., Thummel C. S. and Tennessen J. M. (2017). Metabolomic studies in Drosophila. Genetics 206, 1169-1185. 10.1534/genetics.117.200014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniele S., Giacomelli C., Zappelli E., Granchi C., Trincavelli M. L., Minutolo F. and Martini C. (2015). Lactate dehydrogenase-A inhibition induces human glioblastoma multiforme stem cell differentiation and death. Sci. Rep. 5, 15556 10.1038/srep15556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan D. M., Kiefel P. and Duncan I. (2017). Mutants for Drosophila Isocitrate Dehydrogenase 3b are defective in mitochondrial function and larval cell death. G3 7, 789-799. 10.1534/g3.116.037366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantin V. R., St-Pierre J. and Leder P. (2006). Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 9, 425-434. 10.1016/j.ccr.2006.04.023 [DOI] [PubMed] [Google Scholar]
- Flores A., Schell J., Krall A. S., Jelinek D., Miranda M., Grigorian M., Braas D., White A. C., Zhou J. L., Graham N. A. et al. (2017). Lactate dehydrogenase activity drives hair follicle stem cell activation. Nat. Cell Biol. 19, 1017-1026. 10.1038/ncb3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg E. B. and Colowick S. P. (1965). The role of glycolysis in the growth of tumor cells. 3. Lactic dehydrogenase as the site of action of oxamate on the growth of cultured cells. J. Biol. Chem. 240, 2786-2790. [PubMed] [Google Scholar]
- Goldberg E. B., Nitowsky H. M. and Colowick S. P. (1965). The role of glycolysis in the growth of tumor cells. IV. The basis of glucose toxicity in oxamate-treated, cultured cells. J. Biol. Chem. 240, 2791-2796. [PubMed] [Google Scholar]
- Gratz S. J., Cummings A. M., Nguyen J. N., Hamm D. C., Donohue L. K., Harrison M. M., Wildonger J. and O'Connor-Giles K. M. (2013). Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics 194, 1029-1035. 10.1534/genetics.113.152710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley B. R., Brooks A. N., Carlson J. W., Duff M. O., Landolin J. M., Yang L., Artieri C. G., van Baren M. J., Boley N., Booth B. W. et al. (2011). The developmental transcriptome of Drosophila melanogaster. Nature 471, 473-479. 10.1038/nature09715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havula E. and Hietakangas V. (2018). Sugar sensing by ChREBP/Mondo-Mlx-new insight into downstream regulatory networks and integration of nutrient-derived signals. Curr. Opin. Cell Biol. 51, 89-96. 10.1016/j.ceb.2017.12.007 [DOI] [PubMed] [Google Scholar]
- Hoekstra L. A. and Montooth K. L. (2013). Inducing extra copies of the Hsp70 gene in Drosophila melanogaster increases energetic demand. BMC Evol. Biol. 13, 68 10.1186/1471-2148-13-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra L. A., Siddiq M. A. and Montooth K. L. (2013). Pleiotropic effects of a mitochondrial-nuclear incompatibility depend upon the accelerating effect of temperature in Drosophila. Genetics 195, 1129-1139. 10.1534/genetics.113.154914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno T., Sudo K., Takeuchi I., Kanda S., Honda N., Nishimura Y. and Oyama K. (1980). Hereditary deficiency of lactate dehydrogenase M-subunit. Clin. Chim. Acta 108, 267-276. 10.1016/0009-8981(80)90013-3 [DOI] [PubMed] [Google Scholar]
- Kanno T., Sudo K., Maekawa M., Nishimura Y., Ukita M. and Fukutake K. (1988). Lactate dehydrogenase M-subunit deficiency: a new type of hereditary exertional myopathy. Clin. Chim. Acta 173, 89-98. 10.1016/0009-8981(88)90359-2 [DOI] [PubMed] [Google Scholar]
- Li H. and Tennessen J. M. (2017). Methods for studying the metabolic basis of Drosophila development. Wiley Interdiscip. Rev. Dev. Biol. 6, e280 10.1002/wdev.280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. and Tennessen J. M. (2018). Preparation of Drosophila larval samples for gas chromatography-mass spectrometry (GC-MS)-based metabolomics. J. Vis. Exp. 136, e57847 10.3791/57847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Padmanabha D., Gentile L. B., Dumur C. I., Beckstead R. B. and Baker K. D. (2013). HIF- and non-HIF-regulated hypoxic responses require the estrogen-related receptor in Drosophila melanogaster. PLoS Genet. 9, e1003230 10.1371/journal.pgen.1003230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Chawla G., Hurlburt A. J., Sterrett M. C., Zaslaver O., Cox J., Karty J. A., Rosebrock A. P., Caudy A. A. and Tennessen J. M. (2017). Drosophila larvae synthesize the putative oncometabolite L-2-hydroxyglutarate during normal developmental growth. Proc. Natl. Acad. Sci. USA 114, 1353-1358. 10.1073/pnas.1614102114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Hurlburt A. J. and Tennessen J. M. (2018). A Drosophila model of combined D-2- and L-2-hydroxyglutaric aciduria reveals a mechanism linking mitochondrial citrate export with oncometabolite accumulation. Dis. Model. Mech. 11, dmm035337 10.1242/dmm.035337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhur A., Chawla G., Wu Y.-C., Li J. and Sokol N. S. (2014). Drosha-independent DGCR8/Pasha pathway regulates neuronal morphogenesis. Proc. Natl. Acad. Sci. USA 111, 1421-1426. 10.1073/pnas.1318445111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhur A., Buddika K., Ariyapala I. S., Chen S. and Sokol N. S. (2017). Opposing post-transcriptional control of InR by FMRP and LIN-28 adjusts stem cell-based tissue growth. Cell Rep. 21, 2671-2677. 10.1016/j.celrep.2017.11.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt S. Y. and Vander Heiden M. G. (2011). Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 27, 441-464. 10.1146/annurev-cellbio-092910-154237 [DOI] [PubMed] [Google Scholar]
- MacNeil L. T., Watson E., Arda H. E., Zhu L. J. and Walhout A. J. M. (2013). Diet-induced developmental acceleration independent of TOR and insulin in C. elegans. Cell 153, 240-252. 10.1016/j.cell.2013.02.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa M., Sudo K., Kanno T. and Li S. S.-L. (1990). Molecular characterization of genetic mutation in human lactate dehydrogenase-A (M) deficiency. Biochem. Biophys. Res. Commun. 168, 677-682. 10.1016/0006-291X(90)92374-9 [DOI] [PubMed] [Google Scholar]
- Mandal S., Guptan P., Owusu-Ansah E. and Banerjee U. (2005). Mitochondrial regulation of cell cycle progression during development as revealed by the tenured mutation in Drosophila. Dev. Cell 9, 843-854. 10.1016/j.devcel.2005.11.006 [DOI] [PubMed] [Google Scholar]
- Matoo O. B., Julick C. R. and Montooth K. L. (2019). Genetic variation for ontogenetic shifts in metabolism underlies physiological homeostasis in Drosophila. Genetics 212, 537-552. 10.1534/genetics.119.302052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mccabe C. T. and Bursell E. (1975). Interrelationships between amino acid and lipid metabolism in the tsetse fly, Glossina Morsitans. Insect Biochem. 5, 781-789. 10.1016/0020-1790(75)90022-0 [DOI] [Google Scholar]
- Meiklejohn C. D., Holmbeck M. A., Siddiq M. A., Abt D. N., Rand D. M. and Montooth K. L. (2013). An Incompatibility between a mitochondrial tRNA and its nuclear-encoded tRNA synthetase compromises development and fitness in Drosophila. PLoS Genet. 9, e1003238 10.1371/journal.pgen.1003238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyajima H., Takahashi Y. and Kaneko E. (1995). Characterization of the glycolysis in lactate dehydrogenase-A deficiency. Muscle Nerve 18, 874-878. 10.1002/mus.880180812 [DOI] [PubMed] [Google Scholar]
- Miyazawa H. and Aulehla A. (2018). Revisiting the role of metabolism during development. Development 145, dev131110 10.1242/dev.131110 [DOI] [PubMed] [Google Scholar]
- O'Brien S. J. and MacIntyre R. J. (1972). The α-glycerophosphate cycle in Drosophila melanogaster. I. Biochemical and developmental aspects. Biochem. Genet. 7, 141-161. 10.1007/BF00486085 [DOI] [PubMed] [Google Scholar]
- Oginuma M., Moncuquet P., Xiong F., Karoly E., Chal J., Guevorkian K. and Pourquié O. (2017). A gradient of glycolytic activity coordinates FGF and Wnt signaling during elongation of the body axis in amniote embryos. Dev. Cell 40, 342-353.e310. 10.1016/j.devcel.2017.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira M. T. and Kaguni L. S. (2011). Reduced stimulation of recombinant DNA polymerase gamma and mitochondrial DNA (mtDNA) helicase by variants of mitochondrial single-stranded DNA-binding protein (mtSSB) correlates with defects in mtDNA replication in animal cells. J. Biol. Chem. 286, 40649-40658. 10.1074/jbc.M111.289983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaconstantinou J. and Colowick S. P. (1961). The role of glycolysis in the growth of tumor cells. I. Effects of oxamic acid on the metabolism of Ehrlich ascites tumor cells in vitro. J. Biol. Chem. 236, 278-284. [PubMed] [Google Scholar]
- Pearce E. L., Poffenberger M. C., Chang C.-H. and Jones R. G. (2013). Fueling immunity: insights into metabolism and lymphocyte function. Science 342, 1242454 10.1126/science.1242454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletcher R. C., Hardman S. L., Intagliata S. F., Lawson R. L., Page A. and Tennessen J. M. (2019). A genetic screen using the Drosophila melanogaster TRiP RNAi collection to identify metabolic enzymes required for eye development. G3 (Bethesda) 9, 2061-2070. 10.1101/582940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qing G., Skuli N., Mayes P. A., Pawel B., Martinez D., Maris J. M. and Simon M. C. (2010). Combinatorial regulation of neuroblastoma tumor progression by N-Myc and hypoxia inducible factor HIF-1alpha. Cancer Res. 70, 10351-10361. 10.1158/0008-5472.CAN-10-0740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. (2017). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. [Google Scholar]
- Rechsteiner M. C. (1970). Drosophila lactate dehydrogenase and alpha-glycerolphosphate dehydrogenase: distribution and change in activity during development. J. Insect Physiol. 16, 1179-1192. 10.1016/0022-1910(70)90208-8 [DOI] [PubMed] [Google Scholar]
- Schell J. C., Wisidagama D. R., Bensard C., Zhao H., Wei P., Tanner J., Flores A., Mohlman J., Sorensen L. K., Earl C. S. et al. (2017). Control of intestinal stem cell function and proliferation by mitochondrial pyruvate metabolism. Nat. Cell Biol. 19, 1027-1036. 10.1038/ncb3593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebo Z. L., Lee H. B., Peng Y. and Guo Y. (2014). A simplified and efficient germline-specific CRISPR/Cas9 system for Drosophila genomic engineering. Fly (Austin) 8, 52-57. 10.4161/fly.26828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skuse G. R. and Sullivan D. T. (1985). Developmentally regulated alternate modes of expression of the Gpdh locus of Drosophila. EMBO J. 4, 2275-2280. 10.1002/j.1460-2075.1985.tb03926.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teesalu M., Rovenko B. M. and Hietakangas V. (2017). Salt-inducible kinase 3 provides sugar tolerance by regulating NADPH/NADP(+) Redox balance. Curr. Biol. 27, 458-464. 10.1016/j.cub.2016.12.032 [DOI] [PubMed] [Google Scholar]
- Tennessen J. M. and Thummel C. S. (2011). Coordinating growth and maturation - insights from Drosophila. Curr. Biol. 21, R750-R757. 10.1016/j.cub.2011.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen J. M., Baker K. D., Lam G., Evans J. and Thummel C. S. (2011). The Drosophila estrogen-related receptor directs a metabolic switch that supports developmental growth. Cell Metab. 13, 139-148. 10.1016/j.cmet.2011.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen J. M., Barry W. E., Cox J. and Thummel C. S. (2014a). Methods for studying metabolism in Drosophila. Methods 68, 105-115. 10.1016/j.ymeth.2014.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennessen J. M., Bertagnolli N. M., Evans J., Sieber M. H., Cox J. and Thummel C. S. (2014b). Coordinated metabolic transitions during Drosophila embryogenesis and the onset of aerobic glycolysis. G3 4, 839-850. 10.1534/g3.114.010652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tixier V., Bataille L., Etard C., Jagla T., Weger M., Daponte J. P., Strahle U., Dickmeis T. and Jagla K. (2013). Glycolysis supports embryonic muscle growth by promoting myoblast fusion. Proc. Natl. Acad. Sci. USA 110, 18982-18987. 10.1073/pnas.1301262110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden M. G., Cantley L. C. and Thompson C. B. (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029-1033. 10.1126/science.1160809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Marquardt C. and Foker J. (1976). Aerobic glycolysis during lymphocyte proliferation. Nature 261, 702-705. 10.1038/261702a0 [DOI] [PubMed] [Google Scholar]
- Wang L., Lam G. and Thummel C. S. (2010). Med24 and Mdh2 are required for Drosophila larval salivary gland cell death. Dev. Dyn. 239, 954-964. 10.1002/dvdy.22213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. (1956). On the origin of cancer cells. Science 123, 309-314. 10.1126/science.123.3191.309 [DOI] [PubMed] [Google Scholar]
- Warburg O., Posener K. and Negelein E. (1924). The metabolism of cancer cells. Biochem Z 152, 319-344. [Google Scholar]
- Warton D. I., Wright I. J., Falster D. S. and Westoby M. (2006). Bivariate line-fitting methods for allometry. Biol. Rev. Camb. Philos. Soc. 81, 259-291. 10.1017/S1464793106007007 [DOI] [PubMed] [Google Scholar]
- Watson E., MacNeil L. T., Ritter A. D., Yilmaz L. S., Rosebrock A. P., Caudy A. A. and Walhout A. J. M. (2014). Interspecies systems biology uncovers metabolites affecting C. elegans gene expression and life history traits. Cell 156, 759-770. 10.1016/j.cell.2014.01.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson E., Olin-Sandoval V., Hoy M. J., Li C.-H., Louisse T., Yao V., Mori A., Holdorf A. D., Troyanskaya O. G., Ralser M. et al. (2016). Metabolic network rewiring of propionate flux compensates vitamin B12 deficiency in C. elegans. eLife 5, e17670 10.7554/eLife.17670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H., Hanai J., Ren J.-G., Kats L., Burgess K., Bhargava P., Signoretti S., Billiard J., Duffy K. J., Grant A. et al. (2014). Targeting lactate dehydrogenase--a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab. 19, 795-809. 10.1016/j.cmet.2014.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikuni K., Tagami H., Yamada M., Sudo K. and Kanno T. (1986). Erythematosquamous skin lesions in hereditary lactate dehydrogenase M-subunit deficiency. Arch. Dermatol. 122, 1420-1424. 10.1001/archderm.1986.01660240084023 [DOI] [PubMed] [Google Scholar]
- Zdralevic M., Brand A., Di Ianni L., Dettmer K., Reinders J., Singer K., Peter K., Schnell A., Bruss C., Decking S.-M. et al. (2018). Double genetic disruption of lactate dehydrogenase A and B is required to ablate the ‘Warburg effect’ restricting tumor growth to oxidative metabolism. J. Biol. Chem. 293, 15947-15961. 10.1074/jbc.RA118.004180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zebe E. C. and McShan W. H. (1957). Lactic and alpha-glycerophosphate dehydrogenases in insects. J. Gen. Physiol. 40, 779-790. 10.1085/jgp.40.5.779 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.