

Abstract
Our previous studies showed that the combination of atorvastatin (ATV) and single injection of ATV‐pretreated mesenchymal stem cells (MSCs) (ATV‐MSCs) at 1 week post‐acute myocardial infarction (AMI) promoted MSC recruitment and survival. This study aimed to investigate whether the combinatorial therapy of intensive ATV with multiple injections of ATV‐MSCs has greater efficacy at different stages to better define the optimal strategy for MSC therapy in AMI. In order to determine the optimal time window for MSC treatment, we first assessed stromal cell‐derived factor‐1 (SDF‐1) dynamic expression and inflammation. Next, we compared MSC recruitment and differentiation, cardiac function, infarct size, and angiogenesis among animal groups with single, dual, and triple injections of ATV‐MSCs at early (Early1, Early2, Early3), mid‐term (Mid1, Mid2, Mid3), and late (Late1, Late2, Late3) stages. Compared with AMI control, intensive ATV significantly augmented SDF‐1 expression 1.5∼2.6‐fold in peri‐infarcted region with inhibited inflammation. ATV‐MSCs implantation with ATV administration further enhanced MSC recruitment rate by 3.9%∼24.0%, improved left ventricular ejection fraction (LVEF) by 2.0%∼16.2%, and reduced infarct size in all groups 6 weeks post‐AMI with most prominent improvement in mid groups and still effective in late groups. Mechanistically, ATV‐MSCs remarkably suppressed inflammation and apoptosis while increasing angiogenesis. Furthermore, triple injections of ATV‐MSCs were much more effective than single administration during early and mid‐term stages of AMI with the best effects in Mid3 group. We conclude that the optimal strategy is multiple injections of ATV‐MSCs combined with intensive ATV administration at mid‐term stage of AMI. The translational potential of this strategy is clinically promising. stem cells translational medicine 2019;8:1068–1083
Keywords: Mesenchymal stem cells, Atorvastatin, Timing, Multiple administrations, Acute myocardial infarction
1.
Significance Statement.
Over the past 2 decades, the optimal time window period of mesenchymal stem cell (MSC) transplantation remains controversial because of challenges in augmenting cell recruitment and survival. This study concluded that the optimal strategy is the use of multiple injections of atorvastatin (ATV)‐pretreated MSCs (ATV‐MSCs) combined with intensive ATV administration at mid‐term stage of acute myocardial infarction (AMI). Furthermore, the intravenous administration of ATV‐MSCs in the current study has the unique advantage over traditional methods like intracoronary or intramyocardium injection, due to clinical convenience and feasibility for repeated transplantations at different time points of different stages of AMI. The translational potential of this study is promising, straightforward, and enormous.
Introduction
Stem cell‐based therapy has been recognized as a promising strategy to restore the functional myocardium in acute myocardial infarction (AMI) for decades 1, 2. Multiple stem cell types have been used in both preclinical experiments and clinical trials, all having pros and cons 3, 4, 5, 6, 7. There is accumulating evidence that bone marrow‐derived mesenchymal stem cells (MSCs) presented an ideal candidate in the fight against ischemic heart diseases due to their distinct advantages over other types of stem cells in terms of potency and availability 8, 9, 10. By activating endogenous tissue repair process through paracrine signaling and immunomodulatory properties 11, 12, 13, MSC‐based therapies displayed remarkable clinical promise. However, low engraftment rate and poor survival of transplanted MSCs continued to pose major challenges on the therapeutic efficacy 14. Under this circumstance, optimized strategies have been explored either to modify stem cells or to improve inflammatory microenvironment, thus facilitating the recruitment and survival of implanted MSCs in ischemic myocardium 15, 16.
Our previous studies have shown that in swine AMI model pretreatment with high dose of intensive statins, a type of broadly used clinical lipid‐lowing medicine could augment the cardioprotective effect of MSC therapy. This is possibly through ameliorating the hazardous microenvironment and reducing the apoptosis of transplanted MSCs 17, 18, due to their pleiotropic effects of anti‐inflammation, antioxidation, and antiapoptosis 19. Atorvastatin (ATV) treatment was also reported to have the potential to enhance natural stromal cell‐derived factor‐1 (SDF‐1) expression in infarct region and extend its peak expression from 1 day to 1 week post‐AMI 20, 21. SDF‐1 is a crucial stem cell homing factor, with its G‐protein coupled receptor CXC chemokine receptor 4 (CXCR4), playing a pivotal role in recruitment of transplanted MSCs 22. Furthermore, we also found that ATV pretreatment could not only prevent MSCs from apoptosis induced by hypoxia and serum deprivation 23, but also enhance the expression of CXCR4 on the surface of MSCs, thus remarkably improving cardiac performance through targeted homing 24.
Most importantly, the optimal time window for stem cell transplantation, which depends on the balance of SDF‐1 expression and inflammatory severity in the peri‐infarct region, is still uncertain. Early stage transplantation within 1 week post‐AMI might provide greater scope for function preservation due to the high expression level of SDF‐1 for the cell recruitment; however, the inflammatory microenvironment is too rigorous for the implanted cells to survive. In contrast, late‐stage transplantation of 3–4 weeks post‐AMI is clinically safer for the patients and may offer a better opportunity for transplanted cells to survive due to ameliorative microenvironment, but the SDF‐1 expression level decreases as time progresses. In addition, adverse ventricular remodeling and scar formation are the other main hindrance for efficacy resulted from late‐stage transplantation. Preclinical studies have also shown controversial results on the time window. One earlier study directly compared the timing of MSC administration immediately after coronary ligation, at 1 and 2 weeks post‐AMI in a rat model 25, indicating that transplanting MSCs at 1 week post‐AMI achieved the highest MSC engraftment and survival rate. By contrast, other studies showed that deferred treatment (2–4 weeks post‐AMI) brought greater benefits than early therapy 26, 27.
Herein, this study first aims to explore the dynamic changes of SDF‐1 expression by intensive ATV over the entire 4‐week period of AMI; and then to perform direct comparison of allogeneic rat ATV‐MSCs transplantation at different time windows including early, mid‐term, and late stages of AMI with single, dual, and triple injections to identify the optimal strategy for MSCs therapy on AMI.
Materials and Methods
Ethical Statement
The experiment project and procedures were approved by the Care of Experiment Animals Committee of Fuwai Hospital, and all the animals have received humane care.
Animal Preparation and Grouping
Sprague–Dawley rats (SD rats) were used both in MSCs extraction and AMI models. MSCs were obtained from male SD rats (3–4 weeks of age, 60–80 g) and the AMI models used female SD rats (6–8 weeks of age, 200–220 g). The animals were obtained from the Animal Department of Fuwai Hospital, where animals were housed and fed in a temperature‐controlled room at 25°C.
In the first part of experiment (Part 1) exploring the SDF‐1 expression over the time course of entire 4‐week period of AMI by intensive ATV treatment, a total of 150 SD rats were randomly divided into three groups of the sham operation (Sham) (n = 50), the AMI control (AMI) (n = 50), and the ATV treatment (ATV) (n = 50). In the ATV group, high dose of intensive ATV (Pfizer Pharmaceutical Company, 10 mg/kg per day) was given by gastric gavage from 2 hours after the AMI model procedure to the euthanized harvest. The dose of oral ATV treatment was determined according to the practice guide of conversion between animals and human 28, 29 and previous studies 30, 31, 32, 33, 34 based on the pharmacokinetics of ATV 35, 36. At 1 day (d1), 1 week (d7), 2 weeks (d14), 3 weeks (d21), and 4 weeks (d28) post‐AMI, hearts in different groups were harvested to analyze the expression of SDF‐1and inflammatory cytokines in the peri‐infarcted myocardium.
In the second part of experiment (Part 2) exploring the optimal strategy of MSCs transplantation, a total of 215 female SD rats were randomized into the following 11 groups of the sham (n = 15), the AMI (n = 20), single, dual, and triple injections of ATV‐MSCs in early stage of AMI at Day 1, 3, and 7 (Early1, 2, and 3 groups), mid‐term stage at Day 7, 11, and 14 (Mid1, 2, and 3 groups) and late stage at Day 21, 25, and 28 (Late1, 2, and 3 groups) with each group of 20 SD rats. Fifty‐four (25.1%) rats died during the AMI model procedure and MSC injection process, and 16 (7.4%) died within first week post‐AMI. Another 42 rats were also excluded from the experiment because their baseline echocardiography data (3 days post‐AMI) did not meet the prespecified criteria for successful induction of AMI model (LVEF <60% and abnormal motion of the left ventricular wall). In all the groups (except the Sham and AMI groups), intensive ATV (10 mg/kg per day) was given by gastric gavage 2 hours after the AMI model procedure and continued for 6 weeks. For cell delivery, 2 × 106 1,1′‐dioctadecyl‐3,3,3′,3′‐tetramethylindocarbocyanine perchlorate (CM‐Dil) labeled ATV‐MSCs were transplanted through vein tail in each injection (Fig. 1).
Figure 1.
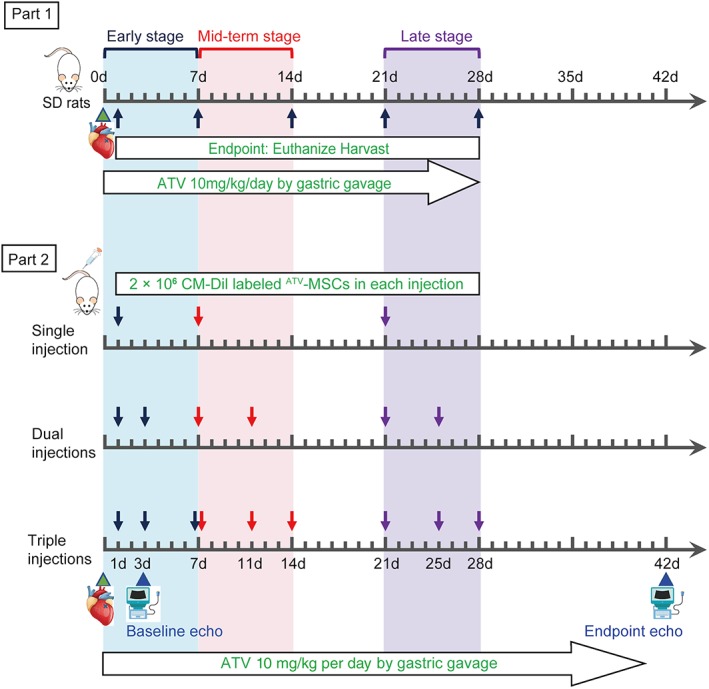
Experiment flowchart. Part 1: Rats were subjected to acute myocardial infarction (AMI) model procedure, followed by high dose of intensive atorvastatin (ATV, 10 mg/kg per day) intragastrically until different endpoints of 1 day (d1), 1 week (d7), 2 weeks (d14), 3 weeks (d21), and 4 weeks (d28) post‐AMI. Part 2: Rats were subjected to AMI model procedure, followed by intensive ATV intragastrically for 6 weeks. Cell delivery in nine transplantation groups was conducted according to the time point on the schematic. 2 × 106 CM‐Dil labeled ATV pretreated‐MSCs were transplanted through vein tail in each injection. Baseline and endpoint echocardiography were assessed on 3 days and 6 weeks post‐AMI, respectively. Abbreviation: SD rats, Sprague–Dawley rats.
MSCs Isolation, Pretreatment, and Transplantation
Bone marrow from the tibia and femur of male SD rats was harvested and cultured in T‐75 cell culture flasks with complete medium (Iscove's Modified Dulbecco's Medium [IMDM, Gibco, Grand Island, NY, http://www.invitrogen.com], 10% fetal bovine serum [Gibco, USA], and 1% penicillin‐streptomycin [Gibco, USA]) in humidified air with 5% CO2 at 37°C. When the adherent cells reached 70%–80% confluence, they were digested with 0.25% trypsin‐EDTA (Gibco, USA) and then reseeded at a 1:2 dilution. All the MSCs used for transplantation were passage three (P3) and were further identified by the surface marker probing as CD90+, CD29+, CD45−, CD11− with the use of flow cytometry (Fig. S1).
On the basis of our previous experiment results 24, all the P3 MSCs were pretreated with 1 μM ATV (Sigma‐Aldrich, St. Louis, MI, http://www.sigmaaldrich.com) for 24 hours before transplantation. For cell transplantation procedure, the MSCs were incubated with 1 μM ATV for 24 hours and then labeled with CellTracker CM‐Dil (Molecular Probe, Invitrogen, Carlsbad, CA, http://www.invitrogen.com) before the transplantation. At each time point of injection, 2 × 106 CM‐Dil labeled ATVMSCs in a total volume of 0.5 ml phosphate‐buffered saline (PBS) were transplanted through the vein tail.
AMI Model Establishment
The AMI model in female SD rats has been previously described 24. In brief, rats were anesthetized by an intraperitoneal injection of pentobarbital sodium (50 mg/kg) before the surgical procedure. Then, the chest was opened gently by left thoracotomy and pericardium was removed to reach the left ventricular (LV) free wall. A 6‐0 polyester suture was used to ligate the proximal left anterior descending coronary artery (LAD) 1–2 mm from the tip of the left atrial appendage. Successful ligation of LAD was confirmed by visual blanching of distal site and also by baseline echocardiography 3 days post‐AMI. Rats with left ventricular ejection fraction (LVEF) above 60% at 3 days after AMI were excluded from the study. For Sham group, the suture was passed around LAD and removed without ligation.
SDF‐1 Expression by Intensive ATV with Immunofluorescence
At the different endpoints (1d, 1w, 2w, 3w, and 4w), after euthanasia, the hearts of the rats used in the SDF‐1 Experiment were perfused retrogradely with heparinized PBS and fixed in 10% formalin for 24 hours and finally subjected to tissue processing and paraffin embedding. And then the wax blocks were cut into 4‐μm‐thick paraffin sections at the mid‐LV level. After deparaffinization with xylene and hydration with a degraded series of ethanol solution, the sections were subjected to antigen retrieval in hot citric acid buffer. After cooling to room temperature, the sections were blocked with 10% normal goat serum in PBS with 0.3% Triton X‐100 for 90 minutes. Then, the sections were coincubated overnight with the primary mouse anti‐SDF‐1 antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA, http://www.scbt.com, sc‐74271, 1:300 dilution) and primary rabbit anti‐sarcomeric alpha actinin antibody (Abcam, Cambridge, MA, http://www.abcam.com, ab68168, 1:300 dilution) at 4°C, followed by coincubation with the goat anti‐mouse crossadsorbed Alexa Fluor 488 secondary antibody (Thermo Fisher Scientific, Hampton, NH, http://www.fisherscientific.com, A‐11001, 1:200 dilution) and goat anti‐rabbit cross‐adsorbed Alexa Fluor 594 secondary antibody (Cell Signaling Technology, Danvers, MA, #8889, 1:200 dilution) for 1 hour at room temperature. After washing, the nuclei were stained with 4′6‐diamidino‐2‐phenylindole dihydrochloride (DAPI). The sections were then observed under a laser scanning confocal microscope (Leica, Germany, http://www.leica.com) at ×400 magnification and four randomly chosen high‐power fields in the peri‐infarcted region were analyzed per animal. The excitation wavelengths used for detection of SDF‐1, α‐actinin, and DAPI were 488, 594, and 405 nm, respectively. The results of SDF‐1 were described as a percentage: the area of SDF‐1 expression/the area of DAPI × 100%, using Image‐Pro‐Plus software.
SDF‐1 Expression and Inflammatory Cytokines with Enzyme Lined Immunosorbent Assay (ELISA) Analyses
In the first part of experiment (Part 1), rat SDF‐1 ELISA kit (elabscience, China, https://www.elabscience.com, E‐EL‐R0922c) and rat interleukin (IL)‐6 and tumor necrosis factor (TNF)‐α ELISA kits (eBioscience, USA, https://www.thermofisher.com, BMS625 and BMS622) were used to quantitatively detect the expression of SDF‐1 and inflammatory cytokines IL‐6 and TNF‐α in the peri‐infarcted myocardium at d1, d7, d14, d21, and d28 post‐AMI. Procedures were performed based on the manufacturer's user guide. Heart tissue homogenates in different groups were prepared and the protein concentration was adjusted to 10 mg/ml. Optical density of each sample was detected using an enzyme‐labeling measuring instrument at the wavelength of 450 nm. All results were converted to SDF‐1/IL‐6/ TNF‐α activity using a rat recombinant SDF‐1/IL‐6/ TNF‐α standard provided within the kits.
In the second part of experiment (Part 2), rat IL‐6 and TNF‐α ELISA kits (eBioscience, BMS625 and BMS622) were also used to detect the expression of inflammatory cytokines IL‐6 and TNF‐α in the peri‐infarcted myocardium at 6 weeks post‐AMI.
MSCs Recruitment and Survival
At 6 weeks post‐AMI, all rats were sacrificed and hearts were harvested. The protocol of the histological procedure has been described above. DAPI was used to stain the nuclei. The sections were analyzed by a laser scanning confocal microscope (Leica). The excitation wavelengths used for detection of CM‐Dil and DAPI were 561 and 405 nm, respectively. The number of CM‐Dil‐labeled MSCs was quantified within the infarct and border zone by an independent blinded researcher in 10 randomized high‐power fields (×400) per rat. The results were described as follows: the number of CM‐Dil‐labeled cells/the number of cells stained with DAPI × 100%.
Cardiac Function and Remodeling by Echocardiography and Catheterization
Transthoracic echocardiography was performed to assess cardiac function and remodeling at 3 days (baseline) and 6 weeks (endpoint) post‐AMI with a small animal echocardiography system (Sonos 7500, Phillips) equipped with a 12‐MHz phased‐array transducer. All the measurements were performed by an experienced technician who was blinded to control and transplantation groups. The data were measured in a two‐dimensional and M‐mode from the parasternal long axis view at the level of papillary muscle. The left ventricular end‐diastolic diameter (LVEDd) and end‐systolic diameter (LVESd) were measured and averaged for three consecutive cardiac cycles. LVEF and left ventricular fractional shortening (LVFS) were calculated with following equations: LVEF (%) = (LVEDd)3 − (LVESd)3]/(LVEDd)3 × 100%, and LVFS (%) = (LVEDd − LVESd)/LVEDd × 100%.
Left heart catheterization was conducted 6 weeks post‐AMI to assess the cardiac function as well at the endpoint. Briefly, just before euthanasia, rats were anesthetized and a catheter (22 GA × 1.00 IN BD Angiocath, USA) filled with heparin solution was inserted into the rat's LV via the right carotid artery. The position of the catheter carefully adjusted until typical signals were acquired. When the recording of the left ventricular pressure was stable, the data of the left ventricular end‐diastolic pressure (LVEDP) and the left ventricular pressure maximal rate of rise and fall (±dp/dtmax) was measured. All data were analyzed by a researcher blinded to the treatment using the LabChart 7 software.
Histological Analysis for Infarct Size and Fibrotic Area
The paraffin sections were stained with H&E staining, Masson's trichrome staining, picrosirius red staining, and immunohistochemistry. H&E staining was used to roughly observe the inflammation and fibrosis in the peri‐infarcted region. To further evaluate the level of inflammatory cells infiltration, CD45 immunohistochemistry was performed. The sections were processed as aforementioned histological procedure and incubated overnight with primary rabbit anti‐CD45 antibody (Abcam, ab10558, 1:1,000 dilution) at 4°C followed by incubation with the goat anti‐rabbit IgG (Beyotime, China, 1:200 dilution) secondary antibody and color reaction with the DAB kit. The stained sections were examined under microscope at ×200 magnification in four randomly chosen fields. The results of inflammatory cells infiltration were described as follows: the number of CD45+ cells/total cells × 100%.
Masson's trichrome staining was used to semiquantify the infarct size with the use of Image‐Pro‐Plus software. The infarct size was described as a percentage of the total left ventricular size (infarct size/total left ventricular size × 100%). From picrosirius red‐stained images acquired under polarized light microscopy, fibrotic area and myocardial collagen content in infarct region were clearly demonstrated. The fibrotic area was also semiquantified using the percentage of the total LV area (fibrotic area/total LV area × 100%). At least three sections from each heart were stained and measured.
Assessment of Apoptosis with TUNEL Assay
To assess the apoptosis in infarcted heart, TdT‐mediated dUTP nick‐end labeling (TUNEL) In Situ Cell Death Detection kit (Roche, Mannheim, Germany, 11772465001) was used according to the manufacturer's protocol. The sections were stained with TUNEL kit and nuclei were stained DAPI. The stained sections were examined under fluorescence microscope at ×400 magnification in four randomly chosen fields. Normal nuclei were presented as blue color, whereas apoptotic nuclei were green. All results were described as the percentage of apoptotic cardiomyocytes/total cells.
Angiogenesis, c‐Kit+ Cells Mobilization, and Differentiation with Immunofluorescence Microscopy
For immunofluorescence staining, the paraffin sections were coincubated with the first antibodies at 4°C overnight, followed by goat anti‐rabbit (Thermo Fisher Scientific, A‐11034, 1:200 dilution) or goat anti‐mouse (Thermo Fisher Scientific, A‐11001, 1:200 dilution) highly cross‐adsorbed Alexa Fluor 488 secondary antibodies for 1 hour at room temperature. After washing, the nuclei were stained with DAPI. The sections were then observed under a laser scanning confocal microscope (Leica) at ×400 magnification and four randomly chosen high‐power fields were analyzed per animal. The results of angiogenesis, reflected by α‐smooth muscle actin (α‐SMA), CD31 staining, were described as vessel density: the area of vessels/the area of DAPI × 100%. And the results of c‐Kit+ cells mobilization were calculated with the equation: the number of c‐Kit+ cells/the number of cells stained with DAPI × 100%. Finally, the differentiation of MSCs was evaluated by the number of CM‐Dil‐DAPI‐c‐TnT+ merged cells per high‐power field.
To determine the angiogenesis process, rabbit anti‐α‐SMA (Abcam, 1:200 dilution), rabbit anti‐CD31 (Abcam, 1:300 dilution) were used as first antibodies. Rabbit anti‐c‐Kit+ (Cell Signaling Technology, Danvers, MA, 1:1000 dilution) antibody was used to assess the number of c‐Kit+ cells in the peri‐infarcted region. The differentiation of implanted MSCs in post‐infarction hearts was evaluated by mouse anti‐c‐TnT (Abcam, 1:200 dilution) staining.
Statistical Analysis
Statistical analysis was performed with SPSS 22.0 software and data were expressed as mean ± SD. All data were analyzed with one‐way ANOVA followed by post hoc LSD‐test (variance homogeneity) or Dunnett's T3 test (variance nonhomogeneity). Graphs were assembled in GraphPad Prism 7. Two‐sided p‐values were used, and a value of p < .05 was considered statistically significant.
Results
Enhancement of SDF‐1 Expression in Different Stages of AMI with Suppressed Inflammation by intensive ATV
As shown in Figure 2 and Table S1, we found that the SDF‐1 expression in AMI group in the infarcted myocardium at each time point with both immunofluorescence (IF) (Fig. 2A, 2B) and ELISA analyses (Fig. 2C) was significantly increased in comparison with that in the Sham group ($ p < .05), which peaks at 1 day post‐AMI followed by a rapid decline to a very low level at 2 weeks and thereafter post‐AMI (α p < .05, μ p < .05, # p < .05, γ p < .05). In contrast, intensive ATV not only postponed the peak of SDF‐1 expression from 1 day to 1 week, but also significantly upregulated SDF‐1 expression over the entire 4‐week period post‐AMI (*p < .05), with the SDF‐1 level still high in the 2nd week and maintained relatively high till 4 weeks post‐AMI compared to the AMI group (3.3‐ and 1.8‐fold high for IF and ELISA, respectively, *p < .05), suggesting a much wider time window for MSC transplantation by ATV treatment. Based on these results, in the following experiments, we focused on three stages of AMI for optimal time window of MSCs transplantation: early (d1 to d7), mid‐term (d7 to d14), and late (d21 to d28) stages after AMI.
Figure 2.
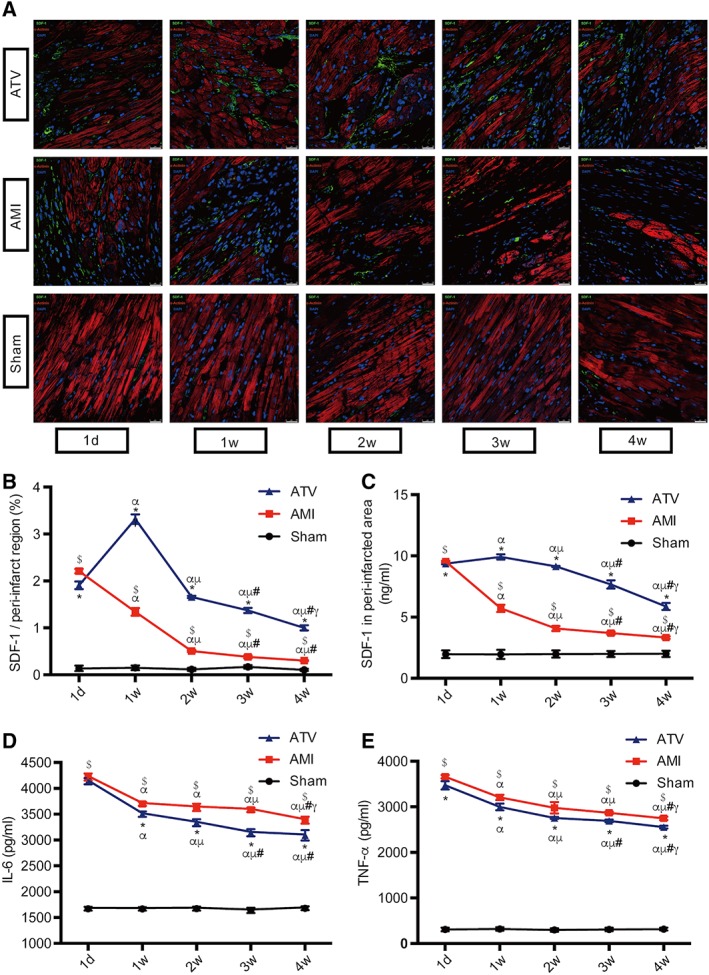
Changes of SDF‐1 expression and inflammation in peri‐infarcted region over the time course. (A): Representative immunofluorescence images of SDF‐1 in different groups at 1 day (d1), 1 week (d7), 2 weeks (d14), 3 weeks (d21), and 4 weeks (d28) post‐AMI. Cardiomyocytes, SDF‐1, α‐actinin, and nuclei were stained green, red, and blue, respectively. Scale bar = 25 μm. (B): Quantitative data of SDF‐1 immunofluorescence, described as the percentage of the area of SDF‐1 expression. (C–E): Enzyme Lined Immunosorbent Assay assessment of SDF‐1 expression, IL‐6, and TNF‐α in peri‐infarcted myocardium during the time course. n = 8–10 for each time point in each group. $ p < .05 vs. Sham group at each time point; *p < .05 vs. AMI group at each time point; α p < .05 vs. d1 time point in each group; μ p < .05 vs. d7 time point in each group; # p < .05 vs. d14 time point in each group; γ p < .05 vs. d21 time point in each group. All data are expressed as means ± SD. Abbreviations: AMI, acute myocardial infarction; ATV, atorvastatin; IL, interleukin; SDF, stromal cell‐derived factor; TNF, tumor necrosis factor.
Meanwhile, as shown in Figure 2D, 2E, both AMI and ATV group demonstrated intense inflammatory responses at each time point compared with the Sham group (p < .05), although the levels of IL‐6 and TNF‐α were both significantly reduced in the ATV group at all time points compared to AMI group (*p < .05) with d1 and d7 showing more and d14 to d28 less alleviative. Taken together, these results suggested that based on the intensive ATV treatment, the optimal transplantation window may be in late‐early and mid‐term stage of AMI because of the high level of SDF‐1 expression with the concurrently attenuated inflammatory microenvironment. Additionally, deferred transplantation in the late stage of AMI might bring cardioprotective benefits as well.
Augmentation of MSCs Recruitment in Different Stages of AMI by ATV‐MSCs Implantations with Intensive ATV
As shown in Figure 3A, 3B, and Table S2, compared with the AMI group at the endpoint, the recruitment rates of ATV‐MSCs labeled with CM‐Dil in the infarcted myocardium were significantly increased in all the nine transplantation groups (& p < .05) with most prominent increases in mid‐term stage groups (14.02% ± 0.95%∼23.96% ± 2.06%, & p < .05), the highest rate in Mid3 group (23.96% ± 2.06%, * p < .05 vs. Mid1) and steep liner cumulative effect by triple injections in both early and mid‐term groups. Although single injection of ATV‐MSCs in both early and late stage groups showed a limited recruitment rate, and triple injections in late stage groups had no cumulative effects (* p > .05).
Figure 3.
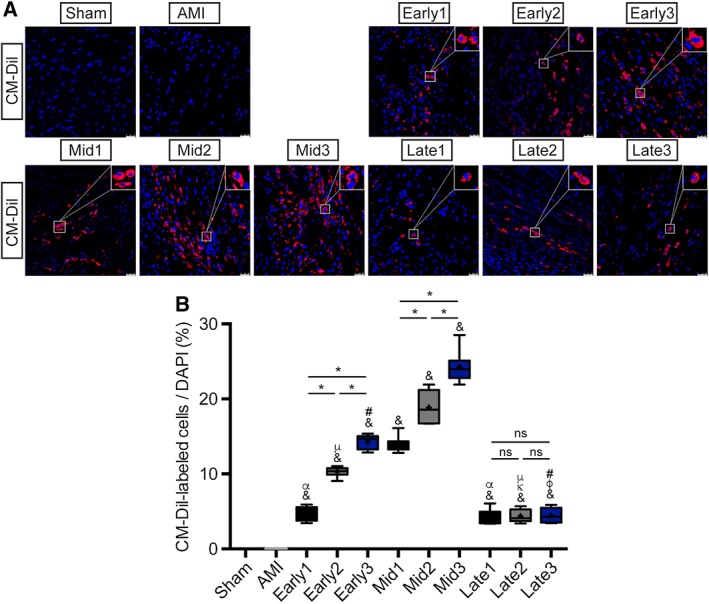
Assessment of mesenchymal stem cells recruitment. (A): Recruitment of CM‐Dil‐labeled cells (red) in the peri‐infarcted myocardium at 6 weeks after myocardial infarction. Nuclei were dyed with DAPI (blue). Scale bar = 25 μm. (B): Quantitation of CM‐Dil‐labeled cells in the peri‐infarcted myocardium per high power field in each group. n = 8–11 for each group. $ p < .05 vs. Sham group; & p < .05 vs. AMI group; γ p < .05 vs. Early1; κ p < .05 vs. Early2; φ p < .05 vs. Early3; α p < .05 vs. Mid1; μ p < .05 vs. Mid2; # p < .05 vs. Mid3; *p < .05 intragroup comparison in different periods. All data are expressed as mean ± SD. Abbreviations: AMI, acute myocardial infarction; CM‐Dil, 1,1′‐dioctadecyl‐3,3,3′,3′‐tetramethylindocarbocyanine perchlorate; DAPI, 4′6‐diamidino‐2‐phenylindole dihydrochloride.
Enhancement of Cardiac Function and Remodeling Attenuation in Different Stages of AMI by ATV‐MSCs Implantations with Intensive ATV
As shown in Figure 4A, echocardiography images were obtained at baseline (3 days) and endpoint (6 weeks) after AMI. The baseline parameters of echocardiography in all AMI groups showed no significant difference (Table S3), validating the reliability and stability of AMI model establishment. In comparison with AMI group, the change values of LVEF and LVFS at the endpoint both showed a significant elevation in all nine transplantation groups (& p < .05) with the prominent trend favoring for mid‐term stage groups (Fig. 4B, 4C). Furthermore, there was also a trend of favoring cumulative effects of triple injections for functional recovery in mid‐term stages (ΔLVEF: 8.97% ± 10.83%∼16.23% ± 9.77%; ΔLVFS: 4.81% ± 6.57%∼8.53% ± 5.90%) with the best performance in Mid3 group (Table S3).
Figure 4.
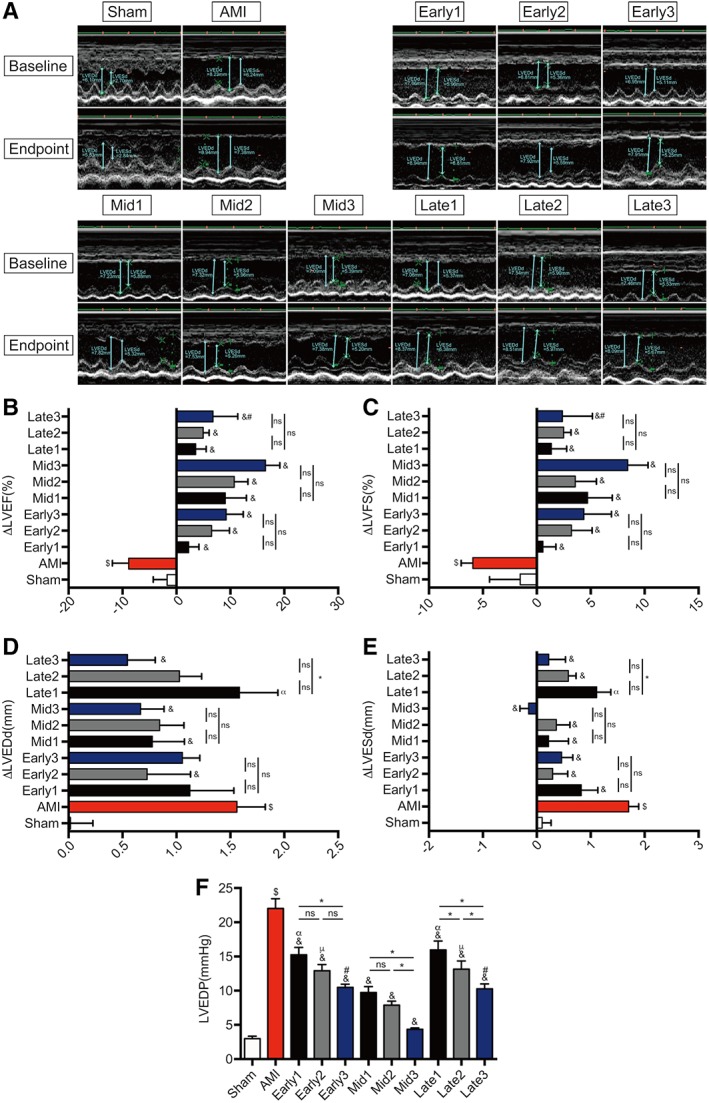
Echocardiography and left heart catheterization assessment of cardiac function. (A): Representative images of M‐mode echocardiograms were shown at baseline (3 days after myocardial infarction) and endpoint (6 weeks after myocardial infarction). (B–E): The variation (=endpoint‐baseline) of left ventricular ejection fraction (ΔLVEF), left ventricular fractional shortening (ΔLVFS), left ventricular end‐diastolic dimension (ΔLVEDd), and left ventricular end‐systolic dimension (ΔLVESd). (F): The values of left ventricular end‐diastolic pressure (LVEDP. n = 8–11 for each group. $ p < .05 vs. Sham group; & p < .05 vs. AMI group; γ p < .05 vs. Early1; κ p < .05 vs. Early2; φ p < .05 vs. Early3; α p < .05 vs. Mid1; μ p < .05 vs. Mid2; # p < .05 vs. Mid3; *p < .05 intragroup comparison in different periods. All data are expressed as mean ± SD. Abbreviations: AMI, acute myocardial infarction; LVEDd, left ventricular end‐diastolic dimension; LVESd, left ventricular end‐systolic dimension; LVEF, left ventricular ejection fraction; LVFS, left ventricular fractional shortening.
Meanwhile, the change values of LVEDd (Fig. 4D) were significantly reduced only in Mid‐term stage groups (& p < .05) compared with AMI group (Table S3), and those of LVESd (Fig. 4E) were significantly decreased in all but Late1 groups (& p < .05) with prominent effect favoring for mid‐term stage groups and the best in Mid3 (ΔLVEDd: 0.67 ± 0.68 vs. 1.57 ± 0.76 mm; ΔLVESd: −0.14 ± 0.55 vs. 1.71 ± 0.54 mm, & p < .05). It was noticeable that cumulative benefit of triple treatment in preventing from ventricular dilatation occurred only in late stage groups with the significance in Late3 (ΔLVEDd: 0.55 ± 0.74 vs. 1.59 ± 1.04 mm; ΔLVESd: 0.23 ± 0.91 vs. 1.11 ± 0.77 mm, *p < .05 vs. Late 1), indicating that ventricular remodeling could even be attenuated in late stage of AMI by multiple ATV‐MSCs transplantation.
Left heart catheterization was also performed at the endpoint to further assess the cardiac function. As shown in Figure 4F, Figure S2, and Table S4, compared with AMI group, all nine MSCs groups elicited functional benefits of significantly reduced LVEDP (& p < .05) and elevated +dp/dtmax except late stage groups (& p < .05), with prominent in mid‐term stage groups in LVEDP reduction (4.29 ± 0.85∼9.66 ± 2.77 mmHg vs. 21.97 ± 4.43 mmHg, & p < .05) and +dp/dtmax improvement (& p < .05). Furthermore, triple injections showed strikingly cumulative effects on LVEDP decrease in all three stage groups (*p < .05), and on +dp/dtmax improvement in both early and mid‐term stage groups (*p < .05), with the best effectiveness also in Mid3 group (LVEDP: 4.29 ± 0.85 vs. 9.66 ± 2.77 mmHg, *p < .05 vs. Mid1; +dp/dtmax: 4529.86 ± 340.82 vs. 3865.50 ± 210.20 mmHg/s, −dp/dtmax: −3298.55 ± 316.27 vs. −2935.97 ± 416.19 mmHg/s; *p < .05 vs. Mid1).
Reduction of Infarct Size and Fibrotic Area at Different Stages of AMI by ATV‐MSC Implantation with Intensive ATV Administration
As shown in Figure 5A, 5B, 5C, and Table S5, compared to Sham group, AMI group showed a significantly increase in both infarct size and fibrotic area ($ p < .05) with a remarkable thinning of the anterior wall and dilatation of LV chamber. In comparison with AMI group, both infarct size and fibrotic area in all nine transplantation groups were significantly decreased (& p < .05), with most dramatic reductions in three mid‐term stage groups (infarct size: 16.05% ± 1.73%∼27.50% ± 0.98% vs. 49.81% ± 2.67%, & p < .05; fibrotic area: 16.18% ± 1.70%∼27.30% ± 0.90% vs. 49.84% ± 2.74%, & p < .05). The cumulative reductions of both infarct size and fibrotic area by triple injections were observed at steeply linear rate in early and mid‐term stages groups (*p < .05), with both being strikingly reduced to 16.05% ± 1.73% and 16.18% ± 1.70%, respectively, in Mid3 group, indicating the predominant benefits of multiple ATV‐MSCs transplantations.
Figure 5.
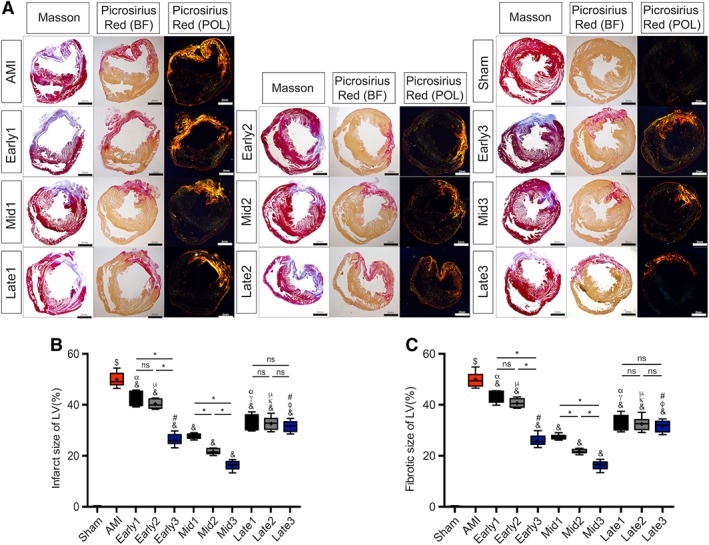
Analyses of infarct size and fibrotic area. (A): Representative images for Masson's trichrome staining and picrosirius red staining at 6 weeks after myocardial infarction. In Masson, red, myocardium; blue, infarcted myocardium. In picrosirius red staining bright field (BF), red, Type I collagen fiber; yellow, normal myocardium. In picrosirius red staining polarization field (POL), bright orange, Type I collagen fiber. Scale bar = 2 mm. (B): Quantitative data for the left ventricular infarct size. (C): Quantitative data for the left ventricular fibrotic area. n = 8–11 for each group. $ p < .05 vs. Sham group; & p < .05 vs. AMI group; γ p < .05 vs. Early1; κ p < .05 vs. Early2; φ p < .05 vs. Early3; α p < .05 vs. Mid1; μ p < .05 vs. Mid2; # p < .05 vs. Mid3; *p < .05 intragroup comparison in different periods. All data are expressed as mean ± SD. Abbreviations: AMI, acute myocardial infarction; LV, left ventricular.
Suppression of Myocardial Inflammation at Different Stages of AMI by ATV‐MSC Implantations with Intensive ATV Administration
As consistently shown in H&E (Fig. 6A, 6B, and Table S5), the rate of CD45+ inflammatory cells in AMI group was significantly increased compared with Sham group (19.39% ± 0.54% vs. 0.43% ± 0.03%, $ p < .05), and the rates in all the nine MSCs treatment groups except Early1 were significantly decreased in comparison with AMI group (& p < .05), with prominent in Early3 and three mid‐term stage groups (10.22% ± 0.33%∼12.78% ± 0.97% vs. 19.39% ± 0.54%, & p < .05). Triple injections also had cumulative effects in early and mid‐term stage groups with the lowest in Mid3 (*p < .05).
Figure 6.
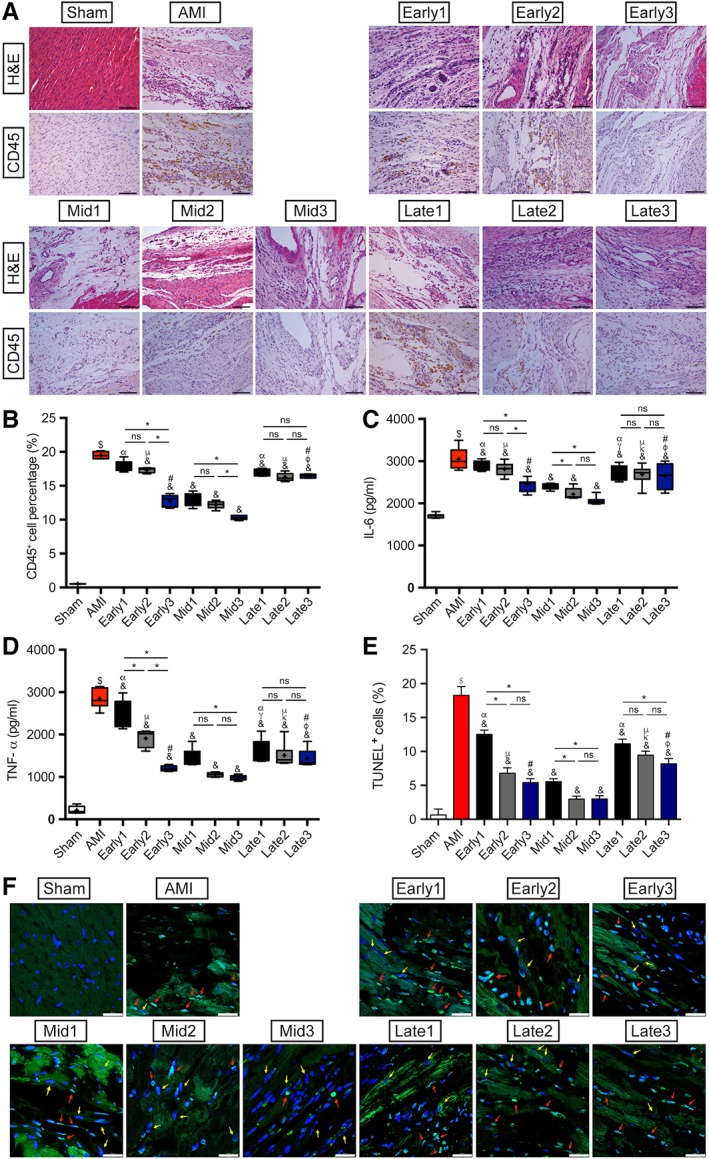
Assessment of myocardial inflammation and cardiomyocytes apoptosis. (A): Representative images of H&E staining and CD45 immunohistochemistry in each group. Scale bar = 100 μm. (B): Quantitation of CD45+ inflammatory cells infiltration in the peri‐infarcted myocardium. n = 5 for each group. (C, D): ELISA assay results of IL‐6 and TNF‐α expressions in the peri‐infarcted myocardium in each group. n = 8 for each group. (E): Quantitative data for the ratio of apoptotic cells in each group. n = 5 for each group. (F): Representative images of TUNEL staining with autofluorescence of cardiomyocytes at 6 weeks after myocardial infarction in each group. Apoptotic cells were stained green and nuclei were blue, and the red arrows showed the apoptotic nuclei of cardiomyocytes whereas the yellow arrows were the normal cardiomyocytes nuclei. Scale bar = 25 μm. $ p < .05 vs. Sham group; & p < .05 vs. AMI group; γ p < .05 vs. Early1; κ p < .05 vs. Early2; φ p < .05 vs. Early3; α p < .05 vs. Mid1; μ p < .05 vs. Mid2; # p < .05 vs. Mid3; *p < .05 intragroup comparison in different periods. All data are expressed as mean ± SD. Abbreviations: AMI, acute myocardial infarction; IL, interleukin; TNF, tumor necrosis factor; TUNEL, terminal‐deoxynucleotidyl transferase‐mediated dUTP nick end labeling.
As shown in Figure 6C, 6D, the two cytokines of IL‐6 and TNF‐α determined by ELISA assay were also significantly increased in the AMI group compared to the Sham group ($ p <.05). Compared to AMI group, the expressions of both IL‐6 and TNF‐α were significantly reduced in all the nine transplantation groups, with the sharpest decreasing rate of TNF‐α in early stage groups and the lowest level in Mid3 (969.98 ± 55.03 vs. 2839.76 ± 233.23 pg/ml, & p < .05). It also demonstrated that both the strongest inflammation status and cumulative effects of anti‐inflammation by triple injections of ATV‐MSCs in early stage suggested the weakest efficacy of single ATV‐MSCs administration in the stage.
Inhibition of Cardiomyocytes Apoptosis at Different Stages of AMI by ATV‐MSC Implantations with Intensive ATV Administration
As shown in Figure 6E, 6F, and Table S5, the antiapoptotic effects of MSCs therapy were evaluated and semiquantified by TUNEL staining with autofluorescence of cardiomyocytes. The apoptotic nuclei of cardiomyocytes showed long nuclei and merged with cardiomyocytes (as shown by red arrows). The apoptosis index of the peri‐infarcted region in AMI group was significantly increased in comparison with Sham group (18.25% ± 1.29% vs. 0.63% ± 0.88%, $ p < .05), whereas AMI‐induced apoptosis of cardiomyocytes was dramatically suppressed in all the nine treatment groups (& p < .05), with similarly very low apoptosis index in Early3 and mid‐term stage groups (2.97% ± 0.44%∼5.54% ± 0.43%, & p < .05). There was only cumulative antiapoptotic effect of triple injections in the early stage between Early1 and Early2 groups, indicating strong antiapoptotic effects of the combination protocol of ATV‐MSCs implantations with intensive ATV treatment.
Enhanced Angiogenesis at Different Stages of AMI by ATV‐MSCs Implantations with Intensive ATV Administration
The vessel densities of both arteries (α‐SμΑ staining) and capillaries (CD31 staining) in peri‐infarcted region as shown in Figure 7A–7C, which is calculated as the total area of vessels per high‐powered field (magnification ×400), were significantly increased in all the nine transplantation groups compared to AMI group, with mostly prominent in three mid‐term stage groups (CD31: 4.23% ± 0.28%∼10.19% ± 0.37% vs. 1.21% ± 0.10%, & p < .05; α‐SμΑ: 10.06% ± 0.56%∼16.06% ± 1.12% vs. 4.44% ± 0.33%, & p < .05), indicating the great ability of angiogenesis in peri‐infarcted myocardium. In consistent with the recruitment of ATV‐MSCs, multiple injections in both early and mid‐term stages groups showed a steeply liner cumulative rise in vessel density (*p < .05), with the highest in Mid3 group (CD31: 10.19% ± 0.37% vs. 4.23% ± 0.28%, *p < .05 vs. Mid1; α‐SμΑ: 16.06% ± 1.12% vs. 10.06% ± 0.56%, *p < .05 vs. Mid1). In contrast, the vessel density of three groups in late stage demonstrated a cumulative rise at a much slower rate.
Figure 7.
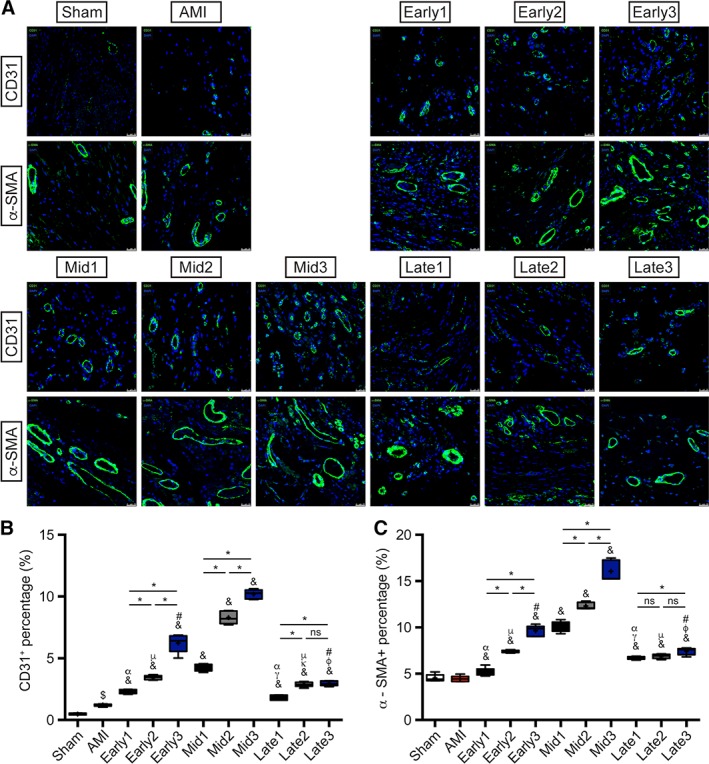
Angiogenesis evaluated by α‐SMA and CD31 staining. (A): Representative images of α‐SMA and CD31 staining in peri‐infarcted region at the endpoint in each group. Vessels formation was stained green and nuclei were dyed with DAPI (blue). Scale bar = 25 μm. (B): Quantitative analyses of CD31 and (C) α‐SMA staining with vessel density per HPF in each group. n = 5 for each group. $ p < .05 vs. Sham group; & p < .05 vs. AMI group; γ p < .05 vs. Early1; κ p < .05 vs. Early2; φ p < .05 vs. Early3; α p < .05 vs. Mid1; μ p < .05 vs. Mid2; # p < .05 vs. Mid3; *p < .05 intragroup comparison in different periods. All data are expressed as mean ± SD. Abbreviations: α‐SMA, α‐smooth muscle actin; AMI, acute myocardial infarction.
Enhancement of c‐Kit+ Cell Mobilization Without Further Differentiation at Different Stages of AMI by ATV‐MSC Implantations with Intensive ATV Administration
Apart from the implanted ATV‐MSCs' recruitment, the mobilization of c‐Kit+ cells in peri‐infarcted region was also investigated (Fig. S3 and Table S2) in particular in consideration of the recent controversies and discussions 37, 38, 39, 40, 41, 42, 43. AMI group presented a slight insignificant increase in the number of c‐Kit+ cells compared to Sham group ($ p < .05). However, all the nine MSCs groups had a significant increase in the number of c‐Kit+ cells compared with AMI group (& p < .05), with noticeable increases in both early and mid‐term stage groups except Early1 (10.01% ± 1.37%∼22.48% ± 1.41% vs. 1.48% ± 0.08%, & p < .05), which was in conformity with the results of ATV‐MSCs recruitment. Similar to the pattern of ATV‐MSCs recruitment, the cumulative effect cell mobilization by triple injections was significantly increased at steeply linear rate in early and mid‐term stages groups (*p < .05) with the highest in Mid3 group (22.48% ± 1.41% vs. 10.17% ± 1.57%, *p< .05 vs. Mid1), and at flat rate in late stage groups. Besides, CM‐Dil and c‐TnT were co‐stained and observed to investigate the differentiation of ATV‐MSCs. As shown in Figure S4 and Table S2, in all the nine transplantation groups, the CM‐Dil positive cells are rarely positive for c‐TnT staining. Quantified results also showed that compared to AMI group, the numbers of CM‐Dil‐DAPI‐c‐TnT+ merged cells failed to reach any statistical significance (& p > .05), indicating that the improvement of cardiac function and morphometrics was not mainly ascribed to the differentiation of implanted MSCs in the peri‐infarcted region. Importantly, we did not observe any further differentiation of these c‐Kit+ cells into CMs in spite of the higher recruitment via MSC injection, consistent with recent reports from many other labs 37, 38, 39, 40, 41, 42, 43. Therefore, the beneficial effects, if any that come from the increased number of c‐Kit+ cells, are likely through secreted cytokines and small molecules.
Discussion
With the combination protocol of high dose of intensive ATV treatment and ATV‐MSCs transplantation, the current study presented a direct comparison of the therapeutic efficacy among the different time window periods of early, mid‐term, and late stages of AMI and different times of single, dual, and triple intravenous (iv) ATV‐MSCs administrations, to identify the optimal strategy to further improve the efficacy of MSCs therapy. The main findings are as follows: (a) intensive ATV treatment dramatically augmented the SDF‐1 expression in peri‐infarcted myocardium over the entire period of AMI with simultaneous inhibiting the inflammation; (b) the mid‐term stage (the 2nd week) of AMI was the optimal time window period for ATV‐MSCs transplantation, thought the benefits were still existing to some degree in late stage (the 4th week); (c) triple ATV‐MSCs transplantations yielded the remarkably cumulative therapeutic efficacy with the best in Mid3, whereas single injection in both Early1 and Late1 was weak in effectiveness; (d) the underlying mechanisms mainly attributed to the marked enhancement of ATV‐MSCs recruitment and survival, and angiogenesis in the infarcted region instead of myocardium regeneration. We found that with the combination protocol, at least triple iv injections of ATV‐MSCs in mid‐term stage (the 2nd week) of AMI was the optimal strategy for cell therapy but still effective even in late stage (the 4th week). The translational potential of this strategy is clinically promising, straightforward, and enormous, which is worthy of further clinical verifying.
Over the past two decades, the optimal time window period of cell transplantation remains controversial 25, 26, 27 because of challenges in augmenting cell recruitment and survival. In the majority of preclinical and clinical studies 25, 44, 45, 46, 47, 48, 49, 50, the most commonly used transplantation time window period is within 1 week post‐AMI 17, 23, 51, 52, 53, although the therapeutic efficacy of MSCs is still weak with only 2%–3% LVEF elevation 6, 54. It has been reported that SDF‐1/CXCR4 axis plays a paramount role in the recruitment of implanted MSCs for cardiac repair 55, 56, the natural soaring upregulation of SDF‐1 expression in infarcted myocardium in the early stage (the 1st week) of AMI can recruit more CXCR4‐expressed MSCs 57, although the severe inflammatory response and oxidative stress 21 may lead to apoptosis and necrosis of the implanted MSCs. On the contrary, in the late stage (the 4th week) of AMI, the inflammation alleviates, making it easier for MSCs to survive, whereas lack of SDF‐1‐driven cell recruitment with scar formation in the infarcted region may impede the potential benefits of MSCs transplantation 26, 27, 58, 59, 60. Many strategies have been investigated to enhance and extend the expression of SDF‐1 61, focusing on pharmacological 62 and genetic manipulations 63 like overexpression of SDF‐1 15, 57, although only ATV treatment was currently clinical feasible with augmenting SDF‐1 expression, extending its peak expression time from 1 to 7 days and subsiding gradually in 2 weeks post‐AMI 20, 21, 22, 64, 65. Whether ATV treatment could also increase the SDF‐1 expression in late stage of AMI has not been clarified. The present study has first ever investigated the dynamic changes of both SDF‐1 expression and inflammatory cytokines in infarcted region over the entire 4‐week period of AMI by high dose of intensive ATV treatment, and found that intensive ATV can not only enhance the SDF‐1 expression 2.5‐ and 3.3‐folds in 1–2 weeks but also 3.3‐ and 3.6‐folds in even 3–4 weeks after AMI with concurrently inhibiting the inflammation.
Most importantly, with the combination protocol of intensive ATV treatment and iv ATV‐MSCs administration, which has powerful targeted homing, anti‐inflammatory, and anti‐apoptotic effects 17, 18, 21, 23, 24, the present study has shown that the mid‐term stage (the 2nd week) rather than the early stage (the 1st week) of AMI is the most appropriate or optimal time window period for transplantation indicated by remarkable ATV‐MSCs recruitment and survival, strong inflammation suppression and anti‐apoptosis, marked augmentation of angiogenesis and c‐Kit+ cells mobilization, striking reduction of both infarcted size and fibrotic area as well as prominent cardiac functional improvement and ventricular remodeling attenuation. The surprising increases in both ATV‐MSCs recruitment and survival in current study, which are the fundamental and bottle‐neck for therapeutic efficacy in cell therapy, were mainly ascribed to the prominent effects of SDF‐1‐driven targeted homing and strong antiapoptotic properties of ATV‐MSCs 23, 24 by the combination protocol. Another contributing factor for ATV‐MSCs survival was the powerful ameliorating of intense inflammatory harsh microenvironment in the infarcted region by intensive ATV treatment with strong anti‐inflammatory and antiapoptotic effects, as in the current study, our previous studies 17, 18, 23, 24, 51, 52, and the studies of others 21, 22, 57, 61. To the best of our knowledge, this is the first report to find the remarkable enhancement of SDF‐1 expression in infarcted myocardium by intensive ATV treatment over the entire 4‐week period of AMI, and the mid‐term stage (the 2nd week) of AMI was the optimal time window period of ATV‐MSCs transplantation with the clinically feasible combination protocol in preclinical AMI model.
It is worth noting that ATV‐MSCs transplantation in late stage (the 4th week) of AMI in the present study did have also exhibited therapeutic effect to some degree, which is clinically relevant and in agreement with some animal studies 26, 27 but not with clinical studies 47, 59, 60, 66, 67. This result mainly ascribes to the remaining 3.6‐folds high level of SDF‐1 expression and subsided inflammatory harsh microenvironment in infarcted myocardium as it has been shown in the current study, although the insufficient level of SDF‐1 expression and scar formation in the late stage of AMI may be the main limitation of further improvement in efficacy. No similar reports have been found in the literature so far either.
On the other hand, an alternative strategy for further improvement in efficacy of cell therapy has been sought to overcome the poor recruitment rate by repeatedly administering stem cells 68, 69, 70, 71. Bolli and his research team have conducted a series of preclinical experiments in rodents, demonstrating that repeated cell therapy was much more effective than single‐dose therapy, either using c‐Kit+ cells or MSCs 68. These results suggested that repeated administrations of stem cells achieved more desirable effects because they produced repetitive and sustainable bursts of paracrine factor release, resulting in cumulative paracrine effects 72, 73. The present study with the combination protocol also demonstrated that triple implants of ATV‐MSCs could dramatically achieve cumulative therapeutic effects in early and mid‐term stages of AMI with the best benefits obtained in Mid3 in respect of 5.4‐fold high of ATV‐MSCs recruitment and survival, 8.4‐fold increase of angiogenesis, and 15.2‐fold rising of c‐Kit+ cell mobilization in the peri‐infarcted region, as well as infarct size decreasing to 16.1% (vs. 49.8% in AMI), LVEDP reducing to 4.3 mmHg (vs. 22 mmHg of AMI) and LVEF elevating by 16.2%. Interestingly, the cardiac performance of Early3 group improved dramatically compared to Early2 group whereas it showed no significant difference in comparison with Mid1 group, which indirectly indicated that the transplantation of MSCs at 1 week contributed effectively to the improvement of heart function. In contrast, single injection of ATV‐MSCs did yield very weak therapeutic effect in both early and late stages (the first and 4th week) post‐AMI. In addition, the late stage of AMI also has weak cumulative effects by triple injections of ATV‐MSCs in terms of angiogenesis, c‐Kit+ cell mobilization, heart function improvement of LVEDP decreasing, and ventricular remodeling attenuation. From the differences of accumulative effect in three stages, we could conclude that only in the optimal transplantation time interval, the MSCs' cumulative paracrine effects of myocardial protection can be fully reflected. These results were not only consistent with the reports on the strategy of repeated cell therapy by Bolli et al. 68, 69, 70, 71, but further verified the best efficacy in mid‐term stages of AMI obtained only by triple injections of ATV‐MSCs, which is originally associated with strong ATV‐MSCs targeted homing and antiapoptotic effects by the combination protocol of ATV‐MSCs implantations with intensive ATV treatment. No similar reports have been found in the literature as well. Furthermore, the iv administration of ATV‐MSCs in the current study has the unique advantage over traditional methods like intracoronary or intramyocardium injection, due to clinical convenience and feasibility for repeated transplantations at different time points of different stages of AMI.
Regarding the mechanism, MSCs were originally believed to exert cardioprotective effects via differentiation into cardiomyocytes and vascular lineage cells 74, 75. Later, cell fusion of implanted MSCs with resident cardiomyocytes was proposed as a potential mechanism 76. Nevertheless, studies have shown that the differentiation rate of MSCs and the frequency of cell infusion were both too low to explain the significant improvement of MSCs transplantation therapy 77. The present study once again confirmed that the underlying mechanism of remarkably therapeutic effects of ATV‐MSCs were angiogenesis instead of myocardium regeneration according to the findings of striking enhancement of ATV‐MSCs targeted homing, angiogenesis but unevenly rare differentiation. This is not only in agreement with, but also further confirms the reports of no myocardial regeneration 37, 38, 39, 40, 41, 42, 43 even in the presence of notably mobilized c‐Kit+ cells. As to the existence, fate, and function of c‐Kit+ cells, many investigators have conducted different experiments using multiple biological techniques 42, 43, and have confirmed the existence of c‐Kit+ cells. However, whether they participate in and how much contribution they make to the heart repair is still needed to be explored 78. To date, compelling evidence indicated that all these beneficial effects of MSCs, including antiapoptosis 63, 79, anti‐inflammation 12, 80, 81, and pro‐angiogenesis effect 58, 82, 83, should be ascribed to the paracrine mechanism. Therefore, with the combination protocol, the prominent enhancement of cardiac function improvements, inflammatory suppression, antiapoptosis, and pro‐angiogenesis in the current study was more likely attributed to the strong paracrine effects of implanted ATV‐MSCs rather than their differentiation capacity.
Conclusion
The present study revealed that with the combination protocol of ATV‐MSCs transplantations with intensive high dose of ATV treatment, the optimal time window period of cell therapy was mid‐term stage (the 2nd week) post‐AMI with late stage (the 4th week) still existing effect due to augmented SDF‐1 expression and subsided inflammation in the infarct region. Triple administrations of ATV‐MSCs were much more effective than the single administration with the best benefits obtained in Mid3. Therefore, the optimal strategy for further improving the efficacy of MSCs therapy was at least triple iv administrations of ATV‐MSCs in mid‐term stage of AMI combined with intensive ATV treatment. The translational potential of this study is promising, straightforward, and enormous.
Author Contributions
J.X.: conception and design, provision of study material or patients, data analysis and interpretation, manuscript writing, final approval of manuscript; Y.‐Y.X., M.‐J.H., J.‐Y.X.: collection and/or assembly of data, final approval of manuscript; Q.L: provision of study material or patients, collection and/or assembly of data, final approval of manuscript; P.‐S.H.: collection and/or assembly of data, data analysis and interpretation, final approval of manuscript; X.‐Q.T.: provision of study material or patients, final approval of manuscript; C.J.: administrative support, final approval of manuscript; J.‐D.L., L.Q.: manuscript writing, final approval of manuscript; Y.‐J.Y.: conception and design, financial support, provision of study material or patients, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Supporting information
Figure S1. Mesenchymal stem cells (MSCs) identification. (A) Flow cytometry results of the surface markers of implanted MSCs, probing as CD90+, CD29+, CD45‐, CD11‐. (B‐C) Representative images of MSCs. Scale bar = 500μm (B) and 200μm (C).
Figure S2. Left heart catheterization assessment of cardiac function. The values of left ventricular pressure maximal rate of rise and fall (±dp/dtmax) were detected at the endpoint (6 weeks after myocardial infarction). n = 8‐11 for each group. $ p <0.05 vs. Sham group; & p <0.05 vs. AMI group; γ p < 0.05 vs. Early1; κ p < 0.05 vs. Early2; φ p < 0.05 vs. Early3; α p < 0.05 vs. Mid1; μ p < 0.05 vs. Mid2; # p < 0.05 vs. Mid3; *p < 0.05 intra‐group comparison in different periods. All data are mean ± SD.
Figure S3. Assessment of c‐Kit+ cells mobilization. (A) Mobilization of c‐Kit+ cells (green) in the peri‐infarcted myocardium at 6 weeks after myocardial infarction. Nuclei were dyed with DAPI (blue). Scale bar = 25μm. (B) The percentage of c‐Kit+ cells in the peri‐infarcted region per HPF in each group. n = 5 for each group. $ p <0.05 vs. Sham group; & p <0.05 vs. AMI group; γ p < 0.05 vs. Early1; κ p < 0.05 vs. Early2; φ p < 0.05 vs. Early3; α p < 0.05 vs. Mid1; μ p < 0.05 vs. Mid2; # p < 0.05 vs. Mid3; *p < 0.05 intra‐group comparison in different periods. All data are mean ± SD.
Figure S4. Assessment of differentiation of MSCs into cardiomyocytes. (A) Representative images of CM‐Dil and c‐TnT co‐staining in the peri‐infarcted regions at the endpoint. Cardiomyocytes, CM‐Dil‐labeled cells and nuclei were stained green, red and blue, respectively. Scale bar = 25μm. (B) Quantitative analyses with the number of CM‐Dil‐DAPI‐ c‐TnT+ merged cells per HPF in each group. n = 5 for each group. $ p <0.05 vs. Sham group; & p <0.05 vs. AMI group; α p < 0.05 vs. Mid1; μ p < 0.05 vs. Mid2; # p < 0.05 vs. Mid3; *p < 0.05 intra‐group comparison in different periods. All data are mean ± SD.
Table S1. Changes of SDF‐1 and inflammatory cytokines in peri‐infarcted region over time course assessed by immunofluorescence and ELISA
Table S2. Quantitation of CM‐Dil‐labeled cells, c‐Kit+ cells, angiogenesis and differentiation in the peri‐infarcted myocardium
Table S3. LV Function and Geometry Parameters by Echocardiography
Table S4. Left heart catheterization assessment of cardiac function
Table S5. Quantitation of infarct and fibrotic size, CD45+ cell infiltration, TUNEL+ cell percentage and measurement of IL‐6, TNF‐α expressions in the peri‐infarcted myocardium
Acknowledgments
This work was supported by the CAMS Innovation Fund for Medical Sciences (CIFMS, 2016‐12M‐1‐009), Innovative Research Foundation of Peking Union Medical College [2017‐1002‐1‐05], grants from 863 Program of China [2013AA020101], National Natural Science Foundation of China [81200107, 81370223, 81573957, 81874461]. We thank Peihe Wang and Fu‐Liang Luo in the Experimental Animal Center of Fuwai Hospital for their technical assistance.
Contributor Information
Li Qian, Email: li_qian@med.unc.edu.
Yue‐Jin Yang, Email: yangyjfw@126.com.
References
- 1. Lee CY, Kim R, Ham O et al. Therapeutic potential of stem cells strategy for cardiovascular diseases. Stem Cells Int 2016;2016:4285938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sanganalmath SK, Bolli R. Cell therapy for heart failure. Circ Res 2013;113:810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bolli R, Ghafghazi S. Cell therapy needs rigorous translational studies in large animal models. J Am Coll Cardiol 2015;66:2000–2004. [DOI] [PubMed] [Google Scholar]
- 4. Dixit P, Katare R. Challenges in identifying the best source of stem cells for cardiac regeneration therapy. Stem Cell Res Ther 2015;6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fisher SA, Doree C, Mathur A et al. Meta‐analysis of cell therapy trials for patients with heart failure. Circ Res 2015;116:1361–1377. [DOI] [PubMed] [Google Scholar]
- 6. Fisher SA, Zhang H, Doree C et al. Stem cell treatment for acute myocardial infarction. Cochrane Database Syst Rev 2015;9:Cd006536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zwetsloot PP, Vegh AM, Jansen of Lorkeers SJ et al. Cardiac stem cell treatment in myocardial infarction: A systematic review and meta‐analysis of preclinical studies. Circ Res 2016;118:1223–1232. [DOI] [PubMed] [Google Scholar]
- 8. Sanina C, Hare JM. Mesenchymal stem cells as a biological drug for heart disease: Where are we with cardiac cell‐based therapy? Circ Res 2015;117:229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zachar L, Bacenkova D, Rosocha J. Activation, homing, and role of the mesenchymal stem cells in the inflammatory environment. J Inflamm Res 2016;9:231–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Afzal MR, Samanta A, Shah ZI et al. Adult bone marrow cell therapy for ischemic heart disease: Evidence and insights from randomized controlled trials. Circ Res 2015;117:558–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Barron CC, Lalu MM, Stewart DJ et al. Assessment of safety and efficacy of mesenchymal stromal cell therapy in preclinical models of acute myocardial infarction: A systematic review protocol. Syst Rev 2017;6:226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Epstein SE, Luger D, Lipinski MJ. Paracrine‐Mediated Systemic Anti‐Inflammatory Activity of Intravenously Administered Mesenchymal Stem Cells. Circ Res 2017;121:1044. [DOI] [PubMed] [Google Scholar]
- 13. Luger D, Lipinski MJ, Westman PC et al. Intravenously delivered mesenchymal stem cells: Systemic anti‐inflammatory effects improve left ventricular dysfunction in acute myocardial infarction and ischemic cardiomyopathy. Circ Res 2017;120:1598–1613. [DOI] [PubMed] [Google Scholar]
- 14. Barbash IM, Chouraqui P, Baron J et al. Systemic delivery of bone marrow‐derived mesenchymal stem cells to the infarcted myocardium: Feasibility, cell migration, and body distribution. Circulation 2003;108:863–868. [DOI] [PubMed] [Google Scholar]
- 15. Haider H, Jiang S, Idris NM et al. IGF‐1‐overexpressing mesenchymal stem cells accelerate bone marrow stem cell mobilization via paracrine activation of SDF‐1alpha/CXCR4 signaling to promote myocardial repair. Circ Res 2008;103:1300–1308. [DOI] [PubMed] [Google Scholar]
- 16. Zimmermann JA, Hettiaratchi MH, McDevitt TC. Enhanced immunosuppression of T cells by sustained presentation of bioactive interferon‐gamma within three‐dimensional mesenchymal stem cell constructs. Stem Cells Trans Med 2017;6:223–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang YJ, Qian HY, Huang J et al. Atorvastatin treatment improves survival and effects of implanted mesenchymal stem cells in post‐infarct swine hearts. Eur Heart J 2008;29:1578–1590. [DOI] [PubMed] [Google Scholar]
- 18. Yang YJ, Qian HY, Huang J et al. Combined therapy with simvastatin and bone marrow‐derived mesenchymal stem cells increases benefits in infarcted swine hearts. Arterioscler Thromb Vasc Biol 2009;29:2076–2082. [DOI] [PubMed] [Google Scholar]
- 19. Liao JK, Laufs U. Pleiotropic effects of statins. Ann Rev Pharmacol Toxicol 2005;45:89–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ma J, Ge J, Zhang S et al. Time course of myocardial stromal cell‐derived factor 1 expression and beneficial effects of intravenously administered bone marrow stem cells in rats with experimental myocardial infarction. Basic Res Cardiol 2005;100:217–223. [DOI] [PubMed] [Google Scholar]
- 21. Qiu R, Cai A, Dong Y et al. SDF‐1alpha upregulation by atorvastatin in rats with acute myocardial infarction via nitric oxide production confers anti‐inflammatory and anti‐apoptotic effects. J Biomed Sci 2012;19:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zaruba MM, Franz WM. Role of the SDF‐1‐CXCR4 axis in stem cell‐based therapies for ischemic cardiomyopathy. Expert Opin Biol Ther 2010;10:321–335. [DOI] [PubMed] [Google Scholar]
- 23. Dong Q, Yang Y, Song L et al. Atorvastatin prevents mesenchymal stem cells from hypoxia and serum‐free injury through activating AMP‐activated protein kinase. Int J Cardiol 2011;153:311–316. [DOI] [PubMed] [Google Scholar]
- 24. Li N, Yang YJ, Qian HY et al. Intravenous administration of atorvastatin‐pretreated mesenchymal stem cells improves cardiac performance after acute myocardial infarction: role of CXCR4. Am J Transl Res 2015;7:1058–1070. [PMC free article] [PubMed] [Google Scholar]
- 25. Hu X, Wang J, Chen J et al. Optimal temporal delivery of bone marrow mesenchymal stem cells in rats with myocardial infarction. Eur J Cardiothorac Surg 2007;31:438–443. [DOI] [PubMed] [Google Scholar]
- 26. Chen Y, Teng X, Chen W et al. Timing of transplantation of autologous bone marrow derived mesenchymal stem cells for treating myocardial infarction. Sci China Life Sci 2014;57:195–200. [DOI] [PubMed] [Google Scholar]
- 27. Richardson JD, Bertaso AG, Psaltis PJ et al. Impact of timing and dose of mesenchymal stromal cell therapy in a preclinical model of acute myocardial infarction. J Card Fail 2013;19:342–353. [DOI] [PubMed] [Google Scholar]
- 28. Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Phar 2016;7:27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sharma V, McNeill JH. To scale or not to scale: The principles of dose extrapolation. Br J Pharmacol 2009;157:907–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Atar S, Ye Y, Lin Y et al. Atorvastatin‐induced cardioprotection is mediated by increasing inducible nitric oxide synthase and consequent S‐nitrosylation of cyclooxygenase‐2. Am J Physiol Heart Circ Physiol 2006;290:H1960–H1968. [DOI] [PubMed] [Google Scholar]
- 31. Hamzeh M, Hosseinimehr SJ, Khalatbary AR et al. Atorvastatin mitigates cyclophosphamide‐induced hepatotoxicity via suppression of oxidative stress and apoptosis in rat model. Res Pharm Sci 2018;13:440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li T, Yao W. Therapeutic effect of irbesartan combined with atorvastatin calcium in the treatment of rats with coronary heart disease. Exp Ther Med 2018;16:4119–4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Subramani C, Rajakannu A, Gaidhani S et al. Glutathione‐redox status on hydro alcoholic root bark extract of Premna integrifolia Linn in high fat diet induced atherosclerosis model. J Ayurveda Integr Med 2019. pii: S0975‐9476(17)30306‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Uretsky BF, Schwarz ER, Rosanio S et al. Prostaglandins mediate the cardioprotective effects of atorvastatin against ischemia‐reperfusion injury. Cardiovasc Res 2005;65:345–355. [DOI] [PubMed] [Google Scholar]
- 35. Birnbaum Y, Ye Y, Lin Y et al. Augmentation of myocardial production of 15‐epi‐lipoxin‐a4 by pioglitazone and atorvastatin in the rat. Circulation 2006;114:929–935. [DOI] [PubMed] [Google Scholar]
- 36. Verpooten GA, Stolear JC, Matthys KE et al. Pharmacokinetics of atorvastatin and its metabolites after single and multiple dosing in hypercholesterolaemic haemodialysis patients. Nephrol Dial Transplant 2003;18:967–976. [DOI] [PubMed] [Google Scholar]
- 37. He L, Li Y, Huang X et al. Genetic lineage tracing of resident stem cells by DeaLT. Nat Protoc 2018;13:2217–2246. [DOI] [PubMed] [Google Scholar]
- 38. van Berlo JH, Kanisicak O, Maillet M et al. c‐kit+ cells minimally contribute cardiomyocytes to the heart. Nature 2014;509:337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhou B, Wu SM. Reassessment of c‐Kit in cardiac cells: A complex interplay between expression, fate, and function. Circ Res 2018;123:9–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Murry CE, Soonpaa MH, Reinecke H et al. Haematopoietic stem cells do not transdifferentiate into cardiac myocytes in myocardial infarcts. Nature 2004;428:664–668. [DOI] [PubMed] [Google Scholar]
- 41. Sultana N, Zhang L, Yan J et al. Resident c‐kit(+) cells in the heart are not cardiac stem cells. Nat Commun 2015;6:8701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu Q, Yang R, Huang X et al. Genetic lineage tracing identifies in situ Kit‐expressing cardiomyocytes. Cell Res 2016;26:119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gude NA, Firouzi F, Broughton KM et al. Cardiac c‐Kit biology revealed by inducible transgenesis. Circ Res 2018;123:57–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hirsch A, Nijveldt R, van der Vleuten PA et al. Intracoronary infusion of mononuclear cells from bone marrow or peripheral blood compared with standard therapy in patients after acute myocardial infarction treated by primary percutaneous coronary intervention: Results of the randomized controlled HEBE trial. Eur Heart J 2011;32:1736–1747. [DOI] [PubMed] [Google Scholar]
- 45. Janssens S, Dubois C, Bogaert J et al. Autologous bone marrow‐derived stem‐cell transfer in patients with ST‐segment elevation myocardial infarction: Double‐blind, randomised controlled trial. Lancet 2006;367:113–121. [DOI] [PubMed] [Google Scholar]
- 46. Schachinger V, Erbs S, Elsasser A et al. Improved clinical outcome after intracoronary administration of bone‐marrow‐derived progenitor cells in acute myocardial infarction: Final 1‐year results of the REPAIR‐AMI trial. Eur Heart J 2006;27:2775–2783. [DOI] [PubMed] [Google Scholar]
- 47. Traverse JH, Henry TD, Pepine CJ et al. Effect of the use and timing of bone marrow mononuclear cell delivery on left ventricular function after acute myocardial infarction: the TIME randomized trial. JAMA 2012;308:2380–2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wollert KC, Meyer GP, Lotz J et al. Intracoronary autologous bone‐marrow cell transfer after myocardial infarction: The BOOST randomised controlled clinical trial. Lancet 2004;364:141–148. [DOI] [PubMed] [Google Scholar]
- 49. Traverse JH, Henry TD, Pepine CJ et al. TIME trial: Effect of timing of stem cell delivery following ST‐elevation myocardial infarction on the recovery of global and regional left ventricular function: Final 2‐year analysis. Circ Res 2018;122:479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu B, Duan CY, Luo CF et al. Impact of timing following acute myocardial infarction on efficacy and safety of bone marrow stem cells therapy: A network meta‐analysis. Stem Cells Int 2016;2016:1031794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Song L, Yang YJ, Dong QT et al. Atorvastatin enhance efficacy of mesenchymal stem cells treatment for swine myocardial infarction via activation of nitric oxide synthase. PLoS One 2013;8:e65702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang Q, Wang H, Yang YJ et al. Atorvastatin treatment improves the effects of mesenchymal stem cell transplantation on acute myocardial infarction: The role of the RhoA/ROCK/ERK pathway. Int J Cardiol 2014;176:670–679. [DOI] [PubMed] [Google Scholar]
- 53. Kanelidis AJ, Premer C, Lopez J et al. Route of delivery modulates the efficacy of mesenchymal stem cell therapy for myocardial infarction: A meta‐analysis of preclinical studies and clinical trials. Circ Res 2017;120:1139–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang Z, Wang L, Su X et al. Rational transplant timing and dose of mesenchymal stromal cells in patients with acute myocardial infarction: A meta‐analysis of randomized controlled trials. Stem Cell Res Ther 2017;8:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu YS, Ou ME, Liu H et al. The effect of simvastatin on chemotactic capability of SDF‐1alpha and the promotion of bone regeneration. Biomaterials 2014;35:4489–4498. [DOI] [PubMed] [Google Scholar]
- 56. Cui X, Chopp M, Zacharek A et al. Chemokine, vascular and therapeutic effects of combination Simvastatin and BMSC treatment of stroke. Neurobiol Dis 2009;36:35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mayorga ME, Kiedrowski M, McCallinhart P et al. Role of SDF‐1:CXCR4 in impaired post‐myocardial infarction cardiac repair in diabetes. Stem Cells Transl Med 2018;7:115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gnecchi M, Zhang Z, Ni A et al. Paracrine mechanisms in adult stem cell signaling and therapy. Circ Res 2008;103:1204–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Surder D, Manka R, Lo Cicero V et al. Intracoronary injection of bone marrow‐derived mononuclear cells early or late after acute myocardial infarction: Effects on global left ventricular function. Circulation 2013;127:1968–1979. [DOI] [PubMed] [Google Scholar]
- 60. Traverse JH, Henry TD, Ellis SG et al. Effect of intracoronary delivery of autologous bone marrow mononuclear cells 2 to 3 weeks following acute myocardial infarction on left ventricular function: The LateTIME randomized trial. JAMA 2011;306:2110–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Askari AT, Unzek S, Popovic ZB et al. Effect of stromal‐cell‐derived factor 1 on stem‐cell homing and tissue regeneration in ischaemic cardiomyopathy. Lancet 2003;362:697–703. [DOI] [PubMed] [Google Scholar]
- 62. Tong J, Ding J, Shen X et al. Mesenchymal stem cell transplantation enhancement in myocardial infarction rat model under ultrasound combined with nitric oxide microbubbles. PLoS One 2013;8:e80186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gnecchi M, He H, Noiseux N et al. Evidence supporting paracrine hypothesis for Akt‐modified mesenchymal stem cell‐mediated cardiac protection and functional improvement. FASEB J 2006;20:661–669. [DOI] [PubMed] [Google Scholar]
- 64. Abbott JD, Huang Y, Liu D et al. Stromal cell‐derived factor‐1alpha plays a critical role in stem cell recruitment to the heart after myocardial infarction but is not sufficient to induce homing in the absence of injury. Circulation 2004;110:3300–3305. [DOI] [PubMed] [Google Scholar]
- 65. Hu X, Sun A, Xie X et al. Rosuvastatin changes cytokine expressions in ischemic territory and preserves heart function after acute myocardial infarction in rats. J Cardiovasc Pharmacol Ther 2013;18:162–176. [DOI] [PubMed] [Google Scholar]
- 66. Delewi R, Hirsch A, Tijssen JG et al. Impact of intracoronary bone marrow cell therapy on left ventricular function in the setting of ST‐segment elevation myocardial infarction: A collaborative meta‐analysis. Eur Heart J 2014;35:989–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schutt RC, Trachtenberg BH, Cooke JP et al. Bone marrow characteristics associated with changes in infarct size after STEMI: A biorepository evaluation from the CCTRN TIME trial. Circ Res 2015;116:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bolli R. Repeated cell therapy: A paradigm shift whose time has come. Circ Res 2017;120:1072–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Guo Y, Wysoczynski M, Nong Y et al. Repeated doses of cardiac mesenchymal cells are therapeutically superior to a single dose in mice with old myocardial infarction. Basic Res Cardiol 2017;112:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tang XL, Nakamura S, Li Q et al. Repeated administrations of cardiac progenitor cells are superior to a single administration of an equivalent cumulative dose. J Am Heart Assoc 2018;7(4). pii: e007400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tokita Y, Tang XL, Li Q et al. Repeated administrations of cardiac progenitor cells are markedly more effective than a single administration: A new paradigm in cell therapy. Circ Res 2016;119:635–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kelkar AA, Butler J, Schelbert EB et al. Mechanisms contributing to the progression of ischemic and nonischemic dilated cardiomyopathy: Possible modulating effects of paracrine activities of stem cells. J Am Coll Cardiol 2015;66:2038–2047. [DOI] [PubMed] [Google Scholar]
- 73. Wysoczynki M, Khan A, Bolli R. New paradigms in cell therapy. Circ Res 2018;123:138–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Orlic D, Kajstura J, Chimenti S et al. Bone marrow cells regenerate infarcted myocardium. Nature 2001;410:701–705. [DOI] [PubMed] [Google Scholar]
- 75. Pittenger MF, Martin BJ. Mesenchymal stem cells and their potential as cardiac therapeutics. Circ Res 2004;95:9–20. [DOI] [PubMed] [Google Scholar]
- 76. Alvarez‐Dolado M, Pardal R, Garcia‐Verdugo JM et al. Fusion of bone‐marrow‐derived cells with Purkinje neurons, cardiomyocytes and hepatocytes. Nature 2003;425:968–973. [DOI] [PubMed] [Google Scholar]
- 77. Nygren JM, Jovinge S, Breitbach M et al. Bone marrow‐derived hematopoietic cells generate cardiomyocytes at a low frequency through cell fusion, but not transdifferentiation. Nat Med 2004;10:494–501. [DOI] [PubMed] [Google Scholar]
- 78. Davis DR. Cardiac stem cells in the post‐Anversa era. Eur Heart J 2019;40:1039–1041. [DOI] [PubMed] [Google Scholar]
- 79. Gnecchi M, He H, Liang OD et al. Paracrine action accounts for marked protection of ischemic heart by Akt‐modified mesenchymal stem cells. Nat Med 2005;11:367–368. [DOI] [PubMed] [Google Scholar]
- 80. Cho DI, Kim MR, Jeong HY et al. Mesenchymal stem cells reciprocally regulate the M1/M2 balance in mouse bone marrow‐derived macrophages. Exp Mol Med 2014;46:e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Peng Y, Pan W, Ou Y et al. Extracardiac‐lodged mesenchymal stromal cells propel an inflammatory response against myocardial infarction via paracrine effects. Cell Transplant 2016;25:929–935. [DOI] [PubMed] [Google Scholar]
- 82. Kinnaird T, Stabile E, Burnett MS et al. Marrow‐derived stromal cells express genes encoding a broad spectrum of arteriogenic cytokines and promote in vitro and in vivo arteriogenesis through paracrine mechanisms. Circ Res 2004;94:678. [DOI] [PubMed] [Google Scholar]
- 83. Chen GH, Xu J, Yang YJ. Exosomes: Promising sacks for treating ischemic heart disease? Am J Physiol Heart Circ Physiol 2017;313:H508–h523. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Mesenchymal stem cells (MSCs) identification. (A) Flow cytometry results of the surface markers of implanted MSCs, probing as CD90+, CD29+, CD45‐, CD11‐. (B‐C) Representative images of MSCs. Scale bar = 500μm (B) and 200μm (C).
Figure S2. Left heart catheterization assessment of cardiac function. The values of left ventricular pressure maximal rate of rise and fall (±dp/dtmax) were detected at the endpoint (6 weeks after myocardial infarction). n = 8‐11 for each group. $ p <0.05 vs. Sham group; & p <0.05 vs. AMI group; γ p < 0.05 vs. Early1; κ p < 0.05 vs. Early2; φ p < 0.05 vs. Early3; α p < 0.05 vs. Mid1; μ p < 0.05 vs. Mid2; # p < 0.05 vs. Mid3; *p < 0.05 intra‐group comparison in different periods. All data are mean ± SD.
Figure S3. Assessment of c‐Kit+ cells mobilization. (A) Mobilization of c‐Kit+ cells (green) in the peri‐infarcted myocardium at 6 weeks after myocardial infarction. Nuclei were dyed with DAPI (blue). Scale bar = 25μm. (B) The percentage of c‐Kit+ cells in the peri‐infarcted region per HPF in each group. n = 5 for each group. $ p <0.05 vs. Sham group; & p <0.05 vs. AMI group; γ p < 0.05 vs. Early1; κ p < 0.05 vs. Early2; φ p < 0.05 vs. Early3; α p < 0.05 vs. Mid1; μ p < 0.05 vs. Mid2; # p < 0.05 vs. Mid3; *p < 0.05 intra‐group comparison in different periods. All data are mean ± SD.
Figure S4. Assessment of differentiation of MSCs into cardiomyocytes. (A) Representative images of CM‐Dil and c‐TnT co‐staining in the peri‐infarcted regions at the endpoint. Cardiomyocytes, CM‐Dil‐labeled cells and nuclei were stained green, red and blue, respectively. Scale bar = 25μm. (B) Quantitative analyses with the number of CM‐Dil‐DAPI‐ c‐TnT+ merged cells per HPF in each group. n = 5 for each group. $ p <0.05 vs. Sham group; & p <0.05 vs. AMI group; α p < 0.05 vs. Mid1; μ p < 0.05 vs. Mid2; # p < 0.05 vs. Mid3; *p < 0.05 intra‐group comparison in different periods. All data are mean ± SD.
Table S1. Changes of SDF‐1 and inflammatory cytokines in peri‐infarcted region over time course assessed by immunofluorescence and ELISA
Table S2. Quantitation of CM‐Dil‐labeled cells, c‐Kit+ cells, angiogenesis and differentiation in the peri‐infarcted myocardium
Table S3. LV Function and Geometry Parameters by Echocardiography
Table S4. Left heart catheterization assessment of cardiac function
Table S5. Quantitation of infarct and fibrotic size, CD45+ cell infiltration, TUNEL+ cell percentage and measurement of IL‐6, TNF‐α expressions in the peri‐infarcted myocardium