

Abstract
The drug–drug interaction profile of atorvastatin confirms that disposition is determined by cytochrome P450 (CYP) 3A4 and organic anion transporting polypeptides (OATPs). Drugs that affect gastric emptying, including dulaglutide, also affect atorvastatin pharmacokinetics (PK). Atorvastatin is a carboxylic acid that exists in equilibrium with a lactone form in vivo. The purpose of this work was to assess gastric acid–mediated lactone equilibration of atorvastatin and incorporate this into a physiologically‐based PK (PBPK) model to describe atorvastatin acid, lactone, and their major metabolites. In vitro acid‐to‐lactone conversion was assessed in simulated gastric fluid and included in the model. The PBPK model was verified with in vivo data including CYP3A4 and OATP inhibition studies. Altering the gastric acid–lactone equilibrium reproduced the change in atorvastatin PK observed with dulaglutide. The model emphasizes the need to include gastric acid–lactone conversion and all major atorvastatin‐related species for the prediction of atorvastatin PK.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Atorvastatin disposition involves organic anion transporting polypeptides and cytochrome P450 (CYP) 3A4 in vivo. Delayed gastric emptying also affects atorvastatin pharmacokinetics (PK). Atorvastatin lactone has similar plasma exposure to atorvastatin following dosing of atorvastatin, although the mechanism of formation has not been fully explained in vivo.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ What is the role of pH‐dependent acid‐lactone conversion in atorvastatin PK and drug–drug interactions?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Significant gastric acid–lactone conversion of atorvastatin is necessary to describe atorvastatin PK, alone and with concomitant drugs. The results observed with CYP3A4 inhibitors can be explained via the incorporation of atorvastatin lactone absorption and back conversion to atorvastatin. Changes in atorvastatin PK observed with delayed gastric emptying can be explained by increased gastric conversion to atorvastatin lactone.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ This model introduces a primary mechanism by which atorvastatin lactone is formed in vivo and its role in the PK of atorvastatin. This model emphasizes the need to consider gastric lactone formation for statins administered in acid form.
Atorvastatin is a 3‐hydroxy‐3‐methylglutaryl‐Coenzyme A reductase inhibitor widely prescribed for hypercholesterolemia. Atorvastatin is administered in the active acid form and in vivo is metabolized by cytochrome P450 (CYP) 3A4 to o‐hydroxyatorvastatin and p‐hydroxyatorvastatin, which demonstrate pharmacologic potency equivalent to parent. Plasma exposure of o‐hydroxyatorvastatin is similar to that of atorvastatin, whereas p‐hydroxyatorvastatin represents <10% exposure of the total active species.1, 2 Following the administration of atorvastatin, inactive lactone metabolites are also present in plasma, as atorvastatin lactone and the corresponding CYP3A4‐mediated metabolites, o‐hydroxyatorvastatin lactone and p‐hydroxyatorvastatin lactone; plasma exposures of atorvastatin lactone and o‐hydroxyatorvastatin lactone are equal to or greater than that of the respective acid forms.
The hydroxylation of atorvastatin in vitro is catalyzed by CYP3A4, consistent with the in vivo interactions reported with itraconazole, clarithromycin, erythromycin, and grapefruit juice.3, 4, 5, 6 In addition, there is clear in vitro and in vivo evidence of the role of organic anion transporting polypeptides (OATPs) in the disposition of the acid forms; both coadministration of OATP inhibitors as well as polymorphisms in the solute carrier organic anion transporter family member 1B1 gene (SLCO1B1) significantly increase plasma exposure.1, 7, 8 Conversely, although atorvastatin lactone exists at an approximately 1:1 ratio with atorvastatin acid in plasma, mechanisms regarding its formation in vivo are not well understood. In vitro, atorvastatin lactone can be formed via hepatic uridine diphosphate glucuronosyltransferase (UGT)–mediated metabolism of atorvastatin acid.9 However, the role of this pathway in vivo has not been clearly defined given the lack of selective, potent inhibitors of UGTs and that genetic variants reveal minimal and inconsistent effects on the pharmacokinetics (PK) of both atorvastatin acid and lactone.10, 11 On the other hand, it has been demonstrated in vitro that atorvastatin acid and lactone can interconvert nonenzymatically. Kearney et al.12 reported that, in buffer, conversion both from acid to lactone and vice versa is rapid at low pH (<2), and conversion from lactone to acid is predominant at pH > 6 (acid to lactone conversion does not occur at this pH). The instability of the lactone forms at physiologic pH and in plasma has been demonstrated.13 In addition, as atorvastatin lactone has high affinity for CYP3A4, it has been suggested that the effects of CYP3A4 inhibitors on atorvastatin PK in vivo may be misinterpreted given the interconversion of the acid and lactone demonstrated in vitro.14
Additional studies have been carried out with glucagon‐like peptide‐1 receptor agonists (GLP1RAs) to assess the effect of delayed gastric emptying induced by these agents on atorvastatin PK.15, 16, 17 Albeit of different magnitudes, the results of each study are consistent in demonstrating significant decreases in the maximum plasma concentration (Cmax) and delayed time to maximum plasma concentration (Tmax) of atorvastatin, with little change in the area under the concentration‐time curve (AUC). In the case of dulaglutide, of all the substrates studied in combination with the GLP1RA, these changes were the greatest for atorvastatin, resulting in a reduction of Cmax by 70%, with a delay in the median Tmax from 0.5–3 hours.15 Although the directional effects on Cmax and Tmax are consistent with delayed gastric emptying, given these specific changes in atorvastatin PK, we hypothesize an additional concurrent mechanism, namely, the increased conversion of atorvastatin acid to atorvastatin lactone as a result of the increased residence time in the acidic gastric environment.
The purpose of this work was to assess and incorporate the gastric conversion of atorvastatin acid to atorvastatin lactone into a physiologically‐based PK (PBPK) model to describe all major circulating atorvastatin‐related species and to verify this model using the metabolic, transporter, and delayed gastric emptying interactions observed in vivo.
Methods
Materials
Atorvastatin, atorvastatin lactone, o‐hydroxyatorvastatin, atorvastatin‐d5, rifamycin SV, mineral oil, silicone oil, and simulated gastric fluid (without pepsin, pH 1–1.4) were purchased from Thermo Fisher Scientific (Waltham, MA). Human hepatocytes, InVitroGRO hepatocyte thawing medium, and Kreb's Henseleit Buffer were purchased from BioreclamationIVT (Baltimore, MD).
In vitro studies
Conversion of atorvastatin to atorvastatin lactone over time (2 hours) was evaluated in simulated gastric fluid. In vitro hepatocyte uptake of atorvastatin and o‐hydroxyatorvastatin was assessed in human hepatocytes. In both experiments, atorvastatin, o‐hydroxyatorvastatin, and/or atorvastatin lactone concentrations were determined via liquid chromatography with tandem mass spectrometry (LC/MS‐MS). In vitro and LC/MS‐MS methods are listed in Supplementary Material S1.
Clinical data
To evaluate the dose‐dependent PK of atorvastatin, published clinical data were compiled from studies in which atorvastatin was administered as a single dose in White or mixed‐ethnicity populations under fasted conditions and atorvastatin plasma concentrations determined directly using LC/MS‐MS methods (as opposed to enzymatic 3‐hydroxy‐3‐methylglutaryl‐Coenzyme A reductase activity assays). In studies in which polymorphisms of enzymes or transporters were investigated, parameters were included from groups with reference alleles. Data from a total of 21 studies were used reporting atorvastatin plasma PK at 0.1 mg,18 10 mg,19, 20, 21 20 mg,4, 8, 19, 22, 23, 24 40 mg,1, 2, 3, 5, 15, 17, 19, 23 and/or 80 mg.19, 25, 26, 27, 28, 29, 30 To verify the PK of all major atorvastatin‐related species, data were used from studies in which 40 mg was administered as a single dose in White or mixed‐ethnicity populations under fasted conditions, and the PK parameters of atorvastatin, atorvastatin lactone, o‐hydroxyatorvastatin, and o‐hydroxyatorvastatin lactone were reported using LC/MS‐MS methods.1, 2, 3, 5 Similarly, for verification of drug–drug interactions (DDIs), the PK data of atorvastatin administered with itraconazole and rifampicin were used from studies in which atorvastatin 40 mg was given with the respective inhibitor and all four species reported.1, 3 Previously published in‐house data for atorvastatin and o‐hydroxyatorvastatin in the presence of dulaglutide were employed.15
PBPK modeling
All simulations were performed using Simcyp version 17 (Certara, Princeton, NJ). Noncompartmental analyses were carried out using Phoenix WinNonlin version 6.4 (Certara, Princeton, NJ). All major atorvastatin‐related species (atorvastatin, o‐hydroxyatorvastatin, atorvastatin lactone, and o‐hydroxyatorvastatin lactone) were included in the model. To incorporate the gastric conversion of atorvastatin to atorvastatin lactone, two model files were created, one for atorvastatin and one for atorvastatin lactone, to represent lactone formation in the stomach. The fraction of atorvastatin absorbed as acid/lactone at each atorvastatin dose was determined as described in Model Files below. The species included in each model file and the inputs are summarized in Table 1. Figure 1 depicts the generalized disposition of all species included in the current model and the overall modeling strategy.
Table 1.
Inputs for atorvastatin and atorvastatin lactone substrate files
| Atorvastatin | O‐hydroxyatorvastatin | Atorvastatin lactone | O‐hydroxyatorvastatin lactone | |||||
|---|---|---|---|---|---|---|---|---|
| Value | Source | Value | Source | Value | Source | Value | Source | |
| Model file(s) | 1 and 2 | 1 and 2 | 2 | 2 | ||||
| Compound (file) | Sub (1)/Pri Met 1 (2) | Pri Met 1 (1)/Sec Met (2) | Sub (2) | Pri Met 2 (2) | ||||
| MW | 559 | ChemAxon | 574 | ChemAxon | 541 | ChemAxon | 556 | ChemAxon |
| logP | 5.39 | ChemAxon | 5.39 | Assumedb | 6.05 | ChemAxon | 6.05 | Assumedb |
| pK a (type) | 4.33 (Monoprotic acid) | ChemAxon | 4.33 (Monoprotic acid) | Assumedb | Neutral | ChemAxon | Neutral | Assumedb |
| B:P | 0.55 | Default (acid) | 0.55 | Default (acid) | 1 | Default (base) | 1 | Default (base) |
| fup | 0.022 | In house | 0.022 | Assumedb | 0.012 | In silico predicted | 0.012 | Assumedb |
| Absorption | ADAM | ADAM | ||||||
| Papp type | CaCo‐2; pH 6.5:7.4 | Wu et al. (2000)32 | — | — | MDCK | In house | — | — |
| Papp (cm/sx × 10−6) | 28.4 | — | — | 33 | — | — | ||
| Peff (10−4 cm/second) | 4.49 | Simcyp predicted | — | — | 8.34 | Simcyp predicted | — | — |
| Formulation | Solution with precipitation | — | — | Solution with precipitation | — | — | ||
| pH/solubility (mg/mL) | 2.1/0.0212 3.1/0.0321 4.1/0.0796 5/0.127 5.4/0.227 6/1.22 | Kearney et al. (1993)12 | — | — | 0.00134 (not pH dependent) | Kearney et al. (1993)12 | — | — |
| Stomach degradation (1/hour) | 50 | Optimizedc | — | — | — | — | — | — |
| Distribution | Full | Full (minimal as Sec Meta) | Minimal | Minimal | ||||
| Vdss (L/kg) | Simcyp predicted | Simcyp predicted | Simcyp predicted | Simcyp predicted | ||||
| Kp scalar | 2 | Optimized to reproduce Cmax at 40 mg | 1 | Default | 0.02 | Optimized to reproduce observed half‐life | 0.008 | Optimized to reproduce observed half‐life |
| Elimination | Enzyme kinetics (metabolite formed in parentheses) | |||||||
| CYP3A4 (Vmax/Km, μL/minute/mg) | 1,353/33 (o‐OHATV) | Jacobsen et al. (2000)14 | — | — | 1,397/1.6 (o‐OHATV lactone) | Jacobsen et al. (2000)14 | — | — |
| CYP3A4 (Vmax/Km, μL/minute/mg) | 1,048/34.8 | Jacobsen et al. (2000)14 | — | — | 3229/1.8 | Jacobsen et al. (2000)14 | — | — |
| UGT1A3 (μL/minute/mg) | 6.2 | Goosen et al. (2007)9 | — | — | — | — | — | — |
| Other HLM (μL/minute/mg) | 65 | Optimizedc | (500 as Sec Meta) | ECM | 1,500 | Optimizedc | 1,200 | Optimizedc |
| Plasma esterase (t1/2, minute) | — | — | — | — | 4 (atorvastatin) | Optimizedc | — | — |
| User ES (μL/minute/mg) | — | — | — | — | — | — | 7,000 (o‐OHATV) | Optimizedc |
| Transport (μL/106 cell second/minute) | ||||||||
| OATP1B1 (SF) | 31.5 (30) | In house/optimizedc | 25 (30)a | In house/optimizedc | — | — | — | — |
| OATP1B3 (SF) | 31.5 (30) | In house/optimizedc | 25 (30)a | In house/optimizedc | — | — | — | — |
| CLpd | 13 | In house | 5a | In house | — | — | — | — |
| CLbile | — | — | 10a | Optimized to reproduce 40 mg AUC | — | — | — | — |
| CLefflux | — | — | 15a | Optimized to reproduce Cmax/Tmax | — | — | — | — |
Model file 1 refers to that of atorvastatin (acid), and model file 2 refers to that of atorvastatin lactone.
ADAM, advanced dissolution, absorption, and metabolism; AUC, area under plasma concentration‐time curve; B:P, blood‐to‐plasma ratio; Cmax, maximum plasma concentration; CLbile, intrinsic biliary clearance; CLefflux, intrinsic sinusoidal efflux clearance; CLpd, intrinsic passive clearance; CYP3A4, cytochrome P450 3A4; ECM, extended clearance model; ES, esterase; Fup, fraction unbound in plasma; HLM, human liver microsomal clearance (non‐CYP3A4); logP, log octanol:water partition coefficient; Km, concentration at half maximal rate of metabolism; Kp, tissue partition coefficient; MW, molecular weight; o‐, ortho; OATP1B1 and 1B3, organic anion transporting polypeptides 1B1 and 1B3; OHATV, hydroxyatorvastatin; Papp, apparent in vitro permeability; Peff, effective in vivo permeability; pKa, log acid dissociation constant; Pri Met 1 and Pri Met 2, included as primary metabolite in the substrate file (value in parentheses indicates which model file); Sec Met, included as the secondary metabolite in the substrate file (value in parentheses indicates which model file); SF, scaling factor; Sub, included as substrate in file (value in parentheses indicates which model file); t1/2, half‐life; Tmax, time to maximum plasma concentration.; UGT1A3, urine diphosphate glucuronosyltransferase 1B3; Vdss, steady‐state volume of distribution; Vmax, mamimal rate of metabolism.
Change made for incorporation into lactone model file (file 2).
Assumed to be similar to parent when data not available.
Optimized as explained in MethodsMethods2.
Figure 1.
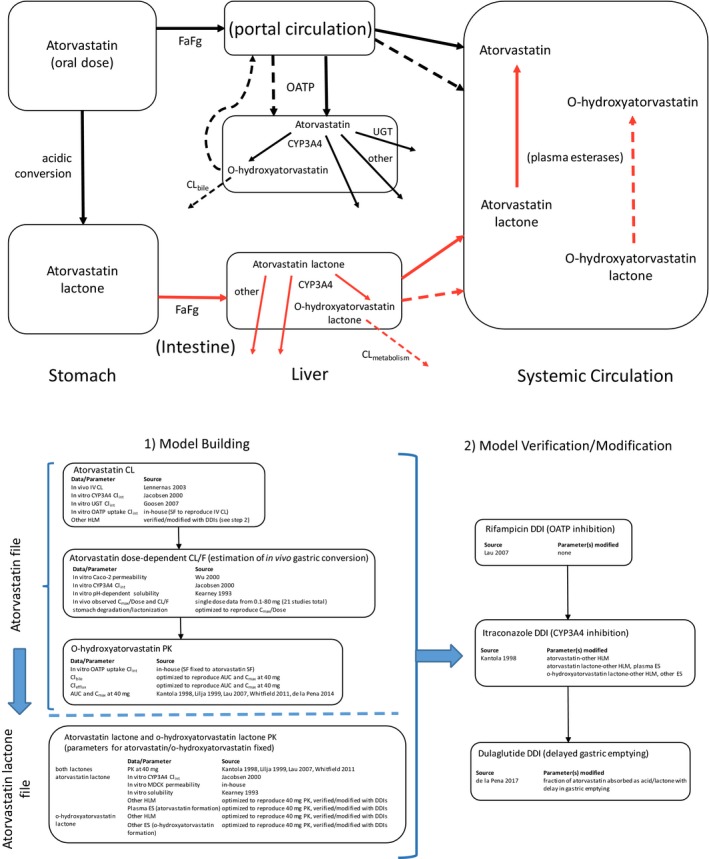
Schematic of atorvastatin disposition and modeling strategy. In the disposition schematic, black lines represent clearance processes related to the acid forms, and red arrows represent those of lactone forms. Solid arrows represent the clearance processes of parent acid/lactone, and the dotted lines represent those of the respective hydroxy metabolite. AUC, area under the plasma concentration‐time curve, CL, clearance; CL/F, oral clearance; CLbile, intrinsic biliary clearance; CLefflux, intrinsic sinusoidal efflux clearance; CLint, intrinsic clearance; Cmax, maximal plasma concentration; CYP3A4, cytochrome P450 3A4; DDI, drug–drug interaction; ES, esterase; Fa, fraction absorbed; Fg, fraction escaping interestinal first‐pass metabolism; HLM, human liver microsomal clearance (non‐CYP3A4); IV, intravenous; OATP, organic anion transporting polypeptide; PK, pharmacokinetics; SF, scaling factor; UGT, uridine diphosphate glucuronosyltransferase.
Model files
The first step in building the atorvastatin file was to reproduce hepatic clearance by inputting in vitro parameters for CYP3A4‐mediated metabolism and OATP‐mediated uptake. The reported intravenous clearance of atorvastatin is 37.5 L/hour, which is considered all hepatic because of negligible renal clearance.31 Scaling factors were applied to the in vitro hepatocyte intrinsic active uptake clearance to match in vivo hepatic clearance (the intrinsic passive uptake clearance was included as measured in vitro). To accurately reproduce the effect of itraconazole on atorvastatin disposition, non‐CYP hepatic microsomal clearance was included while maintaining the observed systemic clearance.
The oral absorption of atorvastatin and atorvastatin lactone were modeled using the advanced dissolution, absorption, and metabolism model implemented in Simcyp. As conversion of acid to lactone in the stomach is not a function of this modeling platform, the stomach degradation function in Simcyp was employed in the atorvastatin model file to represent the first step in acid‐to‐lactone conversion. Given the rapid absorption of atorvastatin (Tmax ≤ 1 hour), it was presumed that the plasma Cmax was primarily that of absorbed atorvastatin acid and not that later formed systemically from atorvastatin lactone. Furthermore, given the poor solubility of atorvastatin at low pH (~ 0.02 mg/mL at pH 2)12 at doses at or greater than 10 mg, the concentration of atorvastatin in the stomach (10 mg/250 mL or 0.04 mg/mL) would exceed the solubility. As only the soluble drug would be subject to conversion, it is predicted that the gastric conversion of atorvastatin acid to atorvastatin lactone would depend on dose; therefore, at higher doses of atorvastatin less gastric conversion occurs. This would lead to a supraproportional increase in Cmax of atorvastatin with an increasing dose, which is indeed observed in the clinical data (shown in Results). Therefore, to determine the degradation/lactonization rate of atorvastatin in the stomach, a sensitivity analysis was performed varying the first‐order degradation rate constant to determine the rate that best reproduced the dose‐dependent change in Cmax (shown in Figure S1 a). In vitro permeability data for atorvastatin predict the intrinsic fraction absorbed (Fa) of atorvastatin to be 1. Therefore, the Simcyp Fa output from this sensitivity analysis (we will currently denote as Fa’) was used to determine the fraction absorbed as acid at each dose (shown in Figure S1 b). The fraction absorbed as lactone could then be determined by the fraction of the atorvastatin acid dose that was degraded (converted to lactone) and could be calculated as 1‐Fa’. For example, at 40 mg, 25% of the atorvastatin dose was predicted to be absorbed in acid form (Fa’ = 0.25); therefore, 10 mg was predicted to be absorbed as atorvastatin acid, and atorvastatin lactone was correspondingly dosed at 30 mg. The intrinsic Fa and fraction escaping metabolism in the gut (Fg) for atorvastatin and atorvastatin lactone were estimated using Simcyp by means of the in vitro measured permeability values and intrinsic clearance values for CYP3A4 metabolism.14, 32
Atorvastatin and atorvastatin lactone hepatic metabolism included CYP3A4 and non‐CYP pathways. The CYP3A4‐mediated metabolic intrinsic clearances of atorvastatin and atorvastatin lactone were input from reported in vitro data.14 Additional non‐CYP microsomal intrinsic clearances were included for atorvastatin and atorvastatin lactone and were adjusted to reproduce the in vivo interaction with itraconazole. It is reported that atorvastatin lactone can be formed via UGT‐mediated metabolism of atorvastatin acid in the liver, and the reported in vitro intrinsic clearance value for atorvastatin acid lactonization in human liver microsomes (6.2 μL/minute/mg protein) was incorporated as part of the non‐CYP hepatic metabolism of atorvastatin.9 However, this value is insignificant both with respect to the fraction of all atorvastatin clearance pathways (4%, after accounting for other pathways in the model) and the rapid CYP3A4 intrinsic clearance of atorvastatin lactone (given in Table 1), so that atorvastatin lactone was not included as a hepatic metabolite of atorvastatin.
O‐hydroxyatorvastatin was modeled as the primary CYP3A4‐mediated metabolite of atorvastatin and was presumed to be eliminated completely in the bile.31 As o‐hydroxyatorvastatin is also an OATP substrate, o‐hydroxyatorvastatin present in plasma was also susceptible to active uptake in the atorvastatin model file. In the atorvastatin lactone model file, OATP‐mediated uptake could not be incorporated as the full PBPK model could not be used for a secondary metabolite, and use of the minimal PBPK model does not allow hepatic uptake transporter kinetics. Hence, the elimination was input as human liver microsome intrinsic clearance, and the value was determined from the extended clearance equation33 using the same optimized o‐hydroxyatorvastatin parameters from the atorvastatin model file. O‐hydroxyatorvastatin lactone was modeled as the primary CYP3A4‐mediated metabolite of atorvastatin lactone, which was presumed to undergo subsequent non‐CYP metabolism in the liver as well as conversion to o‐hydroxyatorvastatin.
Systemic conversion of both lactone forms (atorvastatin lactone and o‐hydroxyatorvastatin lactone) back to the respective acid forms was included, as the lactones are unstable in plasma.12, 13 These rates were optimized to reproduce the PK of atorvastatin and o‐hydroxyatorvastatin at 40 mg (i.e., account for the AUC that could not be attributed to that of drug absorbed as atorvastatin acid) and verified with studies assessing the in vivo interactions of atorvastatin with itraconazole and dulaglutide. As these in vivo interconversion rates could not be directly identified from in vitro data, sensitivity analyses on these parameters, as well as other non‐CYP3A4 intrinstic clearance, were performed to verify their identifiability in the model.
DDIs
For DDI simulations, atorvastatin acid was modeled as a solution using the fixed ratio of dosed atorvastatin acid and lactone determined for a 40‐mg dose (25% acid). For the itraconazole DDI simulations, itraconazole was assumed to inhibit gut and hepatic CYP3A4 by 90%.34 For rifampicin DDI simulations, the Simcyp verified (sim vivo, single dose) SV‐Rifampicin‐SD file was used (details in Table S1 ). Although an interaction with rifampicin exists for both atorvastatin and o‐hydroxyatorvastatin, because the minimal PBPK model was employed for o‐hydroxyatorvastatin in the lactone model file, the interaction with rifampicin could currently only be assessed on atorvastatin.
The effects on gastric emptying have been assessed at doses of dulaglutide from 0.05 to 8 mg using acetaminophen and/or scintigraphy. The results of these studies indicate that when compared with placebo, a 1.5 mg dose of dulaglutide increases gastric emptying time by approximately threefold.35 Therefore, for the DDI with dulaglutide, the default gastric mean residence time (MRT) in Simcyp (0.27 hour) was increased threefold (0.84 hour). As increased residence time in the acidic gastric environment could also lead to increased conversion of atorvastatin to atorvastatin lactone, a sensitivity analysis around the fraction absorbed as acid was performed.
Data analysis
To obtain individual PK parameters for atorvastatin and o‐hydroxyatorvastatin, the individual concentration profiles from dosing both atorvastatin and atorvastatin lactone were added together, before non‐compartmental analysis. The PK parameters for atorvastatin lactone and o‐hydroxyatorvastatin lactone were used directly from Simcyp output.
Results
Figure 2 shows that conversion of atorvastatin to atorvastatin lactone in simulated gastric fluid is rapid, and lactone is present within minutes. Human hepatocyte uptake experiments with atorvastatin and o‐hydroxyatorvastatin indicated uptake of both species was inhibited by the OATP inhibitor, rifamycin SV, by at least 80%. The active and passive uptake clearances determined in hepatocytes were used directly as model inputs and are listed in Table 1.
Figure 2.
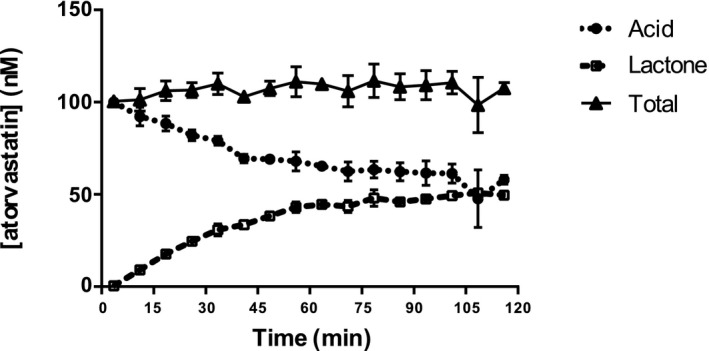
In vitro conversion of atorvastatin acid to atorvastatin lactone in simulated gastric fluid. Total represents acid + lactone.
Shown in Figure 3 are reported dose‐normalized Cmax and oral clearance values and results of the simulations across oral doses of atorvastatin. As shown in Figure 3 a, a greater than dose‐dependent increase in Cmax with increasing dose is observed from literature data, accompanied with a decrease in oral clearance with increasing dose, shown in Figure 3 b. Incorporating solubility‐limited degradation into the atorvastatin absorption model allowed the model to capture these dose‐dependent observations. Shown in Figure 3 c are the fractions of atorvastatin dose administered that are predicted by the model to be absorbed as atorvastatin acid vs. lactone. At all doses, a considerable fraction of the atorvastatin dose is estimated to be absorbed in lactone form. The Fa of both species is predicted to be 1; hence, total atorvastatin absorbed reaches 100% at all doses. For atorvastatin and atorvastatin lactone, the geometric mean–predicted Fg values are 0.9 and 0.55, respectively.
Figure 3.
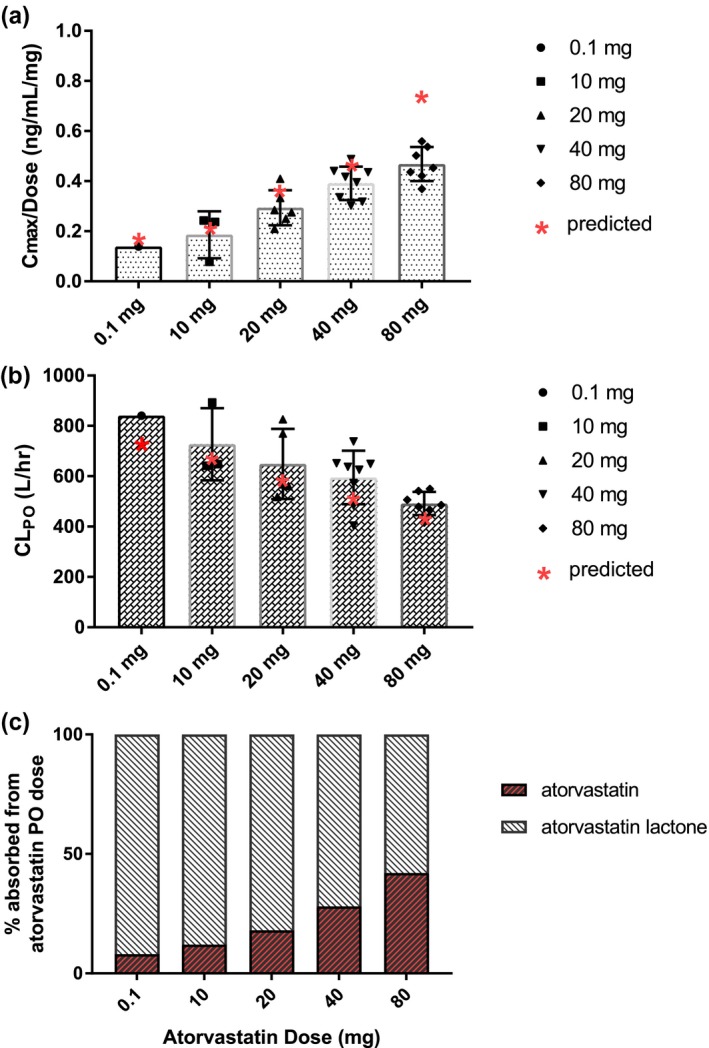
Dose‐dependent pharmacokinetics of atorvastatin. (a and b) Black symbols represent literature reported mean values. Red symbols represent model‐predicted geometric mean. (c) Model‐predicted percentage of dose absorbed as acid/lactone with incorporation of solubility‐limited stomach degradation in the atorvastatin model. CLPO, oral clearance; Cmax, maximim plasma concentration; hr, hour; p.o., oral.
As observed in Figure 4, the model was able to reproduce the concentration‐time profiles of all four major atorvastatin‐related species following oral administration of atorvastatin 40 mg as well as the AUC, Cmax, and Tmax, with the mean observed and predicted PK parameters shown in Table 2. Also shown in Table 2 are the observed and predicted ratios of each species (where available) with itraconazole and rifampicin. The model reproduced these interactions with predicted/observed ratios within approximately twofold for each species. Sensitivity analyses on varying lactone‐to‐acid conversion and other non‐CYP3A4 intrinsic clearances on these DDIs are shown in Figures S2 and S3 . These results demonstrate that the parameters included in the current models are identifiable and describe the clinical data as a whole.
Figure 4.
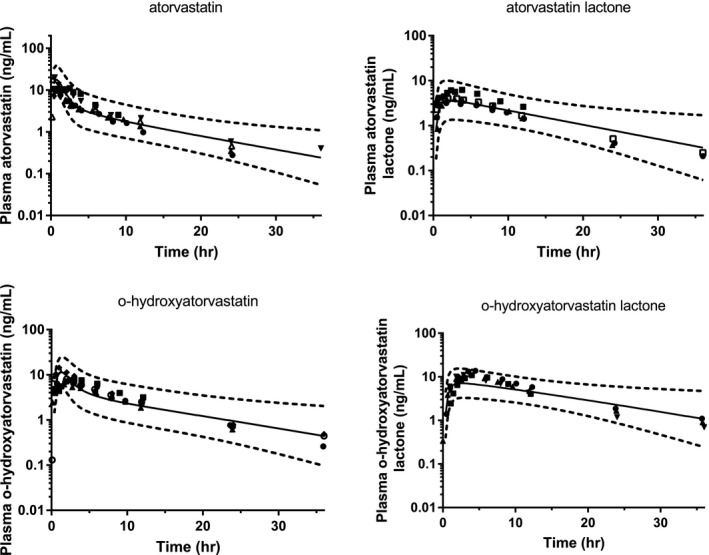
Predicted and observed concentration profiles for the four major atorvastatin‐related species following oral administration of 40 mg atorvastatin in the fasted state. Different symbols represent mean data from different literature studies. Solid lines represent the geometric mean–predicted concentrations, and dotted lines represent the predicted 90% confidence intervals. hr, hour.
Table 2.
Observed and predicted pharmacokinetic parameters and effect of perpetrators on all major atorvastatin‐related species following oral administration of atorvastatin 40 mg
| 40 mg atorvastatin | 40 mg + itraconazole | 40 mg + rifampicin | 40 mg + dulaglutide | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Obs | Pred | Pred/Obs | Obs ratio | Pred ratio | Pred/Obs | Obs ratio | Pred ratio | Pred/Obs | Obs ratio | Pred ratio | Pred/Obs | |
| Atorvastatin | ||||||||||||
| AUC (ng/mL • hour) | 54.2–99 | 78.6 (48) | 0.79–1.45 | 3.3 | 2.56 (2.47–2.67) | 0.78 | 7.3 | 7.66 (7.20, 8.15) | 1.05 | 0.786 (0.752, 0.821) | 0.758 (0.736–0.780) | 0.96 |
| Cmax (ng/mL) | 12.7–19.5 | 18.2 (49) | 0.93–1.43 | 1.2 | 1.33 (1.30–1.35) | 1.1 | 10.5 | 9.42 (9.03–9.84) | 0.90 | 0.296 (0.246, 0.355) | 0.22 (0.21–0.23) | 0.74 |
| Tmax (hour) | 0.5–1 | 0.72 (0.36–1.08) | 0.72–1.44 | — | — | — | — | — | — | 2.5 | 1.08 | 0.43 |
| O‐hydroxyatorvastatin | ||||||||||||
| AUC (ng/mL • hour) | 77.5–102 | 93.5 (52) | 0.92–1.21 | 0.83 | 0.85 (0.81–0.89) | 1.02 | — | — | — | 0.937 (0.887, 0.990) | 0.941 (0.916–0.965) | 1 |
| Cmax (ng/mL) | 7.7–14.4 | 11.9 (48) | 0.83–1.55 | 0.17 | 0.22 (0.21–0.23) | 1.3 | — | — | — | 0.393 (0.330, 0.467) | 0.361 (0.336–0.388) | 0.92 |
| Tmax (hour) | 1–3 | 1.26 (1.08–2.16) | 0.42–1.26 | — | — | — | — | — | — | 5 | 1.8 | 0.36 |
| Atorvastatin lactone | ||||||||||||
| AUC (ng/mL • hour) | 50.7–78.2 | 64.4 (47) | 0.82–1.27 | 4.0 | 3.44 (3.26–3.63) | 0.68 | — | — | — | — | — | — |
| Cmax (ng/mL) | 3.8–8 | 3.75 (60) | 0.47–0.99 | 2.3 | 2.78 (2.64–2.92) | 1.2 | — | — | — | — | — | — |
| Tmax (hour) | 2–3 | 1.93 (1.03–3.86) | 0.64–0.97 | — | — | — | — | — | — | — | — | — |
| O‐hydroxy atorvastatin lactone | ||||||||||||
| AUC (ng/mL • hour) | 128–187 | 162 (39) | 0.87–1.27 | 0.71 | 1.18 (1.14–1.21) | 1.7 | — | — | — | — | — | — |
| Cmax (ng/mL) | 10.5–14.5 | 7.3 (50) | 0.50–0.70 | 0.29 | 0.61 (0.58–0.64) | 2.1 | — | — | — | — | — | — |
| Tmax (hour) | 3–4 | 2.25 (1.2–9.33) | 0.56–0.75 | — | — | — | — | — | — | — | — | — |
Data for atorvastatin alone are from five studies reporting all four major atorvastatin‐related species.1, 2, 3, 5 Observed mean values are the means from all studies, predicted values for AUC and Cmax are the reported as geometric mean (CV%), and the predicted Tmax is reported as median (range). Data for atorvastatin with itraconazole, rifampicin, and dulaglutide are from single studies with each inhibitor.1, 3, 14 Observed ratio values are reported means or geometric means (90% confidence interval) in the case of dulaglutide, predicted values for AUC and Cmax ratios are geometric means (90% confidence interval), predicted Tmax ratio is reported as median difference (presence ‐absence of dulaglutide). AUC, area under the plasma concentration‐time curve; Cmax, maximum plasma concentration; Obs, observed; Pred, predicted; Tmax, time to maximum plasma concentration.
For the interaction between atorvastatin and dulaglutide, it was estimated that the mean fraction of atorvastatin absorbed as acid at a 40 mg dose decreased from 25% in the absence of dulaglutide to 5% with dulaglutide. Simulations using this fraction of acid absorbed (along with increased MRT) were able to accurately reproduce the concentration‐time profiles for atorvastatin and o‐hydroxyatorvastatin, shown in Figure 5. Shown in Table 2, the model captured the minimal change in atorvastatin AUC while demonstrating a substantial reduction in Cmax and delay in Tmax. Furthermore, the model accurately reproduced these changes for o‐hydroxyatorvastatin, predicting an increase in the metabolite ratio, from 1.2 to 1.4, as observed.15 The concentration‐time profiles from sensitivity analyses using the estimated mean gastric MRT implemented with dulaglutide (0.84 hour) and varying fractions of acid absorbed are shown in Figure S4, overlaid with the observed data from the dulaglutide clinical study. From these simulations it can be observed that small changes in the fraction of atorvastatin absorbed as acid result in a wide range in the Cmax and Tmax for a given dose of atorvastatin, which is consistent with the large interindividual variability in the PK data observed in the clinical study.
Figure 5.
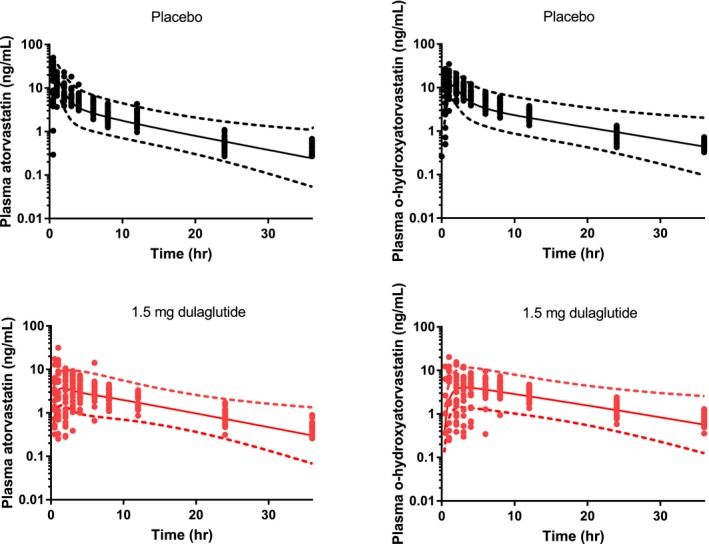
Predicted and observed concentration profiles for atorvastatin and o‐hydroxyatorvastatin following oral administration of 40 mg atorvastatin in the fasted state with and without 1.5‐mg dulaglutide. Symbols represent observed data, solid lines represent predicted geometric mean concentrations, and dotted lines represent the predicted 90% confidence intervals. hr, hour.
Discussion
For many of the statins, interconversion between acid and lactone has been observed in vitro; however, for most of these compounds, the quantitative contribution of this process to the PK is absent. Mechanistic population PK models have been developed incorporating this interconversion for simvastatin36 however, simvastatin is administered in lactone form, and conversion must occur to elicit its pharmacodynamic effects. Other PBPK models for atorvastatin have been described but either do not include lactone forms in the analysis37 or have included atorvastatin lactone as a hepatic metabolite of atorvastatin.38 As of yet, no mechanistic modeling to understand gastric acid/lactone conversion, back conversion in plasma, or quantitative evaluation of the effect of delayed gastric emptying on atorvastatin has been reported. In this analysis, we demonstrate the necessity of including gastric acid to lactone conversion and the inclusion of all major atorvastatin‐related species to explain atorvastatin PK alone and during DDIs. This PBPK approach provides a framework for modeling this gastric conversion for other statins or compounds found to be unstable at gastric pH.
The in vitro data reported here are in agreement with previous data, demonstrating that atorvastatin acid conversion to atorvastatin lactone is rapid at low pH.12 The necessity of considering this mechanism is driven by the clinical data; it is apparent from the current compilation of numerous clinical studies that there is a greater than dose‐dependent increase in the Cmax of atorvastatin acid. Although a dose‐dependent change in Cmax could be attributable to the saturation of other presystemic processes, such as intestinal metabolism or an efflux transporter, the Fa·Fg for atorvastatin is predicted to be high (0.9) at all doses, leaving little room for an effect of saturation of transporters or enzymes. Furthermore, these mechanisms should lead to a similar fold change in both Cmax and oral clearance, which is not observed in the clinical data (i.e., from the lowest to highest dose in the given range, Cmax changes 3.4‐fold, whereas oral clearance changes only 1.7‐fold). These data suggest the involvement of a process more complicated than simply a difference in bioavailability with increasing dose. This dose‐dependent observation, however, can be explained by the current model by incorporating solubility‐limited conversion of dosed atorvastatin acid to the lactone form. This leads to the dose‐dependent changes observed in Cmax, and, as some of the acid that converts to lactone in the stomach back converts to atorvastatin acid in the plasma, this results in a change in oral clearance of smaller magnitude than the change in Cmax.
The need for the incorporation of presystemic acid to lactone conversion is also evident when modeling DDIs. Although atorvastatin is indeed a substrate of CYP3A4, a minimal effect of hepatic CYP3A4 inhibition on the plasma clearance of atorvastatin is expected, as atorvastatin hepatic clearance is uptake rate limited by OATPs.39 A change in atorvastatin Cmax and AUC is observed with intravenous rifampicin administration, and the model is able to accurately capture the magnitude of this interaction. Of course, this does not rule out an effect of CYP3A4 inhibitors on atorvastatin metabolism in the gut. However, the Fg of atorvastatin is predicted to be high from in vitro CYP clearance data, and in vivo a minimal change in Cmax is consistently reported in the presence CYP3A4 inhibitors (ratio of 1–1.6), whereas the change in AUC is larger (ratio of 2.3–3.3).3, 5, 40, 41 In contrast, the AUC and Cmax ratios are both significant (greater than twofold) for atorvastatin lactone in the same studies, indicating both a change in bioavailability and systemic CYP3A4‐mediated clearance, which is consistent with a lower predicted Fg for atorvastatin lactone of ~ 0.5. It follows that a change in the AUC of atorvastatin, with minimal change in Cmax, can predominantly be explained by a change in the systemically formed atorvastatin acid from the lactone form. The use of two models was helpful to elucidate this phenomenon; given the mechanisms of atorvastatin acid clearance discussed, the effect of itraconazole on the atorvastatin absorbed as atorvastatin acid resulted in an AUC ratio of only 1.3 compared with the observed AUC ratio of greater than threefold. To reach an AUC ratio in the order of that observed, the majority of the change in atorvastatin AUC with itraconazole coadministration comes from an initial interaction with atorvastatin lactone.
Confirmation of the importance of atorvastatin lactone presystemic formation comes from significant changes in the Cmax and Tmax of atorvastatin with little change in atorvastatin AUC or AUC of o‐hydroxyatorvastatin following administration of GLP1RAs.15, 16, 17 Although atorvastatin is rapidly absorbed in the intestine (based on the very rapid T max), both Cmax and Tmax are expected to be sensitive to changes in gastric emptying; however, simply changing the mean gastric MRT from ~15 to ~50 minutes does not quantitatively explain the large reduction in Cmax or delay in Tmax observed in the clinical data with dulaglutide coadministration. Given the time‐dependent acid‐lactone conversion data reported here and elsewhere, along with the rapid conversion rate necessary for reproduction of clinical data of atorvastatin alone, it seemed important to consider a change in the atorvastatin acid–lactone equilibrium given the increased residence time at acidic pH. Indeed, a decrease in the fraction of atorvastatin absorbed as acid from 25 to 5% was able to reproduce the dramatic reduction in Cmax, delay in Tmax, and minimal change in AUC. Furthermore, sensitivity analyses around the fraction absorbed as acid indicate that even with similar gastric MRT, the large variability in Cmax and Tmax observed with atorvastatin when given with dulaglutide can be explained by just small differences in the gastric acid–lactone equilibrium between individuals. Dulaglutide also affected a change in the metabolite:parent ratio for o‐hydroxyatorvastatin, which further supports the involvement of a mechanism more complex than simply an increase in gastric MRT. To reproduce the AUC and metabolite:parent ratios with dulaglutide, a greater proportion of o‐hydroxyatorvastatin was generated during lactone absorption via first formation of o‐hydroxyatorvastatin lactone. Information on the lactone forms of atorvastatin in these studies would have been informative but was not measured (or at least reported) with any of the GLP1RAs.15, 16, 17 Data do, however, exist on the interaction of simvastatin with the GLP1RA, albiglutide, in which both acid and lactone forms were reported; a decrease in simvastatin lactone AUC was observed with a corresponding increase in the AUC of simvastatin acid.42 Based on in vitro data, acid–lactone conversion may occur in either direction at low pH, therefore in the case of simvastatin, dosed in lactone form, increased acid production could occur with delayed gastric emptying, leading to the opposing changes observed in acid and lactone forms. We propose the opposite for atorvastatin, increased lactone production, as it is the acid form of atorvastatin that is administered.
A clear limitation of the current model is the inability to truly incorporate reversible metabolism in the current modeling platform (Simcyp v17). As such, although in vitro data do suggest that atorvastatin lactone is a hepatic metabolite of atorvastatin, as well as a nonspecific degradant, both processes could not be incorporated in the current model. However, as previously noted, including atorvastatin lactone as a hepatic metabolite was investigated but because of the reasons discussed previously, the resulting contribution to the lactone AUC from this route was negligible. Other limitations exist in the data available in atorvastatin clinical studies given that in many earlier studies the total atorvastatin‐related species (using enzymatic assays) were reported and not individual analytes. Although this may be of therapeutic relevance, it does not allow mechanistic interpretation. In addition, there exists limited information on other DDIs with atorvastatin involving changes in gastric pH that would allow further confirmation of pH‐dependent conversion.
In conclusion, it is necessary to consider the gastric conversion of atorvastatin acid to atorvastatin lactone to reproduce the observed PK and DDI of atorvastatin. The current model performs well in describing interactions involving CYP3A4/OATP inhibition and delayed gastric emptying using well‐characterized perpetrator compounds. In future clinical studies, at various doses or with other concomitant interacting drugs, the measurement of both active and inactive atorvastatin‐related species could provide valuable information on the quantitative incorporation of these processes. The application of the current strategy may be useful to describe other statins administered in acid form, which would need to be assessed individually both in vitro and through modeling.
Funding
This work was funded by Eli Lilly and Company.
Conflict of Interest
All authors are paid employees of and hold stock ownership in Eli Lilly and Company.
Author Contributions
B.L.M., M.M.P., J.R., K.M.H., S.D.H., and G.L.D. wrote the manuscript. B.L.M., J.J.A., M.M.P., A.K., L.S.T., C.L., K.M.H., S.D.H., and G.L.D. designed the research. B.L.M., J.J.A., A.K., L.S.T., and C.L. performed the research. B.L.M., J.J.A., and A.K. analyzed the data. B.L.M., J.J.A., and A.K. contributed new reagents/analytical tools.
Supporting information
Figure S1. Sensitivity analysis on the effect of stomach degradation/lactone conversion on (a) atorvastatin acid maximum plasma concentration (Cmax) and (b) fraction absorbed as atorvastatin acid (Fa’).
Figure S2. Sensitivity analysis on non‐cytochrome P450 human liver microsomal intrinsic clearance (HLM Clint) of (a,b) atorvastatin lactone, (c,d) o‐hydroxyatorvastatin lactone, and (e,f) atorvastatin.
Figure S3. Sensitivity analysis on (a–d) atorvastatin lactone‐to‐acid plasma half‐life and (e–h) o‐hydroxyatorvastatin lactone‐to‐acid intrinsic clearance (Clint).
Figure S4. Sensitivity analysis around the fraction of atorvastatin absorbed as acid (Fa’).
Table S1. SV‐Rifampicin‐SD (sim vivo, single dose) Inputs.
Supplementary Methods.
Model files.
References
- 1. Lau, Y.Y. , Huang, Y. , Frassetto, L. & Benet, L.Z. Effect of OATP1B transporter inhibition on the pharmacokinetics of atorvastatin in healthy volunteers. Clin. Pharmacol. Ther. 81, 194–204 (2007). [DOI] [PubMed] [Google Scholar]
- 2. Whitfield, L.R. et al Effect of gemfibrozil and fenofibrate on the pharmacokinetics of atorvastatin. J. Clin. Pharmacol. 51, 378–388 (2011). [DOI] [PubMed] [Google Scholar]
- 3. Kantola, T. , Kivisto, K.T. & Neuvonen, P.J. Effect of itraconazole on the pharmacokinetics of atorvastatin. Clin. Pharmacol. Ther. 64, 58–65 (1998). [DOI] [PubMed] [Google Scholar]
- 4. Shin, J. et al Effect of cytochrome P450 3A5 genotype on atorvastatin pharmacokinetics and its interaction with clarithromycin. Pharmacotherapy 31, 942–950 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lilja, J.J. , Kivisto, K.T. & Neuvonen, P.J. Grapefruit juice increases serum concentrations of atorvastatin and has no effect on pravastatin. Clin. Pharmacol. Ther. 66, 118–127 (1999). [DOI] [PubMed] [Google Scholar]
- 6. Siedlik, P.H. , Olson, S.C. , Yang, B.B. & Stern, R.H. Erythromycin coadministration increases plasma atorvastatin concentrations. J. Clin. Pharmacol. 39, 501–504 (1999). [PubMed] [Google Scholar]
- 7. Pasanen, M.K. , Fredrikson, H. , Neuvonen, P.J. & Niemi, M. Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvastatin. Clin. Pharmacol. Ther. 82, 726–733 (2007). [DOI] [PubMed] [Google Scholar]
- 8. Lin, W. et al Evaluation of drug‐drug interaction potential between sacubitril/valsartan (LCZ696) and statins using a physiologically based pharmacokinetic model. J. Pharm. Sci. 106, 1439–1451 (2017). [DOI] [PubMed] [Google Scholar]
- 9. Goosen, T.C. et al Atorvastatin glucuronidation is minimally and nonselectively inhibited by the fibrates gemfibrozil, fenofibrate, and fenofibric acid. Drug Metab. Dispos. 35, 1315–1324 (2007). [DOI] [PubMed] [Google Scholar]
- 10. Riedmaier, S. et al UDP‐glucuronosyltransferase (UGT) polymorphisms affect atorvastatin lactonization in vitro and in vivo. Clin. Pharmacol. Ther. 87, 65–73 (2010). [DOI] [PubMed] [Google Scholar]
- 11. Stormo, C. et al UGT1A1*28 is associated with decreased systemic exposure of atorvastatin lactone. Mol Diagn Ther. 17, 233–237 (2013). [DOI] [PubMed] [Google Scholar]
- 12. Kearney, A.S. , Crawford, L.F. , Mehta, S.C. & Radebaugh, G.W. The interconversion kinetics, equilibrium, and solubilities of the lactone and hydroxyacid forms of the HMG‐CoA reductase inhibitor, CI‐981. Pharm. Res. 10, 1461–1465 (1993). [DOI] [PubMed] [Google Scholar]
- 13. Partani, P. , Verma, S.M. , Gurule, S. , Khuroo, A. & Monif, T. Simultaneous quantitation of atorvastatin and its two active metabolites in human plasma by liquid chromatography/(‐) electrospray tandem mass spectrometry. J. Pharm. Anal. 4, 26–36 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jacobsen, W. et al Lactonization is the critical first step in the disposition of the 3‐hydroxy‐3‐methylglutaryl‐CoA reductase inhibitor atorvastatin. Drug Metab. Dispos. 28, 1369–1378 (2000). [PubMed] [Google Scholar]
- 15. de la Pena, A. , Cui, X. , Geiser, J. & Loghin, C. No dose adjustment is recommended for digoxin, warfarin, atorvastatin or a combination oral contraceptive when coadministered with dulaglutide. Clin. Pharmacokinet. 56, 1415–1427 (2017). [DOI] [PubMed] [Google Scholar]
- 16. Hausner, H. et al Effect of semaglutide on the pharmacokinetics of metformin, warfarin, atorvastatin and digoxin in healthy subjects. Clin. Pharmacokinet. 56, 1391–1401 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Malm‐Erjefalt, M. , Ekblom, M. , Vouis, J. , Zdravkovic, M. & Lennernas, H. Effect on the gastrointestinal absorption of drugs from different classes in the biopharmaceutics classification system, when treating with liraglutide. Mol. Pharm. 12, 4166–4173 (2015). [DOI] [PubMed] [Google Scholar]
- 18. Prueksaritanont, T. et al Validation of a microdose probe drug cocktail for clinical drug interaction assessments for drug transporters and CYP3A. Clin. Pharmacol. Ther. 101, 519–530 (2017). [DOI] [PubMed] [Google Scholar]
- 19. Vargo, R. , Adewale, A. , Behm, M.O. , Mandema, J. & Kerbusch, T. Prediction of clinical irrelevance of PK differences in atorvastatin using PK/PD models derived from literature‐based meta‐analyses. Clin. Pharmacol. Ther. 96, 101–109 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gerber, J.G. et al Effect of efavirenz on the pharmacokinetics of simvastatin, atorvastatin, and pravastatin: results of AIDS Clinical Trials Group 5108 Study. J. Acquir. Immune Defic. Syndr. 39, 307–312 (2005). [DOI] [PubMed] [Google Scholar]
- 21. Chung, M. , Calcagni, A. , Glue, P. & Bramson, C. Bioavailability of amlodipine besylate/atorvastatin calcium combination tablet. J. Clin. Pharmacol. 46, 1030–1037 (2006). [DOI] [PubMed] [Google Scholar]
- 22. Keskitalo, J.E. et al ABCG2 polymorphism markedly affects the pharmacokinetics of atorvastatin and rosuvastatin. Clin. Pharmacol. Ther. 86, 197–203 (2009). [DOI] [PubMed] [Google Scholar]
- 23. Backman, J.T. , Luurila, H. , Neuvonen, M. & Neuvonen, P.J. Rifampin markedly decreases and gemfibrozil increases the plasma concentrations of atorvastatin and its metabolites. Clin. Pharmacol. Ther. 78, 154–167 (2005). [DOI] [PubMed] [Google Scholar]
- 24. Ando, H. et al Effects of grapefruit juice on the pharmacokinetics of pitavastatin and atorvastatin. Br. J. Clin. Pharmacol. 60, 494–497 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Herrera‐Gonzalez, S. et al Effect of AGTR1 and BDKRB2 gene polymorphisms on atorvastatin metabolism in a Mexican population. Biomed Rep. 7, 579–584 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chung, M. , Calcagni, A. , Glue, P. & Bramson, C. Effect of food on the bioavailability of amlodipine besylate/atorvastatin calcium combination tablet. J. Clin. Pharmacol. 46, 1212–1216 (2006). [DOI] [PubMed] [Google Scholar]
- 27. Woo, H.I. , Kim, S.R. , Huh, W. , Ko, J.W. & Lee, S.Y. Association of genetic variations with pharmacokinetics and lipid‐lowering response to atorvastatin in healthy Korean subjects. Drug Des. Devel. Ther. 11, 1135–1146 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Teng, R. , Mitchell, P.D. & Butler, K.A. Pharmacokinetic interaction studies of co‐administration of ticagrelor and atorvastatin or simvastatin in healthy volunteers. Eur. J. Clin. Pharmacol. 69, 477–487 (2013). [DOI] [PubMed] [Google Scholar]
- 29. Daka, A. et al Effects of single nucleotide polymorphisms and haplotypes of the SLCO1B1 gene on the pharmacokinetic profile of atorvastatin in healthy Macedonian volunteers. Pharmazie 70, 480–488 (2015). [PubMed] [Google Scholar]
- 30. Di Spirito, M. , Morelli, G. , Doyle, R.T. , Johnson, J. & McKenney, J. Effect of omega‐3‐acid ethyl esters on steady‐state plasma pharmacokinetics of atorvastatin in healthy adults. Expert Opin. Pharmacother. 9, 2939–2945 (2008). [DOI] [PubMed] [Google Scholar]
- 31. Lennernas, H. Clinical pharmacokinetics of atorvastatin. Clin. Pharmacokinet. 42, 1141–1160 (2003). [DOI] [PubMed] [Google Scholar]
- 32. Wu, X. , Whitfield, L.R. & Stewart, B.H. Atorvastatin transport in the Caco‐2 cell model: contributions of P‐glycoprotein and the proton‐monocarboxylic acid co‐transporter. Pharm. Res. 17, 209–215 (2000). [DOI] [PubMed] [Google Scholar]
- 33. Shitara, Y. et al Clinical significance of organic anion transporting polypeptides (OATPs) in drug disposition: their roles in hepatic clearance and intestinal absorption. Biopharm. Drug Dispos. 34, 45–78 (2013). [DOI] [PubMed] [Google Scholar]
- 34. Olkkola, K.T. , Backman, J.T. & Neuvonen, P.J. Midazolam should be avoided in patients receiving the systemic antimycotics ketoconazole or itraconazole. Clin. Pharmacol. Ther. 55, 481–485 (1994). [DOI] [PubMed] [Google Scholar]
- 35. Tham, L.S.S.K. , Geiser, J.S. , Posada, M.M. & Dickinson, G.L. Integration of population exposure‐response and physiological based pharmacokinetic modeling approaches to evaluate gastric‐empyting induced drug interaction risks for dulaglutide Ninth American Conference on Pharmacometrics, San Diego, CA, October 7–10, 2018. [Google Scholar]
- 36. Tsamandouras, N. et al Development and application of a mechanistic pharmacokinetic model for simvastatin and its active metabolite simvastatin acid using an integrated population PBPK approach. Pharm. Res. 32, 1864–1883 (2015). [DOI] [PubMed] [Google Scholar]
- 37. Duan, P. , Zhao, P. & Zhang, L. Physiologically based pharmacokinetic (PBPK) modeling of pitavastatin and atorvastatin to predict drug‐drug interactions (DDIs). Eur. J. Drug Metab. Pharmacokinet. 42, 689–705 (2017). [DOI] [PubMed] [Google Scholar]
- 38. Zhang, T. Physiologically based pharmacokinetic modeling of disposition and drug‐drug interactions for atorvastatin and its metabolites. Eur. J. Pharm. Sci. 77, 216–229 (2015). [DOI] [PubMed] [Google Scholar]
- 39. Patilea‐Vrana, G. & Unadkat, J.D. Transport vs. metabolism: what determines the pharmacokinetics and pharmacodynamics of drugs? Insights from the extended clearance model. Clin. Pharmacol. Ther. 100, 413–418 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mazzu, A.L. et al Itraconazole alters the pharmacokinetics of atorvastatin to a greater extent than either cerivastatin or pravastatin. Clin. Pharmacol. Ther. 68, 391–400 (2000). [DOI] [PubMed] [Google Scholar]
- 41. European Medicines Agency . Eperzan Product Information. Available from https://www.ema.europa.eu/documents/product-information/eperzan-epar-product-information_en.pdf. 2017. Accessed Jan 17, 2019.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Sensitivity analysis on the effect of stomach degradation/lactone conversion on (a) atorvastatin acid maximum plasma concentration (Cmax) and (b) fraction absorbed as atorvastatin acid (Fa’).
Figure S2. Sensitivity analysis on non‐cytochrome P450 human liver microsomal intrinsic clearance (HLM Clint) of (a,b) atorvastatin lactone, (c,d) o‐hydroxyatorvastatin lactone, and (e,f) atorvastatin.
Figure S3. Sensitivity analysis on (a–d) atorvastatin lactone‐to‐acid plasma half‐life and (e–h) o‐hydroxyatorvastatin lactone‐to‐acid intrinsic clearance (Clint).
Figure S4. Sensitivity analysis around the fraction of atorvastatin absorbed as acid (Fa’).
Table S1. SV‐Rifampicin‐SD (sim vivo, single dose) Inputs.
Supplementary Methods.
Model files.