

ABSTRACT
Developmental defects affecting the heart and aortic arch arteries are a significant phenotype observed in individuals with 22q11 deletion syndrome and are caused by a microdeletion on chromosome 22q11. TBX1, one of the deleted genes, is expressed throughout the pharyngeal arches and is considered a key gene, when mutated, for the arch artery defects. Pax9 is expressed in the pharyngeal endoderm and is downregulated in Tbx1 mutant mice. We show here that Pax9-deficient mice are born with complex cardiovascular malformations that affect the outflow tract and aortic arch arteries with failure of the 3rd and 4th pharyngeal arch arteries to form correctly. Transcriptome analysis indicated that Pax9 and Tbx1 may function together, and mice double heterozygous for Tbx1/Pax9 presented with a significantly increased incidence of interrupted aortic arch when compared with Tbx1 heterozygous mice. Using a novel Pax9Cre allele, we demonstrated that the site of this Tbx1-Pax9 genetic interaction is the pharyngeal endoderm, therefore revealing that a Tbx1-Pax9-controlled signalling mechanism emanating from the pharyngeal endoderm is required for crucial tissue interactions during normal morphogenesis of the pharyngeal arch artery system.
KEY WORDS: Pharyngeal endoderm, Arch artery development, Tbx1, Pax9, Neural crest, 22q11 deletion syndrome
Summary: A strong genetic interaction between Tbx1 and Pax9 that leads to 4th PAA-derived defects in double heterozygous mice is cell-autonomous within the pharyngeal endoderm.
INTRODUCTION
Conotruncal heart malformations, which affect the outflow tract and aortic arch arteries, occur in 30% of all cases of congenital heart defects (Thom et al., 2006). Approximately 20% of foetuses identified with conotruncal defects by ultrasound are diagnosed with 22q11 deletion syndrome (22q11DS) (Boudjemline et al., 2001), the most common microdeletion syndrome with an incidence of 1:4000 live births (Scambler, 2000). Patients typically have a 3 Mb deletion on chromosome 22 that encompasses 45 protein-coding genes (Morrow et al., 2018) and ∼80% of patients present with some form of congenital cardiovascular defect (Momma, 2010), of which one of the most common observed is interruption of the aortic arch (IAA) (Unolt et al., 2018). Although clinically rare in the general population, ∼50% of all cases of IAA occur in 22q11DS patients (Boudjemline et al., 2001; Lewin et al., 1997; Van Mierop and Kutsche, 1986). IAA is a consequence of the left 4th pharyngeal arch artery (PAA) failing to form correctly during embryonic development. There are five pairs of PAAs that arise within a series of repeated protuberances on either side of the developing pharynx known as the pharyngeal arches. These arches consist of paraxial mesoderm- and neural crest cell (NCC)-derived mesenchyme, and are externally enclosed by ectoderm and internally lined by endoderm (Graham, 2003). The initially bilaterally symmetrical PAAs develop sequentially in a cranial to caudal sequence during embryogenesis but undergo a complex remodelling process, which is conserved in mammals, to form the asymmetrical mature aortic arch configuration (Bamforth et al., 2013; Hiruma et al., 2002). Of the genes deleted in 22q11DS, hemizygosity of TBX1 is considered to be the cause of the cardiovascular defects, with point mutations in TBX1 also identified in individuals with 22q11DS-like phenotypes (Yagi et al., 2003; Zweier et al., 2007). Tbx1 is expressed throughout the pharyngeal arches within the ectoderm, endoderm and mesoderm cells, and is crucial for pharyngeal arch and PAA development (Jerome and Papaioannou, 2001; Lindsay et al., 2001; Merscher et al., 2001). Complete loss of Tbx1 from the mouse results in a failure of the pharyngeal arches to form correctly, resulting in hypoplasia of the second arch and aplasia of the remaining caudal arches, leading to an absent thymus and common arterial trunk (Jerome and Papaioannou, 2001; Merscher et al., 2001). In contrast, heterozygous deletion of Tbx1 predominantly affects development of the 4th PAA resulting in IAA in a minority of mice with aberrant right-subclavian artery (A-RSA) observed more frequently (Lindsay et al., 2001). Tbx1 heterozygotes may, however, also present with VSD, overriding aorta, double aortic arch and an abnormal thymus (Merscher et al., 2001; Zhang and Baldini, 2008). Although almost all 22q11DS patients are hemizygous for the deletion, the phenotypic spectrum is highly variable (Unolt et al., 2018); thus, it has been proposed that genes outside of the deleted region may impact on the clinical phenotype (Guo et al., 2011b). Several genes have been identified as potential modifiers of 22q11DS, as determined by genetic interaction studies and transcriptome analyses in Tbx1 mutant mice (Aggarwal and Morrow, 2008; Ivins et al., 2005; Liao et al., 2008; Papangeli and Scambler, 2013). One such candidate gene is Pax9 the expression of which in the pharyngeal endoderm was found to be significantly reduced in Tbx1-deficient embryos (Ivins et al., 2005; Liao et al., 2008). Pax9 belongs to a family of Pax genes encoding transcription factors involved in various cellular roles during embryonic development (Chi and Epstein, 2002; Mansouri et al., 1996) and is specifically expressed in the endoderm of all four pharyngeal pouches of the mouse by E9.5 (Neubüser et al., 1995). At later stages in mouse development, Pax9 is expressed in the oesophagus, somites and limbs, and within the neural crest contributing to craniofacial development (Peters et al., 1998). Embryos deficient for Pax9 exhibit craniofacial defects, including a cleft secondary palate as well as absent teeth, skeletal defects and lack derivatives of the 3rd and 4th pharyngeal pouches, i.e. the thymus, parathyroids and ultimobranchial bodies (Peters et al., 1998), yet the role of Pax9 in cardiovascular development has not been determined.
In this study, we have investigated cardiovascular development in Pax9-deficient mice, and show that loss of Pax9 leads to complex cardiovascular defects affecting the outflow tract and aortic arch arteries. We also uncover a strong genetic interaction between Tbx1 and Pax9 that leads to 4th PAA-derived defects in double heterozygous mice; this interaction is cell-autonomous within the pharyngeal endoderm.
RESULTS
Pax9-deficient embryos have cardiovascular defects
Mice lacking Pax9 die perinatally and exhibit craniofacial, pharyngeal gland and skeletal defects (Peters et al., 1998). As the cause of death was unclear, we carefully re-analysed Pax9–/– neonates from a Pax9+/– intercross and found that all Pax9–/– neonates had a patterning defect of their aortic arch arteries that included an IAA with A-RSA (Fig. 1B; Table 1). To investigate these defects in more detail, we processed Pax9–/– embryos for histology and magnetic resonance imaging (MRI), and found that Pax9–/– embryos presented with a wide range of cardiovascular abnormalities (Fig. 1C-Q; Table 1). These defects included double outlet right ventricle with interventricular communication (Fig. 1F,J,K), IAA and A-RSA (Fig. 1G,K). The common carotid arteries were also frequently absent, resulting in unilateral or bilateral internal and external carotids arising directly from the dorsal aorta and the A-RSA (Fig. 1H,J-L). Pax9–/– embryos also had a hypoplastic aorta (Fig. 1N-O) and bicuspid aortic valves (Fig. 1Q).
Fig. 1.

Loss of Pax9 results in cardiovascular developmental defects. (A) Arch arteries of control neonates were normal (n=8). (B) Pax9–/– neonates (n=5) displayed IAA-B, absent right subclavian artery (presumed retro-oesophageal), and atypical right and left carotid arteries (LC and RC). Scale bars: 1 mm. (C-E) Pax9+/+ embryos at E14.5 show normal outflow tract, arch arteries and thymus. (F-H) In Pax9–/–embryos (n=4), the aorta (Ao) arises aberrantly from the right ventricle producing a double outlet right ventricle (DORV; F). Hypoplastic aorta, IAA-B and an A-RSA are present (G). More cranially, the thymus is absent and aberrant internal and external carotid arteries (iRCA, eRCA, iLCA, eLCA) are seen (H). (I-L) 3D reconstructions of E15.5 hearts from MRI datasets. (I) Pax9+/+ embryo with normal aortic arch arteries. (J-L) In Pax9–/–embryos (n=15), defects seen are DORV with interventricular communication (IVC), IAA (type B or C), A-RSA (retro-esophageal in J,K and isolated in L) and aberrant carotid arteries. Scale bars: 500 μm. (M-O) The ascending aorta is significantly smaller in Pax9–/– embryos (n=5) compared with Pax9+/– control embryos (n=7). Data are mean±s.e.m. ****P<0.0001 (two-tailed unpaired t-test). (P) Pax9+/– control embryos have three normal aortic valve leaflets (n=6): the right (RC), left (LC) and non-coronary (NC). (Q) Pax9–/– embryos (n=5) have bicuspid aortic valve (BAV). Scale bar: 100 μm. AoA, aortic arch; AD, arterial duct; BC, brachiocephalic; dAo, dorsal aorta; LCC, left common carotid artery; LSA, left subclavian artery; LV, left ventricle; RCC, right common carotid artery; RSA, right subclavian artery; RV, right ventricle.
Table 1.
Number of cardiovascular defects observed in mutant embryos (>E13.5) and neonates

Developmental defects of the arch arteries occur when the PAAs fail to form or remodel correctly. To investigate the morphogenesis of the PAAs, we performed high-resolution episcopic microscopy (HREM) on E10.5 and E11.5 Pax9–/– embryos and manually segmented the datasets to create 3D images of the PAA. Embryos were stage-matched by somite counting to exclude developmental delay as a cause of observed defects. Analysis of E10.5 coronal sections revealed that the 3rd and 4th pharyngeal arches of Pax9–/– embryos were smaller compared with Pax9+/+ controls, as previously reported (Peters et al., 1998), but did show segmentation to individual arches (Fig. 2A-C). At E10.5, the developing PAAs are bilaterally symmetrical, having formed in a cranial to caudal sequence. The 1st and 2nd PAAs have normally regressed by this stage, and the 3rd, 4th and 6th PAAs are present (Bamforth et al., 2013; Hiruma et al., 2002) (Fig. 2A). In Pax9–/– embryos, the 1st and 2nd PAAs were abnormally persistent, the 3rd PAAs were either absent, hypoplastic or interrupted, and the 4th PAAs were absent (Fig. 2B,C; Table 2). The aortic sac was also hypoplastic in Pax9–/– embryos compared with the controls (Fig. 2B,C). Intra-cardiac injection of India ink at E10.5-E11.0 showed that, in controls, the caudal PAAs were patent to ink and of equivalent size (Fig. 2D). In Pax9–/– embryos, however, persisting 1st and 2nd PAAs were patent to ink, and the 3rd PAAs were hypoplastic (Fig. 2J-L) or non-patent to ink, and therefore presumed absent. The majority of the 4th PAAs were bilaterally non-patent to ink (Fig. 2J-L; Table 2). Immunohistochemical staining for Pecam1 showed that the endothelium within the 3rd and 4th pharyngeal arches had formed lumenised PAAs at E10.5 in control embryos (Fig. 2G), but in Pax9–/– embryos the 3rd PAAs were visibly smaller in size and only isolated endothelial cells were seen within the 4th pharyngeal arch (Fig. 2H,I).
Fig. 2.

Pax9 loss causes aberrations in pharyngeal arch artery formation. (A-C) Coronal views of control (Pax9+/+) and mutant (Pax9–/–) embryos at E10.5 (30-40 s), examined by high-resolution episcopic microscopy. (A) In Pax9+/+ control embryos (n=5), the 3rd, 4th and 6th PAAs are of equal size and bilaterally symmetrical. (B,C) In Pax9–/– embryos (n=4), the 4th PAAs were bilaterally absent (asterisk), the 3rd PAAs and aortic sac (as) were hypoplastic, and the 1st and/or 2nd PAAs abnormally persisted. (D-F) Intracardiac ink injection into E10.5-E11.0 (31-42 s) embryos. (D) In control embryos (n=12), PAAs 3-6 are patent to ink, are of equivalent diameter and are bilaterally symmetrical. (E,F) In Pax9–/– embryos (n=12), the 3rd PAAs are hypoplastic and the 4th PAAs are non-patent to ink (asterisk). (F) The 1st and 2nd PAAs are persisting anomalously. (G-I) Immunostaining using anti-Pecam1 antibody at E10.5 (31-36 s). (G) Control embryos (n=3) have a ring of Pecam1-positive endothelium lining the 3rd and 4th PAAs. (H,I) In Pax9–/– embryos (n=5), the 3rd PAAs are visibly smaller and disorganized endothelial cells are within the 4th pharyngeal arch (pa; arrows). Somite counts (s) are indicated. Scale bars: 100 μm. The somite numbers given in the legend reflect the range analysed for the whole study. The figure contains representative images only.
Table 2.
Number of pharyngeal arch artery defects

During E11.5 of mouse development, the PAA system begins to remodel and becomes asymmetric in appearance (Bamforth et al., 2013; Hiruma et al., 2002). In Pax9 control embryos at this stage, the right 6th PAAs appeared thinner and the distal outflow tract was septated. The carotid duct, the region of the dorsal aorta between the 3rd and 4th PAAs, had begun to involute, but the 3rd, 4th and 6th PAAs remained connected to the dorsal aortae (Fig. 3A). In Pax9–/– embryos at E11.5, presumed persisting 1st or 2nd PAAs were observed extending anteriorly from the aortic sac, and the 3rd PAAs were also affected, either unilaterally or bilaterally, being either absent, hypoplastic or interrupted and not connected to the dorsal aortae (Fig. 3B-D). The carotid ducts were maintained, and this, coupled with the abnormal detachment of the 3rd PAAs from the dorsal aorta and the persistence of the 1st or 2nd PAAs, resulted in an absent common carotid artery and the internal and external carotid arteries arising directly from the aortic arch arteries, as observed at E15.5 (Fig. 1). Additionally, the distal outflow tract was unseptated and the 4th PAAs were bilaterally absent (Fig. 3B-D; Table 2).
Fig. 3.

Pax9–/– embryos display aberrant rotation of the outflow tract and abnormal PAA remodelling. Image datasets were acquired by high-resolution episcopic microscopy. (A-D) 3D reconstructions of the PAAs at E11.5 (45-50 s). (A) In Pax9+/+ control embryos (n=5), the outflow tract is septated, the right 6th PAA has thinned and the carotid duct (cd) has begun to involute. (B-D) In Pax9–/– embryos (n=4), the 4th PAAs are absent and the 3rd PAAs are aberrantly connected or detached from the dorsal aorta (da; asterisks). The carotid duct has failed to involute and septation of the outflow tract is delayed. Persisting 1st or 2nd PAAs are observed coursing anteriorly from the abnormal 3rd PAAs (B,D) or from the aortic sac (as; C). (E,F) Looping of the heart tube at E10.5 was not affected in Pax9–/– embryos (n=4). (G-L) Outflow tract rotation from E10.5 to E11.5 (39-49 s). (G-I) In control embryos, the major outflow tract cushions (parietal in green; septal in yellow) rotated in an anti-clockwise direction, resulting in the cushions being aligned side by side. (J-L) In Pax9–/– embryos, the cushions rotated in a clockwise direction, resulting in the parietal cushion being anterior to the septal cushion. at, aortic trunk; pa, pulmonary arteries; pt, pulmonary trunk. Somite (s) counts indicated. Scale bars: 100 μm.
Looping of the heart tube at E10.5 was not affected in Pax9–/– embryos (Fig. 3E,F).
In normal mouse development, the outflow tract cushions rotate through an anticlockwise direction (Bajolle et al., 2006), resulting in the major septal and parietal cushions being positioned side by side (Fig. 3G-I). In Pax9–/– embryos, however, by the end of the E11.5 stage these cushions were aberrantly positioned, with the parietal cushion more anterior to the septal cushion, indicating that a clockwise rotation of the outflow tract cushions had occurred (Fig. 3J-L).
Pharyngeal endoderm signalling influences neural crest cell differentiation
During normal embryogenesis, a subset of NCCs, the cardiac neural crest cells, migrate into the caudal pharyngeal arches to provide support to the developing PAAs and also contribute to outflow tract septation (Hutson and Kirby, 2003). Pax9 is predominantly expressed in the pharyngeal endoderm (Fig. S1), but is also known to be expressed within the NCC and contributes to craniofacial development (Kist et al., 2007), although expression in cardiac NCC has not been described. We therefore looked to see whether a cell-autonomous loss of Pax9 from NCC resulted in cardiovascular defects. Conditional deletion of Pax9 using Wnt1Cre transgenic mice (Danielian et al., 1998), and the Pax9flox allele (Kist et al., 2007), however, did not result in cardiovascular abnormalities, although cleft palate was observed as expected (Fig. 4A-D). We next looked to see whether the migration of NCCs was affected in global Pax9–/– embryos using Wnt1Cre and eYFP (Srinivas et al., 2001) reporter mice to create embryos where the NCC could be lineage traced. Live fluorescence imaging of Pax9+/+;Wnt1Cre;eYFP embryos at E9.5 and E10.5 revealed NCCs present in the head and dorsal root ganglia, as well as migrating towards and filling the pharyngeal arches (Fig. 4E,G). A similar pattern was observed in Pax9–/–;Wnt1Cre;eYFP embryos (Fig. 4F,H). Counting of NCCs revealed no significant change in their number in the 3rd pharyngeal arch at E9.5, but a significant reduction was found in the 3rd and 4th arches at E10.5 (Fig. 4K). Immunostaining for BrdU incorporation specifically in NCCs showed no significant difference in NCC proliferation in the caudal pharyngeal arches (Fig. 4L-N), and very little apoptosis was seen in both control and Pax9–/– embryos (Fig. 4O-Q). These observations indicated that the migration of NCCs per se to the pharyngeal arches was not affected in Pax9–/– embryos. Given that NCCs differentiate into the smooth muscle cells (SMCs) that support the remodelling PAAs (Waldo et al., 1996), we looked to see whether SMC recruitment to the 3rd PAAs was affected in Pax9–/– embryos. Sections from E10.5 and E11.5 embryos were immunostained using an anti-alpha smooth muscle actin antibody to visualise SMCs around the PAAs. Evidence of SMC recruitment to the 3rd PAAs was seen in each control embryo examined (Fig. 4R-U); however, very little SMC recruitment to the 3rd PAAs in Pax9–/– embryo sections was observed (Fig. 4V-Y). These results indicate that the 3rd PAAs in Pax9–/– embryos form but collapse from a failure of SMC recruitment.
Fig. 4.

Failure in smooth muscle cell recruitment causes the 3rd PAAs to collapse. (A-D) E15.5 embryos with a Pax9 conditional deletion in neural crest cells (NCCs) were examined by MRI. (A) Control embryos had a normal palate (P). (B) Pax9f/f;Wnt1Cre mutant embryos (n=6) had cleft palate (CP). (C,D) No cardiovascular defects were observed in control or mutant embryos. Ao, aorta; AD, arterial duct; LCC, left common carotid artery; LSA, left subclavian artery; LV, left ventricle; RCC, right common carotid artery; RSA, right subclavian artery; RV, right ventricle. (E-J) NCCs in Pax9+/+ (E,G,I) or Pax9–/– (F,H,J) embryos were labelled using Wnt1Cre and eYFP alleles. (E-H) Fluorescence imaging showed no difference in NCC migration into the pharyngeal arches (arrowheads) at E9.5 (n≥3, 23-25 s) and E10.5 (n≥5, 32-36 s). (I,J) Fluorescent embryos were sectioned coronally and immunostained using anti-GFP and anti-Pax9 antibodies. (K) NCCs were counted from immunostained sections. *P<0.05, ***P<0.001 (two-tailed unpaired t-test). (L,M) Embryo sections were immunostained with anti-BrdU and anti-GFP antibodies to detect proliferation in NCCs in Pax9+/+ (L) and Pax9–/– (M) embryos. (N) No significant difference in the rate of proliferation was found between control and Pax9–/– NCCs in the caudal arches at E9.5 (n=3, 23-28 s) or E10.5 (n≥4, 31-39 s). (O,P) Embryo sections immunostained using an anti-caspase 3 antibody to detect apoptosis in the caudal pharyngeal arches of Pax9+/+ (N,O) and Pax9–/– (P) embryos. (Q) No significant difference in the rate of apoptosis was found between control and Pax9–/– embryos at E9.5 (n=3, 24-28 s) or E10.5 (n=3, 30-35 s). Two-tailed unpaired t-test. (R-Y) Embryo sections were immunostained using anti-αSMA antibody for smooth muscle (R,R″-U,V,V″-Y,S,W), anti-Pecam1 (R,R′,V,V′) or -ERG (T,U,X,Y) antibodies for endothelium, or anti-GFP to label NCCs in Wnt1Cre;eYFP embryos (S,W). In all control embryos, SMC surrounded the 3rd PAAs [E10.5, n=7, 31-36 s (R,S); and E11.5, n=3, 43-45 s (T,U); red arrowhead]. In Pax9–/– embryos, the 3rd PAAs had limited recruitment of SMCs [E10.5, n=6; 32-35 s (V,W); and E11.5, n=3, 42-45 s (X,Y); white arrowheads]. da, dorsal aorta; en, endoderm; mc, mesenchyme; ns, not significant; pa, pharyngeal arch. Somite counts (s) are indicated. Scale bars: 500 μm in A-D; 100 μm in E-J,L,M,O,P; 50 µm in R-Y. The somite numbers given in the legend reflect the range analysed for the whole study. The figure contains representative images only.
Tbx1 and Pax9 interact in 4th PAA formation
To further investigate the role of Pax9 in cardiovascular development, we looked at the effect of Pax9 loss on the pharyngeal arch transcriptome at E9.5 (Fig. 5A-C). Analysis of the RNA-seq data identified 3863 significant differentially expressed genes (Table S1). Gene set enrichment analysis revealed several significantly downregulated pathways, including ‘extracellular matrix organisation’ and ‘axon guidance’ (Fig. 5D). Interestingly, Tbx1 was significantly reduced in Pax9–/– embryos at E9.5, and this was confirmed by qPCR (Fig. 5I). We also confirmed that Pax9 expression was reduced in Tbx1-null embryos (Fig. S2). As Tbx1 expression is crucial for 4th PAA morphogenesis (Lindsay et al., 2001; Zhang et al., 2005), these data suggested that Pax9 may function together with Tbx1 rather than being downstream of it. We therefore compiled a list of all the genes reported to be significantly differentially expressed in Tbx1-null embryos from published microarray (Ivins et al., 2005; Liao et al., 2008; van Bueren et al., 2010) and mouse genetic interaction studies, giving 1476 unique Tbx1-related genes. These were then compared with all genes significantly changed in Pax9–/– embryo pharyngeal arch tissue, giving 342 genes that were common to both datasets (Fig. 5E-G; Fig. S3; Table S1) and suggesting that Tbx1 and Pax9 may share a genetic network. Among these common genes were Chd7 and Gbx2, which have been shown to interact with Tbx1 in mice (Calmont et al., 2009; Randall et al., 2009), and were also reduced in Pax9-null tissue by qPCR and in situ hybridisation (Fig. 5H-S).
Fig. 5.

Pax9 shares a genetic network with Tbx1. (A) The pharyngeal arch region of control and Pax9–/– embryos at E9.5 (n=3 per genotype; 26-27 s) was dissected (indicated by dashed lines). Scale bar: 500 µm. (B) Total RNA was extracted, converted to cDNA and checked by qPCR. (C) Principle component analysis of RNA-seq data showed a clear separation between control and mutant samples (shown on PC2) and only slight biological variation across replicates (shown on PC1). (D) Gene set enrichment analysis identified signalling pathways predicted to be significantly downregulated in Pax9–/– embryos. All pathways shown have a familywise-error rate of P<0.001. (E) Tbx1-related genes (n=1476) were compared with those genes significantly differentially expressed (DE) in Pax9–/– embryo tissue (n=3863, adjusted P≤0.1) giving 342 genes common to both datasets (*P=0.0029, as assessed by a hypergeometric distribution test). Examples of genes in each cohort are shown. (F) Volcano plot highlighting Tbx1-interacting genes significantly DE in Pax9–/– embryos (based on adjusted P≤0.1). (G) Heat map showing colour-coded (red, up; blue, down) expression levels of DE genes between Pax9+/+ (wt) and Pax9–/– (mt) embryos that are common to Tbx1 DE genes (adjusted P≤0.05 are shown). (H-K) qPCR analysis confirmed that Pax9 (H), Tbx1 (I), Chd7 (J) and Gbx2 (K) were significantly reduced in Pax9–/– pharyngeal tissue at E9.5 (n=5 embryos per genotype). **P<0.01; two-tailed unpaired t-test (Tbx1 and Chd7) and two-tailed Mann–Whitney (Pax9 and Gbx2). (L-S) RNAscope in situ hybridisation showed a reduced expression of Pax9 (M), Tbx1 (O), Chd7 (Q) and Gbx2 (S) in the pharyngeal endoderm in Pax9–/– embryos at E9.5 (n=3 per genotype, 23-28 s). en, endoderm; me, mesoderm; pa, pharyngeal arch. Somite counts (s) are indicated. Scale bars: 50 µm.
To investigate a potential genetic interaction in vivo between Tbx1 and Pax9, we confirmed that both genes were co-expressed in the pharyngeal endoderm (Fig. 6A-G). We then crossed Tbx1+/– mice (Jerome and Papaioannou, 2001) with Pax9+/– mice to create Tbx1+/–;Pax9+/– double heterozygotes. Genotype analysis of 168 pups at weaning revealed that only two Tbx1+/–;Pax9+/– mice were alive, a highly significant deviation from the 42 expected of each genotype from this cross (P=1.4×10−12; Table S2). To identify the timing of lethality, neonates were observed from the day of birth with all Tbx1+/–;Pax9+/– mutants dying during the first 24 h. Examination of neonatal aortic arch arteries revealed that all controls were normal (Fig. 6H), while Tbx1+/– neonates frequently presented with A-RSA (Fig. 6I; Table 1). All dead Tbx1+/–;Pax9+/– neonates had 4th PAA-derived defects, including IAA (Fig. 6J,K; Table 1).
Fig. 6.

Tbx1;Pax9 double heterozygous embryos have 4th PAA defects. Tbx1 and Pax9 expression in the pharyngeal arches was examined at E9.5. (A-D) RNAScope in situ hybridisation expression of Tbx1 in the 2nd and 3rd pharyngeal arch (pa), endoderm (en) and mesoderm (me; A), and Pax9 expression in the endoderm only (B). The signals overlap in the endoderm (C, and higher power in C′). (D) DAPI staining. (E-G) Immunostaining for Tbx1 and Pax9 (E) demonstrates protein colocalisation within cells of the pharyngeal endoderm. Single staining for Tbx1 (F) and Pax9 (G) is shown. (H-K) Neonates from a Tbx1+/– and Pax9+/– intercross were recovered and the aortic arches examined. (H) Arch arteries were normal in all Pax9+/– neonates (n=10). (I) Tbx1+/– neonates (n=6) often presented with A-RSA, inferred by the absence of the brachiocephalic and right subclavian arteries. (J,K) All Tbx1+/–; Pax9+/– neonates (n=9) died within 24 h of birth with IAA and/or A-RSA. (L-O) 3D reconstructions of E15.5 embryo hearts from MRI datasets. (L) Pax9+/– hearts were normal (n=15). (M) Tbx1+/– hearts (n=19) frequently displayed 4th PAA defects such as A-RSA. (N,O) All Tbx1+/–; Pax9+/– embryos (n=20) presented with a 4th PAA-derived defect such as IAA-B, A-RSA and/or cervical right subclavian artery (cRSA). (P-S) Intracardiac ink injections into E10.5 embryos (27-38 s). (P) PAAs in control embryos were normal (n=10). (Q) In Tbx1+/– embryos (n=8) the 4th PAAs were often hypoplastic. (R,S) In Tbx1+/–;Pax9+/– embryos (n=9), the 4th PAAs were frequently bilaterally absent. Ao, aorta; AoA, aortic arch; AD, arterial duct; BC, brachiocephalic; LCC, left common carotid artery; LSA, left subclavian artery; LV, left ventricle; RCC, right common carotid artery; RSA, right subclavian artery; RV, right ventricle. Somite counts (s) are indicated. Scale bars: 2 mm in H-K; 500 μm in L-O; 100 μm in A-D,P-S; 50 µm in E-G. (T-W) qPCR analysis in wild-type, Tbx1+/–, Pax9+/– and Tbx1+/–;Pax9+/– embryos (n≥3 per genotype, somite range 23-27). (T) Tbx1 levels were significantly reduced in Tbx1+/– and Tbx1+/–;Pax9+/– embryos. (U) Pax9 levels were significantly reduced in Pax9+/– and Tbx1+/–;Pax9+/– embryos. There was no significant difference in Tbx1 (T), Pax9 (U), Chd7 (V) or Gbx2 (W) levels between single heterozygotes and Tbx1+/–;Pax9+/– embryos. One-way ANOVA with Tukey's multiple comparison test. ns, not significant. *P<0.05, **P<0.01, ****P<0.0001.
Having established postnatal cardiovascular defects, we next analysed E15.5 Tbx1+/–;Pax9+/– embryos by MRI (Fig. 6L-O). All wild-type and Pax9+/– embryos were normal. Of the Tbx1+/– embryos examined, a high proportion had some form of 4th PAA-derived defect, including IAA and A-RSA (Fig. 6M; Table 1). All Tbx1+/–;Pax9+/– embryos examined, however, had some form of 4th PAA-derived defect (Fig. 6N,O; Table 1) with the incidence of IAA being significantly increased compared with Tbx1+/– embryos (P<0.001). The incidence of perimembranous VSD was also significantly increased (P=0.04), but none of the embryos examined displayed outflow tract defects or bicuspid aortic valve, although an abnormal thymus was frequently observed (Fig. S4; Table S3). Tbx1+/–;Pax9+/– embryos at E10.5 were injected intracardially with ink (Fig. 6P-S) and the patency of the developing 4th PAAs was then compared between Tbx1+/– and Tbx1+/–;Pax9+/– embryos (Table 2). All Tbx1+/– embryos displayed a predominantly hypoplastic 4th PAA defect, with 50% of embryos having a unilateral, and 50% a bilateral, defect. In Tbx1+/–;Pax9+/– embryos, a significant increase in bilateral aplasia of the 4th PAAs (P=0.004) was found, but no defects of PAAs 1, 2, 3 or 6 were seen. RNA analysis showed that Tbx1, Pax9, Chd7 and Gbx2 RNA levels were not significantly changed between the respective single heterozygotes and Tbx1+/–;Pax9+/– pharyngeal arches (Fig. 6T-W).
Tbx1 and Pax9 interact cell autonomously in the pharyngeal endoderm for 4th PAA formation
The presumed site of the Tbx1-Pax9 interaction is the pharyngeal endoderm, and to demonstrate a cell-autonomous interaction in this tissue, we crossed Tbx1flox mice (Xu et al., 2004) with a novel Pax9Cre transgenic mouse (Fig. 7A-C). This Pax9Cre strain has the Cre recombinase gene inserted into the Pax9-coding region and effectively results in a Pax9-null allele. The Pax9Cre allele functions as expected by activating R26RlacZ expression specifically in the pharyngeal endoderm from E9.5 to E11.5 (Fig. 7D-G; Fig. S5), and Pax9Cre;Pax9flox neonates (i.e. Pax9-null) die at birth with the typical Pax9–/– phenotype (Fig. 7H-Q). To test the specificity of the Pax9Cre allele, the pharyngeal arches of Pax9Cre;eYFP E9.5 embryos (Fig. 8A) were dissociated into single cells and flow-sorted into eYFP-positive and -negative populations. The eYFP-positive population was significantly enriched for eYFP and Pax9 transcripts (Fig. 8B,C). Tbx1flox and Pax9Cre mice were intercrossed to create Tbx1+/ flox;Pax9Cre embryos, which are double heterozygous for each gene but only in the Pax9 expression domain. A significant reduction in Tbx1 RNA levels was observed in eYFP-positive flow-sorted pharyngeal endoderm cells from conditionally deleted E9.5 mutant embryos compared with Pax9Cre;eYFP controls (Fig. 8D). Litters from a Tbx1+/flox and Pax9Cre cross were collected on the day of birth; a significant reduction in the expected number of Tbx1+/flox;Pax9Cre mutants was found (P=2.4×10−5; Table S4) and 62.5% of these mutants displayed an arch artery defect (Fig. 8F; Table 1). We then collected embryos (E13.5-E15.5) and analysed them by MRI or µCT, which revealed that 38.5% of mutants presented with an arch artery defect (Fig. 8H; Table 1) and an abnormal thymus (Fig. S4; Table S3). We also collected embryos at E10.5 and injected them intracardially with ink to visualise the PAAs. All Tbx1+/flox control embryos analysed had normal PAAs, with all three vessels being patent to ink and of a similar diameter, while the majority of Tbx1+/flox;Pax9Cre mutant embryos presented with some form of 4th PAA defect (Fig. 8J; Table 2).
Fig. 7.

Generation of the Pax9Cre mouse. (A) A Pax9Cre targeting vector was created with a Cre cassette inserted into the SmaI restriction sites found in exons 2 and 3 of the Pax9 gene. For transformation, the vector was linearised with SfiI. (B) Correctly targeted ES cells were identified by Southern blotting on XbaI-digested genomic DNA. (C) The derived Pax9Cre-Neo mice were crossed with FLPe-expressing mice to remove the neomycin cassette and create Pax9Cre mice, which were genotyped using the indicated primers. (D-G) Pax9Cre mice were mated with R26RlacZ reporter mice and embryos were stained with X-Gal at E8.5 (n=3), E9.5 (n=7), E10.5 (n=6) and E11.5 (n=5). Positive staining in regions known to express Pax9 was observed (alb, anterior limb bud; np, nasal process; pp, pharyngeal pouch; vc, vertebral column). (H-Q) Pax9Cre and Pax9flox/flox mice were crossed to create Pax9+/flox (control) and Pax9Cre/flox (mutant) mice. All Pax9Cre/flox neonates died on the day of birth and presented with the typical Pax9-null phenotype (n=5), including a bloated abdomen (I, arrow) and pre-axial digit duplication (d) in the hind limb (K, arrow). Sections of E14.5-E15.5 embryos (n=6) showed cleft palate (CP; M), absent thymus and cardiovascular defects (O,Q). AoA, aortic arch; BC, brachiocephalic; Th, thymus. Somite counts (s) are indicated. Scale bars: 500 µm.
Fig. 8.

Conditional deletion of Tbx1 from the pharyngeal endoderm. (A) Pax9Cre drives expression of the eYFP reporter gene in the pharyngeal endoderm at E9.5. Scale bar: 500 µm. (B-D) Flow sorting and qPCR of eYFP-positive and -negative pharyngeal arch cells from Pax9Cre;eYFP control and Tbx1+/flox;Pax9Cre E9.5 embryos (n=5 of each; dissected as shown in A). From control embryos, the eYFP-positive cells (yellow circles) were significantly enriched for eYFP (B) and Pax9 (C) compared with eYFP-negative cells (black squares). (D) There was a significant reduction in Tbx1 levels in Tbx1+/flox;Pax9Cre (mutant) eYFP-positive cells (yellow squares) compared with controls (yellow circles). *P<0.05, **P<0.01 (two-tailed unpaired t-test). (E-J) Cardiovascular defects in Tbx1+/flox;Pax9Cre mice. (E,F) Neonates were collected shortly after birth and the aortic arch arteries examined. (E) Control neonates (n=39 wild-type, n=43 Pax9Cre and n=33 Tbx1+/flox) all had normal arch arteries. (F) Tbx1+/flox;Pax9Cre mutants (n=8) had cardiovascular defects such as a right-sided aorta (R-Ao) and A-RSA. Scale bars: 1 mm. (G,H) Embryos at E13.5-E15.5 were analysed by imaging (n=26 mutants). (G) Control embryo with normal cardiovascular system. (H) In the mutant there is a vascular ring formed by a R-Ao and an A-RSA. Scale bars: 500 µm. (I,J) Embryos collected at E10.5 (36-43 s) were injected intra-cardially with ink. (I) In Tbx1+/flox control embryos (n=15), PAAs 3-6 were patent to ink. (J) In Tbx1+/flox;Pax9Cre mutant embryos (n=17), the 4th PAA was frequently absent (asterisk). Somite counts (s) are indicated. Scale bars: 100 µm. Ao, aorta; AD, arterial duct; LCC, left common carotid artery; LSA, left subclavian artery; LV, left ventricle; RCC, right common carotid artery; RSA, right subclavian artery; RV, right ventricle. The somite numbers given in the legend reflect the range analysed for the whole study. The figure contains representative images only.
In summary, our data show that Pax9 is crucial for cardiovascular development, and when lost the PAAs and outflow tract fail to develop correctly, resulting in postnatal death. The 4th PAAs do not form and the 3rd PAAs collapse with reduced recruitment from vascular SMC. Our data also demonstrate that Tbx1 and associated genes are deregulated in Pax9-null embryos, and Tbx1 and Pax9 function together in the pharyngeal endoderm for 4th PAA morphogenesis.
DISCUSSION
Loss of Pax9 results in complex cardiovascular defects
Pax9 plays a key role in cardiovascular development, as Pax9–/– neonates and embryos display a range of cardiovascular defects. Gene mutations and chromosomal deletions that include PAX9 have been identified in individuals with cardiovascular abnormalities and other conditions (Table S5), suggesting that PAX9 has potential importance in human cardiovascular development. Of these, one individual with a 105 kb hemizygous deletion of 14q13 presented with IAA, a hypoplastic aorta, bicuspid aortic valve and a ventricular septal defect (Santen et al., 2012), all of which appear with high penetrance in mouse Pax9-null embryos. This small deletion removed only PAX9, NKX2-1 and NKX2-8, although deletion of either Nkx2 gene in mice does not result in any cardiovascular defects (Kimura et al., 1996; Pabst et al., 2003). It is therefore possible that hemizygous deletion of PAX9 in humans can recapitulate, in part, the mouse knockout phenotype.
Tbx1 and Pax9 interact in 4th PAA development
Published microarray studies investigating the transcriptome of Tbx1-null embryos have implicated Pax9 as a downstream target of Tbx1 (Ivins et al., 2005; Liao et al., 2008). Further evidence of crosstalk between Tbx1 and Pax9 has been suggested via the interaction of Ripply3 (Okubo et al., 2011). This protein, which is a Tbx1 repressor, has been shown to prevent Tbx1 activation of luciferase from a Pax9 promoter construct, and Pax9 expression in the pharyngeal endoderm is increased in Ripply3-null embryos suggesting that Tbx1 may be upstream of Pax9. Ripply3 levels were not significantly changed in our Pax9-null pharyngeal arch transcriptome data, but Tbx1 levels were significantly reduced, indicating that perhaps Pax9 and Tbx1 may function together rather than work in a hierarchal pathway. This is further supported by analysis of Tbx1 and Pax9 RNA levels in Tbx1+/–;Pax9+/– embryos, which were not significantly changed compared with levels seen in the single heterozygotes. Further analysis identified that many genes differentially expressed in Tbx1-null embryos, or shown to interact with Tbx1 in mouse models, were also significantly differentially expressed in Pax9-null embryos, e.g. Chd7 and Gbx2. Mice heterozygous for Chd7 have abnormal 4th PAA derivatives and a combined heterozygosity with Tbx1 results in an increased incidence of 4th PAA defects (Randall et al., 2009). Gbx2-null embryos display cardiovascular defects (Byrd and Meyers, 2005), and Tbx1 and Gbx2 have also been shown to genetically interact in PAA development (Calmont et al., 2009). Although both these Tbx1 targets have been demonstrated to interact in the pharyngeal ectoderm, they are also expressed in the pharyngeal endoderm and could therefore interact with Pax9 either directly or indirectly in this tissue. As our data strongly suggested that Pax9 may interact with Tbx1 and its related genes in the morphogenesis of the PAAs, we therefore looked for a genetic interaction between Tbx1 and Pax9 in mice. We found that all mice examined (embryos and neonates) double heterozygous for each gene (i.e. Tbx1+/–;Pax9+/–) displayed some defect associated with abnormal development of the 4th PAA, most notably IAA-B. Although hypoplasia or absence of the 4th PAAs is a feature in mouse embryos heterozygous for Tbx1 (Lindsay et al., 2001), this phenotype was significantly more prevalent in Tbx1+/–;Pax9+/– mice, and much higher than observed in Tbx1+/– embryos. It therefore appears that combining one Pax9-null allele with one Tbx1-null allele exacerbates the Tbx1+/– phenotype.
We used our novel Pax9Cre mouse to conditionally delete Tbx1 from the pharyngeal endoderm, while simultaneously creating double haploinsufficiency for the two genes in this tissue. A highly significant reduction in the number of Tbx1+/flox;Pax9Cre neonates born was observed, indicating that heterozygous loss of Tbx1 and Pax9 from the pharyngeal endoderm has a major impact on postnatal survival, with almost two-thirds of the recovered neonates examined having identifiable cardiovascular defects. A similar proportion of affected mutants was found at foetal stages. A vascular ring structure was observed in Tbx1+/flox;Pax9Cre mutants (Fig. 8H), similar to that described in Lgdel/+ mice, which are chromosomally engineered to be hemizygous for Tbx1 (Merscher et al., 2001), and also in Six1;Eya1 mutant embryos (Guo et al., 2011a). Eya1 was significantly reduced in Pax9-null pharyngeal arch tissue. When embryos were examined at mid-embryogenesis, however, we found that almost all displayed 4th PAA defects. When compared with our Tbx1+/–;Pax9+/– embryo data it was apparent that there was no significant difference in the incidence of bilateral 4th PAA defects between Tbx1+/flox;Pax9Cre and Tbx1+/–;Pax9+/– embryos. This indicates that the conditional heterozygous deletion of Tbx1 from the pharyngeal endoderm, in the context of Pax9 heterozygosity, has the same effect on 4th PAA morphogenesis as seen in the global Tbx1+/–;Pax9+/– embryos at E10.5. Although the ink injection data from Tbx1+/flox;Pax9Cre embryos matches that seen in Tbx1+/–;Pax9+/– embryos, and shows that heterozygous loss of Tbx1 from the pharyngeal endoderm severely affects PAA morphogenesis, there is a discrepancy in the cardiovascular phenotype observed at foetal and neonatal stages that does not reflect that seen at mid-embryogenesis, nor that observed in Tbx1+/–;Pax9+/– embryos. It is well documented, however, that the Tbx1+/– 4th PAA phenotype does recover during development (see Table S6). It therefore appears that a conditional deletion of Tbx1 from the pharyngeal endoderm in the context of Pax9 haploinsufficiency replicates the Tbx1 heterozygous arch artery phenotype in terms of a highly penetrant 4th PAA defect at mid-embryogenesis that partially recovers by the foetal stage. Our data, however, do raise the question of whether we are seeing the effect of a pharyngeal endoderm-specific loss of Tbx1 with Pax9Cre or a genetic interaction occurring between Tbx1 and Pax9 in the pharyngeal endoderm? Conditional deletion experiments have been performed in transgenic mice to identify the role of Tbx1 in specific tissues and have examined the effect of Tbx1 heterozygosity in the pharyngeal mesoderm and/or epithelium for 4th PAA development (see Table S7). When compared, our data clearly show that the 4th PAA phenotype is much more penetrant than that seen in embryos only heterozygous for Tbx1 in the pharyngeal epithelia. We therefore conclude that we are showing a strong genetic and cell-autonomous interaction between Tbx1 and Pax9 within the pharyngeal endoderm that impacts on 4th PAA morphogenesis. Moreover, and in support of a phenotype extending beyond what is expected from a Tbx1 heterozygous phenotype, our Tbx1flox;Pax9Cre mice display a high incidence of perinatal lethality, a feature not previously described in Tbx1+/– mice. Interestingly, the homozygous deletion of Tbx1 from the pharyngeal endoderm using Sox17Cre mice resulted in pharyngeal arch aplasia but not in a common arterial trunk, although IAA was observed (Jackson et al., 2014). This phenotype is similar to that seen in our Tbx1;Pax9 mutants but is distinct from either Tbx1 and Pax9 constitutive null mutants.
Pax9 signalling from the pharyngeal endoderm is required for 3rd PAA maintenance
The mammalian PAAs develop symmetrically in a cranial to caudal sequence, and then subsequently remodel to form the typical asymmetric aortic arch artery system seen in the adult (Bamforth et al., 2013; Hiruma et al., 2002). In normal PAA development, the 1st artery forms first, followed by the 2nd, but these two vessels rapidly remodel and contribute to capillary beds and the mandibular and hyoid arteries. In some Pax9–/– embryos, these vessels aberrantly persisted into E10.5 and E11.5, and may contribute to the anomalous arrangement of the aortic arch arteries when the 3rd PAAs collapse. In normal development, the 3rd PAAs will form part of the common and proximal internal carotid arteries, which elongate as the embryo grows. The carotid duct involutes and the dorsal aorta anterior to this segment will form the distal internal carotid artery, with the external carotid artery derived from the proximal parts of the 1st and 2nd PAA (Hiruma et al., 2002). The persistence of the 1st and/or 2nd PAAs in Pax9–/– embryos, coupled with the failure of the 3rd PAAs to be maintained and the carotid duct to involute, results in the external carotid arteries rising directly from the aorta, and the internal carotid arteries from the anterior dorsal aortae. This phenomenon has been described clinically (Roberts and Gerald, 1978). Although reduced numbers of NCCs were found in the caudal pharyngeal arches at E10.5, migration, apoptosis or proliferation of NCCs did not appear to be impaired in Pax9-null embryos. It is well recognised that NCCs differentiate into the SMCs that are important for the stabilisation of the remodelled PAAs (Hutson and Kirby, 2003) and it is likely that the failure of SMCs to envelop the 3rd PAAs in Pax9-null embryos results in the collapse of this vessel. This has also been described in Hoxa3-null embryos (Kameda, 2009) and reduced NCC migration in genetic mouse and surgical chick models also leads to defects of the 3rd PAAs (Bradshaw et al., 2009; Epstein et al., 2000; Franz, 1989; Nishibatake et al., 1987). We were unable to look for a similar occurrence in the 4th PAAs, as this fails to form in the majority of Pax9-null embryos. Signalling between the pharyngeal endoderm and NCC does occur (Graham et al., 2005) so it is therefore possible that signals emanating from the pharyngeal endoderm under Pax9 control are necessary to influence the differentiation of NCCs into SMCs; without these signals, the 3rd PAAs are not supported during the remodelling phase of arch artery development and they collapse. The top two GSEA pathways identified from the RNA-seq data were ‘extracellular matrix organisation’ and ‘axon guidance’, and these could be relevant for a NCC-derived phenotype, particularly as the extracellular matrix is known to be crucial for the migration of NCCs (Henderson and Copp, 1997; Perris and Perissinotto, 2000) and the slit, ephrin, semaphorin and plexin genes listed under ‘axon guidance’ are usually associated with NCC expression or migration in cardiovascular development (Epstein et al., 2015; Kirby and Hutson, 2010; Sun et al., 2018). There is, however, an alternative explanation for the collapse of the 3rd PAAs. Haemodynamic force plays a major role in artery development (Culver and Dickinson, 2010; Yashiro et al., 2007), so it is possible that the 3rd PAA defect is a consequence of increased blood flow to the persistent 1st/2nd PAAs, which prevents sufficient blood flow to the 3rd PAAs, resulting in impairment of flow-induced arteriogenesis. This may also be an explanation for the failure of the 4th PAAs to form. A reduced haemodynamic force would also explain the reduction in SMC recruitment as this is also dependent on blood flow (Padget et al., 2019). Although endothelial cells were present within the 4th pharyngeal arch of Pax9-null embryos, they apparently failed to condense into tubes, a feature that has also been described in both Tbx1-null and Gbx2-null embryos (Byrd and Meyers, 2005; Calmont et al., 2009).
The double outlet right ventricle phenotype observed at E15.5 in Pax9-null embryos probably arises from the marked outflow tract rotation defect due to a change in the aortic to pulmonary valve axis angle (Bostrom and Hutchins, 1988; Lomonico et al., 1986), which is also observed in Pax3 and Pitx2c mutant hearts (Bajolle et al., 2006).
In summary, Pax9 expression in the pharyngeal endoderm has regional (anterior/posterior) roles in regulating the morphogenesis of the PAAs (Fig. 9). In the 3rd pharyngeal arch, Pax9 is important for controlling NCC differentiation into the SMCs that support the remodelling vessel, whereas in the 4th pharyngeal arch endoderm, the interaction of Pax9 with Tbx1 and its targets is crucial for the formation of the 4th PAAs.
Fig. 9.
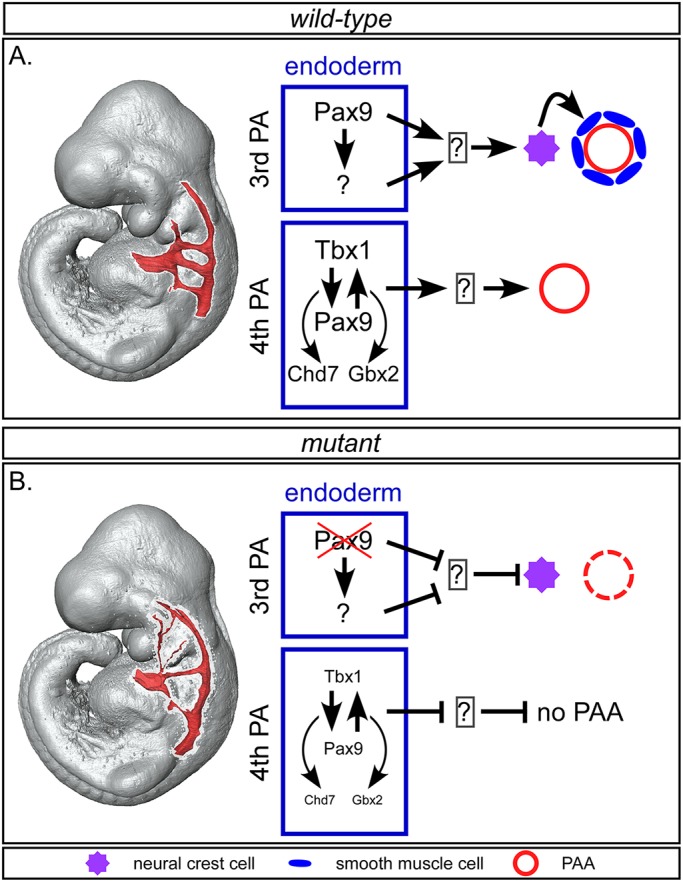
Proposed model for role of Pax9-mediated signalling during pharyngeal arch artery development. (A) In wild-type embryos, Pax9 expression in the 3rd pharyngeal arch endoderm induces neural crest cells (NCCs), either directly or indirectly, to differentiate into the SMCs that form around the endothelium of the remodelling 3rd PAAs. In the 4th arch endoderm, Tbx1 and Pax9 function together to, most likely indirectly, control formation of the 4th PAAs, potentially through collaboration with Chd7 and Gbx2. (B) In Pax9-deficient embryos, signals from the 3rd pharyngeal arch endoderm are compromised and NCC differentiation into SMCs is impaired, resulting in the collapse of the 3rd PAAs by E11.5. In Tbx1-Pax9 double heterozygous embryos, reduced levels of Tbx1 and Pax9 in the 4th pharyngeal arch endoderm are insufficient to allow the 4th PAAs to form.
MATERIALS AND METHODS
Mice
The mice used in this study have previously been described: Pax9+/– (Peters et al., 1998), Pax9flox (Kist et al., 2007), Tbx+/– (Jerome and Papaioannou, 2001), Tbx1flox (Xu et al., 2004), Wnt1Cre (Danielian et al., 1998), R26ReYFP (Srinivas et al., 2001), R26RlacZ (Soriano, 1999) and FLPe (Dymecki, 1996). All mice were maintained on a C57Bl/6J genetic background. All studies involving animals were performed in accordance with UK Home Office Animals (Scientific Procedures) Act 1986.
Generation of Pax9Cre mice
The Pax9Cre targeting construct was designed to replace a 1.06 kb section of the Pax9 gene, spanning the second half of exon two, containing the start codon, and the first half of the 3rd exon, containing the paired box and octapeptide motif, with a promoterless Cre recombinase gene preceded by a consensus Kozak sequence, in the pPNT4 targeting vector (Conrad et al., 2003), using standard molecular biology methods (Fig. 7A). The 1.3 kb right homology arm, generated as previously described (Kist et al., 2005), was inserted between the FRT-flanked PGK-Neomycin cassette and the HSV-Thymidine Kinase cassette of pPNT4. The unwanted loxP site of pPNT4 was removed by XbaI partial digestion, followed by Klenow treatment and re-ligation. The left homology arm was generated as a 7.4 kb SalI to SmaI fragment of the Pax9 genomic region that included exon 1 and the first half of the second exon, and was inserted 5′ of the Cre recombinase gene. The targeting vector was linearized with SfiI and mouse ES cells were targeted and selected as previously described (Kearns et al., 2013). Southern blot analysis was performed according to standard methods following XbaI digest of genomic DNA. An external southern probe was used to verify integration of the targeting cassette in the 5′ region (Fig. 7B) and PCR was used to validate integration of the 3′ region (not shown). The FRT-flanked PGK-neomycin cassette was removed by crossing the derived Pax9Cre-Neo mice with FLPe mice (Dymecki, 1996) to create Pax9Cre mice, which were then backcrossed to strain C57Bl/6J for over six generations. Genotyping by multiplex PCR [primers: F1-ACTCAAGCCTCTTTCAGCCC (common), R1-TTGTTCTCACTGAGCCGGCCTGT (Pax9 exon 2) and R2-GTTGCATCGACCGGTAATGC (Cre)] were used to simultaneously identify the wild-type and Pax9Cre alleles (Fig. 7C).
Breeding
Male and female mice were mated and the detection of a vaginal plug the next morning considered to be E0.5. Pregnant females were culled on the required day and embryos collected. Embryos at E9.5-E11.5 were staged by somite counting.
Imaging
Magnetic resonance imaging (MRI) was performed using a 9.4T MR system (Varian, US) as previously described (Bamforth et al., 2012; Schneider et al., 2004) on embryos at E15.5. High Resolution Episcopic Microscopy (HREM) and micro-computed tomography (µCT) techniques have been described in detail elsewhere (Degenhardt et al., 2010; Geyer et al., 2009). All MRI, HREM and µCT images were converted into a volume dataset and segmented using Amira software (Thermo Fisher Scientific) to create 2- and 3-dimensional (3D) images. Structures were manually outlined using the label field function of Amira and surface rendered to produce the 3D images. Intra-cardiac ink injections were performed as described previously (Calmont et al., 2009). Aorta measurements were taken from the region just proximal to the point at which the ascending aorta becomes the aortic arch. Haematoxylin and Eosin staining, X-gal staining and Pax9 whole-mount in situ hybridisation were performed using standard techniques. mRNA expression on sections was examined by in situ hybridisation using RNAscope Multiplex Fluorescent v2 Assay (Advanced Cell Diagnostics) following the manufacturer's instructions. Probe details given in Table S8. For proliferation assays, BrdU (Sigma-Aldrich) dissolved in PBS was administered by intraperitoneal injection at a dose of 50 mg/kg to pregnant dams 1 h before embryo collection. Immunohistochemistry was performed using standard techniques (antibody details are in Table S8) on paraformaldehyde-fixed embryo sections and fluorescently imaged on an Axioimager (Zeiss). To assess an apoptotic index within the pharyngeal arches, control and Pax9–/– embryos at E9.5 (n=3 per genotype; 24-28 somites) and E10.5 (n=3 per genotype; 30-35 somites) were examined following immunostaining with the anti-caspase 3 antibody. To assess a proliferative index within the NCC in the pharyngeal arches, control and Pax9–/– embryos expressing Wnt1Cre activated eYFP at E9.5 (n=3 per genotype; 23-28 somites) and E10.5 (n≥4 for each genotype; 31-39 somites) were examined following immunostaining using anti-BrdU and anti-GFP antibodies. All positively stained and DAPI-positive cells within the pharyngeal arches were counted and the ratio of positively stained cells over the total number of cells calculated. The average number of NCCs within the caudal pharyngeal arches was calculated at E9.5 (n=3 per genotype, 23-28 somites) and E10.5 (n=4 per genotype, 32-34 somites). For all cell counting experiments, at least three sections per embryo were assessed.
Transcriptome analysis
Wild-type (Pax9+/+) and Pax9-null (Pax9−/−) embryos at E9.5 (n=3 of each genotype, stage-matched for 26-27 somites) were collected in ice-cold DEPC-PBS and the pharyngeal region, from the junction between the first and second pharyngeal arch to the base of the heart, was dissected free in RNAlater (Sigma-Aldrich). Genotypes were identified by PCR from the embryo tails and/or yolk sac material. Total RNA was extracted using the QIAGEN Plus Micro kit with genomic DNA Eliminator columns, and total RNA eluted in 30 μl RNase-free water. RNA concentration and purity were assessed by NanoDrop spectrophotometry and Agilent Bioanalyzer with all samples producing a RIN of 10. Genomic DNA contamination was excluded by PCR for intronic regions (sensitivity 5 pg/μl). To confirm that the correct genotypes were processed, cDNA was made from 100 ng total RNA and a qPCR for Pax9 performed (Fig. 5B). The total RNA samples were shipped on dry-ice to EMBLM-GeneCore for RNA-seq first-strand specific library preparation using Illumina TruSeq, and 100 bp paired end reads were generated from an Illumina HiSeq 2000 Sequencer. Quality control by FastQC suggested a small amount of adaptor contamination in each sample, so raw reads were trimmed of adaptor sequence with Trimmomatic (Bolger et al., 2014). Reads shorter than 36 bp after trimming were discarded, and reads whose pair had been discarded were also removed. Trimmed reads were further filtered to keep only uniquely aligned and non-redundant reads to remove a vast majority of pseudogenes caused by multi-mapping. In addition, to avoid bias due to imbalanced library sizes among conditions, all samples were sub-sampled towards the smallest sample (mt-663_1). The resulting reads were aligned to the Mouse Genome (Ensembl release 81, GRCm38) using the splice aware read aligner STAR 2 (star-2.5.2a-0, 20/01/2017) (Dobin et al., 2013). Finally, aligned reads were assigned to Ensembl genes using featureCounts (Liao et al., 2014) with default parameters. These count tables were imported into R and analysed using the package LIMMA (Ritchie et al., 2015) and voom function from Bioconductor (Gentleman et al., 2004). A gene set enrichment analysis tool (Subramanian et al., 2005) was used to predict signalling pathways that were affected when gene expression in the wild-type and the Pax9 mutant samples was compared. All resulting pathways were filtered for a familywise-error rate (P<0.001) to correct for multiple hypotheses testing.
Flow cytometry
The pharyngeal arch region of E9.5 Pax9Cre;eYFP control (n=5; 23-27 somites) and mutant Tbx1+/flox;Pax9Cre;eYFP (n=5; 24-29 somites) embryos was dissected free as described above and dissociated to single cells with Accumax (eBioscience) by incubating at 37°C for 30 min. The reaction was stopped by the addition of 10% foetal calf serum (FCS), and cells were washed in PBS and resuspended in 10% FCS. Cells were stained with propidium iodide. Fluorescence-activated cell sorting was performed on a Becton Dickinson FACS Aria II using a 100 µm nozzle and a sheath pressure of 20 psi. Single cells were gated using FSC-A versus SSC-A followed by FSC-A versus FSC-H and FSC-A versus SSC-W to remove any doublets. Live single cells were gated using propidium iodide versus FSC-A, and this population was finally gated on eYFP-positive and -negative cells and sorted into collection tubes.
Quantitative real-time RT-PCR (qPCR)
RNA, from either dissociated pharyngeal arch cells or flow-sorted cells, was extracted using Trizol reagent (Invitrogen) combined with a Purelink RNA Mini Kit (Ambion) and on-column DNase1 treatment. RNA was eluted with 30 µl RNase-free water. Total RNA was converted to cDNA using a High-Capacity cDNA Reverse transcription kit (Applied Biosystems) and random hexamers. qPCR was performed using SYBR Green JumpStart Taq ReadyMix (Sigma-Aldrich) using the primers listed in Table S9. All qPCR reactions were performed in triplicate on a QuantStudio 7 Real-Time PCR System (Thermo Fisher Scientific). Data were analysed using the comparative Ct method (Schmittgen and Livak, 2008).
Statistical analysis
A chi-squared test was used to compare genotype frequencies of litters, and Pearson's chi-squared test for associations was used to compare defect frequencies between the different genotypes. For analysis of qPCR data, aorta measurements and cell counts, datasets were tested for variance using the Shapiro-Wilk test (Prism 8.01 software, GraphPad). Two-tailed unpaired t-tests were performed on normally distributed data, and a two-tailed Mann–Whitney U non-parametric test was used for not normally distributed data. One-way ANOVA with Tukey's multiple comparison test was used to analyse qPCR data from four embryo genotypes. A hypergeometric distribution test was used to calculate a P-value for observing an overlap between two gene lists in R. Groups were considered significantly different when P<0.05 or P≤0.1 for RNA-seq data. No statistical methods were used to predetermine sample size, which were chosen based on previous experience to obtain statistical significance and reproducibility. No data points were excluded and all data collected from each individual experiment were used for analysis.
Supplementary Material
Acknowledgements
We thank Jessica Addison, Kathleen Allinson and Divya Venkatesh for technical assistance, Sushma Grellscheid for facilitating the RNA-seq experiment, and Nicoletta Bobola for critically reading the manuscript. Tbx1+/– mice were obtained from Robert Kelly and Virginia Papaioannou. We acknowledge the Newcastle University Flow Cytometry Core Facility (FCCF) for assistance with the generation of flow cytometry data.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: H.M.P., S.D.B.; Methodology: K.S., T.B., M.N., R.M., R.K., H.P., S.D.B.; Validation: S.D.B.; Formal analysis: A.I.K., S.J.C., S.D.B.; Investigation: C.A.S., W.M.S.Q., J.A.B.-L., R.R.K., C.O., R.S., S.M., R.D., T.B., J.E.S., T.J.M., R.K., S.D.B.; Resources: H.M.P., K.S., T.B., M.N., J.E.S., T.J.M., R.M., R.K., H.P., S.D.B.; Writing - original draft: S.D.B.; Writing - review & editing: H.M.P., R.K., H.P.; Visualization: A.I.K., S.D.B.; Supervision: H.M.P., R.K., H.P., S.D.B.; Project administration: H.M.P., S.D.B.; Funding acquisition: S.D.B.
Funding
This work was funded by a British Heart Foundation Intermediate Basic Science Research Fellowship (FS/08/016/24741 to S.D.B.), a British Heart Foundation project grant (PG/16/39/32115 to S.D.B.), a British Heart Foundation PhD Non-Clinical PhD Studentship (FS/16/8/31984 to S.D.B.) and a grant from the Newcastle upon Tyne Hospitals NHS Foundation Trust (BH120404 to S.D.B.). J.A.B.-L. received a PhD Scholarship from the Consejo Nacional de Ciencia y Tecnología (CONACyT). R.R.K. received a PhD Scholarship from Yarmouk University, Jordan. Deposited in PMC for immediate release.
Data availability
The RNA-seq data have been deposited in GEO under accession number GSE128087.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.177618.supplemental
References
- Aggarwal V. S. and Morrow B. E. (2008). Genetic modifiers of the physical malformations in velo-cardio-facial syndrome/DiGeorge syndrome. Dev. Disabil. Res. Rev. 14, 19-25. 10.1002/ddrr.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajolle F., Zaffran S., Kelly R. G., Hadchouel J., Bonnet D., Brown N. A. and Buckingham M. E. (2006). Rotation of the myocardial wall of the outflow tract is implicated in the normal positioning of the great arteries. Circ. Res. 98, 421-428. 10.1161/01.RES.0000202800.85341.6e [DOI] [PubMed] [Google Scholar]
- Bamforth S. D., Schneider J. E. and Bhattacharya S. (2012). High-throughput analysis of mouse embryos by magnetic resonance imaging. Cold Spring Harb. Protoc. 2012, 93-101. 10.1101/pdb.prot067538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamforth S. D., Chaudhry B., Bennett M., Wilson R., Mohun T. J., Van Mierop L. H. S., Henderson D. J. and Anderson R. H. (2013). Clarification of the identity of the mammalian fifth pharyngeal arch artery. Clin. Anat. 26, 173-182. 10.1002/ca.22101 [DOI] [PubMed] [Google Scholar]
- Bolger A. M., Lohse M. and Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114-2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostrom M. P. and Hutchins G. M. (1988). Arrested rotation of the outflow tract may explain double-outlet right ventricle. Circulation 77, 1258-1265. 10.1161/01.CIR.77.6.1258 [DOI] [PubMed] [Google Scholar]
- Boudjemline Y., Fermont L., Le Bidois J., Lyonnet S., Sidi D. and Bonnet D. (2001). Prevalence of 22q11 deletion in fetuses with conotruncal cardiac defects: a 6-year prospective study. J. Pediatr. 138, 520-524. 10.1067/mpd.2001.112174 [DOI] [PubMed] [Google Scholar]
- Bradshaw L., Chaudhry B., Hildreth V., Webb S. and Henderson D. J. (2009). Dual role for neural crest cells during outflow tract septation in the neural crest-deficient mutant Splotch(2H). J. Anat. 214, 245-257. 10.1111/j.1469-7580.2008.01028.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd N. A. and Meyers E. N. (2005). Loss of Gbx2 results in neural crest cell patterning and pharyngeal arch artery defects in the mouse embryo. Dev. Biol. 284, 233-245. 10.1016/j.ydbio.2005.05.023 [DOI] [PubMed] [Google Scholar]
- Calmont A., Ivins S., Van Bueren K. L., Papangeli I., Kyriakopoulou V., Andrews W. D., Martin J. F., Moon A. M., Illingworth E. A., Basson M. A. et al. (2009). Tbx1 controls cardiac neural crest cell migration during arch artery development by regulating Gbx2 expression in the pharyngeal ectoderm. Development 136, 3173-3183. 10.1242/dev.028902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi N. and Epstein J. A. (2002). Getting your Pax straight: Pax proteins in development and disease. Trends Genet. 18, 41-47. 10.1016/S0168-9525(01)02594-X [DOI] [PubMed] [Google Scholar]
- Conrad M., Brielmeier M., Wurst W. and Bornkamm G. W. (2003). Optimized vector for conditional gene targeting in mouse embryonic stem cells. BioTechniques 34, 1136-1138, 1140 10.2144/03346bm03 [DOI] [PubMed] [Google Scholar]
- Culver J. C. and Dickinson M. E. (2010). The effects of hemodynamic force on embryonic development. Microcirculation 17, 164-178. 10.1111/j.1549-8719.2010.00025.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielian P. S., Muccino D., Rowitch D. H., Michael S. K. and McMahon A. P. (1998). Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr. Biol. 8, 1323-1326. 10.1016/S0960-9822(07)00562-3 [DOI] [PubMed] [Google Scholar]
- Degenhardt K., Wright A. C., Horng D., Padmanabhan A. and Epstein J. A. (2010). Rapid 3D phenotyping of cardiovascular development in mouse embryos by micro-CT with iodine staining. Circ. Cardiovasc. Imaging 3, 314-322. 10.1161/CIRCIMAGING.109.918482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M. and Gingeras T. R. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15-21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dymecki S. M. (1996). Flp recombinase promotes site-specific DNA recombination in embryonic stem cells and transgenic mice. Proc. Natl. Acad. Sci. USA 93, 6191-6196. 10.1073/pnas.93.12.6191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein J. A., Li J., Lang D., Chen F., Brown C. B., Jin F., Lu M. M., Thomas M., Liu E., Wessels A. et al. (2000). Migration of cardiac neural crest cells in Splotch embryos. Development 127, 1869-1878. [DOI] [PubMed] [Google Scholar]
- Epstein J. A., Aghajanian H. and Singh M. K. (2015). Semaphorin signaling in cardiovascular development. Cell Metab. 21, 163-173. 10.1016/j.cmet.2014.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz T. (1989). Persistent truncus arteriosus in the Splotch mutant mouse. Anat. Embryol. 180, 457-464. 10.1007/BF00305120 [DOI] [PubMed] [Google Scholar]
- Gentleman R. C., Carey V. J., Bates D. M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J. et al. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80 10.1186/gb-2004-5-10-r80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer S. H., Mohun T. J. and Weninger W. J. (2009). Visualizing vertebrate embryos with episcopic 3D imaging techniques. Sci. World J. 9, 1423-1437. 10.1100/tsw.2009.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham A. (2003). Development of the pharyngeal arches. Am. J. Med. Genet. A 119A, 251-256. 10.1002/ajmg.a.10980 [DOI] [PubMed] [Google Scholar]
- Graham A., Okabe M. and Quinlan R. (2005). The role of the endoderm in the development and evolution of the pharyngeal arches. J. Anat. 207, 479-487. 10.1111/j.1469-7580.2005.00472.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C., Sun Y., Zhou B., Adam R. M., Li X. K., Pu W. T., Morrow B. E., Moon A. and Li X. (2011a). A Tbx1-Six1/Eya1-Fgf8 genetic pathway controls mammalian cardiovascular and craniofacial morphogenesis. J. Clin. Invest. 121, 1585-1595. 10.1172/JCI44630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T., McDonald-McGinn D., Blonska A., Shanske A., Bassett A. S., Chow E., Bowser M., Sheridan M., Beemer F., Devriendt K. et al. (2011b). Genotype and cardiovascular phenotype correlations with TBX1 in 1,022 velo-cardio-facial/DiGeorge/22q11.2 deletion syndrome patients. Hum. Mutat. 32, 1278-1289. 10.1002/humu.21568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson D. J. and Copp A. J. (1997). Role of the extracellular matrix in neural crest cell migration. J. Anat. 191, 507-515. 10.1046/j.1469-7580.1997.19140507.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiruma T., Nakajima Y. and Nakamura H. (2002). Development of pharyngeal arch arteries in early mouse embryo. J. Anat. 201, 15-29. 10.1046/j.1469-7580.2002.00071.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutson M. R. and Kirby M. L. (2003). Neural crest and cardiovascular development: a 20-year perspective. Birth Defects Res. C Embryo Today 69, 2-13. 10.1002/bdrc.10002 [DOI] [PubMed] [Google Scholar]
- Ivins S., Lammerts van Beuren K., Roberts C., James C., Lindsay E., Baldini A., Ataliotis P. and Scambler P. J. (2005). Microarray analysis detects differentially expressed genes in the pharyngeal region of mice lacking Tbx1. Dev. Biol. 285, 554-569. 10.1016/j.ydbio.2005.06.026 [DOI] [PubMed] [Google Scholar]
- Jackson A., Kasah S., Mansour S. L., Morrow B. and Basson M. A. (2014). Endoderm-specific deletion of Tbx1 reveals an FGF-independent role for Tbx1 in pharyngeal apparatus morphogenesis. Dev. Dyn. 243, 1143-1151. 10.1002/dvdy.24147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerome L. A. and Papaioannou V. E. (2001). DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat. Genet. 27, 286-291. 10.1038/85845 [DOI] [PubMed] [Google Scholar]
- Kameda Y. (2009). Hoxa3 and signaling molecules involved in aortic arch patterning and remodeling. Cell Tissue Res. 336, 165-178. 10.1007/s00441-009-0760-7 [DOI] [PubMed] [Google Scholar]
- Kearns N. A., Genga R. M. J., Ziller M., Kapinas K., Peters H., Brehm M. A., Meissner A. and Maehr R. (2013). Generation of organized anterior foregut epithelia from pluripotent stem cells using small molecules. Stem Cell Res. 11, 1003-1012. 10.1016/j.scr.2013.06.007 [DOI] [PubMed] [Google Scholar]
- Kimura S., Hara Y., Pineau T., Fernandez-Salguero P., Fox C. H., Ward J. M. and Gonzalez F. J. (1996). The T/ebp null mouse: thyroid-specific enhancer-binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev. 10, 60-69. 10.1101/gad.10.1.60 [DOI] [PubMed] [Google Scholar]
- Kirby M. L. and Hutson M. R. (2010). Factors controlling cardiac neural crest cell migration. Cell Adh. Migr. 4, 609-621. 10.4161/cam.4.4.13489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kist R., Watson M., Wang X., Cairns P., Miles C., Reid D. J. and Peters H. (2005). Reduction of Pax9 gene dosage in an allelic series of mouse mutants causes hypodontia and oligodontia. Hum. Mol. Genet. 14, 3605-3617. 10.1093/hmg/ddi388 [DOI] [PubMed] [Google Scholar]
- Kist R., Greally E. and Peters H. (2007). Derivation of a mouse model for conditional inactivation of Pax9. Genesis 45, 460-464. 10.1002/dvg.20295 [DOI] [PubMed] [Google Scholar]
- Lewin M. B., Lindsay E. A., Jurecic V., Goytia V., Towbin J. A. and Baldini A. (1997). A genetic etiology for interruption of the aortic arch type B. Am. J. Cardiol. 80, 493-497. 10.1016/S0002-9149(97)00401-3 [DOI] [PubMed] [Google Scholar]
- Liao J., Aggarwal V. S., Nowotschin S., Bondarev A., Lipner S. and Morrow B. E. (2008). Identification of downstream genetic pathways of Tbx1 in the second heart field. Dev. Biol. 316, 524-537. 10.1016/j.ydbio.2008.01.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G. K. and Shi W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923-930. 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- Lindsay E. A., Vitelli F., Su H., Morishima M., Huynh T., Pramparo T., Jurecic V., Ogunrinu G., Sutherland H. F., Scambler P. J. et al. (2001). Tbx1 haploinsufficiency in the DiGeorge syndrome region causes aortic arch defects in mice. Nature 410, 97-101. 10.1038/35065105 [DOI] [PubMed] [Google Scholar]
- Lomonico M. P., Moore G. W. and Hutchins G. M. (1986). Rotation of the junction of the outflow tract and great arteries in the embryonic human heart. Anat. Rec. 216, 544-549. 10.1002/ar.1092160412 [DOI] [PubMed] [Google Scholar]
- Mansouri A., Hallonet M. and Gruss P. (1996). Pax genes and their roles in cell differentiation and development. Curr. Opin. Cell Biol. 8, 851-857. 10.1016/S0955-0674(96)80087-1 [DOI] [PubMed] [Google Scholar]
- Merscher S., Funke B., Epstein J. A., Heyer J., Puech A., Lu M. M., Xavier R. J., Demay M. B., Russell R. G., Factor S. et al. (2001). TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell 104, 619-629. 10.1016/S0092-8674(01)00247-1 [DOI] [PubMed] [Google Scholar]
- Momma K. (2010). Cardiovascular anomalies associated with chromosome 22q11.2 deletion syndrome. Am. J. Cardiol. 105, 1617-1624. 10.1016/j.amjcard.2010.01.333 [DOI] [PubMed] [Google Scholar]
- Morrow B. E., McDonald-McGinn D. M., Emanuel B. S., Vermeesch J. R. and Scambler P. J. (2018). Molecular genetics of 22q11.2 deletion syndrome. Am. J. Med. Genet. A 176, 2070-2081. 10.1002/ajmg.a.40504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubüser A., Koseki H. and Balling R. (1995). Characterization and developmental expression of Pax9, a paired-box-containing gene related to Pax1. Dev. Biol. 170, 701-716. 10.1006/dbio.1995.1248 [DOI] [PubMed] [Google Scholar]
- Nishibatake M., Kirby M. L. and Van Mierop L. H. (1987). Pathogenesis of persistent truncus arteriosus and dextroposed aorta in the chick embryo after neural crest ablation. Circulation 75, 255-264. 10.1161/01.CIR.75.1.255 [DOI] [PubMed] [Google Scholar]
- Okubo T., Kawamura A., Takahashi J., Yagi H., Morishima M., Matsuoka R. and Takada S. (2011). Ripply3, a Tbx1 repressor, is required for development of the pharyngeal apparatus and its derivatives in mice. Development 138, 339-348. 10.1242/dev.054056 [DOI] [PubMed] [Google Scholar]
- Pabst O., Rummelies J., Winter B. and Arnold H.-H. (2003). Targeted disruption of the homeobox gene Nkx2.9 reveals a role in development of the spinal accessory nerve. Development 130, 1193-1202. 10.1242/dev.00346 [DOI] [PubMed] [Google Scholar]
- Padget R. L., Mohite S. S., Hoog T. G., Justis B. S., Green B. E. and Udan R. S. (2019). Hemodynamic force is required for vascular smooth muscle cell recruitment to blood vessels during mouse embryonic development. Mech. Dev. 156, 8-19. 10.1016/j.mod.2019.02.002 [DOI] [PubMed] [Google Scholar]
- Papangeli I. and Scambler P. (2013). The 22q11 deletion: DiGeorge and velocardiofacial syndromes and the role of TBX1. Wiley Interdiscip. Rev. Dev. Biol. 2, 393-403. 10.1002/wdev.75 [DOI] [PubMed] [Google Scholar]
- Perris R. and Perissinotto D. (2000). Role of the extracellular matrix during neural crest cell migration. Mech. Dev. 95, 3-21. 10.1016/S0925-4773(00)00365-8 [DOI] [PubMed] [Google Scholar]
- Peters H., Neubuser A., Kratochwil K. and Balling R. (1998). Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev. 12, 2735-2747. 10.1101/gad.12.17.2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall V., McCue K., Roberts C., Kyriakopoulou V., Beddow S., Barrett A. N., Vitelli F., Prescott K., Shaw-Smith C., Devriendt K. et al. (2009). Great vessel development requires biallelic expression of Chd7 and Tbx1 in pharyngeal ectoderm in mice. J. Clin. Invest. 119, 3301-3310. 10.1172/JCI37561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W. and Smyth G. K. (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts L. K. and Gerald B. (1978). Absence of both common carotid arteries. Am. J. Roentgenol. 130, 981-982. 10.2214/ajr.130.5.981 [DOI] [PubMed] [Google Scholar]
- Santen G. W. E., Sun Y., Gijsbers A. C. J., Carré A., Holvoet M., Haeringen A., Lesnik Oberstein S. A. J., Tomoda A., Mabe H., Polak M. et al. (2012). Further delineation of the phenotype of chromosome 14q13 deletions: (positional) involvement of FOXG1 appears the main determinant of phenotype severity, with no evidence for a holoprosencephaly locus. J. Med. Genet. 49, 366-372. 10.1136/jmedgenet-2011-100721 [DOI] [PubMed] [Google Scholar]
- Scambler P. J. (2000). The 22q11 deletion syndromes. Hum. Mol. Genet. 9, 2421-2426. 10.1093/hmg/9.16.2421 [DOI] [PubMed] [Google Scholar]
- Schmittgen T. D. and Livak K. J. (2008). Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101-1108. 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
- Schneider J. E., Böse J., Bamforth S. D., Gruber A. D., Broadbent C., Clarke K., Neubauer S., Lengeling A. and Bhattacharya S. (2004). Identification of cardiac malformations in mice lacking Ptdsr using a novel high-throughput magnetic resonance imaging technique. BMC Dev. Biol. 4, 16 10.1186/1471-213X-4-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. (1999). Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70-71. 10.1038/5007 [DOI] [PubMed] [Google Scholar]
- Srinivas S., Watanabe T., Lin C.-S., William C. M., Tanabe Y., Jessell T. M. and Costantini F. (2001). Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1, 4 10.1186/1471-213X-1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S. et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545-15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q., Liu S., Liu K. and Jiao K. (2018). Role of Semaphorin signaling during cardiovascular development. J. Am. Heart Assoc. 7, e008853 10.1161/JAHA.118.008853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thom T., Haase N., Rosamond W., Howard V. J., Rumsfeld J., Manolio T., Zheng Z. J., Flegal K., O'Donnell C., Kittner S. et al. (2006). Heart disease and stroke statistics--2006 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 113, e85-e151. 10.1161/circulationaha.105.171600 [DOI] [PubMed] [Google Scholar]
- Unolt M., Versacci P., Anaclerio S., Lambiase C., Calcagni G., Trezzi M., Carotti A., Crowley T. B., Zackai E. H., Goldmuntz E. et al. (2018). Congenital heart diseases and cardiovascular abnormalities in 22q11.2 deletion syndrome: From well-established knowledge to new frontiers. Am. J. Med. Genet. A 176, 2087-2098. 10.1002/ajmg.a.38662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bueren K. L., Papangeli I., Rochais F., Pearce K., Roberts C., Calmont A., Szumska D., Kelly R. G., Bhattacharya S. and Scambler P. J. (2010). Hes1 expression is reduced in Tbx1 null cells and is required for the development of structures affected in 22q11 deletion syndrome. Dev. Biol. 340, 369-380. 10.1016/j.ydbio.2010.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Mierop L. H. S. and Kutsche L. M. (1986). Cardiovascular anomalies in digeorge syndrome and importance of neural crest as a possible pathogenetic factor. Am. J. Cardiol. 58, 133-137. 10.1016/0002-9149(86)90256-0 [DOI] [PubMed] [Google Scholar]
- Waldo K. L., Kumiski D. and Kirby M. L. (1996). Cardiac neural crest is essential for the persistence rather than the formation of an arch artery. Dev. Dyn. 205, 281-292. [DOI] [PubMed] [Google Scholar]
- Xu H., Morishima M., Wylie J. N., Schwartz R. J., Bruneau B. G., Lindsay E. A. and Baldini A. (2004). Tbx1 has a dual role in the morphogenesis of the cardiac outflow tract. Development 131, 3217-3227. 10.1242/dev.01174 [DOI] [PubMed] [Google Scholar]
- Yagi H., Furutani Y., Hamada H., Sasaki T., Asakawa S., Minoshima S., Ichida F., Joo K., Kimura M., Imamura S.-I. et al. (2003). Role of TBX1 in human del22q11.2 syndrome. Lancet 362, 1366-1373. 10.1016/S0140-6736(03)14632-6 [DOI] [PubMed] [Google Scholar]
- Yashiro K., Shiratori H. and Hamada H. (2007). Haemodynamics determined by a genetic programme govern asymmetric development of the aortic arch. Nature 450, 285-288. 10.1038/nature06254 [DOI] [PubMed] [Google Scholar]
- Zhang Z. and Baldini A. (2008). In vivo response to high-resolution variation of Tbx1 mRNA dosage. Hum. Mol. Genet. 17, 150-157. 10.1093/hmg/ddm291 [DOI] [PubMed] [Google Scholar]
- Zhang Z., Cerrato F., Xu H., Vitelli F., Morishima M., Vincentz J., Furuta Y., Ma L., Martin J. F., Baldini A. et al. (2005). Tbx1 expression in pharyngeal epithelia is necessary for pharyngeal arch artery development. Development 132, 5307-5315. 10.1242/dev.02086 [DOI] [PubMed] [Google Scholar]
- Zweier C., Sticht H., Aydin-Yaylagül I., Campbell C. E. and Rauch A. (2007). Human TBX1 missense mutations cause gain of function resulting in the same phenotype as 22q11.2 deletions. Am. J. Hum. Genet. 80, 510-517. 10.1086/511993 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.