

Abstract
Teriparatide and abaloparatide are parathyroid hormone receptor 1 (PTHR1) analogs with unexplained differential efficacy for the treatment of osteoporosis. Therefore, we compared the effects of abaloparatide and teriparatide on bone structure, turnover, and levels of receptor activator of nuclear factor‐kappa B ligand (RANKL) and osteoprotegerin (OPG). Wild‐type (WT) female mice were injected daily with vehicle or 20–80 µg/kg/day of teriparatide or abaloparatide for 30 days. Femurs and spines were examined by microcomputed tomography scanning and serum levels of bone turnover markers, RANKL, and OPG, were measured by ELISA. Both analogs similarly increased the distal femoral fractional trabecular bone volume, connectivity, and number, and reduced the structure model index (SMI) at 20–80 µg/kg/day doses. However, only abaloparatide exhibited a significant increase (13%) in trabecular thickness at 20 µg/kg/day dose. Femoral cortical evaluation showed that abaloparatide caused a greater dose‐dependent increase in cortical thickness than teriparatide. Both teriparatide and abaloparatide increased lumbar 5 vertebral trabecular connectivity but had no or modest effect on other indices. Biochemical analysis demonstrated that abaloparatide promoted greater elevation of procollagen type 1 intact N‐terminal propeptide, a bone formation marker, and tartrate‐resistant acid phosphatase 5b levels, a bone resorption marker, and lowered the RANKL/OPG ratio. Furthermore, PTHR1 signaling was compared in cells treated with 0–100 nmol/L analog. Interestingly, abaloparatide had a markedly lower EC50 for cAMP formation (2.3‐fold) and β‐arrestin recruitment (1.6‐fold) than teriparatide. Therefore, abaloparatide‐improved efficacy can be attributed to enhanced bone formation and cortical structure, reduced RANKL/OPG ratio, and amplified Gs‐cAMP and β‐arrestin signaling.
Keywords: Abaloparatide, osteoporosis, PTH, signaling, teriparatide
Introduction
Bone is continuously remodeled to preserve skeletal integrity and maintain calcium (Ca2+) homeostasis. The process of bone remodeling is highly coordinated and is initiated by osteoclast, multinucleated cells of hematopoietic origin, responsible for bone resorption. Release of local factors at the remodeling site stimulates the recruitment and differentiation of osteoblast lineage cells, cells of mesenchymal origin, to synthesize new bone matrix with subsequent mineralization (Martin and Sims 2005; Henriksen et al. 2009; Martin et al. 2009; Sims and Vrahnas 2014). Predominance of bone resorption over bone formation in old age results in a marked decline in bone mineral density (BMD) and biomechanical properties causing senile osteoporosis. Therefore, antiresorptive and bone anabolic agents that inhibit osteoclast function and stimulate osteoblast maturation, respectively, have been widely used to treat osteoporosis.
Parathyroid hormone (PTH) and PTH‐related peptide (PTHrP) are two important regulators of bone remodeling. PTH is a single‐chain 84 amino acid polypeptide hormone secreted by the parathyroid glands in response to hypocalcemia. The primary function of the endogenous PTH is to act directly on the kidney and bone, and indirectly on the intestine by the action of vitamin D to maintain Ca2+ and inorganic phosphate (Pi) homeostasis. On the other hand, PTHrP, as was initially discovered, is only detectible in the circulation under certain types of malignancy that secrete high levels of PTHrP contributing to bone breakdown and paraneoplastic hypercalcemia (Honda et al. 1988; Kukreja et al. 1988; Nagata et al. 1989; Gaich and Burtis 1990; Tsuchihashi et al. 1990; Grill et al. 1991; Burtis et al. 1992). Thus, under normal conditions, PTHrP is produced only locally by a variety of normal fetal and adult tissues and acts in an autocrine and paracrine fashion. The locally secreted PTHrP acts to promote placental Ca2+ transport from the mother to the fetus, and to regulate chondrocyte differentiation and development of the growth plate, postnatal bone remodeling, vascular smooth muscle function, mammary gland development, tooth eruption, beta‐cell proliferation, and keratinocyte differentiation (Noda et al. 1994; Lee et al. 1996; Vortkamp et al. 1996; Chung et al. 1998; Foley et al. 1998; Philbrick et al. 1998; Wysolmerski et al. 1998; Lanske et al. 1999; Foley et al. 2001; Cebrian et al. 2002; Miao et al. 2005; Hens et al. 2007; Meziani et al. 2008; Hirai et al. 2011; Williams et al. 2011; Raison et al. 2013; Boras‐Granic et al. 2014; Mozar et al. 2016). PTHrP exists in three isoforms of 139, 141, or 173 amino acids due to alternative promoter activity and mRNA splicing.
Both PTH and PTHrP act by activating a common receptor known as the PTH/PTHrP receptor or PTH receptor 1 (PTHR1), a G protein‐coupled receptor that activates multiple downstream pathways and effectors (Abou‐Samra et al. 1992; Sneddon et al. 2000; Tawfeek et al. 2001; Tawfeek et al. 2002; Ahmed et al. 2003; Sneddon et al. 2004; Tawfeek and Abou‐Samra 2004; Singh et al. 2005; Gesty‐Palmer et al. 2006; Rey et al. 2006; Sneddon and Friedman 2007; Sneddon et al. 2007; Tawfeek and Abou‐Samra 2008; Garrido et al. 2009; Tawfeek and Abou‐Samra 2012). PTH effects on bone remodeling involve multiple molecules and complex interaction between osteoblast progenitors, osteoblasts, osteocytes, osteoclasts, and T cells (Locklin et al. 2003; Lotinun et al. 2004; Bellido et al. 2005; Bouxsein et al. 2005; Bellido 2006; Jilka et al. 2009; Terauchi et al. 2009; Bedi et al. 2010; Tawfeek et al. 2010; Jilka et al. 2010; Bohinc and Gesty‐Palmer 2011; Robling et al. 2011; Bedi et al. 2012; Hanyu et al. 2012). However, osteoblast lineage cells and their PTHR1 remain central for PTH and PTHrP effects on bone formation and resorption. The latter is due to PTH or PTHrP stimulation of osteoclastogenic factors produced by the osteoblast lineage cells.
The effects of PTH and PTHrP on bone remodeling are of high clinical interest. This interest emerges from the fact that both PTH and PTHrP cause bone loss when continuously secreted during hyperparathyroidism and hypercalcemia of malignancy, respectively. In contrast, the amino‐terminal fragments of these two proteins (PTH1‐34, PTHrP1‐34, and PTHrP1‐36) offer a treatment for osteoporosis if administered intermittently. Specifically, repeated transient elevation of PTH or PTHrP (Plotkin et al. 1998; Stewart et al. 2000; Horwitz et al. 2003; Horwitz et al. 2010; Horwitz et al. 2013) achieved by daily administration to animals or humans stimulates predominantly bone formation and increases primarily trabecular bone mass.
For these reasons, a daily regimen of PTH (1–34) and PTHrP (1–34) derivative peptide analogs, teriparatide and abaloparatide, respectively, have attained Food and Drug Administration (FDA) approval for the treatment of severe postmenopausal osteoporosis (Neer et al. 2001; Leder et al. 2015; Cosman et al. 2017; Bilezikian et al. 2018). Abaloparatide shares 41% homology to teriparatide and 71% to the parent PTHrP1‐34 but with modifications of five amino acids between residues 22 and 34. Abaloparatide demonstrated more favorable effects on the skeleton in animals and humans with more potent anabolic activity and limited bone resorptive and calcemic response when compared to teriparatide (Leder et al. 2015; Bahar et al. 2016; Miller et al. 2016; Cosman et al. 2017; Varela et al. 2017; Chandler et al. 2018; Doyle et al. 2018). The mechanisms underlying the superior actions of abaloparatide over teriparatide remain elusive. In this study, we used an in vivo and in vitro approach to evaluate, in parallel, the effects of teriparatide and abaloparatide on bone structure and microarchitecture, bone turnover, and PTHR1 signaling.
Materials and Methods
Reagents
Abaloparatide and teriparatide were chemically synthesized (LifeTein, NJ) and purified by reverse‐phase chromatography (>95% pure). The molecular mass of the peptide was verified by mass spectrometry, and the net peptide content and composition were confirmed by amino acid analysis. It is not possible to use the commercially available teriparatide (Forteo, Lilly) and abaloparatide (Tymlos, Radius) in animals due to the low concentration formulation for human use and the prohibitively high cost of the commercial products for human use. Tissue culture media were purchased from GE Healthcare Life Sciences HyClone Laboratories (Logan, Utah). Fetal bovine serum (FBS) (Cat number 900–108, lot number A47D) was from Geminibio‐Products (West Sacramento, CA). Penicillin‐streptomycin (PS, Cat number MT‐30‐001‐CI), Trypsin (0.05%, Cat number 25‐300‐054), and phosphate‐buffered saline (PBS, Cat number 10‐010‐072) were from Lifescience Technologies (Grand Island, NY). 3‐isobutyl‐1‐methylxanthine (IBMX) (Cat number I5879) was from Sigma‐Aldrich (St. Louis, MO). Tissue culture plates and other supplies were from Corning (Oneonta, NY) and Fisher Scientific (Pittsburgh, PA).
Animals
All animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee at James J. Peters Veterans Affairs Medical Center. All experiments were conducted on 16‐week‐old wild‐type (WT) female C57BL/6J mice (Stock number 664) from Jackson Laboratories (Kingston, NY). Vehicle (0.9% NaCl/10 mmol/L acetic acid) or 20–80 µg/kg/day teriparatide or abaloparatide was injected subcutaneously (SC) daily (except Sunday) and continued for 30 days. No peptide injection was performed on the day of animal sacrifice.
Cell culture
MC3T3‐E1 subclone 4 osteoblast cell line (Cat number ATCC® CRL‐2593) was purchased from American Type Culture Collection (ATCC) (Manassas, VA). All cell cultures were performed in alpha‐modified Eagle's medium (alpha‐MEM) (Cat number SH30265.02) from GE Healthcare Life Science Hyclone Laboratories (Logan, Utah) supplemented with 10% FBS and 1% PS. Cells were incubated at 37°C using a humidified atmosphere containing 95% air and 5% CO2. Cell passage was performed every 7 days (1 million cells/75 cm flask) and cells were not used beyond passage 8.
Micro‐CT measurements of trabecular and cortical bone structure
Animals were euthanized using isoflurane inhalation (to effect) anesthesia followed by exsanguination by cardiac puncture and cervical dislocation. Harvested femurs were fixed in 10% neutral‐buffered formalin for 4 h at RT and 24 h at 4°C, rinsed extensively in tap water, and preserved in 70% ethanol at 4°C until analysis. Bones were scanned using a microcomputed tomographic (micro‐CT) instrument (VivaCT‐40, Scanco Medical AG, Bassersdorf, Switzerland). The VivaCT‐40 is calibrated weekly using a phantom provided by Scanco. Trabecular bone volume fraction and microarchitecture in the lumbar 5 vertebra (L5) and distal femoral region and cross‐sectional geometry at the femoral mid‐shaft were assessed. Scanning for the distal femoral trabecular region was performed in the secondary spongiosa starting approximately at 0.6 mm proximal to the growth plate and extending proximally 1.5 mm. Scans for the cortical region were performed at the midpoint of each femur and an isotropic pixel size of 10.5 µm and slice thickness of 10.5 µm were used to calculate the average total cross‐sectional area (mm2), bone area (mm2), and marrow area (mm2). The bones were scanned at high resolution with energy level of 70 kVp, and intensity of 114 μA and 8 W at 10.5 μm voxel size, and integration time 200 msec. Trabecular 150 consecutive slices at 10.5‐µm interval were evaluated with a threshold of 165 density units. Cortical 50 consecutive slices were evaluated with a threshold of 700 density units. Standard nomenclature and guidelines for assessment of bone microstructure were followed, as recommended by the American Society for Bone and Mineral Research (Bouxsein et al. 2010). The main bone parameters are TV (total volume, mm3), BV (bone volume, mm3), BV/TV (relative volume of calcified tissue in the selected volume of interest (VOI)), Tb. Th (thickness of the trabecular structure, mm), Tb. N (number of trabeculae/mm), Tb. Sp (trabecular separation or spacing; a measurement of the thickness of the spaces between the trabeculae, mm), Conn. D (connectivity density; 3‐D connectivity index; a measure of the degree to which a structure is multiply connected, normed by TV, 1/mm3), TRI‐SMI (three‐dimensional structure model index (SMI), related to the architecture of the structure and ranges between 0 and 3, SMI toward 0 signifies that the structure is mainly concave plates, whereas a value of 3 means only cylindrical rods), Ct. Th (cortical thickness, mm), Ct. Po (Cortical porosity in a given cortical region, the volume of pores (Po.V, mm3)/total volume of cortical bone compartment (Ct.V, mm3).
Measurement of serum markers of bone turnover
Blood was drawn from isoflurane‐anesthetized animals using cardiac puncture as a terminal procedure. Blood was collected in BD Microtainer™ Capillary Blood Collector and BD Microgard™ Closure from Fisher Scientific (Fisher Cat number 02‐675‐185 and BD manufacturer Cat number BD 365967). Serum was separated after spinning in a microcentrifuge for 10 min at 4°C and then frozen at −80°C. Serum markers of bone turnover were measured, as described previously (Gao et al. 2008; Terauchi et al. 2009; Tawfeek et al. 2010) and by following the manufacturer's instructions. Serum procollagen type 1 intact N‐terminal propeptide (P1NP), a specific marker of bone formation, collagen type I C‐telopeptide or carboxy‐terminal collagen crosslinks (CTX), a marker of bone resorption, and tartrate‐resistant acid phosphatase (TRAcP‐5b), a specific serum marker of osteoclastic activity, were measured using rodent‐specific ELISA assay kits (Cat numbers AC33F1, AC‐06F1, SB‐TR103, respectively) from Immunodiagnostic Systems (Scottsdale, AZ). Serum levels of receptor activator of nuclear factor‐kappa B ligand (RANKL) and osteoprotegerin (OPG) were measured using Mouse TRANCE/RANK L/TNFSF11 (Cat number MTR00) and mouse osteoprotegerin/TNFRSF11B (Cat number MOP00) quantikine ELISA kits, respectively, from R&D Systems, Inc. (Minneapolis, MN).
Measurement of serum Ca2+ and Pi
Blood was collected and serum was separated as described above. Serum Ca2+ and Pi concentrations were measured using colorimetric Ca2+ and Pi assay kits (Cat numbers K380 and K410, respectively) as indicated by the manufacturer (BioVision, Milpitas, CA).
Measurement of intracellular cAMP generation
MC3T3‐E1 cells were seeded at 40,000 cells/well of a 24‐well plate containing 500‐µL alpha‐MEM supplemented with 10% FBS and 1% PS. After culture for 1 week, the medium was removed and replaced with 250 µL of stimulation medium (alpha‐MEM containing 0.05% FBS, 0.1% BSA, 5 mmol/L hepes buffer, and 0.5 mmol/L IBMX) for 15 min. IBMX is a phosphodiesterase inhibitor that prevents degradation of the generated cAMP. Vehicle, abaloparatide, and teriparatide were then added in 250 µL stimulation medium to achieve final concentrations of 0, 0.01, 0.1, 1, 10, and 100 nmol/L/well. Incubation continued for 40 min at 37°C before the medium was removed and the plates were snap frozen in liquid N3 and stored at −80°C. For extraction of intracellular cAMP, 100 mmol/L Hcl was added and cells were incubated at room temperature for 1 h. Intracellular cAMP was assayed using a cAMP competitive ELISA kit (Cat number EMSCAMPL) from Thermo Fisher Scientific (Waltham, MA) and following the manufacturer protocol and instructions.
PathHunter® eXpress PTHR1 CHO‐K1 β‐arrestin GPCR assay
To assess the effects of abaloparatide and teriparatide stimulation of PTHR1 on β‐arrestin recruitment to the cell membrane, a PathHunter eXpress PTHR1 Chinese Hamster Ovary‐K1 (CHO‐K1) β‐arrestin GPCR Assay from Eurofins DiscoverX (Fremont, CA) (Cat Number 93‐0315E2CP0S) was used. The assay takes advantage of Enzyme Fragment Complementation technology. The PTHR1 is fused in frame with a small enzyme donor fragment ProLink™ (PK) and co‐expressed in CHO‐K1 cells stably expressing a fusion protein of β‐arrestin and the larger, N‐terminal deletion mutant of β‐galactosidase (called enzyme acceptor or EA). Activation of the PTHR1 stimulates binding of β‐arrestin to the PK‐tagged GPCR and forces complementation of the two enzyme fragments, resulting in the formation of an active β‐galactosidase enzyme. An increase in enzyme activity is then measured using chemiluminescent PathHunter Detection Reagents. Cell seeding, incubation, and detection were performed as instructed by the manufacturer. Briefly, cells were seeded in a clear bottom white 96‐well plate and incubated for 48 h at 37°C CO2 incubator. Cells were treated with vehicle, teriparatide, or abaloparatide for 60 min at 37°C in a CO2 incubator. At the end of the incubation, β‐gal enzyme substrate was added for 60 min at room temperature in the dark. Light generation (Relative Light Units, RLU), an indication of β‐gal enzyme fragment complementation and β‐Arrestin/ PTHR1 interaction, was measured using BMG Labtech PHERAstar FS luminescence plate reader.
PathHunter® eXpress PTHR1 activated GPCR internalization assay
To determine PTHR1 internalization, we used PathHunter eXpress PTHR1 U2OS Activated GPCR Internalization Assay (Cat number 93‐0770E3CP0S) from Eurofins DiscoverX (Fremont, CA). PathHunter® PTHR1 Activated GPCR Internalization U2OS cell lines are engineered to co‐express an untagged PTHR1, an EA‐tagged β‐arrestin, and a PK tag localized to the endosomes. Activation of the untagged PTHR1 induces β‐arrestin recruitment, followed by internalization of the GPCR‐β‐arrestin‐EA complex in PK‐tagged endosomes. Similar to the β‐arrestin assay format, this internalization forces complementation of the two β‐gal enzyme fragments, forming functional enzyme that hydrolyzes substrate to generate a chemiluminescent signal. U2OS osteoblastic cell line seeding, incubation, and detection were performed as instructed by the manufacturer. Cells were treated with vehicle, teriparatide, or abaloparatide for 60 min at 37°C in a CO2 incubator. At the end of the incubation, β‐gal enzyme substrate was added for 60 min at room temperature in the dark. Light generation (RLU), an indication of β‐gal enzyme fragment complementation and β‐arrestin/endosome/PTHR1 formation, was measured using BMG Labtech PHERAstar FS luminescence plate reader.
Statistical analysis
As per experimental protocol, animals were randomized between vehicle, teriparatide, and abaloparatide groups and the analyses were performed blindly regarding the control and test groups using unique animal identification codes. For in vivo studies, five mice/group were used. For in vitro cell biological and biochemical studies, each experiment was repeated three to four times and in each experiment, every experimental condition was performed in duplicate or triplicate. Statistical analyses were performed using GraphPad Prism and Microsoft excel. EC50 represents the molar concentration at which a substance exerts half of its maximal response and is an indication of drug potency. EC50 was calculated using nonlinear regression analyses of normalized cAMP dose–response curves of abaloparatide and teriparatide. The results are presented as the means ± SD. Unpaired Student t‐test was used for comparison of EC50 between two teriparatide and abaloparatide groups. For carrying out multiple group comparisons (all in vivo experiments), one‐way analysis of variance (ANOVA) was performed. When ANOVA indicated unequal means, Tukey's honestly significant difference (HSD) posthoc test was performed to determine which means were different to the level of significance. Statistical significance was considered when P value was less than 0.05 (P < 0.05).
Results
Abaloparatide and teriparatide similarly improve trabecular bone architecture
To assess the effects of intermittent abaloparatide and teriparatide administration on trabecular bone structure, WT female C57BL/6J mice were injected daily (except Sunday) SC with vehicle or 20–80 µg/kg/day of teriparatide or abaloparatide for 30 days. Mice were then sacrificed and the distal metaphyseal regions of the isolated femurs and L5 vertebrae were analyzed by micro‐CT. Abaloparatide and teriparatide administration at 20 or 80 µg/kg/day similarly increased fractional trabecular bone volume (BV/TV) and trabecular connectivity (Conn. D), and reduced the three‐dimensional SMI (Fig. 1A, C, and D). There was no detectible changes in trabecular number (Tb. N) or spacing (Tb. Sp) among all treatment groups (Fig. 1E and F). However, Tb. Th was the only trabecular index that was marginally responsive to abaloparatide treatment at 20 (113%) or 80 µg/kg/day (108%) when comparing either dose group to vehicle‐treated controls (Fig. 1B). Qualitative changes in femoral trabecular bone structure are shown in micro‐CT images of the distal femoral regions (Fig. 1G).
Figure 1.
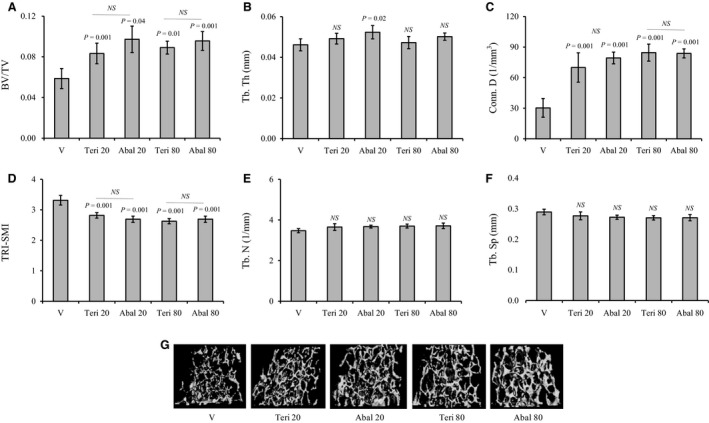
Effects of abaloparatide and teriparatide administration on femoral trabecular bone architecture: Female C57BL/6J WT mice were SC injected daily, except Sunday, with vehicle or 20–80 µg/kg/day teriparatide or abaloparatide for 30 days. Femurs were isolated, fixed, and distal femur metaphysis regions were analyzed by micro‐CT. (A) BV/TV is trabecular bone volume/total volume. (B) Tb. Th is trabecular thickness. (C) Conn. D is trabecular connectivity density. (D) SMI is trabecular three‐dimensional structure model index. (E) Tb. N is trabecular number. (F) Tb. Sp is trabecular spacing or separation. (G) Images: Examples of distal femur micro‐CT scans. The statistical designation directly listed on the top of each bar indicates the significance level in comparison to vehicle. Statistical significance levels between other experimental groups are shown using the drawn lines. The data in all graph are expressed as the means ± SD. N = 5 mice/group. NS is not significant. V is vehicle, Teri 20 is teriparatide 20 µg/kg/day, Abal 20 is abaloparatide 20 µg/kg/day, Teri 80 is teriparatide 80 µg/kg/day, and Abal 80 is abaloparatide 80 µg/kg/day.
Overall, the effects of teriparatide and abaloparatide on L5 structure and microarchitecture were modest compared to the femur (Fig. 2A–G). While, only animals receiving 80 µg/kg/day teriparatide had a significant (P = 0.006) elevation (42%) in vertebral BV/TV when compared to vehicle, this effect was not significant when compared to 80 µg/kg/day abaloparatide counterparts (Fig. 2A). Furthermore, Conn. D was greatly augmented by 64–100% in response to 20 or 80 µg/kg/day teriparatide or abaloparatide administration (Fig. 2C). Tb. N was equally increased only in 80 µg/kg/day teriparatide‐ and abaloparatide‐treated animals (Fig. 2E). Conversely, L5 Tb. Th, TRI‐SMI, and Tb. Sp were not altered in any animal group irrespective of the treatment (Fig. 2B, D, and F). Micro‐CT images show the qualitative changes in bone structure of L5 vertebrae (Fig. 2G).
Figure 2.
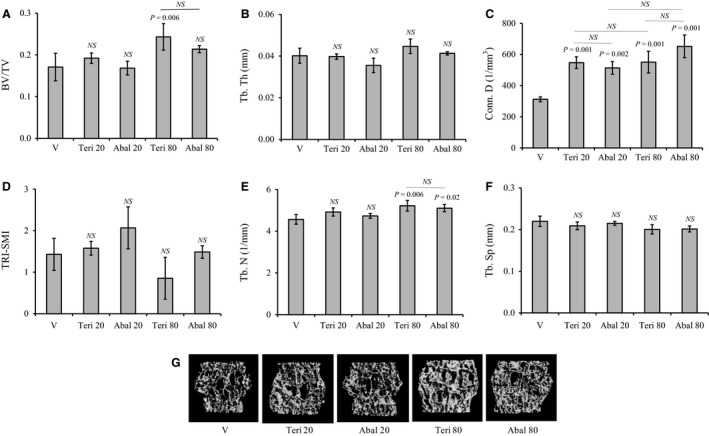
Effects of abaloparatide and teriparatide administration on vertebral trabecular bone architecture: Female C57BL/6J WT mice were SC injected daily, except Sunday, with vehicle or 20–80 µg/kg/day teriparatide or abaloparatide for 30 days. L5 vertebrae were isolated, fixed, and analyzed by micro‐CT. (A) BV/TV is trabecular bone volume/total volume. (B) Tb. Th is trabecular thickness. (C) Conn. D is trabecular connectivity density. (D) SMI is trabecular three‐dimensional structure model index. (E) Tb. N is trabecular number. (F) Tb. Sp is trabecular spacing or separation.(G) Images: Examples of L5 micro‐CT scans. The statistical designation directly listed on the top of each bar indicates the significance level in comparison to vehicle. Statistical significance levels between other experimental groups are shown using the drawn lines. The data in all graph are expressed as the means ± SD. N = 5 mice/group. NS is not significant. V is vehicle, Teri 20 is teriparatide 20 µg/kg/day, Abal 20 is abaloparatide 20 µg/kg/day, Teri 80 is teriparatide 80 µg/kg/day, and Abal 80 is abaloparatide 80 µg/kg/day.
Abaloparatide treatment causes larger increase in cortical bone thickness than teriparatide
To determine the effects of abaloparatide and teriparatide treatment on cortical bone structure, mid‐shaft regions of isolated femurs were analyzed by micro‐CT. Abaloparatide administration efficiently expanded cortical thickness (Ct. Th) at both doses of 20 and 80 µg/kg/day by 17% and 18%, respectively (Fig. 3A). In contrast, teriparatide at 20 µg/kg/day failed to achieve a significant increase in Ct. Th (Fig. 3A). The effects of teriparatide and abaloparatide on Ct. Th at 80 µg/kg/day dose were not statistically different (Fig. 3A). No effects were observed on cortical porosity (Ct. Po) for either abaloparatide or teriparatide treatment at 20 or 80 µg/kg/day dose regimen (Fig. 3B). Qualitative changes in cortical bone structure are shown in micro‐CT images of the femoral mid‐shaft regions (Fig. 3C).
Figure 3.
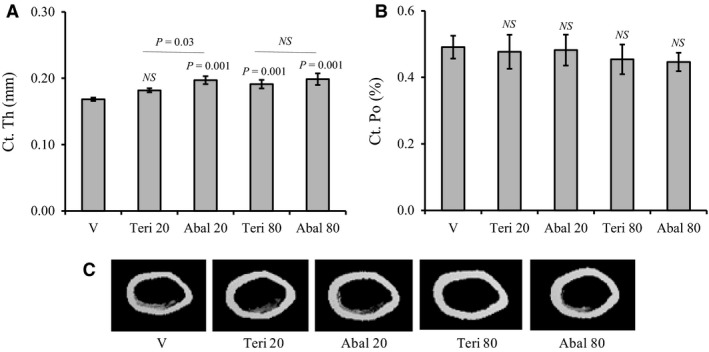
Changes in cortical bone structure in response to abaloparatide and teriparatide administration: Female C57BL/6J WT mice were injected SC daily, except Sunday, with vehicle or 20–80 µg/kg/day teriparatide or abaloparatide for 30 days. Femurs were isolated, fixed, and mid‐shaft regions were analyzed by micro‐CT. (A) Ct. Th is cortical thickness. (B) Ct. Po is cortical porosity. (C) Images: Examples of femur mid‐shaft micro‐CT scans. The statistical designation directly listed on the top of each bar indicates the significance level in comparison to vehicle. Statistical significance levels between other experimental groups are shown using the drawn lines. The data in all graph are expressed as the means ± SD. N = 5 mice/group. NS is not significant. V is vehicle, Teri 20 is teriparatide 20 µg/kg/day, Abal 20 is abaloparatide 20 µg/kg/day, Teri 80 is teriparatide 80 µg/kg/day, and Abal 80 is abaloparatide 80 µg/kg/day.
Abaloparatide produces higher bone formation and lower RANKL/OPG ratio than teriparatide
To determine the effects of teriparatide and abaloparatide administration on the levels of serum markers of bone turnover, OPG, and RANKL, specific ELISA kits were used. Analysis of sera collected from WT female mice after 30 days of vehicle, teriparatide, or abaloparatide treatment showed that only abaloparatide achieved a significant dose‐dependent increase in P1NP, a specific bone formation marker (Fig. 4A). Abaloparatide increased P1NP levels to 227% and 407% at 20 and 80 µg/kg/day, respectively. On the other hand, teriparatide stimulation of P1NP was not statistically significant at 20 µg/kg/day dose but a 291% elevation of P1NP was achieved when teriparatide was used at 80 µg/kg/day dose (P = 0.002) (Fig. 4A). Neither abaloparatide nor teriparatide was able at any dose to alter serum levels of CTX, a bone resorption marker (Fig. 4B). However, measurement of serum levels of TRAcP‐5b, a specific marker of osteoclast activity, demonstrated that abaloparatide treatment efficiently increased TRAcP‐5b levels at both 20 (167%) and 80 µg/kg/day (227%) doses (Fig. 4C). Teriparatide‐treated animals, however, had increased TRAcP‐5b levels (193%) only at 80 µg/kg/day dose (Fig. 4C). Of note, there was no statistical difference between abaloparatide and teriparatide TRAcP‐5b response at 80 µg/kg/day dose or between abaloparatide responses at 20 and 80 µg/kg/day doses.
Figure 4.
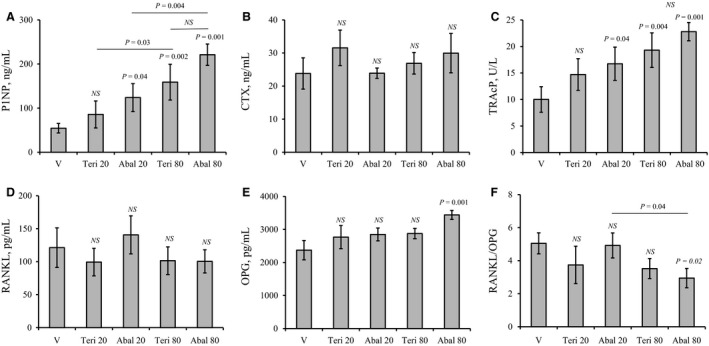
Abaloparatide and teriparatide stimulation of serum markers of bone turnover, RANKL and OPG: Female C57BL/6 WT mice were injected SC daily, except Sunday, with vehicle or 20–80 µg/kg/day teriparatide or abaloparatide for 30 days. Sera were collected and serum levels of bone formation and resorption markers, RANKL, and OPG were determined using specific ELISA kits. (A) Serum levels of P1NP. (B) Serum levels of CTX. (C) Serum levels of TRAcP‐5b. (D) Serum levels of RANKL. (E) Serum levels of OPG. (F) RANKL/OPG ratio. The statistical designation directly listed on the top of each bar indicates the significance level in comparison to vehicle. Statistical significance levels between other experimental groups are shown using the drawn lines. The data in all graph are expressed as the means ± SD. N = 5 mice/group. NS is not significant. V is vehicle, Teri 20 is teriparatide 20 µg/kg/day, Abal 20 is abaloparatide 20 µg/kg/day, Teri 80 is teriparatide 80 µg/kg/day, and Abal 80 is abaloparatide 80 µg/kg/day.
Assessment of serum levels of RANKL revealed that neither abaloparatide nor teriparatide had any significant effects on RANKL levels (Fig. 4D). On the other hand, only abaloparatide at 80 µg/kg/day increased OPG levels by 44% when compared to vehicle (Fig. 4E). Consistently, calculation of relative serum levels of RANKL and OPG further demonstrated that only 80 µg/kg/day abaloparatide treatment intervention was able to lower RANKL/OPG ratio by 42% (Fig. 4F). A reduction of RANKL/OPG ratio by 80 µg/kg/day of teriparatide administration was not statistically significant (P = 0.2).
No significant differences between teriparatide and abaloparatide treatment on mineral homeostasis
Ca2+ and Pi levels were measured, as described in the methods section, in the serum samples collected at the end of the 30‐day injection. No significant differences in serum Ca2+ levels were detected when all animal groups treated with vehicle, abaloparatide, and teriparatide were compared (Fig. 5A). However, a statistically significant (P = 0.006) 24% increase in serum Ca2+ of 20 and 80 µg/kg/day abaloparatide groups was detected when compared to only vehicle controls (Fig. 5A). The effects on serum Pi were observed only in animals receiving 80 µg/kg/day teriparatide or abaloparatide (Fig. 5B). Administration of 80 µg/kg/day of either teriparatide or abaloparatide resulted in a considerable 50% elevation in Pi levels when compared to vehicle‐treated animals (Fig. 5B).
Figure 5.
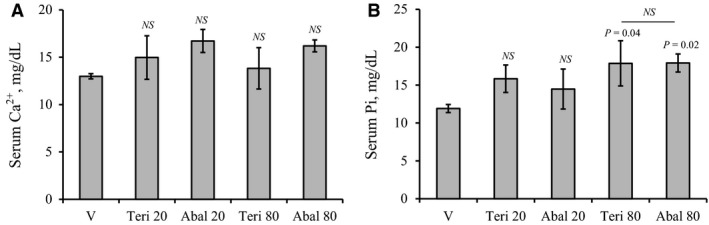
Effects of daily abaloparatide and teriparatide on serum Ca2+ and Pi: Female C57BL/6 WT mice were injected SC daily, except Sunday, with vehicle or 20–80 µg/kg/day teriparatide or abaloparatide for 30 days. Sera were collected at sacrifice (24 h after last injection) and levels of Ca2+ and Pi were measured using specific colorimetric assays. (A) Serum Ca2+ levels. (B) Serum Pi levels. The statistical designation directly listed on the top of each bar indicates the significance level in comparison to vehicle. Statistical significance levels between other experimental groups are shown using the drawn lines. The data in all graph are expressed as the means ± SD. N = 5 mice/group. NS is not significant. V is vehicle, Teri 20 is teriparatide 20 µg/kg/day, Abal 20 is abaloparatide 20 µg/kg/day, Teri 80 is teriparatide 80 µg/kg/day, and Abal 80 is abaloparatide 80 µg/kg/day.
Enhanced Gs/cAMP signaling and β‐arrestin recruitment in abaloparatide‐treated cells
MC3T3‐E1 osteoblast cells were treated with vehicle or 0.01–100 nmol/L of abaloparatide or teriparatide for 40 min at 37°C in the presence of 0.5 mmol/L IBMX. The results revealed that exposure of cells to abaloparatide or teriparatide caused a robust elevation of intracellular cAMP levels (Fig. 6A, upper panel). Increasing doses of abaloparatide caused a shift to the left of the cAMP dose–response curve compared to teriparatide‐treated cell response (Fig. 6A, lower panel). As a result, abaloparatide treatment resulted in a 2.3‐fold decrease in EC50 value for cAMP formation compared to teriparatide (EC50 = 0.3 ± 0.03 nmol/L and 0.7 ± 0.2 nmol/L, respectively, p = 0.02). Maximum cAMP stimulation, however, was not significantly different between the two analogs (Fig. 6A, upper panel).
Figure 6.
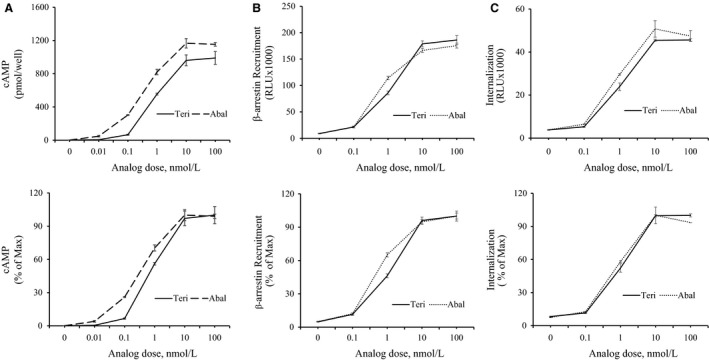
Activation of PTHR1 signaling by abaloparatide and teriparatide treatment: (A) Activation of intracellular cAMP production: MC3T3‐E1 cells were challenged with 0–100 nmol/L of abaloparatide or teriparatide for 40 min at 37°C in the presence of IBMX. The medium was disposed and cells were snap frozen on liquid N3 before storage at 80°C. Intracellular cAMP was extracted and measured using ELISA assay as described by the manufacturer protocol. Each dose treatment was performed in duplicate and the experiment was repeated three times. The EC50 was calculated using results from three independent experiments. Results are the means ± SD. Upper panel: Absolute values are expressed as pmol/well. Lower panel: Values are expressed as % stimulation/maximal response. (B) Activation of β‐arrestin recruitment: PathHunter eXpress PTHR1 CHO‐K1 β‐arrestin GPCR assay was described under the material and methods section. CHO‐K1 cells were treated with 0–100 nmol/L of teriparatide or abaloparatide for 60 min at 37°C. Teriparatide and abaloparatide stimulations of β‐arrestin recruitment were determined by measuring light generation after adding β‐gal enzyme substrate. The EC50 was calculated using results from three independent experiments. Results are the means ± SD. Upper panel: Absolute values are expressed as relative light units (RLU). Lower panel: Values are expressed as % stimulation/maximal response. (C) Stimulation of PTHR1 internalization: PathHunter eXpress PTHR1 U2OS activated GPCR internalization assay was described under the material and methods section. U2OS cells were treated with 0–100 nmol/L of teriparatide or abaloparatide for 60 min at 37°C. PTHR1 internalization was determined by measuring light generation after adding β‐gal enzyme substrate. The EC50 was calculated using results from three independent experiments. Results are the means ± SD. Upper panel: Absolute values are expressed as RLU. Lower panel: Values are expressed as % stimulation/maximal response.
β‐arrestin is an important cytosolic protein that interacts with PTHR1 and mediates PTHR1 internalization, desensitization, and signaling to ERK1/2 MAPK pathway (Ferrari et al. 1999; Gesty‐Palmer et al. 2006; Rey et al. 2006; Sneddon et al. 2007; Tawfeek and Abou‐Samra, 2012). To assess the effects of abaloparatide and teriparatide stimulation of PTHR1 on β‐arrestin recruitment, a PathHunter eXpress PTHR1 CHO‐K1 β‐arrestin GPCR Assay was used as described under the methods section. A dose‐dependent stimulation of β‐arrestin/PTHR1 interaction was demonstrated by abaloparatide and teriparatide analogs (Fig. 6B, upper panel). Compared to teriparatide, abaloparatide treatment caused a shift to the left of the dose–response curve (Fig. 6B, lower panel). Consistently, the calculated the EC50 value for abaloparatide was 1.6‐fold lower than that of teriparatide (EC50 = 0.9 ± 0.2 nmol/L and 1.5 ± 0.3 nmol/l, respectively, P = 0.02). The maximal stimulation for both analogs was reached at equimolar dose of a 100 nmol/L.
To measure PTHR1 internalization, PathHunter® PTHR1 Activated GPCR Internalization U2OS Cell Line was used as described under the methods section. Both abaloparatide and teriparatide efficiently induced a dose‐dependent stimulation of PTHR1 internalization with a dose as low as 0.1 nmol/L and reached maximum stimulation at 100 nmol/L concentration (Fig. 6C, upper panel). There was, however, no significant difference between abaloparatide and teriparatide activation of PTHR1 internalization (Fig. 6C, lower panel). The EC50 values for both abaloparatide (EC50 = 0.8 ± 0.4 nmol/L) and teriparatide (EC50 = 1.1 ± 0.4 nmol/L) stimulation of internalization were not statistically different.
Discussion
In this report, a combination of in vivo and in vitro approaches was used to identify the mechanisms mediating the differential clinical efficacy of the two PTHR1‐related agents, abaloparatide and teriparatide. Using a mouse model, we demonstrate that the skeletal response to abaloparatide treatment was more amplified compared to teriparatide‐treated animals. A daily regimen of abaloparatide administration caused a larger increase in trabecular and cortical bone thickness and higher bone formation than teriparatide. In vitro studies further demonstrated that abaloparatide activation of PTHR1 produces more efficient coupling of PTHR1 to Gs‐cAMP and β‐arrestin signaling mechanisms than that for teriparatide.
Human life expectancy has increased, thanks to the advances in medical research and the subsequent improvement in health care. As a result, there is a growing demand for better management of the common illnesses of the increasing elderly population. One of these illnesses, osteoporosis, is a common world‐wide bone disorder and a major public health threat for 44 million Americans, 68% of them being women. Osteoporosis accounts for an estimated 1.5 million fractures in the US every year costing over 14 billion dollars. Furthermore, the National Osteoporosis Foundation estimates that 50% of women and 25% of men will break a bone in their lifetime due to osteoporosis. The development of drugs that efficiently control bone resorption and/or enhance bone formation will lead to prevention of fractures. In this regard, various antiresorptive drugs, such as bisphosphonates, estrogens, calcitonin, and human monoclonal antibodies (e.g., denosumab to RANKL), have become available. Although these antiresorptive agents prevent further bone loss by inhibiting osteoclast function, they cannot completely restore bone structure. The reduction in fracture risk has also not been satisfactory and some drugs have serious side effects. In light of these drawbacks and the search for anabolic option, animal studies demonstrated that a regimen of daily administration of PTH promotes osteoblast cell differentiation and survival leading to enhanced bone formation and improved bone biomechanical properties (Liu and Kalu 1990; Hirano et al. 1999; Jilka et al. 1999; Bellido et al. 2003; Iida‐Klein et al. 2007; Jilka 2007; Jilka et al. 2010; Kim et al. 2012). Subsequent evaluation of patients with osteoporosis further confirmed that PTH increases BMD in the spine and femur of men and women more efficiently than treatment with bisphosphonates, and decreased the risk of vertebral and nonvertebral fractures by more than 50% (Neer et al. 2001). As a result, the amino‐terminal fragment of PTH (PTH1‐34, teriparatide or Forteo) became the first osteoanabolic treatment approved by the US FDA for treatment of patients with primary (Neer et al. 2001) or secondary osteoporosis due to glucocorticoid therapy who have a high risk of fracture (Saag et al. 2007).
Recently, abaloparatide, a novel modified PTHrP1‐34 and PTHR1 agonist analog, has emerged as another attractive candidate with potent anabolic activity and limited bone resorptive and calcemic response when compared to teriparatide (Leder et al. 2015; Bahar et al. 2016; Miller et al. 2016; Cosman et al. 2017; Varela et al. 2017; Chandler et al. 2018; Doyle et al. 2018). Abaloparatide was therefore recently approved by the US FDA for treatment of severe osteoporosis.
One of these studies was an 18‐month randomized, double‐blind placebo‐controlled Abaloparatide Comparator Trial in Vertebral Endpoints (ACTIVE) trial of more than 2000 women with postmenopausal osteoporosis. Abaloparatide was associated with an 86% reduction in vertebral fracture incidence, the primary end point, compared with placebo (Cosman et al. 2017). Additionally, abaloparatide administration resulted in significantly greater reductions in nonvertebral fractures compared with teriparatide administration and minimal activation of bone resorption (Leder et al. 2015; Cosman et al. 2017; Bilezikian et al. 2018). Our findings suggest that the higher reduction in fracture risk by abaloparatide over teriparatide is likely attributed to its improvement in bone microarchitecture, primarily trabecular and cortical bone thickness. The expansion in Tb. Th produced by the 20 but not the 80 µg/kg/day dose of abaloparatide might be ascribed to stimulation of a catabolic bone mechanism(s) by the higher abaloparatide concentration. Furthermore, the more pronounced effects of abaloparatide than teriparatide on the cortical bone compartment could explain the higher reduction in long bone fracture risk achieved by abaloparatide. The more amplified cortical bone effects of abaloparatide also suggest that periosteal postmitotic pre‐osteoblasts are likely more responsive to abaloparatide stimulation. Periosteal postmitotic pre‐osteoblasts have previously been implicated in daily PTH effects on cortical bone formation due to stimulation of progenitor differentiation and/or survival (Jilka et al. 2009). A decrease in the plate/rod ratio (high SMI) and connectivity are believed to be major contributing factors to fragility fracture in various forms of osteoporosis and with advancing age, while an increase in these parameters induced by treatment with bone anabolic and antiresorptive agents is beneficial for bone quality and strength (Ding and Hvid 2000; Benito et al. 2003; Borah et al. 2004; Rupprecht et al. 2006; Benhamou 2007; Liu et al. 2013; Altman et al. 2014; Stein et al. 2014; Sutter et al. 2014; Zhou et al. 2014; Chang et al. 2015). Our results suggest that both abaloparatide and teriparatide similarly and effectively enhance femoral trabecular connectivity and modify femoral bone structure in favor of concave plate‐like structures. In contrast, the effects of teriparatide and abaloparatide on the spine were predominant on the vertebral trabecular connectivity and number; the actions of the two agents on BV/TV, Tb. Th, Tb. Sp, and SMI were either modest or absent. This disparity in the axial (L5) and appendicular (femur) bone responsiveness to both agents suggests that unloading and weight bearing may play a role in modifying the skeletal response to PTHR1 activation. The comparable responsiveness of the spine to teriparatide and abaloparatide treatment is consistent with those findings reported recently in ovariectomized rats showing parallel improvement in lumbar BMD and strength by both agents (Makino et al. 2018) and further support the reduced vertebral fracture risk in humans.
Surprisingly, we did not observe changes in CTX serum levels, a commonly used bone resorption marker, in response to abaloparatide or teriparatide administration. On the other hand, TRAcP‐5b, a specific serum marker of osteoclast activation, was more sensitive to abaloparatide stimulation than teriparatide. The difference in the response of CTX and TRAcP‐5b markers to teriparatide and abaloparatide stimulation could be explained by the report that TRAcP‐5b is a more specific and sensitive osteoclast and resorption marker than CTX (Halleen et al. 2001). Alternatively, another report suggested that CTX is more reflective of osteoclast activity while TRAcP‐5b of osteoclast number (Rissanen et al. 2008). Other factors that regulate bone resorption are RANKL and its decoy receptor OPG; both are important cytokines produced primarily by the osteoblast lineage cells. Binding of RANKL to its receptor (RANK) in the osteoclast progenitor cells stimulates osteoclast formation and activation, and inhibits apoptosis leading to enhanced bone resorption. As such, the balance between the two cytokines expressed as RANKL/OPG ratio controls the process of bone resorption. In this regard, the only evidence we report for a possible weaker osteoclast activation and bone resorption by abaloparatide is the decrease in RANKL/OPG ratio produced by abaloparatide administration. Thus, the bone turnover and the RANKL/OPG findings suggest that abaloparatide retains a fine‐tuned (due to low RANKL/OPG) bone resorptive property but possesses superior bone formation capacity over teriparatide. Indeed, it appears that the ability of abaloparatide to more efficiently couple bone resorption and formation than teriparatide accounts for the observed more enhanced bone gain. However, we acknowledge that a more comprehensive analysis including longer and shorter time course is needed for careful evaluation of the catabolic and anabolic activity of both abaloparatide and teriparatide. Moreover, whether the catabolic effects of abaloparatide would be modified by the prior status of bone resorption, such as in estrogen‐deficient (ovariectomized) mice, remains to be elucidated.
Hypercalcemia is transient and infrequent event with teriparatide or abaloparatide therapy and has been reported in 6.4% and 3.4% of patients, respectively (Miller et al. 2018). Consistently, we did not observe a significant difference in serum Ca2+ among animals treated with vehicle, abaloparatide, or teriparatide. The significant increase in serum Ca2+ detected when abaloparatide was compared to vehicle‐treated animals is consistent with the amplified PTHR1 signaling by abaloparatide treatment. The elevation in serum Pi induced by the higher doses of teriparatide and abaloparatide is probably related to the increased bone resorption and/or stimulation of Vitamin D‐mediated intestinal Pi resorption. It is important to emphasize that these findings reflect the effects of teriparatide and abaloparatide on mineral homeostasis in response to intermittent treatment and 24 h after the last injection. The classic hypercalcemic and hypophosphatemic actions of PTH have commonly and more accurately been assessed using multiple measurements during the first 0.5–24 h after drug injection (Maeda et al. 2013; Bi et al. 2016). This is, however, beyond the scope of this investigation.
Despite this major progress in clinical development of PTHR1‐based anabolic agents, there are still limitations and unmet needs, primarily the absence of other modes of administration causing lack of treatment adherence, and the restricted 2‐year anabolic window. To overcome these limitations and to design next generation of improved PTHR1 bone therapeutics, a better understanding of the cellular mechanisms that underlie the superior anabolic actions of abaloparatide over teriparatide should prove useful.
PTHR1 is a member of subclass B seven transmembrane spanning GTP‐binding protein (G‐protein) coupled receptors (GPCR) that activates multiple G‐proteins, primarily the Gs but also Gq/11, Gi, and G12/13 G‐proteins. As a result, PTHR1 activation stimulates multiple downstream pathways and events such as Gs/adenylate cyclase/cAMP/protein kinase A, Gq11/phospholipase C/diacylglycerol‐inositol‐triphosphate‐Ca/protein kinase C, phospholipase D, and Src/β‐arrestin/MAPK pathways and PTHR1 phosphorylation and internalization (Abou‐Samra et al. 1992; Sneddon et al. 2000; Tawfeek et al. 2001; Tawfeek et al. 2002; Ahmed et al. 2003; Sneddon et al. 2004; Tawfeek and Abou‐Samra 2004; Singh et al. 2005; Gesty‐Palmer et al. 2006; Rey et al. 2006; Sneddon and Friedman 2007; Sneddon et al. 2007; Tawfeek and Abou‐Samra 2008; Garrido et al. 2009; Tawfeek and Abou‐Samra 2012). Despite the complexity of PTHR1 signaling, only two pathways, namely Gs‐cAMP and β‐arrestin, have been implicated as the major mediators of PTH effects on bone remodeling. The role of Gs‐cAMP pathway was initially suggested using a mouse model of a constitutively active naturally occurring PTHR1 mutant (H223R). This mutant has been shown to enhance basal Gs/cAMP activity when expressed in cells and to subsequently cause great increase in trabecular bone turnover and volume whether expressed in osteoblasts or osteocytes (Calvi and Schipani 2000; Calvi et al. 2001; O'Brien et al. 2008; Rhee et al. 2011). Following studies further demonstrated that postnatal deletion of the G‐protein Gs alpha in the osteoblast lineage cells of male or female mice led to loss of daily PTH‐increased fractional trabecular bone volume and cortical bone thickness (Sinha et al. 2016). The role of β‐arrestin in PTHR1 osteoanabolic effects was suggested by investigations using a D‐Trp12, Tyr34 bovine PTH (7‐34), a signal selective PTH analog; this analog has been shown to stimulate PTHR1 recruitment of β‐arrestin/ERK1/2 signaling but to lack PTHR1 activation of G‐proteins signaling (Gesty‐Palmer et al. 2006). When injected daily to WT mice, D‐Trp12, Tyr34 bovine PTH (7‐34) produced an increase in bone formation and bone gain (Gesty‐Palmer et al. 2009; Bohinc and Gesty‐Palmer 2011). However, these effects were attenuated when the same analog was administered to β‐arrestin 2 knockout mice. Furthermore, Gq/11 pathway in osteoblast cells seems to act as a negative regulator of PTH stimulation of bone mass (Ogata et al. 2007; 2011). In this context, our findings demonstrate that abaloparatide activation of PTHR1 causes a reduction in EC50 values for both cAMP and β‐arrestin stimulation when compared to teriparatide. Accordingly, the greater anabolic effects of abaloparatide over teriparatide can be attributed, at least, in part to the abaloparatide‐improved PTHR1 signaling to both Gs/cAMP and β‐arrestin pathways. Recently, however, Ricarte et al. indicated that PTH (1–34) treatment produced higher stimulation of cAMP and its downstream PKA effectors than PTHrP (1‐36) or abaloparatide (Ricarte et al. 2018). Moreover, PTH was shown to induce larger RANKL gene expression in vivo and in vitro (Ricarte et al. 2018). The discrepancy between our findings and those of Ricarte et al is likely attributed to the different osteoblast cell systems (primary calvarial osteoblast vs. clonal MC3T3‐E1 cell line) and/or the methodology used in the two studies. On the other hand, the difference in RANKL results clearly reflects the complexity of the mechanisms regulating serum RANKL versus RANKL gene expression in osteoblast cells or in cells in bone extract. Alternatively, gene and its protein product levels do not necessarily correlate given that distinct regulatory mechanisms are involved in transcription and translation.
In conclusion, the present report demonstrates that the superior osteoanabolic effects of abaloparatide are likely attributed to the enhanced bone formation and the subsequent improvement of both cortical and trabecular bone thickness. The study further suggests that these favorable effects are mediated by the greater activation of both Gs‐cAMP and β‐arrestin signaling by abaloparatide.
Conflict of Interest
The authors declare no competing financial interests.
Acknowledgments
We thank L. Liaw and T. Henderson at the Center for Molecular Medicine at Maine Medical Center Research Institute (Scarborough, ME) for performing the micro‐CT analysis.
Sahbani Karim Cardozo Christopher P. Bauman William A. Tawfeek Hesham A.. Abaloparatide exhibits greater osteoanabolic response and higher cAMP stimulation and β‐arrestin recruitment than teriparatide. Physiol Rep, 7(19), 2019, e14225, 10.14814/phy2.14225
Funding Information
This work was supported by the Department of Veterans Affairs Rehabilitation Research and Development Service (B2020C to WAB).
References
- Abou‐Samra, A. B. , Juppner H., Force T., Freeman M. W., Kong X. F., Schipani E., et al. 1992. Expression cloning of a common receptor for parathyroid hormone and parathyroid hormone‐related peptide from rat osteoblast‐like cells: a single receptor stimulates intracellular accumulation of both cAMP and inositol trisphosphates and increases intracellular free calcium. Proc. Natl. Acad. Sci. USA 89:2732–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed, I. , Gesty‐Palmer D., Drezner M. K., and Luttrell L. M.. 2003. Transactivation of the epidermal growth factor receptor mediates parathyroid hormone and prostaglandin F2 alpha‐stimulated mitogen‐activated protein kinase activation in cultured transgenic murine osteoblasts. Mol. Endocrinol. 17:1607–1621. [DOI] [PubMed] [Google Scholar]
- Altman, A. R. , Tseng W. J., de Bakker C. M., Huh B. K., Chandra A., Qin L., et al. 2014. A closer look at the immediate trabecula response to combined parathyroid hormone and alendronate treatment. Bone 61:149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar, H. , Gallacher K., Downall J., Nelson C. A., Shomali M., and Hattersley G.. 2016. Six weeks of daily abaloparatide treatment increased vertebral and femoral bone mineral density, microarchitecture and strength in ovariectomized osteopenic rats. Calcif. Tissue Int. 99:489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedi, B. , Li J. Y., Grassi F., Tawfeek H., Weitzmann M. N., and Pacifici R.. 2010. Inhibition of antigen presentation and T cell costimulation blocks PTH‐induced bone loss. Ann. N. Y. Acad. Sci. 1192:215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedi, B. , Li J. Y., Tawfeek H., Baek K. H., Adams J., Vangara S. S., et al. 2012. Silencing of parathyroid hormone (PTH) receptor 1 in T cells blunts the bone anabolic activity of PTH. Proc. Natl. Acad. Sci. USA 109:E725–E733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellido, T. 2006. Downregulation of SOST/sclerostin by PTH: a novel mechanism of hormonal control of bone formation mediated by osteocytes. J. Musculoskelet. Neuronal Interact. 6:358–359. [PubMed] [Google Scholar]
- Bellido, T. , Ali A. A., Plotkin L. I., Fu Q., Gubrij I., Roberson P. K., et al. 2003 Proteasomal degradation of Runx2 shortens parathyroid hormone‐induced anti‐apoptotic signaling in osteoblasts. A putative explanation for why intermittent administration is needed for bone anabolism. J. Biol. Chem. 278:50259–50272. [DOI] [PubMed] [Google Scholar]
- Bellido, T. , Ali A. A., Gubrij I., Plotkin L. I., Fu Q., O'Brien C. A., et al. 2005. Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology 146:4577–4583. [DOI] [PubMed] [Google Scholar]
- Benhamou, C. L. 2007. Effects of osteoporosis medications on bone quality. Joint Bone Spine 74:39–47. [DOI] [PubMed] [Google Scholar]
- Benito, M. , Gomberg B., Wehrli F. W., Weening R. H., Zemel B., Wright A. C., et al. 2003. Deterioration of trabecular architecture in hypogonadal men. J. Clin. Endocrinol. Metab. 88:1497–1502. [DOI] [PubMed] [Google Scholar]
- Bi, R. , Fan Y., Lauter K., Hu J., Watanabe T., Cradock J., et al. 2016. Diphtheria toxin‐ and GFP‐based mouse models of acquired hypoparathyroidism and treatment with a long‐acting parathyroid hormone analog. J. Bone Miner. Res. 31:975–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilezikian, J. P. , Hattersley G., Fitzpatrick L. A., Harris A. G., Shevroja E., Banks K., et al. 2018. Abaloparatide‐SC improves trabecular microarchitecture as assessed by trabecular bone score (TBS): a 24‐week randomized clinical trial. Osteoporos. Int. 29:323–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohinc, B. N. , and Gesty‐Palmer D.. 2011. beta‐arrestin‐biased agonism at the parathyroid hormone receptor uncouples bone formation from bone resorption. Endocrinol. Metab. Immune Disord. Drug Targets 11:112–119. [DOI] [PubMed] [Google Scholar]
- Borah, B. , Dufresne T. E., Chmielewski P. A., Johnson T. D., Chines A., and Manhart M. D.. 2004. Risedronate preserves bone architecture in postmenopausal women with osteoporosis as measured by three‐dimensional microcomputed tomography. Bone 34:736–746. [DOI] [PubMed] [Google Scholar]
- Boras‐Granic, K. , Dann P., Vanhouten J., Karaplis A., and Wysolmerski J.. 2014. Deletion of the nuclear localization sequences and C‐terminus of PTHrP impairs embryonic mammary development but also inhibits PTHrP production. PLoS ONE 9:e90418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouxsein, M. L. , Pierroz D. D., Glatt V., Goddard D. S., Cavat F., Rizzoli R., et al. 2005. beta‐Arrestin2 regulates the differential response of cortical and trabecular bone to intermittent PTH in female mice. J. Bone Miner. Res. 20:635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouxsein, M. L. , Boyd S. K., Christiansen B. A., Guldberg R. E., Jepsen K. J., and Muller R.. 2010. Guidelines for assessment of bone microstructure in rodents using micro‐computed tomography. J. Bone Miner. Res. 25:1468–1486. [DOI] [PubMed] [Google Scholar]
- Burtis, W. J. , Fodero J. P., Gaich G., Debeyssey M., and Stewart A. F.. 1992. Preliminary characterization of circulating amino‐ and carboxy‐terminal fragments of parathyroid hormone‐related peptide in humoral hypercalcemia of malignancy. J. Clin. Endocrinol. Metab. 75:1110–1114. [DOI] [PubMed] [Google Scholar]
- Calvi, L. M. , and Schipani E.. 2000. The PTH/PTHrP receptor in Jansen's metaphyseal chondrodysplasia. J. Endocrinol. Invest. 23:545–554. [DOI] [PubMed] [Google Scholar]
- Calvi, L. M. , Sims N. A., Hunzelman J. L., Knight M. C., Giovannetti A., Saxton J. M., et al. 2001. Activated parathyroid hormone/parathyroid hormone‐related protein receptor in osteoblastic cells differentially affects cortical and trabecular bone. J. Clin. Invest. 107:277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cebrian, A. , Garcia‐Ocana A., Takane K. K., Sipula D., Stewart A. F., and Vasavada R. C.. 2002. Overexpression of parathyroid hormone‐related protein inhibits pancreatic beta‐cell death in vivo and in vitro. Diabetes 51:3003–3013. [DOI] [PubMed] [Google Scholar]
- Chandler, H. , Lanske B., Varela A., Guillot M., Boyer M., Brown J., et al. 2018. Abaloparatide, a novel osteoanabolic PTHrP analog, increases cortical and trabecular bone mass and architecture in orchiectomized rats by increasing bone formation without increasing bone resorption. Bone 120:148–155. [DOI] [PubMed] [Google Scholar]
- Chang, G. , Honig S., Liu Y., Chen C., Chu K. K., Rajapakse C. S., et al. 2015. 7 Tesla MRI of bone microarchitecture discriminates between women without and with fragility fractures who do not differ by bone mineral density. J. Bone Miner. Metab. 33:285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, U. I. , Lanske B., Lee K., Li E., and Kronenberg H.. 1998. The parathyroid hormone/parathyroid hormone‐related peptide receptor coordinates endochondral bone development by directly controlling chondrocyte differentiation. Proc. Natl. Acad. Sci. USA 95:13030–13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosman, F. , Hattersley G., Hu M. Y., Williams G. C., Fitzpatrick L. A., and Black D. M.. 2017. Effects of abaloparatide‐SC on fractures and bone mineral density in subgroups of postmenopausal women with osteoporosis and varying baseline risk factors. J. Bone Miner. Res. 32:17–23. [DOI] [PubMed] [Google Scholar]
- Ding, M. and Hvid I.. 2000. Quantification of age‐related changes in the structure model type and trabecular thickness of human tibial cancellous bone. Bone 26:291–295. [DOI] [PubMed] [Google Scholar]
- Doyle, N. , Varela A., Haile S., Guldberg R., Kostenuik P. J., Ominsky M. S., et al. 2018. Abaloparatide, a novel PTH receptor agonist, increased bone mass and strength in ovariectomized cynomolgus monkeys by increasing bone formation without increasing bone resorption. Osteoporos. Int. 29:685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari, S. L. , Behar V., Chorev M., Rosenblatt M., and Bisello A.. 1999. Endocytosis of ligand‐human parathyroid hormone receptor 1 complexes is protein kinase C‐dependent and involves beta‐arrestin2. Real‐time monitoring by fluorescence microscopy. J. Biol. Chem. 274:29968–29975. [DOI] [PubMed] [Google Scholar]
- Foley, J. , Longely B. J., Wysolmerski J. J., Dreyer B. E., Broadus A. E., and Philbrick W. M.. 1998. PTHrP regulates epidermal differentiation in adult mice. J. Invest. Dermatol. 111:1122–1128. [DOI] [PubMed] [Google Scholar]
- Foley, J. , Dann P., Hong J., Cosgrove J., Dreyer B., Rimm D., et al. 2001. Parathyroid hormone‐related protein maintains mammary epithelial fate and triggers nipple skin differentiation during embryonic breast development. Development 128:513–525. [DOI] [PubMed] [Google Scholar]
- Gaich, G. , and Burtis W. J.. 1990. Measurement of circulating parathyroid hormone‐related protein in rats with humoral hypercalcemia of malignancy using a two‐site immunoradiometric assay. Endocrinology 127:1444–1449. [DOI] [PubMed] [Google Scholar]
- Gao, Y. , Wu X., Terauchi M., Li J. Y., Grassi F., Galley S., et al. 2008. T cells potentiate PTH‐induced cortical bone loss through CD40L signaling. Cell Metab. 8:132–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido, J. L. , Wheeler D., Vega L. L., Friedman P. A., and Romero G.. 2009. Role of phospholipase D in parathyroid hormone type 1 receptor signaling and trafficking. Mol. Endocrinol. 23:2048–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesty‐Palmer, D. , Chen M., Reiter E., Ahn S., Nelson C. D., Wang S., et al. 2006. Distinct beta‐arrestin‐ and G protein‐dependent pathways for parathyroid hormone receptor‐stimulated ERK1/2 activation. J. Biol. Chem. 281:10856–10864. [DOI] [PubMed] [Google Scholar]
- Gesty‐Palmer, D. , Flannery P., Yuan L., Corsino L., Spurney R., Lefkowitz R. J., et al. 2009. A beta‐arrestin‐biased agonist of the parathyroid hormone receptor (PTH1R) promotes bone formation independent of G protein activation. Sci. Transl. Med. 1:1ra1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grill, V. , Ho P., Body J. J., Johanson N., Lee S. C., Kukreja S. C., et al. 1991. Parathyroid hormone‐related protein: elevated levels in both humoral hypercalcemia of malignancy and hypercalcemia complicating metastatic breast cancer. J. Clin. Endocrinol. Metab. 73:1309–1315. [DOI] [PubMed] [Google Scholar]
- Halleen, J. M. , Alatalo S. L., Janckila A. J., Woitge H. W., Seibel M. J., and Vaananen H. K.. 2001. Serum tartrate‐resistant acid phosphatase 5b is a specific and sensitive marker of bone resorption. Clin. Chem. 47:597–600. [PubMed] [Google Scholar]
- Hanyu, R. , Wehbi V. L., Hayata T., Moriya S., Feinstein T. N., Ezura Y., et al. 2012. Anabolic action of parathyroid hormone regulated by the beta2‐adrenergic receptor. Proc. Natl. Acad. Sci. USA 109:7433–7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksen, K. , Neutzsky‐Wulff A. V., Bonewald L. F., and Karsdal M. A.. 2009. Local communication on and within bone controls bone remodeling. Bone 44:1026–1033. [DOI] [PubMed] [Google Scholar]
- Hens, J. R. , Dann P., Zhang J. P., Harris S., Robinson G. W., and Wysolmerski J.. 2007. BMP4 and PTHrP interact to stimulate ductal outgrowth during embryonic mammary development and to inhibit hair follicle induction. Development 134:1221–1230. [DOI] [PubMed] [Google Scholar]
- Hirai, T. , Chagin A. S., Kobayashi T., Mackem S., and Kronenberg H. M.. 2011. Parathyroid hormone/parathyroid hormone‐related protein receptor signaling is required for maintenance of the growth plate in postnatal life. Proc. Natl. Acad. Sci. USA 108:191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano, T. , Burr D. B., Turner C. H., Sato M., Cain R. L., and Hock J. M.. 1999. Anabolic effects of human biosynthetic parathyroid hormone fragment (1–34), LY333334, on remodeling and mechanical properties of cortical bone in rabbits. J. Bone Miner. Res. 14:536–545. [DOI] [PubMed] [Google Scholar]
- Honda, S. , Yamaguchi K., Suzuki M., Sato Y., Adachi I., Kimura S., et al. 1988. Expression of parathyroid hormone‐related protein mRNA in tumors obtained from patients with humoral hypercalcemia of malignancy. Jpn. J. Cancer Res. 79:677–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz, M. J. , Tedesco M. B., Gundberg C., Garcia‐Ocana A., and Stewart A. F.. 2003. Short‐term, high‐dose parathyroid hormone‐related protein as a skeletal anabolic agent for the treatment of postmenopausal osteoporosis. J. Clin. Endocrinol. Metab. 88:569–575. [DOI] [PubMed] [Google Scholar]
- Horwitz, M. J. , Tedesco M. B., Garcia‐Ocana A., Sereika S. M., Prebehala L., Bisello A., et al. 2010. Parathyroid hormone‐related protein for the treatment of postmenopausal osteoporosis: defining the maximal tolerable dose. J. Clin. Endocrinol. Metab. 95:1279–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz, M. J. , Augustine M., Khan L., Martin E., Oakley C. C., Carneiro R. M., et al. 2013. A comparison of parathyroid hormone‐related protein (1–36) and parathyroid hormone (1–34) on markers of bone turnover and bone density in postmenopausal women: the PrOP study. J. Bone Miner. Res. 28:2266–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida‐Klein, A. , Lu S. S., Cosman F., Lindsay R., and Dempster D. W.. 2007. Effects of cyclic vs. daily treatment with human parathyroid hormone (1–34) on murine bone structure and cellular activity. Bone 40:391–398. [DOI] [PubMed] [Google Scholar]
- Jilka, R. L. 2007. Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone 40:1434–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jilka, R. L. , Weinstein R. S., Bellido T., Roberson P., Parfitt A. M., and Manolagas S. C.. 1999. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J. Clin. Invest. 104:439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jilka, R. L. , O'Brien C. A., Ali A. A., Roberson P. K., Weinstein R. S., and Manolagas S. C.. 2009. Intermittent PTH stimulates periosteal bone formation by actions on post‐mitotic preosteoblasts. Bone 44:275–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jilka, R. L. , Almeida M., Ambrogini E., Han L., Roberson P. K., Weinstein R. S., et al. 2010. Decreased oxidative stress and greater bone anabolism in the aged, when compared to the young, murine skeleton with parathyroid hormone administration. Aging Cell 9:851–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jilka, R. L. , O'Brien C. A., Bartell S. M., Weinstein R. S., and Manolagas S. C.. 2010. Continuous elevation of PTH increases the number of osteoblasts via both osteoclast‐dependent and ‐independent mechanisms. J. Bone Miner. Res. 25:2427–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S. W. , Pajevic P. D., Selig M., Barry K. J., Yang J. Y., Shin C. S., et al. 2012. Intermittent parathyroid hormone administration converts quiescent lining cells to active osteoblasts. J. Bone Miner. Res. 27:2075–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukreja, S. C. , Shevrin D. H., Wimbiscus S. A., Ebeling P. R., Danks J. A., Rodda C. P., et al. 1988. Antibodies to parathyroid hormone‐related protein lower serum calcium in athymic mouse models of malignancy‐associated hypercalcemia due to human tumors. J. Clin. Invest. 82:1798–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanske, B. , Amling M., Neff L., Guiducci J., Baron R., and Kronenberg H. M.. 1999. Ablation of the PTHrP gene or the PTH/PTHrP receptor gene leads to distinct abnormalities in bone development. J. Clin. Invest. 104:399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leder, B. Z. , O'Dea L. S., Zanchetta J. R., Kumar P., Banks K., McKay K., et al. 2015. Effects of abaloparatide, a human parathyroid hormone‐related peptide analog, on bone mineral density in postmenopausal women with osteoporosis. J. Clin. Endocrinol. Metab. 100:697–706. [DOI] [PubMed] [Google Scholar]
- Leder, B. Z. , Tsai J. N., Uihlein A. V., Wallace P. M., Lee H., Neer R. M., et al. 2015. Denosumab and teriparatide transitions in postmenopausal osteoporosis (the DATA‐Switch study): extension of a randomised controlled trial. Lancet 386:1147–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, K. , Lanske B., Karaplis A. C., Deeds J. D., Kohno H., Nissenson R. A., et al. 1996. Parathyroid hormone‐related peptide delays terminal differentiation of chondrocytes during endochondral bone development. Endocrinology 137:5109–5118. [DOI] [PubMed] [Google Scholar]
- Liu, C. C. , and Kalu D. N.. 1990. Human parathyroid hormone‐(1–34) prevents bone loss and augments bone formation in sexually mature ovariectomized rats. J. Bone Miner. Res. 5:973–982. [DOI] [PubMed] [Google Scholar]
- Liu, X. S. , Wang J., Zhou B., Stein E., Shi X., Adams M., et al. 2013. Fast trabecular bone strength predictions of HR‐pQCT and individual trabeculae segmentation‐based plate and rod finite element model discriminate postmenopausal vertebral fractures. J. Bone Miner. Res. 28:1666–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locklin, R. M. , Khosla S., Turner R. T., and Riggs B. L.. 2003. Mediators of the biphasic responses of bone to intermittent and continuously administered parathyroid hormone. J. Cell Biochem. 89:180–190. [DOI] [PubMed] [Google Scholar]
- Lotinun, S. , Evans G. L., Bronk J. T., Bolander M. E., Wronski T. J., Ritman E. L., et al. 2004. Continuous parathyroid hormone induces cortical porosity in the rat: effects on bone turnover and mechanical properties. J. Bone Miner. Res. 19:1165–1171. [DOI] [PubMed] [Google Scholar]
- Maeda, A. , Okazaki M., Baron D. M., Dean T., Khatri A., Mahon M., et al. 2013. Critical role of parathyroid hormone (PTH) receptor‐1 phosphorylation in regulating acute responses to PTH. Proc. Natl. Acad. Sci. USA 110:5864–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino, A. , Takagi H., Takahashi Y., Hase N., Sugiyama H., Yamana K., et al. 2018. Abaloparatide exerts bone anabolic effects with less stimulation of bone resorption‐related factors: a comparison with teriparatide. Calcif. Tissue Int. 103:289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, T. J. , and Sims N. A.. 2005. Osteoclast‐derived activity in the coupling of bone formation to resorption. Trends Mol. Med. 11:76–81. [DOI] [PubMed] [Google Scholar]
- Martin, T. , Gooi J. H., and Sims N. A.. 2009. Molecular mechanisms in coupling of bone formation to resorption. Crit. Rev. Eukaryot. Gene Expr. 19:73–88. [DOI] [PubMed] [Google Scholar]
- Meziani, F. , Tesse A., Welsch S., Kremer H., Barthelmebs M., Andriantsitohaina R., et al. 2008. Expression and biological activity of parathyroid hormone‐related peptide in pregnant rat uterine artery: any role for 8‐iso‐prostaglandin F2alpha? Endocrinology 149:626–633. [DOI] [PubMed] [Google Scholar]
- Miao, D. , He B., Jiang Y., Kobayashi T., Soroceanu M. A., Zhao J., et al. 2005. Osteoblast‐derived PTHrP is a potent endogenous bone anabolic agent that modifies the therapeutic efficacy of administered PTH 1–34. J. Clin. Invest. 115:2402–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, P. D. , Hattersley G., Riis B. J., Williams G. C., Lau E., Russo L. A., et al. 2016. Effect of abaloparatide vs placebo on new vertebral fractures in postmenopausal women with osteoporosis: a randomized clinical trial. JAMA 316:722–733. [DOI] [PubMed] [Google Scholar]
- Miller, P. D. , Hattersley G., Lau E., Fitzpatrick L. A., Harris A. G., Williams G. C., et al. 2018. Bone mineral density response rates are greater in patients treated with abaloparatide compared with those treated with placebo or teriparatide: results from the ACTIVE phase 3 trial. Bone 120:137–140. [DOI] [PubMed] [Google Scholar]
- Mozar, A. , Lin H., Williams K., Chin C., Li R., Kondegowda N. G., et al. 2016. Parathyroid hormone‐related peptide (1–36) enhances beta cell regeneration and increases beta cell mass in a mouse model of partial pancreatectomy. PLoS ONE 11:e0158414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata, N. , Yasutomo Y., Kugai N., Matsuura Y., Akatsu T., Sugiyama K., et al. 1989. Parathyroid hormone‐related protein and transforming growth factor activities in an extract from a breast cancer associated with humoral hypercalcemia of malignancy. Jpn. J. Clin. Oncol. 19:353–359. [PubMed] [Google Scholar]
- Neer, R. M. , Arnaud C. D., Zanchetta J. R., Prince R., Gaich G. A., Reginster J. Y., et al. 2001. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N. Engl. J. Med. 344:1434–1441. [DOI] [PubMed] [Google Scholar]
- Noda, M. , Katoh T., Takuwa N., Kumada M., Kurokawa K., and Takuwa Y.. 1994. Synergistic stimulation of parathyroid hormone‐related peptide gene expression by mechanical stretch and angiotensin II in rat aortic smooth muscle cells. J. Biol. Chem. 269:17911–17917. [PubMed] [Google Scholar]
- O'Brien, C. A. , Plotkin L. I., Galli C., Goellner J. J., Gortazar A. R., Allen M. R., et al. 2008. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS ONE 3:e2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata, N. , Kawaguchi H., Chung U. I., Roth S. I., and Segre G. V.. 2007. Continuous activation of G alpha q in osteoblasts results in osteopenia through impaired osteoblast differentiation. J. Biol. Chem. 282:35757–35764. [DOI] [PubMed] [Google Scholar]
- Ogata, N. , Shinoda Y., Wettschureck N., Offermanns S., Takeda S., Nakamura K., et al. 2011. G alpha(q) signal in osteoblasts is inhibitory to the osteoanabolic action of parathyroid hormone. J. Biol. Chem. 286:13733–13740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philbrick, W. M. , Dreyer B. E., Nakchbandi I. A., and Karaplis A. C.. 1998. Parathyroid hormone‐related protein is required for tooth eruption. Proc. Natl. Acad. Sci. USA 95:11846–11851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin, H. , Gundberg C., Mitnick M., and Stewart A. F.. 1998. Dissociation of bone formation from resorption during 2‐week treatment with human parathyroid hormone‐related peptide‐(1–36) in humans: potential as an anabolic therapy for osteoporosis. J. Clin. Endocrinol. Metab. 83:2786–2791. [DOI] [PubMed] [Google Scholar]
- Raison, D. , Coquard C., Hochane M., Steger J., Massfelder T., Moulin B., et al. 2013. Knockdown of parathyroid hormone related protein in smooth muscle cells alters renal hemodynamics but not blood pressure. Am. J. Physiol. Renal Physiol. 305:F333–F342. [DOI] [PubMed] [Google Scholar]
- Rey, A. , Manen D., Rizzoli R., Caverzasio J., and Ferrari S. L.. 2006. Proline‐rich motifs in the parathyroid hormone (PTH)/PTH‐related protein receptor C terminus mediate scaffolding of c‐Src with beta‐arrestin2 for ERK1/2 activation. J. Biol. Chem. 281:38181–38188. [DOI] [PubMed] [Google Scholar]
- Rhee, Y. , Allen M. R., Condon K., Lezcano V., Ronda A. C., Galli C., et al. 2011. PTH receptor signaling in osteocytes governs periosteal bone formation and intracortical remodeling. J. Bone Miner. Res. 26:1035–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricarte, F. R. , Le Henaff C., Kolupaeva V. G., Gardella T. J., and Partridge N. C.. 2018. Parathyroid hormone(1–34) and its analogs differentially modulate osteoblastic Rankl expression via PKA/SIK2/SIK3 and PP1/PP2A‐CRTC3 signaling. J. Biol. Chem. 293:20200–20213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissanen, J. P. , Suominen M. I., Peng Z., and Halleen J. M.. 2008. Secreted tartrate‐resistant acid phosphatase 5b is a Marker of osteoclast number in human osteoclast cultures and the rat ovariectomy model. Calcif. Tissue Int. 82:108–115. [DOI] [PubMed] [Google Scholar]
- Robling, A. G. , Kedlaya R., Ellis S. N., Childress P. J., Bidwell J. P., Bellido T., et al. 2011. Anabolic and catabolic regimens of human parathyroid hormone 1–34 elicit bone‐ and envelope‐specific attenuation of skeletal effects in Sost‐deficient mice. Endocrinology 152:2963–2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupprecht, M. , Pogoda P., Mumme M., Rueger J. M., Puschel K., and Amling M.. 2006. Bone microarchitecture of the calcaneus and its changes in aging: a histomorphometric analysis of 60 human specimens. J. Orthop. Res. 24:664–674. [DOI] [PubMed] [Google Scholar]
- Saag, K. G. , Shane E., Boonen S., Marin F., Donley D. W., Taylor K. A., et al. 2007. Teriparatide or alendronate in glucocorticoid‐induced osteoporosis. N. Engl. J. Med. 357:2028–2039. [DOI] [PubMed] [Google Scholar]
- Sims, N. A. , and Vrahnas C.. 2014. Regulation of cortical and trabecular bone mass by communication between osteoblasts, osteocytes and osteoclasts. Arch. Biochem. Biophys. 561:22–28. [DOI] [PubMed] [Google Scholar]
- Singh, A. T. , Gilchrist A., Voyno‐Yasenetskaya T., Radeff‐Huang J. M., and Stern P. H.. 2005. G alpha12/G alpha13 subunits of heterotrimeric G proteins mediate parathyroid hormone activation of phospholipase D in UMR‐106 osteoblastic cells. Endocrinology 146:2171–2175. [DOI] [PubMed] [Google Scholar]
- Sinha, P. , Aarnisalo P., Chubb R., Poulton I. J., Guo J., Nachtrab G., et al. 2016. Loss of Gsalpha in the postnatal skeleton leads to low bone mass and a blunted response to anabolic parathyroid hormone therapy. J. Biol. Chem. 291:1631–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneddon, W. B. , and Friedman P. A.. 2007. Beta‐arrestin‐dependent parathyroid hormone‐stimulated extracellular signal‐regulated kinase activation and parathyroid hormone type 1 receptor internalization. Endocrinology 148:4073–4079. [DOI] [PubMed] [Google Scholar]
- Sneddon, W. B. , Liu F., Gesek F. A., and Friedman P. A.. 2000. Obligate mitogen‐activated protein kinase activation in parathyroid hormone stimulation of calcium transport but not calcium signaling. Endocrinology 141:4185–4193. [DOI] [PubMed] [Google Scholar]
- Sneddon, W. B. , Magyar C. E., Willick G. E., Syme C. A., Galbiati F., Bisello A., et al. 2004. Ligand‐selective dissociation of activation and internalization of the parathyroid hormone (PTH) receptor: conditional efficacy of PTH peptide fragments. Endocrinology 145:2815–2823. [DOI] [PubMed] [Google Scholar]
- Sneddon, W. B. , Yang Y., Ba J., Harinstein L. M., and Friedman P. A.. 2007. Extracellular signal‐regulated kinase activation by parathyroid hormone in distal tubule cells. Am. J. Physiol. Renal Physiol. 292:F1028–F1034. [DOI] [PubMed] [Google Scholar]
- Stein, E. M. , Kepley A., Walker M., Nickolas T. L., Nishiyama K., Zhou B., et al. 2014. Skeletal structure in postmenopausal women with osteopenia and fractures is characterized by abnormal trabecular plates and cortical thinning. J. Bone Miner. Res. 29:1101–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, A. F. , Cain R. L., Burr D. B., Jacob D., Turner C. H., and Hock J. M.. 2000. Six‐month daily administration of parathyroid hormone and parathyroid hormone‐related protein peptides to adult ovariectomized rats markedly enhances bone mass and biomechanical properties: a comparison of human parathyroid hormone 1–34, parathyroid hormone‐related protein 1–36, and SDZ‐parathyroid hormone 893. J. Bone Miner. Res. 15:1517–1525. [DOI] [PubMed] [Google Scholar]
- Sutter, S. , Nishiyama K. K., Kepley A., Zhou B., Wang J., McMahon D. J., et al. 2014. Abnormalities in cortical bone, trabecular plates, and stiffness in postmenopausal women treated with glucocorticoids. J. Clin. Endocrinol. Metab. 99:4231–4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawfeek, H. A. , and Abou‐Samra A. B.. 2004. Important role for the V‐type H(+)‐ATPase and the Golgi apparatus in the recycling of PTH/PTHrP receptor. Am. J. Physiol. Endocrinol. Metab. 286:E704–E710. [DOI] [PubMed] [Google Scholar]
- Tawfeek, H. A. , and Abou‐Samra A. B.. 2008. Negative regulation of parathyroid hormone (PTH)‐activated phospholipase C by PTH/PTH‐related peptide receptor phosphorylation and protein kinase A. Endocrinology 149:4016–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawfeek, H. A. , and Abou‐Samra A. B.. 2012. Disruption of parathyroid hormone and parathyroid hormone‐related peptide receptor phosphorylation prolongs ERK1/2 MAPK activation and enhances c‐fos expression. Am. J. Physiol. Endocrinol Metab. 302:E1363–E1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawfeek, H. A. , Che J., Qian F., and Abou‐Samra A. B.. 2001. Parathyroid hormone receptor internalization is independent of protein kinase A and phospholipase C activation. Am. J. Physiol. Endocrinol. Metab. 281:E545–E557. [DOI] [PubMed] [Google Scholar]
- Tawfeek, H. A. , Qian F., and Abou‐Samra A. B.. 2002. Phosphorylation of the receptor for PTH and PTHrP is required for internalization and regulates receptor signaling. Mol. Endocrinol. 16:1–13. [DOI] [PubMed] [Google Scholar]
- Tawfeek, H. , Bedi B., Li J. Y., Adams J., Kobayashi T., Weitzmann M. N., et al. 2010. Disruption of PTH receptor 1 in T cells protects against PTH‐induced bone loss. PLoS ONE 5:e12290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terauchi, M. , Li J. Y., Bedi B., Baek K. H., Tawfeek H., Galley S., et al. 2009. T lymphocytes amplify the anabolic activity of parathyroid hormone through Wnt10b signaling. Cell Metab. 10:229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchihashi, T. , Yamaguchi K., Miyake Y., Otsubo K., Nagasaki K., Honda S., et al. 1990. Parathyroid hormone‐related protein in tumor tissues obtained from patients with humoral hypercalcemia of malignancy. J. Natl. Cancer Inst. 82:40–44. [DOI] [PubMed] [Google Scholar]
- Varela, A. , Chouinard L., Lesage E., Smith S. Y., and Hattersley G.. 2017. One year of abaloparatide, a selective activator of the PTH1 receptor, increased bone formation and bone mass in osteopenic ovariectomized rats without increasing bone resorption. J Bone Miner Res. 32:24–33. [DOI] [PubMed] [Google Scholar]
- Vortkamp, A. , Lee K., Lanske B., Segre G. V., Kronenberg H. M., and Tabin C. J.. 1996. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH‐related protein. Science 273:613–622. [DOI] [PubMed] [Google Scholar]
- Williams, K. , Abanquah D., Joshi‐Gokhale S., Otero A., Lin H., Guthalu N. K., et al. 2011. Systemic and acute administration of parathyroid hormone‐related peptide(1–36) stimulates endogenous beta cell proliferation while preserving function in adult mice. Diabetologia 54:2867–2877. [DOI] [PubMed] [Google Scholar]
- Wysolmerski, J. J. , Philbrick W. M., Dunbar M. E., Lanske B., Kronenberg H., and Broadus A. E.. 1998. Rescue of the parathyroid hormone‐related protein knockout mouse demonstrates that parathyroid hormone‐related protein is essential for mammary gland development. Development 125:1285–1294. [DOI] [PubMed] [Google Scholar]
- Zhou, B. , Liu X. S., Wang J., Lu X. L., Fields A. J., and Guo X. E.. 2014. Dependence of mechanical properties of trabecular bone on plate‐rod microstructure determined by individual trabecula segmentation (ITS). J. Biomech. 47:702–708. [DOI] [PMC free article] [PubMed] [Google Scholar]