

Abstract
Collagen type III (COL3) is one of the 3 major collagens in the body, and loss of expression or mutations in the COL3 gene have been associated with the onset of vascular diseases such the Ehlers-Danlos syndrome. Previous work reported a significant reduction of COL3 in tissues such as skin and vessels with aging. In agreement, we found that COL3 was significantly reduced in senescent human mesenchymal stem cells and myofibroblasts derived from patients with Hutchinson-Gilford progeria syndrome, a premature aging syndrome. Most notably, we discovered that ectopic expression of the embryonic transcription factor Nanog homeobox (NANOG) restored COL3 expression by restoring the activity of the TGF-β pathway that was impaired in senescent cells. RNA sequencing analysis showed that genes associated with the activation of the TGF-β pathway were up-regulated, whereas negative regulators of the pathway were down-regulated upon NANOG expression. Chromatin immunoprecipitation sequencing and immunoprecipitation experiments revealed that NANOG bound to the mothers against decapentaplegic (SMAD)2 and SMAD3 promoters, in agreement with increased expression and phosphorylation levels of both proteins. Using chemical inhibition, short hairpin RNA knockdown, and gain of function approaches, we established that both SMAD2 and SMAD3 were necessary to mediate the effects of NANOG, but SMAD3 overexpression was also sufficient for COL3 production. In summary, NANOG restored production of COL3, which was impaired by cellular aging, suggesting novel strategies to restore the impaired extracellular matrix production and biomechanical function of aged tissues, with potential implications for regenerative medicine and anti-aging treatments.—Rong, N., Mistriotis, P., Wang, X., Tseropoulos, G., Rajabian, N., Zhang, Y., Wang, J., Liu, S., Andreadis, S. T. Restoring extracellular matrix synthesis in senescent stem cells.
Keywords: aging, elasticity, ECM, collagen, NANOG
Aging is considered a principal cause of cardiovascular dysfunction. In particular, it is well accepted that smooth muscle cells (SMCs) of the vasculature are affected significantly by aging. SMCs comprise the medial layer of blood vessels and predominantly regulate the vascular tone by contracting and relaxing in response to environmental signals. However, with aging, SMCs exhibit significantly reduced capacity for contractile force generation (1, 2), produce reduced amounts of extracellular matrix (ECM) proteins, and secrete matrix metalloproteinases that degrade the surrounding matrix (3). SMCs in aged arteries become senescent or die due to apoptosis (4) and secrete proinflammatory cytokines that attract immune cells (5) and increase oxidative stress (6). Collectively, age-related SMC dysfunction results in stiffer arteries, which are prone to aneurysms, vascular inflammation, reduced SMC numbers, and loss of the vessel’s repair capacity. As a result, aging contributes to the emergence and progression of cardiovascular diseases such as aortic aneurysms, hypertension, atherosclerosis, and atherosclerotic plaque rupture (7).
Collagen is the main ECM component that is thought to impart strength to mesenchymal tissues such as the arterial wall. The main collagen types present in the vascular wall are type I (70–75%) and type III (20–25%) collagen. Collagen type III (COL3) is found primarily in the media layer of the arterial wall, where it is deposited at the peripheral region of collagen type I (COL1) and is believed to play a role in regulating COL1 fibrillogenesis (8, 9), a process of crucial importance in vascular development (10), bone formation (11), and wound healing (12). Deficiency in COL3 is associated with bicuspid aortic valve disease, aneurysm, and atherosclerotic plaque, resulting in weak vessels that are prone to rupture (13, 14). Indeed, COL3-deficient mice develop a dissecting aneurysm and aortic rupture (9). Similarly, mutations in the human COL3 gene cause vascular Ehlers-Danlos syndrome (15), a disease associated with arterial dilation and rupture (16). Mounting evidence suggests that COL1 primarily regulates the strength, whereas COL3 regulates the elasticity of the collagen meshwork. Thus, the COL3-to-COL1 ratio is thought to be an important indicator of vascular elasticity and adaptive remodeling capacity (10).
Previous work from our laboratory revealed that the proliferation capacity and contractility of mesenchymal stem cell (MSC)-derived SMCs were greatly diminished with donor aging and culture senescence (17). We also discovered that ectopic expression of the pluripotent factor NANOG (Nanog homeobox) was sufficient to reverse cellular aging and restore the diminished contractile capacity of aged myogenic progenitors using 3 different models of aging: 1) organismal aging; 2) replicative senescence; and 3) Hutchinson-Gilford progeria syndrome (HGPS) or premature aging (18). In addition, we reported that NANOG restored ACTIN polymerization, and induced myocardin-related transcription factor A (MRTF-A) translocation to the nucleus and serum response factor (SRF)-dependent myogenic gene expression (18), ultimately restoring the force generation ability of senescent MSCs and HGPS cells (19). Here we report that NANOG also reversed the impaired production of COL3 in senescent MSCs and HGPS cells by reactivating the impaired TGF-β1 pathway through transcriptional control of several genes, including mothers against decapentaplegic (SMAD)2 and SMAD3.
MATERIALS AND METHODS
Cell lines and cell culture
Human hair follicle–derived MSCs (hHF-MSCs) from a 73-yr-old donor and human neonatal dermal fibroblasts from foreskin tissues were isolated as previously described by Mistriotis and Andreadis (20). hHF-MSCs have been previously characterized with respect to their proliferation and differentiation capacity (21). Human dermal fibroblasts from patients suffering from HGPS (HGADFN167) and from a healthy donor (CNT, HGADFN168, father of HGADFN167) were obtained from the Progeria Research Foundation (Peabody, MA, USA). Early passage (EP) and late passage (LP) or senescent cells were characterized and defined based on their proliferation and differentiation potential as previously described by Mistriotis et al. (18). In general, hHF-MSCs at passage 6–7 were defined as EP and passage 13–16 as LP cells. Human neonatal fibroblast, HGPS, and the parental control fibroblasts were used at passages 18–21.
hHF-MSC and human neonatal fibroblast were cultured in DMEM (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% (v/v) MSC-qualified fetal bovine serum (Thermo Fisher Scientific) and 1 ng/ml basic fibroblast growth factor (ORF Genetics, Kópavogur, Iceland), whereas HGPS fibroblast was maintained in DMEM supplement with 15% (v/v) fetal bovine serum and 2 mM L-glutamine (Thermo Fisher Scientific). For collagen production, cells were cultured in DMEM supplemented with 2 ng/ml (TGF-β1; BioLegend, San Diego, CA, USA). All the media contained 1% (v/v) antibiotic-antimycotic (Thermo Fisher Scientific).
Doxycycline (DOX)-regulated overexpression of NANOG or ZsGreen (control) in the aforementioned cells were previously described by Mistriotis et al. (18). To evaluate the effect of NANOG on COL3A1 production, confluent cell monolayers in growth medium were first induced with 1 µg/ml DOX (MilliporeSigma, Burlington, MA, USA) to overexpress NANOG or ZsGreen for 2 d, before coaxing the cells to differentiate in differentiation medium for 5 d in the continuous presence of DOX. To block the TGF-β pathway, cells in either growth medium or differentiation medium were supplemented with 10 µM SB431542 for 7 d.
Tetracycline-regulatable system
To generate cells expressing NANOG or ZsGreen under a tetracycline-regulatable system, EP-MSCs (passage 6–7) or HGPS, and the parental control fibroblasts (passage 16–17) were cotransduced with 2 separate lentiviral vectors. The first vector combined NANOG or ZsGreen with the selection marker puromycin N-acetyl-transferase (pale cress protein) under the control of the tetracycline response elements (TREs) and a minimal cytomegalovirus sequence (TRE-NANOG-IRES-PURO or TRE-ZsGreen-IRES-PURO). The second vector (FUdeltaGW-rtTA; Addgene, Watertown, MA, USA) encodes for the reverse tetracycline transactivator (rtTA), a fusion protein of the reverse Tet repressor and the VP16 transcription activation domain. This system enables NANOG or ZsGreen expression upon DOX treatment. Transduced cells were selected with puromycin for 6 d and then cultured until they reached senescence (passage 13–16) (Fig. 1A). At that time, NANOG or ZsGreen were induced upon treatment with DOX for the indicated times.
Figure 1.

NANOG reverses senescence of hMSCs and HGPS fibroblasts. A) Schematic of the experiment timeline. EP-MSCs were first cotransduced with virus carrying rtTA, and NANOG or ZsGreen (EPZ). Cells were selected with puromycin for 6 d and cultured until they reached senescence. At that time, 1 µg/ml DOX was added to induce expression of NANOG (LPN) or ZsGreen (LPZ) for 7 d. B, C) Percentage senescent cells as shown by SA-β-gal staining and quantification. D, E) Immunostaining for Ki67 and quantification. F, G) Immunostaining for γ-H2AX and quantification. EPZ, EP+ZsGreen hMSCs; LPZ, HGPS+ZsGreen hMSCs; LPN, LP+NANOG hMSCs (A). HGPS fibroblasts were transduced with virus carrying NANOG or ZSG and rtTA and then selected with puromycin. H, I) Percentage senescent cells as shown by SA-β-gal staining and quantification. J, K) Immunostaining for Ki67 and quantification. L, M) Immunostaining for γ-H2AX and its quantification. CNTZ, CNT+ZsGreen fibroblast; HGPSZ, HGPS+ZsGreen fibroblast; HGPSN, HGPS+NANOG fibroblasts. Data are normalized to LPZ (C, E, G) or HGPSZ (I, K, M) and presented as means ± sd, n = 3 independent experiments. Asterisk designates statistical significance as compared to LPZ or HGPSZ (P < 0.05).
Plasmids constructs and cell transduction
Gene knockdown was performed using the short hairpin left ventricular diastolic pressure vector that was developed in our group (22). Briefly, complementary oligos containing sequences targeting SMAD2, SMAD3, or SMAD4 (Supplemental Table S1) (23, 24) were mixed at 50 µM each and annealed at 62°C for 2 min. The annealed products were cloned downstream of the [1H] promoter in the short hairpin ventricular diastolic pressure (short hairpin lentiviral dual promoter). A scramble sequence without any homology to the human genome [confirmed by the Basic Local Alignment Search Tool (BLAST; https://blast.ncbi.nlm.nih.gov/Blast.cgi)] was used as a control.
For the overexpression experiments, coding sequences for SMAD2 and SMAD3 were obtained from the National Center for Biotechnology Information (NCBI; https://www.ncbi.nlm.nih.gov/) database. Primers were designed using the first and last 20 nt of the coding sequences (Supplemental Table S1), and the PCR products were cloned downstream of the cytomegalovirus promoter in pCS-CP. The pCS-CP was modified from pCS-CG vector (25), where the green fluorescent protein gene was replaced with a puromycin N-acetyltransferase pale cress protein gene.
For lentivirus production, 3 plasmids (lentiviral vector, psPAX2, and pMD2.G) were used to transfect 293T/17 cells using the standard calcium phosphate precipitation method. The virus was harvested 24 h post-transfection, filtered through a 0.45 μm filter (Corning, Corning, NY, USA), and pelleted by centrifugation (50,000 g at 2°C for 2 h). Finally, the pellet was resuspended in fresh DMEM and stored at −80°C until use.
Senescence-associated β-galactosidase assay
Senescence-associated β-galactosidase assay (SA-β-gal) activity was detected using a Senescence Staining Kit (MilliporeSigma) according to the manufacturer’s recommendations. Stained cells were then visualized and photographed using a Zeiss Axio Observer Z1 inverted microscope with an Orca-ER Charge Coupled Device (CCD) camera (Hamamatsu Photonics, Hmamatsu, Japan). The percentage of cells positive for SA-β-gal was determined in duplicate samples from multiple fields of view containing ∼200 cells/sample.
Immunoprecipitation
Cells were washed 3 times with cold PBS, followed by addition of lysis buffer containing complete protease inhibitors (Thermo Fisher Scientific). The cell monolayer was then scraped off from the surface, and lysates were centrifuged at 14,000 g for 20 min at 4°C. The supernatants were incubated with normal IgG (2729S; Cell Signaling Technology, Danvers, MA, USA) or rabbit anti-SMAD2 antibody (5339; Cell Signaling Technology) overnight at 4°C, and the immune complexes were then mixed with Protein A-Sepharose beads (20421; Thermo Fisher Scientific) after overnight incubation at 4°C. Samples were subjected to Western blot analysisusing mouse anti-NANOG (1:1000) (AF1997; R&D Systems, Minneapolis, MN, USA) and rabbit anti-SMAD2/SMAD3 (1:1000) (8685; Cell Signaling Technology).
Western blot analysis
For Western blot analysis, cells were lysed on ice, and lysates were centrifuged at 15,000 g for 10 min at 4°C. Supernatants were then collected, and the amount of protein in each sample was determined by Bradford assay. Subsequently, 20 μg of total protein was loaded onto 4–20% Tris-glycine SDS-PAGE gels. After electrophoresis, proteins were transferred to PVDF membrane (Bio-Rad, Hercules, CA, USA), and the expression level of the indicated proteins was then detected using the antibodies as presented in Supplemental Table S2.
Immunostaining
Immunostaining was performed, according to the manufacturer’s suggested conditions, to examine the expression of γH2aX (9718S; Cell Signaling Technology), Ki67 (16667; Abcam, Cambridge, MA, USA), and collagen 3 (23445; Abcam) in EP, LP+ZsGreen (LPZ), hMSC, and LP+NANOG (LPN), hMSC cells. For examining the expression of p-SMAD2/SMAD3 (8828; Cell Signaling Technology), NANOG was first expressed for 3 d. Afterward, cells were serum starved overnight and then stimulated with 2 ng/ml TGF-β1 for 30 h in the continuous presence of DOX. All primary antibodies were used at 1:200 dilution in 5% (v/v) goat serum (Thermo Fisher Scientific)/PBS. Subsequently, samples were incubated with secondary antibody (A11012, Alexa Fluor 594–conjugated goat anti-rabbit IgG; Thermo Fisher Scientific) at 1:400 dilution in 5% (v/v) goat serum/PBS for 1 h at room temperature. Nuclei were counterstained with 12.5 µg/ml Hoechst 33342 (Thermo Fisher Scientific) for 5 min at room temperature. The results were analyzed with ImageJ (National Institutes of Health, Bethesda, MA, USA) software and expressed as a ratio of fluorescence intensity to the cell number.
RNA extraction, cDNA synthesis, and real-time PCR
Total RNA was extracted using RNeasy Plus Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol. The cDNA was produced with OneStep RT-PCR Kit (Qiagen). Real-time PCR was performed with SYBR Green Supermix (Bio-Rad) using the primer pairs as listed in Supplemental Table S3.
RNA sequencing analysis
RNA sequencing (RNA-Seq) was performed with the Illumina platform to assess the global gene expression profile upon NANOG expression as previously described by Mistriotis et al. (18). Genes that were differentially expressed by >1.5-fold and with an adjusted value of P < 0.05 were analyzed. Data lists of all the genes that satisfied these criteria were analyzed by the Ingenuity Pathway Analysis (IPA) software (Ingenuity Systems, Redwood City, CA, USA; http://www.ingenuity.com).
Chromatin immunoprecipitation sequencing and chromatin immunoprecipitation PCR analysis
Chromatin immunoprecipitation (ChIP) assay to determine the degree of NANOG protein enrichment was performed using the Magna ChIP A/G Chromatin Immunoprecipitation Kit (MilliporeSigma) according to the manufacturer’s instructions. Briefly, LP MSCs expressing NANOG were seeded in 150-mm dishes and grown to confluence before harvesting. Cells were fixed with 1% formaldehyde for 10 min at room temperature. After sonication, chromatin was immunoprecipitated with a goat anti-NANOG antibody (AF-1997; R&D Systems) or IgG (provided in the kit) or anti-RNA polymerase (provided in the kit) at 4°C overnight. The protein/DNA complexes were eluted from the magnetic beads after standard washing steps. The crosslinks were reversed by incubating at 62°C for 2 h and 95°C for 10 min. Final DNA products were purified and used for sequencing as well as PCR. PCR was then performed with Phusion High-Fidelity PCR Master Mix (M0532S; New England Biolabs, Ipswich, MA, USA) using the primer pairs as listed in Supplemental Table S4.
Raw Fastq read sequences that passed quality filter from Illumina real-time analysis were preprocessed by using FastQC for sequencing base quality control. Then the reads were mapped to the human GRCh38.p7 genome sequence using Burrows-Wheeler Aligner (26) and converted to β-barrel assembly machinery (BAM) files. The NANOG binding site [also known as ChIP sequencing (ChIP-Seq) peaks] were called using model-based analysis of ChIP-Seq (MACS2) (27) with input DNA as control samples. ChIPseeker (28), an R package (https://www.r-project.org/), was used to annotate ChIP peaks, to visualize tag densities around transcription start sites (TSSs) region (−3 kbp, +3 kbp), and to perform Kyoto Encyclopedia of Genes and Genomes (KEGG: https://www.genome.jp/kegg/kegg1.html) pathway enrichment analysis in different ChIP experiments with value of P < 0.05 as the cutoff of significance. MEME (motif-based sequence analysis tool)-ChIP (29) was used to identify NANOG cobinding factors using top 500 peaks in TSSs region against the Jaspar database (http://jaspar.genereg.net/). Genes in NANOG binding site at TSSs and differentially expressed upon NANOG expression were identified as potential target genes.
Statistical analysis
The values represent the mean ± sd of ≥3 independent experiments. The statistical significance between controls and treated groups was evaluated by Student’s t test (paired, 2-tailed) and determined by value of P = 0.05.
RESULTS
Ectopic expression of NANOG restores the senescent phenotype of aged cells
We have recently shown that the pluripotency-associated transcription factor NANOG could restore the ability of senescent MSCs to differentiate toward functional and contractile SMCs (18). Here we hypothesize that NANOG may also restore other aspects of cellular aging including significantly impaired synthesis of ECM proteins. To test this hypothesis, we employed 2 widely established models of aging: 1) replicative senescence of human (h)MSCs and human dermal fibroblasts; and 2) myofibroblasts derived from patients with HGPS, who suffer from premature aging. To induce expression of NANOG or ZsGreen (control), we applied a Tet-on system that we previously developed (18), which enables expression of either NANOG or ZsGreen in the presence of DOX (Fig. 1A). The effect of DOX was also determined using nontransduced cells (18), and we found that DOX did not have an effect on COL3 expression (Supplemental Fig. S1).
Senescent cells exhibited growth arrest, DNA damage, altered gene expression, and up-regulation of senescence-specific genes. Specifically, cells that have undergone replicative senescence [LP+ZsGreen (LPZ)] showed significantly increased SA-β-gal staining (Fig. 1B, C), decreased proliferation as shown by Ki67 immunostaining (Fig. 1D, E), and DNA damage as shown by γ-H2AX (Fig. 1F, G). Similar results were observed in cells originating from patient with progeria [HGPS+ZsGreen (HGPSZ)] (Fig. 1H–M). Interestingly, the levels of SA-β-gal and γ-H2AX were greater in LPZ-MSCs as compared to those from patients with HGPS, suggesting that replicative senescence may lead to higher levels of DNA damage. Notably, NANOG (LPN, HGPSN) reversed expression of all 3 markers (Fig. 1B–M), as well as expression of senescence-associated genes (p53, p21) in both cells and in the case of HGPSZ cells also p16 (Supplemental Fig. S2A, B) but had no effect on the levels of progerin (Supplemental Fig. S2C).
NANOG restores COL3 production in senescent cells
Mutations of COL3 have been associated with type III and IV Ehlers-Danlos syndrome and with aortic and arterial aneurysms (14). In addition, aging is accompanied by significant reduction of the COL3 content in tissues such as skin (30) and blood vessels (10), leading to decreased elasticity or increased stiffness. Notably, we found that COL3 protein was significantly reduced in LPZ-MSCs and HGPSZ cells but was completely restored by expression of NANOG (Fig. 2A, B). Interestingly, NANOG did not have a significant effect on COL1A2 but up-regulated the expression of collagen type 4 (only in LPN) and collagen type 6 (LPN, HGPSN) mRNA (Supplemental Fig. S3A–D).
Figure 2.

Ectopic expression of NANOG in senescent hMSCs and HGPS fibroblasts restore COL3 expression in 2-dimensional and 3-dimensional model. A, B) Western blotting analysis and quantification for COL3 in EP+ZsGreen (EPZ), LPZ, and LPN hMSCs (A) as well as in CNT+ZsGreen (CNTZ), HGPS+ZsGreen (HGPSZ), and HGPS+NANOG (HGPSN) fibroblasts (B). To prepare tissue constructs, cells were introduced into fibrin hydrogels that were allowed to polymerize around mandrels. C–F) Immunostaining and quantification of engineered tissues of EPZ, LPZ, and LPN hMSCs (C, D) and CNTZ, HGPSZ, and HGPSN fibroblasts (E, F). EPZ, Early passage (6, 7); LPN/LPZ, late passage (13–16). One microgram per milliliter DOX was added in the medium to express NANOG or ZsGreen. Data are compared to LPZ (A, D) or HGPSZ (B, F) and presented as means ± sd, n = 3 independent experiments. *P < 0.05 compared to LPZ or HGPSZ (n = 3 independent experiments).
Next, we investigated whether NANOG could also restore collagen expression in 3-dimensional vascular tissue constructs prepared with senescent hMSCs or HGPS cells. To this end, cells were embedded into fibrin hydrogels that were allowed to polymerize around mandrels to form cylindrical constructs. After 2 wk in culture, the cells compacted the hydrogels down to ∼5% of their original volume yielding cylindrical constructs with wall thickness of <500 µm. Immunostaining showed significant loss of COL3 content in tissue constructs prepared with senescent LPZ-MSCs or HGPSZ cells, which was reversed by NANOG expression (Fig. 2C–F). Furthermore, hydroxyproline measurements showed increased total collagen content in NANOG-expressing tissue constructs (Supplemental Fig. S3E, F).
Ectopic expression of NANOG restores the activity of the TGF-β pathway
To delineate the molecular mechanisms responsible for the restoration of ECM production by NANOG, we performed RNA sequencing and analyzed gene expression profiles of LPZ-MSCs and LPN-MSCs that were treated with TGF-β1. IPA was performed to identify biologic functions and canonical pathways that were significantly affected by NANOG (see Fig. 3A):
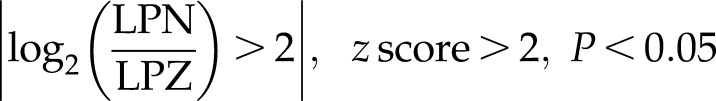 |
IPA showed that ectopic expression of NANOG significantly activated canonical pathways including stem cell pluripotency genes, IFN-γ, and TGF-β signaling pathways. Among them, TGF-β signaling is well known to be essential for ECM production (31–35). Several TGF-β pathway–associated genes including SMAD3, TGF-β2, TGF-βR1, and latent transforming growth factor beta binding protein 1 (LTBP1) were up-regulated (see Fig. 3B, C):
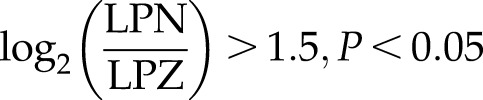 |
whereas negative regulators of the TGF-β pathway, such as SMAD6, SMAD7, and SMAD specific E3 ubiquitin protein ligase 2 (SMURF2) were down-regulated upon NANOG expression (see Fig. 3B, D):
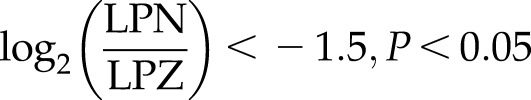 |
Overall, our RNA-Seq data suggested that NANOG might activate the TGF-β pathway, which was impaired in senescent cells (Fig. 3E).
Figure 3.

TGF-β is highly activated by NANOG expression in senescent hMSCs. A) Bar chart represents the top canonical signaling pathways that were affected by NANOG. Red bar represents positively affected pathways, and the blue bar represents negatively affected pathways. P values were determined using Fisher’s exact test with a threshold value of P = 0.05. Ratios represent the number of genes that mapped to a specific canonical pathway divided by the total number of genes that make up the respective pathway. Pathways with z score >2 were selected. B) Heat map of RNA-Seq for TGF-β associated genes in control (LPZ) vs. NANOG-expressing (LPN) cells. C, D) Validation of RNA-Seq through real-time PCR. E) Activation of the canonical TGF-β pathway. The red symbols represent up‐regulated genes, and the green symbols represent down-regulated genes, with the intensity of node color indicating a degree of up- and/or down‐regulation. White symbols indicate genes that do not appear on the list and the shape of each node denotes the function of the gene product. An alignment search of the promoter sequence of SMAD2 (F) or SMAD3 (G) using ChIP-Seq result shows that NANOG directly binds to SMADs promoter in the presence or absence of TGF-β. NM number denotes SMAD2/SMAD3 mRNA in RefSeq (NCBI Reference Sequences) records. Data range of 4–50 is filtered. Cells were treated with TGF-β (2 ng/ml) for 3 d and 1 µg/ml DOX was added in the medium throughout the experiment. *P < 0.05 compared to LPZ hMSCs (n = 3 independent experiments).
NANOG directly binds to the SMAD promoter and protein
Because NANOG is a transcription factor, we examined whether it bound to gene promoters, including those of genes associated with the TGF-β pathway. To this end, we employed ChIP-Seq of LPN cell lysates using the antibody against NANOG. Overall, ∼18,500 peaks were identified, 60% of which were located in promoter regions (−3000 to +3000 bp from the TSSs). Analysis of the ChIP-Seq data showed that NANOG could directly bind to SMAD2 (Fig. 3F) and SMAD3 (Fig. 3G) promoter regions in the presence or absence of TGF-β1. Furthermore, we performed chromatin immunoprecipitation and confirmed that NANOG bound directly to SMAD2 and SMAD3 promoter regions (Supplemental Fig. S5).
Kyoto Encyclopedia of Genes and Genomes (KEGG; https://www.genome.jp/kegg/) pathway analysis of the ChIP-Seq data revealed many pathways that were directly affected by NANOG, including the TGF-β pathway (Supplemental Fig. S4A). Then we used the ChIP-Seq results to filter the genes that were directly affected by NANOG (i.e., the genes whose promoter was bound by NANOG) from our RNA-Seq data. This subset of RNA-Seq data was then reanalyzed using IPA. Again, the TGF-β pathway was among the most highly activated pathways (Supplemental Fig. S4B):
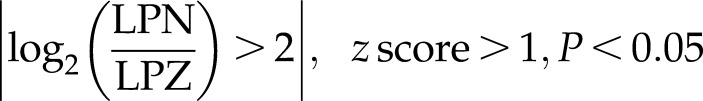 |
Interestingly, NANOG bound to the promoters and affected expression of several genes regulating TGF-β signaling, including bone morphogenetic protein 4 (BMP4), activin A receptor type 1C [ACVR1C(ALK7)], TGF-β2, latent transforming growth factor beta binding protein 1 (LTBP1) (up) or SMURF2, paired-like homeodomain transcription factor 2 (PITX2), inhibitor of DNA binding (ID)1, ID2, ID4 (down) (Supplemental Fig. S4C)
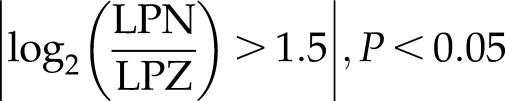 |
Taken together, the combined ChIP-Seq and RNA-Seq analysis suggest that NANOG binds to several promoters and activates the TGF-β pathway.
NANOG activates the TGF-β pathway that is impaired by senescence
To address this hypothesis, we measured the phosphorylation of SMAD2/3, which is known to increase within minutes and diminish within hours after TGF-β1 treatment. Phosphorylation of SMAD2/3 in LPZ cells increased 1 h after TGF-β1 treatment but decreased significantly by 6 h and diminished by 12 h. In contrast, upon addition of DOX (NANOG expression), the level of SMAD2/3 phosphorylation increased and remained elevated for up to 12 h (Fig. 4A, B). Increased phosphorylation was accompanied by nuclear retention of phosphorylated SMAD2/3 for up to 30 h (Fig. 4C, D), suggesting increased transcriptional activity. In addition, total SMAD2/3 decreased significantly by 1 d and diminished by 5 d post–TGF-β1 treatment of LPZ cells. Surprisingly, total SMAD2/3 was up-regulated and maintained at higher levels in the presence of NANOG (Fig. 4A, E–G), suggesting that the increased and sustained phosphorylation of SMAD2/3 might be the result of increased and sustained expression of total SMAD2/3 by NANOG.
Figure 4.

Ectopic expression of NANOG increases and maintains the activation of the TGF-β pathway. A) Western blot analysis for pSMAD2 and SMAD2/3 and (B) quantification of pSMAD2 as a function of the time of TGF-β treatment. C) Immunostaining and (D) quantification for nuclear pSMAD2/SMAD3 as a function of time of TGF-β treatment. E–G) Kinetics of SMAD2/3 protein expression upon TGF-β treatment. H) Immunoprecipitation of SMAD2 and then Western blot for NANOG and SMAD2/3 show direct binding of NANOG with activated SMADs. One microgram per milliliter DOX was added in the medium to express NANOG or ZsGreen. *P < 0.05 compared to LPZ (n = 3 independent experiments). Data are normalized to LPZ 1 h (B, D) or LPZ-TGF-β1 (F, G) and presented as means ± sd, n = 3 independent experiments.
Interestingly, immunoprecipitation with SMAD2 antibody showed that NANOG formed a complex with SMAD2 and SMAD3 proteins, but only in the presence of TGF-β1, suggesting that binding may require phosphorylation of SMAD2/3. Indeed, binding was abolished when SMAD2/3 phosphorylation was inhibited by a chemical inhibitor, SB431542 (SB43) (Fig. 4H).
TGF-β pathway activation is necessary for NANOG-induced COL3 synthesis
Next, we examined whether TGF-β pathway activation was necessary for NANOG-induced COL3 expression. To test this, we first quantified COL3 production in the presence or absence of TGF-β1 (Fig. 5A). Interestingly, TGF-β1 alone increased COL3 by 2.6-fold or 3.5-fold, after 1 or 3 d of treatment, respectively; however, in the presence of NANOG and TGF-β1, COL3 expression increased by 4.5 and 8.5-fold, respectively. In agreement, blocking the TGF-β pathway with chemical inhibitor SB431542 (SB43) eliminated the NANOG-induced COL3 expression (Fig. 5B). Similarly, knocking down of SMAD4, which is known to form a complex with pSMAD2/3, abolished COL3 expression (Fig. 5C) but surprisingly did not prevent translocation of pSMAD2/3 to the nucleus (Supplemental Fig. S6). Collectively, these results suggest that activation of the TGF-β pathway was necessary for NANOG-induced COL3 expression in senescent cells.
Figure 5.

SMAD2 and SMAD3 are necessary for NANOG-induced COL3 expression but SMAD3 overexpression is also sufficient for enhanced COL3 production. A) Protein expression of COL3 in LPZ and LPN hMSCs as a function of the time of TGF-β treatment. B, C) COL3 levels after blocking with SB431542 (SB43) or SMAD4 knockdown in LPZ and LPN hMSCs. D) COL3 levels after SMAD2 or SMAD3 knockdown in LPZ and LPN hMSCs. E) COL3 expression upon overexpression of SMAD2 or SMAD3 in LP hMSCs. DOX (1 μg/ml) was added in the medium to express NANOG or ZsGreen. *P < 0.05 compared to LPZ (n = 3 independent experiments). Data are normalized to LPZ-TGF-β1 (A) or LPZ (B–E) and presented as means ± sd, n = 3 independent experiments.
SMAD2/3 mediate NANOG-induced up-regulation of COL3
Next, we employed loss and gain of function strategies to determine which of the 2, SMAD2 or SMAD3 was responsible for increased COL3 production in response to NANOG. As shown in Fig. 5D, knocking down either SMAD2 or SMAD3 with short hairpin RNA encoding lentivirus eliminated the effect of NANOG on COL3 expression, albeit the effect of SMAD3 seemed to be more severe. Conversely, overexpression of SMAD3, but not SMAD2, increased COL3 expression, to the similar level as NANOG expression (Fig. 5E), suggesting that SMAD2 and SMAD3 are necessary, but SMAD3 is also sufficient for restoring COL3 production. Taken together, our results demonstrate that NANOG restores COL3 expression in senescent MSCs by up-regulating SMAD2/3 and restoring the impaired TGF-β pathway activity (Fig. 6A).
Figure 6.

Schematic representation of the pathways that mediate the effects of NANOG on COL3 expression by senescent cells. NANOG restored COL3 expression by reactivating the TGF-β pathway through transcriptional control of several genes in that pathway. Also, NANOG directly bound to SMAD2/3 promoter and phosphorylated protein, which might further activate the TGF-β pathway and contribute to restoration of COL3 expression.
DISCUSSION
We have previously demonstrated that the pluripotent factor NANOG was sufficient to reverse cellular aging and to completely restore the diminished differentiation capacity of aged MSCs and the contractile function of MSC-derived SMCs (1, 2, 18, 19). Here, we report that NANOG also restored the effects of aging on collagen production in both senescent MSCs and HGPS-derived myofibroblasts as shown in traditional cultures and 3-dimensional vascular tissue mimetics. The effect of NANOG was especially pronounced in COL3 expression, which was facilitated by reactivating the TGF-β pathway in senescent cells. Because loss of collagen is associated with aortic aneurysms (13) and atherosclerotic plaque rupture (36), our results suggest that NANOG may represent a novel multipurpose gene target regulating several aspects of cellular aging and promoting vascular stability and regeneration.
As one of the main pluripotency genes, NANOG is critical in maintaining embryonic stem cell self-renewal and pluripotency (37). Seminal work by the Yamanaka group demonstrated that ectopic expression of pluripotent factors organic cation/carnitine transporter 4 (OCT4), SRY-box transcription factor 2 (SOX2), Krüppel-like factor 4 (KLF4), and MYC (MYC proto-oncogene) was sufficient to reset the cellular age clock and reprogram adult cells into an embryonic-like state (38). More recently, cyclic induction of the Yamanaka factors prolonged the lifespan and reversed the loss of SMCs in the aorta of progeria mice, suggesting that pluripotency factors may regenerate the aged vasculature (39). Here we show that a single pluripotency factor (NANOG) reduced the levels of senescent markers including SA-β-gal, P53, and P21, but increased expression of Ki67, an indicator of cell proliferation. The effect of NANOG on cell proliferation might be driven by the decreased expression of cell cycle inhibitors through DNA methyltransferases (40). Interestingly, NANOG also reduced DNA damage—a critical mediator of aging (41)—as shown by γ-H2AX immunostaining. DNA damage is highly elevated in the aged vasculature (42) and promotes vascular cell senescence and dysfunction (43). Thus, we speculate that NANOG might exert a rejuvenating role in the vascular wall by inhibiting DNA damage. Although it is currently unclear how NANOG reduces DNA damage in senescent cells, 1 possible mechanism might involve restoration of the diminished DNA repair mechanism of aged cells (44).
NANOG also reversed the loss of COL3 expression in senescent cells. COL1 and COL3 are the main collagen subtypes expressed in the vascular wall (13). Although COL1 is responsible for vascular strength, COL3 regulates the elasticity of the collagen meshwork (45). In the healthy vasculature, the ratio of COL1 to COL3 is relatively low but increases significantly in vascular diseases (e.g., atherosclerosis) contributing to elevated vascular wall stiffness and high blood pressure (46). Indeed, COL3 knockout mice display ruptured blood vessels and die within 6 mo after birth (9). Additionally, COL3 gene mutations have been causally linked to Ehlers-Danlos syndrome, which is characterized by vascular aneurysms (15).
Interestingly, NANOG reversed COL3 expression in senescent cells but affected COL1 to a much lower extent, thereby decreasing the COL1 to COL3 ratio. To increase COL3, NANOG restored the activity of the TGF-β pathway, which was attenuated in aged cells (18). Specifically, NANOG expression increased the expression levels and subsequently the phosphorylation and nuclear retention of SMADs (e.g., SMAD2 and SMAD3). Although both SMAD2 and SMAD3 were necessary for NANOG-mediated COL3 up-regulation, only SMAD3 was sufficient for increased COL3 expression. SMAD2 and SMAD3 share 92% amino acid sequence similarity (47), but the additional 30 aa in the MH1 domain of SMAD2 make it less likely to bind to the DNA directly and more likely to bind as part of a complex with SMAD4 (48, 49). On the other hand, SMAD3 can bind to DNA directly through its transactivation domain in the linker region (50), thereby providing a potential explanation for the differential role of SMAD2 vs. SMAD3 in COL3 expression.
We also identified the mechanisms through which NANOG restores the activity of the TGF-β pathway in senescent cells. RNA-Seq and ChIP-Seq suggested that NANOG activated the TGF-β pathway that had been impaired by cellular senescence. NANOG was found to bind to the promoter of TGF-β pathway genes and either up-regulated activators such as SMAD2, SMAD3, TGF-βR1, latent transforming growth factor beta binding protein 1 (LTBP1), and TGF-β2 or down-regulated inhibitors such as SMURF2. Interestingly, we also found that NANOG bound to the p-SMAD2/3 protein, and the complex was colocalized in the nucleus. Although the significance of this interaction is not currently clear, binding to NANOG may prolong nuclear retention of SMAD2/3, thereby extending their transcriptional activity. Indeed, in the presence of NANOG, the level of pSMAD2 in the nucleus was maintained at higher levels even after termination of TGF-β treatment (Fig. 4A–D), in support of this hypothesis.
Collectively, our results reveal for the first time that NANOG restored COL3 expression by reactivating the TGF-β pathway through transcriptional control of several genes in that pathway. It will be interesting to examine whether these results can be extended in vivo using mice prone to vascular diseases such as ApoE-deficient or progeria mice.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
ACKNOWLEDGMENTS
This work was supported by a grant from the U.S. National Institutes of Health, National Heart, Lung, and Blood Institute (R01 HL086582) and New York Stem Cell Science (NYSTEM; C30290GG) to S.T.A. The authors declare no conflicts of interest.
Glossary
- ChIP
chromatin immunoprecipitation
- ChIP-Seq
ChIP sequencing
- COL1
collagen type I
- COL3
collagen type III
- DOX
doxycycline
- ECM
extracellular matrix
- EP
early passage
- HGPS
Hutchinson-Gilford progeria syndrome
- HGPSN
HGPS+NANOG
- HGPSZ
HGPS+ZsGreen
- hHF-MSC
human hair follicle–derived MSC
- IPA
Ingenuity Pathway Analysis
- LP
late passage
- LPN
LP+NANOG
- LPZ
LP+ZsGreen
- MSC
mesenchymal stem cell
- NANOG
Nanog homeobox
- RNA-Seq
RNA sequencing
- rtTA
reverse tetracycline transactivator
- SA-β-gal
senescence-associated β-galactosidase assay
- SMAD
mothers against decapentaplegic
- SMC
smooth muscle cell
- SMURF2
SMAD-specific E3 ubiquitin protein ligase 2
- TSS
transcription start site
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
Experiments were planned and designed by N. Rong, P. Mistriotis, and S. T. Andreadis; experimental data were generated and collected by N. Rong, X. Wang, G. Tseropoulos, and N. Rajabian; data analysis and interpretation involved N. Rong, X. Wang, N. Rajabian, Y. Zhang, J. Wang, and S. Liu; and article draft and further critical revisions of the manuscript were performed by N. Rong, P. Mistriotis, and S. T. Andreadis.
REFERENCES
- 1.Han J., Mistriotis P., Lei P., Wang D., Liu S., Andreadis S. T. (2012) Nanog reverses the effects of organismal aging on mesenchymal stem cell proliferation and myogenic differentiation potential. Stem Cells 30, 2746–2759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mistriotis P., Andreadis S. T. (2017) Vascular aging: molecular mechanisms and potential treatments for vascular rejuvenation. Ageing Res. Rev. 37, 94–116 [DOI] [PubMed] [Google Scholar]
- 3.Zieman S. J., Melenovsky V., Kass D. A. (2005) Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler. Thromb. Vasc. Biol. 25, 932–943 [DOI] [PubMed] [Google Scholar]
- 4.Ragnauth C. D., Warren D. T., Liu Y., McNair R., Tajsic T., Figg N., Shroff R., Skepper J., Shanahan C. M. (2010) Prelamin A acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging. Circulation 121, 2200–2210 [DOI] [PubMed] [Google Scholar]
- 5.Davis R., Pillai S., Lawrence N., Sebti S., Chellappan S. P. (2012) TNF-α-mediated proliferation of vascular smooth muscle cells involves Raf-1-mediated inactivation of Rb and transcription of E2F1-regulated genes. Cell Cycle 11, 109–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manea A., Manea S. A., Gafencu A. V., Raicu M., Simionescu M. (2008) AP-1-dependent transcriptional regulation of NADPH oxidase in human aortic smooth muscle cells: role of p22phox subunit. Arterioscler. Thromb. Vasc. Biol. 28, 878–885 [DOI] [PubMed] [Google Scholar]
- 7.Brassard J. A., Fekete N., Garnier A., Hoesli C. A. (2016) Hutchinson-Gilford progeria syndrome as a model for vascular aging. Biogerontology 17, 129–145 [DOI] [PubMed] [Google Scholar]
- 8.Fleischmajer R., MacDonald E. D., Perlish J. S., Burgeson R. E., Fisher L. W. (1990) Dermal collagen fibrils are hybrids of type I and type III collagen molecules. J. Struct. Biol. 105, 162–169 [DOI] [PubMed] [Google Scholar]
- 9.Liu X., Wu H., Byrne M., Krane S., Jaenisch R. (1997) Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc. Natl. Acad. Sci. USA 94, 1852–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qiu H., Depre C., Ghosh K., Resuello R. G., Natividad F. F., Rossi F., Peppas A., Shen Y. T., Vatner D. E., Vatner S. F. (2007) Mechanism of gender-specific differences in aortic stiffness with aging in nonhuman primates. Circulation 116, 669–676 [DOI] [PubMed] [Google Scholar]
- 11.Volk S. W., Shah S. R., Cohen A. J., Wang Y., Brisson B. K., Vogel L. K., Hankenson K. D., Adams S. L. (2014) Type III collagen regulates osteoblastogenesis and the quantity of trabecular bone. Calcif. Tissue Int. 94, 621–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Volk S. W., Wang Y., Mauldin E. A., Liechty K. W., Adams S. L. (2011) Diminished type III collagen promotes myofibroblast differentiation and increases scar deposition in cutaneous wound healing. Cells Tissues Organs (Print) 194, 25–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsamis A., Krawiec J. T., Vorp D. A. (2013) Elastin and collagen fibre microstructure of the human aorta in ageing and disease: a review. J. R. Soc. Interface 10, 20121004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pepin M., Schwarze U., Superti-Furga A., Byers P. H. (2000) Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N. Engl. J. Med. 342, 673–680; erratum: 344, 392 [DOI] [PubMed] [Google Scholar]
- 15.Schwarze U., Schievink W. I., Petty E., Jaff M. R., Babovic-Vuksanovic D., Cherry K. J., Pepin M., Byers P. H. (2001) Haploinsufficiency for one COL3A1 allele of type III procollagen results in a phenotype similar to the vascular form of Ehlers-Danlos syndrome, Ehlers-Danlos syndrome type IV. Am. J. Hum. Genet. 69, 989–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McDonnell N. B., Gorman B. L., Mandel K. W., Schurman S. H., Assanah-Carroll A., Mayer S. A., Najjar S. S., Francomano C. A. (2006) Echocardiographic findings in classical and hypermobile Ehlers-Danlos syndromes. Am. J. Med. Genet. A. 140, 129–136 [DOI] [PubMed] [Google Scholar]
- 17.Han J., Liu J. Y., Swartz D. D., Andreadis S. T. (2010) Molecular and functional effects of organismal ageing on smooth muscle cells derived from bone marrow mesenchymal stem cells. Cardiovasc. Res. 87, 147–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mistriotis P., Bajpai V. K., Wang X., Rong N., Shahini A., Asmani M., Liang M. S., Wang J., Lei P., Liu S., Zhao R., Andreadis S. T. (2017) NANOG reverses the myogenic differentiation potential of senescent stem cells by restoring ACTIN filamentous organization and SRF-dependent gene expression. Stem Cells 35, 207–221 [DOI] [PubMed] [Google Scholar]
- 19.Shahini A., Mistriotis P., Asmani M., Zhao R., Andreadis S. T. (2017) NANOG restores contractility of mesenchymal stem cell-based senescent microtissues. Tissue Eng. Part A. 23, 535–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mistriotis P., Andreadis S. T. (2013) Hair follicle: a novel source of multipotent stem cells for tissue engineering and regenerative medicine. Tissue Eng. Part B Rev. 19, 265–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bajpai V. K., Mistriotis P., Andreadis S. T. (2012) Clonal multipotency and effect of long-term in vitro expansion on differentiation potential of human hair follicle derived mesenchymal stem cells. Stem Cell Res. (Amst.) 8, 74–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alimperti S., Lei P., Tian J., Andreadis S. T. (2012) A novel lentivirus for quantitative assessment of gene knockdown in stem cell differentiation. Gene Ther. 19, 1123–1132 [DOI] [PubMed] [Google Scholar]
- 23.Phanish M. K., Wahab N. A., Colville-Nash P., Hendry B. M., Dockrell M. E. (2006) The differential role of Smad2 and Smad3 in the regulation of pro-fibrotic TGFbeta1 responses in human proximal-tubule epithelial cells. Biochem. J. 393, 601–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao X., Nicholls J. M., Chen Y.-G. (2008) Severe acute respiratory syndrome-associated coronavirus nucleocapsid protein interacts with Smad3 and modulates transforming growth factor-β signaling. J. Biol. Chem. 283, 3272–3280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyoshi H., Blömer U., Takahashi M., Gage F. H., Verma I. M. (1998) Development of a self-inactivating lentivirus vector. J. Virol. 72, 8150–8157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H., Durbin R. (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., Liu X. S. (2008) Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu G., Wang L. G., He Q. Y. (2015) ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383 [DOI] [PubMed] [Google Scholar]
- 29.Bailey T. L., Boden M., Buske F. A., Frith M., Grant C. E., Clementi L., Ren J., Li W. W., Noble W. S. (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng W., Yan-hua R., Fang-gang N., Guo-an Z. (2011) The content and ratio of type I and III collagen in skin differ with age and injury. Afr. J. Biotechnol. 10, 13 [Google Scholar]
- 31.Okumura N., Kay E. P., Nakahara M., Hamuro J., Kinoshita S., Koizumi N. (2013) Inhibition of TGF-β signaling enables human corneal endothelial cell expansion in vitro for use in regenerative medicine. PLoS One 8, e58000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hinz B. (2015) The extracellular matrix and transforming growth factor-β1: tale of a strained relationship. Matrix Biol. 47, 54–65 [DOI] [PubMed] [Google Scholar]
- 33.Horiguchi M., Ota M., Rifkin D. B. (2012) Matrix control of transforming growth factor-β function. J. Biochem. 152, 321–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roberts A. B., McCune B. K., Sporn M. B. (1992) TGF-β: regulation of extracellular matrix. Kidney Int. 41, 557–559 [DOI] [PubMed] [Google Scholar]
- 35.Chakravarthy A., Khan L., Bensler N. P., Bose P., De Carvalho D. D. (2018) TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat. Commun. 9, 4692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gorenne I., Kavurma M., Scott S., Bennett M. (2006) Vascular smooth muscle cell senescence in atherosclerosis. Cardiovasc. Res. 72, 9–17 [DOI] [PubMed] [Google Scholar]
- 37.Ivanova N., Dobrin R., Lu R., Kotenko I., Levorse J., DeCoste C., Schafer X., Lun Y., Lemischka I. R. (2006) Dissecting self-renewal in stem cells with RNA interference. Nature 442, 533–538 [DOI] [PubMed] [Google Scholar]
- 38.Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 [DOI] [PubMed] [Google Scholar]
- 39.Ocampo A., Reddy P., Martinez-Redondo P., Platero-Luengo A., Hatanaka F., Hishida T., Li M., Lam D., Kurita M., Beyret E., Araoka T., Vazquez-Ferrer E., Donoso D., Roman J. L., Xu J., Rodriguez Esteban C., Nunez G., Nunez Delicado E., Campistol J. M., Guillen I., Guillen P., Izpisua Belmonte J. C. (2016) In vivo amelioration of age-associated hallmarks by partial reprogramming. Cell 167, 1719–1733.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsai C. C., Su P. F., Huang Y. F., Yew T. L., Hung S. C. (2012) Oct4 and Nanog directly regulate Dnmt1 to maintain self-renewal and undifferentiated state in mesenchymal stem cells. Mol. Cell 47, 169–182 [DOI] [PubMed] [Google Scholar]
- 41.López-Otín C., Blasco M. A., Partridge L., Serrano M., Kroemer G. (2013) The hallmarks of aging. Cell 153, 1194–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morgan R. G., Ives S. J., Lesniewski L. A., Cawthon R. M., Andtbacka R. H., Noyes R. D., Richardson R. S., Donato A. J. (2013) Age-related telomere uncapping is associated with cellular senescence and inflammation independent of telomere shortening in human arteries. Am. J. Physiol. Heart Circ. Physiol. 305, H251–H258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Durik M., Kavousi M., van der Pluijm I., Isaacs A., Cheng C., Verdonk K., Loot A. E., Oeseburg H., Bhaggoe U. M., Leijten F., van Veghel R., de Vries R., Rudez G., Brandt R., Ridwan Y. R., van Deel E. D., de Boer M., Tempel D., Fleming I., Mitchell G. F., Verwoert G. C., Tarasov K. V., Uitterlinden A. G., Hofman A., Duckers H. J., van Duijn C. M., Oostra B. A., Witteman J. C., Duncker D. J., Danser A. H., Hoeijmakers J. H., Roks A. J. (2012) Nucleotide excision DNA repair is associated with age-related vascular dysfunction. Circulation 126, 468–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gray K., Kumar S., Figg N., Harrison J., Baker L., Mercer J., Littlewood T., Bennett M. (2015) Effects of DNA damage in smooth muscle cells in atherosclerosis. Circ. Res. 116, 816–826 [DOI] [PubMed] [Google Scholar]
- 45.Silver F. H., Freeman J. W., DeVore D. (2001) Viscoelastic properties of human skin and processed dermis. Skin Res. Technol. 7, 18–23 [DOI] [PubMed] [Google Scholar]
- 46.Plenz G. A., Deng M. C., Robenek H., Völker W. (2003) Vascular collagens: spotlight on the role of type VIII collagen in atherogenesis. Atherosclerosis 166, 1–11 [DOI] [PubMed] [Google Scholar]
- 47.Brown K. A., Pietenpol J. A., Moses H. L. (2007) A tale of two proteins: differential roles and regulation of Smad2 and Smad3 in TGF-beta signaling. J. Cell. Biochem. 101, 9–33 [DOI] [PubMed] [Google Scholar]
- 48.Yang F., Chung A. C., Huang X. R., Lan H. Y. (2009) Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-beta-dependent and -independent Smad pathways: the role of Smad3. Hypertension 54, 877–884 [DOI] [PubMed] [Google Scholar]
- 49.De Kroon L. M. G., Narcisi R., van den Akker G. G. H., Vitters E. L., Blaney Davidson E. N., van Osch G. J. V. M., van der Kraan P. M. (2017) SMAD3 and SMAD4 have a more dominant role than SMAD2 in TGFβ-induced chondrogenic differentiation of bone marrow-derived mesenchymal stem cells. Sci. Rep. 7, 43164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ungefroren H., Groth S., Sebens S., Lehnert H., Gieseler F., Fändrich F. (2011) Differential roles of Smad2 and Smad3 in the regulation of TGF-β1-mediated growth inhibition and cell migration in pancreatic ductal adenocarcinoma cells: control by Rac1. Mol. Cancer 10, 67 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.