

Abstract
Brigatinib, a next‐generation anaplastic lymphoma kinase (ALK) inhibitor, received accelerated approval in the United States for the treatment of patients with metastatic ALK+ non–small‐cell lung cancer who have progressed on or are intolerant to crizotinib. A clinical study was conducted to assess the effect of food on brigatinib pharmacokinetics (PK). Healthy subjects received a single oral dose of brigatinib 180 mg (2 × 90‐mg tablets) after a 10‐hour fast or after a high‐fat meal in a 2‐period, 2‐sequence crossover study. Plasma samples for PK characterization were collected over 168 hours postdose. Twenty‐four subjects were enrolled (mean age 44 years; 58% male), with 21 included in the PK‐evaluable population. Brigatinib peak concentration was reduced by 13% under fed (high‐fat meal) versus fasted conditions, with no effect on area under the concentration‐time curve. The median time to peak concentration of brigatinib was longer under fed conditions (5 hours) than in fasted conditions (2 hours). Treatment‐emergent adverse events were similar under fasted (48%) and fed (46%) conditions and were of mild intensity. Consumption of a high‐fat meal decreased the rate of brigatinib oral absorption but had no impact on the extent of absorption, thereby supporting brigatinib administration without regard to meals. These recommendations are reflected in the US prescribing information for brigatinib.
Keywords: Absorption, anaplastic lymphoma kinase, brigatinib, food effect, non–small‐cell lung cancer
Approximately 3% to 9% of patients with non–small‐cell lung cancer (NSCLC) have oncogenic rearrangements of the anaplastic lymphoma kinase gene (ALK).1, 2 Crizotinib was the first ALK inhibitor approved to treat metastatic ALK+ NSCLC, but most patients treated with crizotinib eventually experience disease progression due to acquired resistance involving mutations in ALK or activation of alternative signaling pathways.3, 4 Second‐generation ALK inhibitors, including ceritinib and alectinib, have been approved for the treatment of patients with crizotinib‐resistant ALK+ NSCLC; however, the long‐term efficacy of these agents is limited by the development of secondary mutations associated with clinical resistance.3, 5
Brigatinib (ARIAD Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited) is a next‐generation, oral ALK inhibitor that potently inhibits a broad range of clinically relevant ALK mutations.6, 7 In nonclinical studies, brigatinib demonstrated potent activity against all ALK resistance mutations tested and overcame mechanisms of resistance to crizotinib, ceritinib, and alectinib at clinically achievable concentrations.8 A pivotal phase 2 trial, ALTA (ALK in Lung Cancer Trial of Brigatinib), found that brigatinib had substantial efficacy with an acceptable safety profile in patients with crizotinib‐refractory ALK+ NSCLC, with a confirmed objective response rate of 56% and median progression‐free survival of 16.7 months at the recommended brigatinib dosing regimen (180 mg orally once daily with a 7‐day lead‐in at 90 mg orally once daily).9, 10, 11 On the basis of ALTA efficacy and safety data, brigatinib received accelerated approval in the United States and approval in Canada for the treatment of patients with metastatic ALK+ NSCLC who have progressed on or are intolerant to crizotinib.
The recommended dosing regimen for brigatinib was selected because it provides the best balance between safety and efficacy. It mitigates the risk of a subset of pulmonary adverse events that occurred within 7 days of treatment initiation with the 180‐mg dose while allowing for the optimal long‐term progression‐free survival and intracranial efficacy associated with the higher dose.9, 10, 12, 13 Brigatinib is formulated as immediate‐release tablets.
Brigatinib is classified as a high‐solubility–high‐permeability drug. The apparent permeability coefficient A‐to‐B values for [14C]brigatinib in C2BBe1 cells were in the range of 3.27 × 10−6 cm/s to 5.16 × 10−6 cm/s, comparable to those of drugs that exhibit moderate to good oral absorption in humans.14 The mean oral bioavailability of brigatinib was 52% in rats7 and has not been determined in humans. Following administration of oral doses of 30 mg to 240 mg, the median time to maximum plasma concentration (Tmax) ranged from 1 to 3 hours postdose.12 Steady‐state systemic exposure of brigatinib was dose proportional over the dose range of 60 mg to 240 mg once daily.12 The mean accumulation ratio after repeat dosing was 1.9 to 2.4.12 Following oral administration of brigatinib 180 mg once daily, the geometric mean apparent oral clearance at steady‐state was 10.6 L/h, and the mean plasma elimination half‐life was 25 hours.15 Brigatinib is metabolized by cytochrome P450 (CYP)2C8 and CYP3A4 in human liver microsomes and hepatocytes, with an N‐demethylated and a cysteine‐conjugated form being the 2 major metabolites.16 Because of potential drug‐drug interactions, concomitant use of brigatinib with strong CYP3A inhibitors and strong CYP3A inducers should be avoided.16 Following the oral administration of a single 180‐mg dose of brigatinib to healthy subjects, the majority (65%) of the administered dose was excreted in the feces, with 25% excreted in the urine.16
Ingestion of food causes several physiological gastrointestinal effects, including delayed gastric emptying, changes in gastrointestinal pH, and increased splanchnic blood flow, that can alter the rate and/or extent of absorption of orally administered antineoplastic agents.17 Of 35 orally administered cancer drugs that were approved by the US Food and Drug Administration (FDA) between January 2011 and April 2017, all but 1 had conducted a food effect study and had included food effects data in their initial submissions.18 A landscape analysis of the food effects and labeling recommendations for 30 oral antineoplastic drugs approved between 2010 and 2016 reported greater diversity in food recommendations for drugs that have increased bioavailability in the fed state as compared with 11 drugs approved in 2000 to 2009, when the drugs were almost always labeled as “taken on empty stomach.”19 Food effects on drug bioavailability and clearance are highly variable among small‐molecule kinase inhibitors.20 Although food does not meaningfully affect crizotinib exposure,21 food increases exposure to ceritinib,22 and ingestion of a high‐fat meal increases combined exposure to alectinib and its major active metabolite M4.23
A food effect study was conducted to assess the pharmacokinetics (PK) of brigatinib administered following a standardized high‐calorie, high‐fat meal, representing the setting of maximum possible perturbation of oral bioavailability in the postprandial state. Based on US FDA guidance, the recommended design for a food effect bioavailability study is a randomized, balanced, single‐dose, 2‐treatment (fed versus fasted), 2‐period, 2‐sequence crossover study, with each dose separated by an adequate washout period.24 An earlier unpublished food effect study in 8 healthy subjects found that consumption of a high‐fat meal within 30 minutes of administration of a single oral dose of brigatinib 180 mg (6 × 30‐mg tablets) reduced brigatinib maximum plasma concentration (Cmax) by approximately 24% and delayed median Tmax by 3.5 hours compared with dosing in the fasted state but had no effect on total exposure (the area under the plasma concentration–time curve from 0 to infinity [AUC0‐∞]) (ARIAD Pharmaceuticals, Inc, data on file). Given these results, the primary objective of this study was to provide a definitive assessment of the effect of a high‐fat meal on the PK of a single oral dose of 180 mg brigatinib in healthy subjects. The secondary objective was to evaluate both the safety and tolerability of brigatinib 180 mg in healthy subjects under fasted and fed conditions.
Methods
Subjects
The protocol and consent form were approved by the institutional review board for the study center (Ontario Institutional Review Board, Aurora, Ontario, Canada) before study initiation. All subjects provided written informed consent. The study was performed at the phase 1 unit of INC Research, Toronto, in accordance with the requirements of the Declaration of Helsinki, the International Council for Harmonisation guidelines for Good Clinical Practice, and other applicable regulatory requirements.
Eligible subjects were healthy nonsmoking men or women, 18 to 55 years of age, with a body mass index of 18 to 30 kg/m2 and a minimum weight of 50 kg at screening. Subjects were excluded if they had a clinically significant abnormality as assessed by physical examination, medical history, 12‐lead ECG, vital signs, or laboratory values; a history of any clinically significant illness; or the presence of an acute disease state (eg, nausea, vomiting, fever, or diarrhea) within 7 days of brigatinib administration. Subjects were prohibited from using nonprescription medication (except acetaminophen up to 2000 mg daily) within 7 days before the first drug administration, and prescription medications and natural health products (except vitamin or mineral supplements, acceptable forms of birth control, and hormone replacement) within 14 days before the first drug administration. Concomitant medications were prohibited throughout the study unless deemed by the investigator to be unlikely to affect study results or subject safety. Local anesthetics and topical medications were not permitted for at least 72 hours before the first study drug administration.
Study Design
This was a phase 1, single‐center, single‐dose, randomized, open‐label, 2‐period, 2‐sequence crossover study conducted in healthy subjects in accordance with US FDA guidelines for the assessment of food effects.24 The study included the screening visit, 2 treatment periods separated by a washout period of at least 16 days between brigatinib doses, and a follow‐up visit within 7 days after the last brigatinib dose. After screening, eligible subjects were randomized to 1 of 2 treatment sequences (fasted→fed or fed→fasted) in a crossover fashion (Figure 1).
Figure 1.
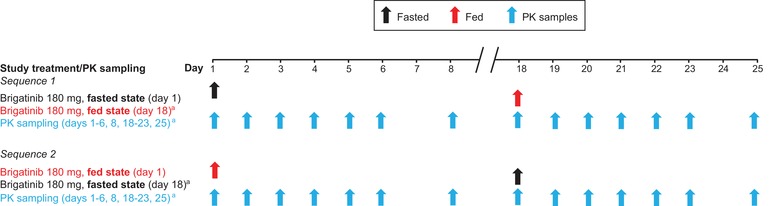
Brigatinib food effect study design. Arrows indicate dosing or sampling days. aThe 2 treatment periods were separated by a washout period of at least 16 days. Day 18 shown here represents day 1 of the second treatment period. PK indicates pharmacokinetics.
During each treatment period, subjects received a single oral dose of 180 mg brigatinib (2 × 90‐mg tablets), administered in the morning under the fasted (overnight fast of at least 10 hours) or fed (overnight fast of at least 10 hours followed by consumption of a standardized high‐fat meal 30 ± 5 minutes prior to brigatinib administration) condition, with approximately 240 mL of water. No food was allowed until 4 hours postdose. Water was provided ad libitum except for 1 hour before and 1 hour after drug administration. The standardized meal conformed to the FDA recommendations for food effects bioavailability studies with a high‐fat (approximately 50% of total caloric content from fat), high‐calorie (approximately 800 to 1000 calories) meal,24 with 158, 231, and 530 calories derived from protein, carbohydrates, and fat, respectively. The composition of the meal was 3 eggs, 2 strips of bacon, 1 slice of bread, 1 tablespoon of butter, 132 grams of potatoes, 237 mL of 2% milk, and 1 tablespoon of vegetable oil. The approximate nutritional content of the meal was 59 grams of fat, 58 grams of carbohydrates, and 40 grams of protein.
Assessments
For each treatment period, blood samples were collected predose and at 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 7, 8, 12, 24, 36, 48, 72, 96, 120, and 168 hours postdose to determine plasma concentrations of brigatinib. Brigatinib was extracted from dipotassium ethylenediaminetetraacetic acid–containing plasma samples by protein precipitation. Chromatographic separation was accomplished using ACE C18 50 × 2.1 mm, 3‐µm columns (Advanced Chromatography Technologies, Aberdeen, Scotland). Mobile phase A consisted of 2 mmol/L ammonium acetate in water, and mobile phase B consisted of 2 mmol/L ammonium acetate in methanol. The gradient method was employed, starting at 5% mobile phase B and increasing to 98% over 2.5 minutes at a flow rate of 0.6 mL/min. Compounds were then ionized using positive ion atmospheric pressure chemical ionization with an API‐5500 mass spectrometer (SCIEX, Redwood City, California) in multiple reaction–monitoring scan mode and m/z mass range parameters (parent/product) of 584.3/418.0 for brigatinib and 588.3/484.2 for the internal standard (AP29109; D4‐brigatinib). Two validated methods were used to analyze brigatinib. The first method was a single‐range method developed to allow analysis of plasma samples with concentrations higher than 500 ng/mL. The lower limit of quantitation for brigatinib was 25 ng/mL, with a calibration range of 25 to 2500 ng/mL using a 50‐µL plasma sample; any sample less than 25 ng/mL was reanalyzed using the second method. The second method, a dual‐range assay, had a lower limit of quantitation of 0.100 ng/mL and an upper limit of 500 ng/mL using a 100‐µL plasma sample. In the single‐range method, intrarun precision for brigatinib in human plasma samples ranged from 1.0% to 12.9% coefficient of variation (CV), with a bias of −11.6% to 5.6%, and interrun precision ranged from 1.7% to 10.1% CV, with a bias of −2.5% to 1.1%. For the dual‐range method, intrarun precision for brigatinib in human plasma samples for the high‐range and low‐range assays ranged from 3.3% to 10.0% and 1.1% to 12.7% CV, respectively, with a bias of −15.8% to 0.3% and −8.7% to −2.3%. Interrun precision ranged from 4.8% to 8.8% and 3.1% to 8.4% CV, with a bias of −6.3% to −1.3% and −7.3% to −4.2%.
Pharmacokinetic parameters for the primary end‐point analysis included Cmax, Tmax, and area under the plasma concentration–time curve from 0 to the time of the last quantifiable concentration (AUC0–t) or to AUC0–∞. Brigatinib PK parameters were estimated by noncompartmental methods using Phoenix WinNonlin, version 6.3 (Certara, Princeton, New Jersey). Safety evaluations included adverse events, vital signs, clinical laboratory assessments (hematology, chemistry, urinalysis), physical examinations, and 12‐lead ECG. Adverse events were assessed throughout the study and up to 7 days after the last brigatinib dose.
Statistical Analyses
In a previous food effect study (ARIAD Pharmaceuticals, Inc, data on file) of brigatinib in healthy subjects, the intrasubject CV for the paired difference in ln(Cmax) was 16.2. Based on this value, with the 90%CI for the geometric mean ratio of fed versus fasted within the limits of 80% to 125%, 21 subjects were required in order to have an 80% power of showing that the fed and fasted subject conditions were bioequivalent. Twenty‐four healthy subjects were randomized into the treatment phase, with the intention that approximately 21 subjects would complete both treatment conditions.
All subjects who received at least 1 dose of brigatinib were included in the safety and PK analyses. The PK‐evaluable population included all subjects in the safety population who had at least 1 evaluable plasma concentration. PK parameters were not calculated for subjects who experienced emesis within the first 6 hours (following dosing under fasting conditions) or 12 hours (following dosing under fed conditions). All subjects who had received both study drug treatments and had no protocol deviations were included in the ANOVA.
All PK analyses were conducted using the PK‐evaluable population. Brigatinib PK parameters were estimated using noncompartmental methods and were summarized using descriptive statistics. Brigatinib concentrations that were below the lower limit of quantification were imputed as 0. The effect of food (high‐fat meal) on the pharmacokinetics of brigatinib was assessed using 90%CIs obtained within the framework of a mixed‐effects model for ln(Cmax), ln(AUC0–∞), and ln(AUC0–t) using SAS version 9.3. The ANOVA model included sequence, treatment, and period as fixed effects and subject nested within sequence as a random effect. Sequence was tested using subject nested within sequence as the error term at a 10% level of significance; all other main effects were tested using the residual error (error mean square). Ratios of least‐square means for the fed condition relative to the fasted condition (reference treatment) were calculated using the exponential of the difference between treatment least‐square mean from ln‐transformed AUC0–t, AUC0–∞, and Cmax. Tmax was not ln‐transformed and was analyzed using nonparametric analysis (Walsh averages and Wilcoxon signed rank).
Safety parameters were summarized using descriptive statistics.
Results
Subjects
A total of 24 subjects were randomized and received at least 1 dose of brigatinib (the safety population). Twenty‐one subjects completed the study, constituting the PK‐evaluable population, and 3 subjects discontinued the study before period 2 (positive carbon monoxide test, n = 2; noncompliance [subject arrived for admission to treatment period 2 with prohibited medications], n = 1). Demographic characteristics of the PK‐evaluable population (n = 21; not shown) were similar to those of the safety population (n = 24; shown in Table 1).
Table 1.
Subject Demographics and Baseline Characteristics
| Characteristic | Overall (N = 24) |
|---|---|
| Mean (SD) age, y | 44 (8) |
| Age range, y | 24‐55 |
| Male, n (%) | 14 (58) |
| Race, n (%) | |
| White | 16 (67) |
| Black | 4 (17) |
| Asian | 4 (17) |
| Ethnicity, n (%) | |
| Non‐Hispanic/non‐Latino | 18 (75) |
| Mean (SD) body weight, kg | 72 (13) |
| Body weight range, kg | 51‐99 |
Pharmacokinetics
Mean brigatinib plasma concentrations over time after consumption of a high‐fat, high‐calorie meal and under fasted conditions are shown in Figure 2. The median Tmax for brigatinib was delayed following dosing in the fed state (5 hours) versus the fasted state (2 hours; Table 2). The geometric mean Cmax was approximately 13% lower in the fed state (604.6 ng/mL) compared with the fasted state (701.3 ng/mL), with a ratio of 0.87 (90%CI, 0.78‐0.97; Table 2). However, total systemic exposure (AUC0–∞) of brigatinib was similar when it was administered under fed and fasted conditions, as the 90%CI (0.89‐1.07) was contained within the 80% to 125% range for establishing equivalence (Table 2). The mean terminal elimination half‐life was also similar under fed (31.1 hours) and fasted (31.0 hours) conditions.
Figure 2.
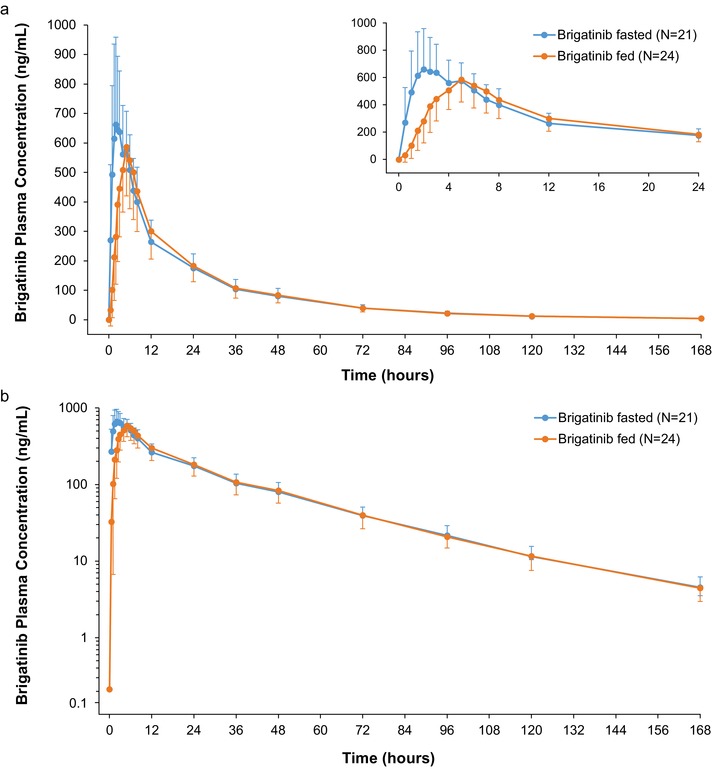
a, Mean (± SD) brigatinib plasma concentration–time profiles after oral administration of 180 mg brigatinib under fasted (N = 21) and fed (N = 24) conditions. The inset shows the mean plasma brigatinib concentrations over the first 24 hours after dosing. No plasma concentration values were below the lower limit of quantification. b, Log‐linear plot of mean (± SD) brigatinib plasma concentration–time profiles after oral administration of 180 mg brigatinib under fasted (N = 21) and fed (N = 24) conditions. No plasma concentration values were below the lower limit of quantification.
Table 2.
Plasma PK Parameters of 180 mg Brigatinib Under Fasted and Fed Conditions
| PK Parameter | Fasted (N = 21) | Fed (N = 24) | Fed vs Fasted, Geometric Mean Ratioa (90%CI) |
|---|---|---|---|
| Cmax, ng/mL | |||
| Mean (SD) | 743.0 (251.9) | 621.9 (145.8) | |
| Geometric mean (% CV) | 701.3 (36.6) | 604.6 (25.2) | 0.87 (0.78‐0.97) |
| AUC0–∞, h•ng/mL | |||
| Mean (SD) | 13 767 (3599) | 13 354 (3408) | |
| Geometric mean (% CV) | 13 261 (29.8) | 12 944 (26.6) | 0.98 (0.89‐1.07) |
| Tmax, h | |||
| Median (minimum‐maximum) | 2.00 (1.00‐6.00) | 5.00 (2.50‐7.00) | |
| t1/2, h | |||
| Mean (SD) | 31.0 (4.2) | 31.1 (3.9) | |
AUC0–∞ indicates area under the plasma concentration–time curve from time 0 to infinity; Cmax, maximum plasma concentration; CV, coefficient of variation; PK, pharmacokinetics; t1/2, terminal elimination half‐life; Tmax, time to maximum plasma concentration.
Geometric mean ratios reported in this column are based on n = 21.
Safety
The incidence of treatment‐emergent adverse events (TEAEs) was similar under fed (46%) and fasted (48%) conditions. The most commonly reported TEAE was cough, occurring in 5 (24%) and 7 (29%) subjects under fasted and fed conditions, respectively. Somnolence, nausea, and headache each occurred in 3 (14%) subjects following study drug administration under fasted conditions and 1 (4%) subject under fed conditions. All TEAEs were mild in intensity, with no severe events reported.
No deaths or serious TEAEs were reported, and no subjects discontinued the study due to TEAEs. There were no clinically significant abnormalities in laboratory findings, vital signs, ECGs, or physical examination findings during the study. Overall, the safety profile for brigatinib was similar after single‐dose administration in the fasted or fed state.
Discussion
This study was conducted to provide a definitive assessment of the effect of food (a high‐fat meal) on the PK of a single oral 180‐mg dose of brigatinib in healthy subjects. The study was designed in accordance with the guidance provided by the US FDA, which recommends the use of a randomized, balanced, 2‐treatment (fed versus fasted), single‐dose, 2‐period, 2‐sequence, crossover study in which each treatment is separated by an adequate washout period.24
This study showed that consumption of a high‐fat meal reduced brigatinib Cmax by 13% and delayed the median Tmax from 2 to 5 hours compared with administration in the fasted state, with no effect on the total exposure (AUC). The effect of food on brigatinib PK was not considered clinically meaningful, consistent with expectations from its high solubility and permeability. As a result, brigatinib may be administered without regard to meals, consistent with the dosing recommendations used in the pivotal ALTA study9 and current dosing recommendations. A single oral dose of 180 mg brigatinib was well tolerated in healthy subjects under both fasted and fed conditions. Importantly, the ALTA study demonstrated efficacy in patients with locally advanced or metastatic ALK+ NSCLC when brigatinib was administered without regard to food intake.
Varying effects of food on the PK of other ALK inhibitors indicated for the treatment of ALK+ NSCLC have been observed.21, 22, 23, 25 Crizotinib exposure (both Cmax and AUC) was 14% lower in healthy subjects following a high‐fat meal.21 The observed decrease in crizotinib exposure with food was not considered clinically meaningful; therefore, crizotinib may be taken without regard to food intake.21 However, ceritinib and alectinib should be taken with food to maximize drug exposure (alectinib) and improve gastrointestinal tolerability (ceritinib) in patients. A food effect study of ceritinib in patients with advanced ALK+ NSCLC found that ceritinib 450 mg taken with food demonstrated Cmax and AUC0–24 values comparable to those observed with 750 mg administered in the fasted state.25 Furthermore, the 450‐mg dose administered with food was associated with a lower incidence of gastrointestinal toxicities compared with 750 mg administered under fasted conditions. Based on the findings in healthy subjects and consistent with observations in patients with advanced ALK+ NSCLC, the originally recommended daily dose (750 mg, administered in the fasted state) was revised to 450 mg daily administered with food to improve the benefit‐risk profile of ceritinib.25 For alectinib, a high‐fat, high‐calorie meal increased the combined exposure (AUC0‐∞) of alectinib plus its major active metabolite M4 by 3.1‐fold (90%CI, 2.7‐3.6) following oral administration of a single 600‐mg dose.23 Consequently, it is recommended that alectinib be administered with food.23
Conclusions
This study showed that consumption of a high‐fat meal had no clinically meaningful effect on the PK of brigatinib, indicating that brigatinib may be administered without regard to meals. The results of this analysis are reflected in the Dosage and Administration section of the US Prescribing Information for brigatinib.
Acknowledgments
This study was funded by ARIAD Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. The authors wish to thank the Principal Investigator of the study, Luis Robles, MD, of INC Research Toronto, Inc, Toronto, Ontario, Canada. Professional medical writing assistance was provided by Lauren Gallagher, PhD, of Peloton Advantage, LLC, Parsippany, New Jersey, USA, and funded by ARIAD Pharmaceuticals, Inc.
Author Disclosures
Meera Tugnait is a former employee and stockholder of ARIAD. Neeraj Gupta is an employee of Millennium Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. Michael J. Hanley is an employee of Millennium Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. Karthik Venkatakrishnan is an employee of Millennium Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. Daryl Sonnichsen is a consultant to ARIAD. David Kerstein is an employee and holds stock and other ownership interests in ARIAD. David Dorer is a former employee and holds stock and other ownership interests in ARIAD. Narayana Narasimhan is an employee and holds stock and other ownership interests in ARIAD.
Data‐Sharing Statement
Takeda makes patient‐level, de‐identified data sets and associated documents available after applicable marketing approvals and commercial availability have been received, an opportunity for the primary publication of the research has been allowed, and other criteria have been met as set forth in Takeda's Data Sharing Policy (see www.TakedaClinicalTrials.com/ for details). To obtain access, researchers must submit a legitimate academic research proposal for adjudication by an independent review panel, who will review the scientific merit of the research and the requestor's qualifications and conflict of interest that can result in potential bias. Once approved, qualified researchers who sign a data‐sharing agreement are provided access to these data in a secure research environment.
References
- 1. Pao W, Girard N. New driver mutations in non‐small‐cell lung cancer. Lancet Oncol. 2011;12(2):175–180. [DOI] [PubMed] [Google Scholar]
- 2. Sholl LM, Aisner DL, Varella‐Garcia M, et al. Multi‐institutional oncogenic driver mutation analysis in lung adenocarcinoma: The Lung Cancer Mutation Consortium experience. J Thorac Oncol. 2015;10(5):768–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wilson FH, Johannessen CM, Piccioni F, et al. A functional landscape of resistance to ALK inhibition in lung cancer. Cancer Cell. 2015;27(3):397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Romanidou O, Landi L, Cappuzzo F, Califano R. Overcoming resistance to first/second generation epidermal growth factor receptor tyrosine kinase inhibitors and ALK inhibitors in oncogene‐addicted advanced non‐small cell lung cancer. Ther Adv Med Oncol. 2016;8(3):176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Katayama R, Lovly CM, Shaw AT. Therapeutic targeting of anaplastic lymphoma kinase in lung cancer: a paradigm for precision cancer medicine. Clin Cancer Res. 2015;21(10):2227–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Katayama R, Khan TM, Benes C, et al. Therapeutic strategies to overcome crizotinib resistance in non‐small cell lung cancers harboring the fusion oncogene EML4‐ALK. Proc Natl Acad Sci U S A. 2011;108(18):7535–7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huang WS, Liu S, Zou D, et al. Discovery of brigatinib (AP26113), a phosphine oxide‐containing, potent, orally active inhibitor of anaplastic lymphoma kinase. J Med Chem. 2016;59(10):4948–4964. [DOI] [PubMed] [Google Scholar]
- 8. Zhang S, Anjum R, Squillace R, et al. The potent ALK inhibitor brigatinib (AP26113) overcomes mechanisms of resistance to first‐ and second‐generation ALK inhibitors in preclinical models. Clin Cancer Res. 2016;22(22):5527–5538. [DOI] [PubMed] [Google Scholar]
- 9. Kim DW, Tiseo M, Ahn MJ, et al. Brigatinib in patients with crizotinib‐refractory anaplastic lymphoma kinase‐positive non‐small‐cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol. 2017;35(22):2490–2498. [DOI] [PubMed] [Google Scholar]
- 10. Camidge DR, Kim DW, Tiseo M, et al. Exploratory analysis of brigatinib activity in patients with anaplastic lymphoma kinase‐positive non‐small‐cell lung cancer and brain metastases in two clinical trials. J Clin Oncol. 2018;36(26):2693–2701. [DOI] [PubMed] [Google Scholar]
- 11. Huber RM, Kim DW, Ahn MJ, et al. Brigatinib (BRG) in crizotinib (CRZ)‐refractory ALK+ non–small cell lung cancer (NSCLC): efficacy updates and exploratory analysis of CNS ORR and overall ORR by baseline (BL) brain lesion status [abstract]. J Clin Oncol. 2018;36(15 Suppl):9061. [Google Scholar]
- 12. Gettinger SN, Bazhenova LA, Langer CJ, et al. Activity and safety of brigatinib in ALK‐rearranged non‐small‐cell lung cancer and other malignancies: a single‐arm, open‐label, phase 1/2 trial. Lancet Oncol. 2016;17(12):1683–1696. [DOI] [PubMed] [Google Scholar]
- 13. Camidge DR, Kim HR, Ahn MJ, et al. Brigatinib versus crizotinib in ALK‐positive non‐small‐cell lung cancer. N Engl J Med. 2018;379:2027–2039. [DOI] [PubMed] [Google Scholar]
- 14. Artursson P, Karlsson J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco‐2) cells. Biochem Biophys Res Commun. 1991;175(3):880–885. [DOI] [PubMed] [Google Scholar]
- 15. Gupta N, Wang X, Prohn M, et al. Population pharmacokinetic (PK) analysis of the ALK inhibitor brigatinib: model‐informed posology decisions and global drug development [abstract 076]. Clin Pharmacol Drug Dev. 2018;7(Suppl 1):61–62. [Google Scholar]
- 16. Hirota T, Muraki S, Ieiri I. Clinical pharmacokinetics of anaplastic lymphoma kinase inhibitors in non‐small‐cell lung cancer. [published online ahead of print June 19, 2018] Clin Pharmacokinet. [DOI] [PubMed] [Google Scholar]
- 17. Singh BN, Malhotra BK. Effects of food on the clinical pharmacokinetics of anticancer agents: underlying mechanisms and implications for oral chemotherapy. Clin Pharmacokinet. 2004;43(15):1127–1156. [DOI] [PubMed] [Google Scholar]
- 18. Faucette S, Wagh S, Trivedi A, Venkatakrishnan K, Gupta N. Reverse translation of US Food and Drug Administration reviews of oncology new molecular entities approved in 2011‐2017: lessons learned for anticancer drug development. Clin Transl Sci. 2018;11(2):123–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deng J, Brar SS, Lesko LJ. To take or not to take with meals? Unraveling issues related to food effects labeling for oral antineoplastic drugs. Clin Pharmacol Drug Dev. 2018;7(5):455–464. [DOI] [PubMed] [Google Scholar]
- 20. Chatelut E, Bruno R, Ratain MJ. Intraindividual pharmacokinetic variability: focus on small‐molecule kinase inhibitors. Clin Pharmacol Ther. 2018;103(6):956–958. [DOI] [PubMed] [Google Scholar]
- 21. Xu H, O'Gorman M, Boutros T, et al. Evaluation of crizotinib absolute bioavailability, the bioequivalence of three oral formulations, and the effect of food on crizotinib pharmacokinetics in healthy subjects. J Clin Pharmacol. 2015;55(1):104–113. [DOI] [PubMed] [Google Scholar]
- 22. Lau YY, Gu W, Lin T, Song D, Yu R, Scott JW. Effects of meal type on the oral bioavailability of the ALK inhibitor ceritinib in healthy adult subjects. J Clin Pharmacol. 2016;56(5):559–566. [DOI] [PubMed] [Google Scholar]
- 23. Morcos PN, Guerini E, Parrott N, et al. Effect of food and esomeprazole on the pharmacokinetics of alectinib, a highly selective ALK inhibitor, in healthy subjects. Clin Pharmacol Drug Dev. 2017;6(4):388–397. [DOI] [PubMed] [Google Scholar]
- 24. US Food and Drug Administration . Guidance for Industry. Food‐effect bioavailability and fed bioequivalence studies. 2002. https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm070241.pdf Accessed December 4, 2018.
- 25. Cho BC, Kim DW, Bearz A, et al. ASCEND‐8: a randomized phase 1 study of ceritinib, 450 mg or 600 mg, taken with a low‐fat meal versus 750 mg in fasted state in patients with anaplastic lymphoma kinase (ALK)‐rearranged metastatic non‐small cell lung cancer (NSCLC). J Thorac Oncol. 2017;12(9):1357–1367. [DOI] [PubMed] [Google Scholar]