

Abstract
Condensin II subunits are known to be expressed and localized to interphase nuclei of eukaryotic cells. Although some studies have shown that condensin II likely exerts axial compaction forces, organizes chromosome territories, and has possible transcriptional modulatory functions, the full range of condensin II interphase activities are not known. In particular, it is not known if condensin II interphase activities are generally genome‐wide or if they have additional local activities unique to specific chromosomal structures such as telomeres. Here, we find that NCAPH2 interacts with TRF1 and these two proteins co‐localize at telomeres. Depletion of NCAPH2 leads to ATR‐dependent accumulation of 53BP1 and γH2AX DNA damage foci, including damage specific to telomeres. Furthermore, depletion of NCAPH2 results in a fragile telomere phenotype and apparent sister‐telomere fusions only days after NCAPH2 depletion. Taken together these observations suggest that NCAPH2 promotes telomere stability, possibly through a direct interaction with the TRF1 shelterin component, and prevents telomere dysfunction resulting from impaired DNA replication. Because proper telomere function is essential for chromosome integrity these observations reveal a previously unappreciated function for NCAPH2 in ensuring genome and telomere stability.
Keywords: condensin, DNA damage, replication, shelterin, telomeres, TRF1
1. INTRODUCTION
Telomeres are nucleoprotein complexes that protect the chromosome ends in two essential ways. First, telomeres protect chromosome ends from replicative shortening associated with the inability of the DNA replication machinery to completely replicate chromosome ends, termed the end‐replication problem (Hug & Lingner, 2006; Watson, 1972). Second, telomeres prevent chromosome ends from being recognized as double‐strand breaks (DSBs; de Lange, 2009; Lydall, 2009). Inappropriate recognition of chromosome ends as DSBs activates DNA damage checkpoints and repair mechanisms that can result in end‐to‐end fusions and genome instability (Harrison & Haber, 2006; Rong, 2008).
In most eukaryotes, the end‐replication problem is solved and telomere length is maintained by the reverse transcriptase telomerase, that adds short GC‐rich tandem repeats to chromosome ends after each cell division (Lingner & Cech, 1998). Chromosome ends are then protected by a multiprotein complex termed shelterin that interacts with the GC‐rich repeat sequences generated by telomerase (Palm & de Lange, 2008). Mammalian shelterin is composed of TRF1, TRF2, POT1, TPP1 TIN2, and hRap1 (de Lange, 2005), all of which localize exclusively to telomeres and each have distinct roles. Myb‐related proteins TRF1 and TRF2 both bind to double‐stranded TTAGGG telomeric repeats (Bianchi, Smith, Chong, Elias, & de Lange, 1997; Broccoli, Smogorzewska, Chong, & de Lange, 1997), but have distinct telomeric functions. TRF1 functions as a negative regulator of telomere length, as its overexpression has been shown to result in telomere shortening (van Steensel & de Lange, 1997). TRF1 also promotes semiconservative replication of telomeres, in conjunction with the BLM helicase and TPP1/POT1 (Sfeir et al., 2009; Zimmermann, Kibe, Kabir, & de Lange, 2014). Though TRF2 also negatively regulates telomere length (Smogorzewska et al., 2000), it functions predominantly in capping and protection of chromosome ends, preventing activation of the ATM‐dependent DNA damage pathway (Karlseder et al., 2004; Sfeir & de Lange, 2012). In addition, a number of conserved shelterin accessory factors, which have localization and function that are not exclusive to telomeres, are also required for eukaryotic telomere maintenance. These include the DNA repair proteins ATM, ATR, ATRIP, Chk2, Ku70/80, MRE11‐RAD50‐NBS1 (MRB) complex, Rad51, as well as heterochromatin protein 1 (HP1) homologs, and the H3 lysine 9 methyltransferase SUV39 (Cenci, Ciapponi, & Gatti, 2005; Jain & Cooper, 2010; Palm & de Lange, 2008).
Evidence from yeast suggests that condensin complex proteins are involved in maintaining telomere stability. Condensins are conserved multisubunit protein complexes that promote mitotic chromosome condensation and segregation (Hirano, Kobayashi, & Hirano, 1997). Most eukaryotes have two condensin complexes, condensin I and condensin II, which differ in their localization and function. Condensin I promotes lateral compaction of chromosomes and is only found on mitotic chromosomes after nuclear envelope breakdown, whereas condensin II promotes axial compaction of chromosomes and localizes to the nucleus throughout the cell cycle (Bauer, Hartl, & Bosco, 2012; Buster et al., 2013; Green et al., 2012; Hirota, Gerlich, Koch, Ellenberg, & Peters, 2004; Ono, Fang, Spector, & Hirano, 2004; Shintomi & Hirano, 2011). Condensin II comprises core structural maintenance of chromosome (SMC) proteins SMC2 and SMC4 as well as non‐SMC chromosome‐associated protein (CAP) subunits CAPH2, CAPD3, and CAPG2 (Neuwald & Hirano, 2000; Ono et al., 2003) and has recently emerged as an important regulator of interphase nuclear organization (Bateman et al., 2012; Bauer et al., 2012; Hartl, Smith, & Bosco, 2008; Joyce, Williams, Xie, & Wu, 2012; Wallace & Bosco, 2013). Several studies have linked condensins with a possible function at telomeres. Yeast condensin plays a role in proper segregation of telomeres, with the proteins Cut3 (SMC4) and Cut14 (SMC2) acting to prevent telomere entanglements in Schizosaccharomyces pombe (Motwani, Doris, Holmes, & Flory, 2010). In addition, ChIP‐seq experiments in chicken DT40 cells have shown that condensin I is enriched at telomeric TTAGGG repeats (Kim et al., 2013), whereas condensin I protein CAP‐H was shown to co‐localize with TRF1 at telomeres of mitotic and meiotic chromosomes in mice (Viera et al., 2007). Still, the particular telomeric function of condensin complexes, remains unknown.
Here, we investigated whether condensin II specifically contributes to telomere stability in human cells. We found that the condensin II protein NCAPH2 localizes to telomeric repeats where it interacts with TRF1. Depletion of NCAPH2 results in accumulation of ATR‐mediated telomeric DNA damage foci. Depletion of NCAPH2 also results in an accumulation of replication protein A (RPA) foci and an increased occurrence of fragile telomeres, suggesting that NCAPH2 is required for proper replication at telomeres.
2. METHODS
2.1. Anti‐NCAPH2 antisera production
Rabbit polyclonal antisera were raised against the following carboxy‐terminal amino acid residues of NCAPH2: KRFQTYAAPSMAQP (593–606) as also previously described (Ono et al., 2003).
2.2. Cell culture and siRNA
RPE‐1, HeLa, HEK293T, and U2OS cells were maintained at 37°C and 5% CO2 in Dulbecco's Modified Eagle Medium containing 10% fetal bovine serum (FBS), 100 μg/ml streptomycin, and 1,000 U/ml penicillin (Life Technologies, Carlsbad, CA). HCT116 cells were maintained at 37°C and 5% CO2 in McCoy's 5a Modified Medium (Sigma, Natick, MA) containing 10% FBS, 100 μg/ml streptomycin, and 1,000 U/ml penicillin (Life Technologies). siGENOME SMARTpool small interfering RNAs (siRNAs) targeting NCAPH2, NCAPD3, SMC2, TRF1, TRF2, ATR, 53BP1, and ON‐TARGETplus SMARTpool siRNAs targeting ATM were purchased from Dharmacon. siGENOME nontargeting siRNA Pool #2 (scrambled) was used as a negative control for all siRNA treatments. The secondary pool of siRNAs targeting NCAPH2 ORF was designed using Dharmacon's siDESIGN Center (http://dharmacon.horizondiscovery.com/design‐center/). Sense sequences are as follows: NCAPH2.1 siRNA ‐ CUGAUGAAAUGGAGAAGAAUU, NCAPH2.2 siRNA ‐ ACAGUAAGAAGGUGGAAUGUU, NCAPH2.3 siRNA ‐ CGGAAGGAUUUCAGGAUGAUU. Cells were electroporated with 300 pmol siRNA (unless otherwise indicated) using the Nucleofector II and Amaxa Cell Line Nucleofector Kit V (Lonza, Portsmouth, NH) and were analyzed 48 hr after nucleofection.
2.3. Chromatin immunoprecipitation and dot‐blot hybridization
Cells were harvested by trypsinization, washed with phosphate‐buffered saline (PBS), and fixed with 1% formaldehyde in PBS for 20 min. Glycine was added to a final concentration of 0.125 M to stop crosslinking. Cells were pelleted, washed two times with cold PBS, and resuspended in lysis buffer (1% sodium dodecyl sulfate [SDS], 10 mM EDTA pH 8.0, and 50 mM Tris‐HCl pH 8.0) supplemented with complete EDTA‐free protease inhibitor cocktail (Roche, Indianapolis, IN). After 15 min on ice, samples were sonicated at 35% amplitude for 10 cycles of 10 s, followed by 30 s on ice with a Branson SLPe Digital Sonifier (Branson Ultrasonics Corporation, North Billerica, MA). Samples were centrifuged for 10 min and immunoprecipitation (IP) dilution buffer (0.01% SDS, 1.1% Triton X‐100, 1.2 mM EDTA, 16.7 mM Tris‐HCl pH 8.0, and 150 mM NaCl) with protease inhibitors was added the equivalent of 2 × 106 cells. Ten percent was set aside for input and IP samples were incubated with rabbit anti‐TRF2 (NB110–57130, 1:25, Novus Biologicals, Centennial, CO), rabbit anti‐NCAPH2 (1:25), or rabbit IgG (1:25) overnight at 4°C. Chromatin/antibody complexes were captured by incubation with Protein G Magnetic Beads (Pierce, Rockford, IL) for 1 hr at 4°C. Beads were washed one time each with buffer A (0.1% SDS, 1% Triton X‐100, 2 mM EDTA pH 8.0, 20 mM Tris‐HCl pH 8.0, and 150 mM NaCl), buffer B (0.1% SDS, 1% Triton X‐100, 2 mM EDTA pH 8.0, 20 mM Tris‐HCl pH 8.0, and 500 mM NaCl), and buffer C (0.25 M LiCl, 1% NP‐40, 1% Na‐deoxycholate, 1 mM EDTA pH 8.0, and 10 mM Tris‐HCl pH 8.0), and TE buffer (10 mM Tris‐HCl pH 8.0 and 1 mM EDTA pH 8.0). Input samples and beads were resuspended in elution buffer (1% SDS, 0.1 M sodium bicarbonate). For crosslink reversal, 20 μl of 5 M NaCl and 40 μg Proteinase K (Boehringer Mannheim, Indianapolis, IN) was added and samples were incubated overnight at 65°C, followed by a 1‐hr treatment at 37°C with 20 μg RNAse A (Invitrogen, Carlsbad, CA). After phenol–chloroform extraction and ethanol precipitation, DNA was dot‐blotted onto nylon membrane in 2X SSC. TTAGGG and Alu probes were labeled and hybridized using the DIG High Prime DNA Labeling and Detection Starter Kit II (Roche) according to the manufacturer's instruction and the hybridized probe was visualized using X‐ray film. Signal quantification was performed by densitometry analysis using the ImageJ software (NIH).
2.4. Constructs and transfections
A complementary DNA (cDNA) fragment encoding NCAPH2 isoform 1 was amplified by polymerase chain reaction (PCR) using pCMV6‐NCAPH2 (Origene: Rockville, MD, NM_152299) as a template and primers 5′‐CGATTGGCCGGCCAATGGAGGACGTGGAGGCGC‐3′ and 5′‐TGACTGGCGCGCCTCAGGGCTGGGCCATGGA‐3′. The PCR product was subcloned into the FseI/AscI sites of the pCS2‐MYC‐FA vector (gift from Ethan Lee, Vanderbilt University) and was verified by DNA sequencing. MYC‐NCAPH2‐AxAxA was made by mutating F192, L194, and P196 residues to alanine using the Agilent QuikChange Site‐Directed Mutagenesis kit (Agilent Santa Clara, CA. pLPC‐FLAG‐TRF1 (#16058) was obtained from Addgene (Watertown, MA), and pCS2‐FLAG was a gift from Ethan Lee. pCDNA3‐NCAPH2‐EGFP was made by cloning isoform 1 NCAPH2 with a 3 × ‐glycine linker into the pCDNA3‐EGFP vector available through Addgene (Watertown, MA, #13031). Transient transfections were performed using the Nucleofector II and Amaxa Cell Line Nucleofector Kit V (Lonza) according to manufacturer's instructions and cells were harvested 24 hr post‐nucleofection.
2.5. Immunoblotting and coimmunoprecipitations
RPE‐1 cells were harvested by trypsinization, suspended in media with serum, washed with PBS, and lysed in nondenaturing lysis buffer (1% Triton X‐100, 50 mM Tris pH 7.5, 150 mM NaCl, 5 mM EDTA, and 0.02% sodium azide), Universal Nuclease (Pierce), and cOmplete Mini Protease Inhibitor Cocktail (Roche).
For coimmunoprecipitations using anti‐FLAG antibodies to pull down FLAG‐TRF1, lysates (500 µg) prepared as described above were incubated overnight with 5 µg of mouse anti‐FLAG antibody (F3165, Sigma) covalently bound to Protein A/G magnetic beads using the Pierce Crosslink Magnetic IP/Co‐IP Kit according to manufacturer's instructions. For reciprocal co‐IPs in which anti‐MYC antibodies were used to pull down MYC‐NCAPH2, nuclear lysates were prepared as described previously (Mendez & Stillman, 2000). Briefly, cells were harvested by trypsinization, suspended in media with serum, and washed with PBS. Cells were resuspended in buffer A (10 mM Hepes, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, and 1 mM DTT), and cOmplete Mini Protease Inhibitor Cocktail (Roche). Cells were lysed by the addition of 0.1% Triton X‐100 and then incubated on ice for 8 min. Nuclei were collected by centrifugation for 5 min at 1,300 g at 4°C and lysed in nondenaturing lysis buffer (1% Triton X‐100, 50 mM Tris pH 7.5, 150 mM NaCl, 5 mM EDTA, and 0.02% sodium azide), Universal Nuclease (Pierce), and cOmplete Mini Protease Inhibitor Cocktail (Roche). Total of 500 µg of nuclear lysate was incubated overnight with 5 µg of mouse anti‐MYC antibody (sc40, Santa Cruz, Santa Cruz, CA) covalently bound to Protein A/G magnetic beads using Pierce Crosslink Magnetic IP/Co‐IP Kit according to manufacturer's instructions.
For immunoblotting, samples were boiled in Laemmli sample buffer, resolved on sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE), transferred to a membrane, and blocked in PBS with 5% milk/0.1%Tween‐20. The membranes were incubated with the following primary antibodies in PBS/2.5% milk/0.1% Tween‐20: rabbit anti‐TRF1 (1449, 1:3,000 gift from Titia de Lange), rabbit anti‐TRF2 (NB110–57130, 1:1,000, Novus Biologicals, Centennial, CO), rabbit anti‐NCAPH2 (1:500), rabbit anti‐SMC2 (A300‐058A, 1:500, Bethyl Laboratories, Montgomery, TX), rabbit anti‐NCAPD3 (ab70349, 1:250, Abcam, Cambridge, MA), rabbit anti‐ATR (2790, 1:200, Cell Signaling, Danvers, MA), rabbit anti‐ATM (ab32420, 1:500, Abcam), rabbit anti‐53BP1 (ab21083, 1:250, Abcam), mouse anti‐RPA32/RPA2 (ab2175, 1:500, Abcam), rabbit anti‐PhosphoRPA (S4/S8) (A300‐245A, Bethyl Laboratories, Montgomery, TX, 1:200), mouse anti‐Chk1 (1:100, G‐4, Santa Cruz), mouse anti‐PhosphoChk1(Ser345) (2341, 1:500, Cell Signaling), rabbit anti‐GAPDH (G9545, 1:5,000, Sigma), rabbit anti‐Kinesin Heavy Chain (AKIN01, 1:800, Cytoskeleton, Denver, CO), mouse anti‐tubulin (DM1ɑ, 1:1,000, Sigma), mouse anti‐MYC (sc40, 1:125, Santa Cruz), and mouse anti‐FLAG (F3165, 1:200, Sigma). After incubation with primary and secondary HRP‐conjugated antibodies, immunoblots were developed with SuperSignal West chemiluminescent substrate (Thermo, Waltham, MA).
2.6. Immunofluorescence
Cells grown on coverslips in 12‐well plates were fixed 100% methanol on ice for 3 min, permeabilized in 0.5% Triton X‐100 in PBS, and washed three times in PBS + 0.1% Triton X‐100 (PBST). Cells were blocked in 5% bovine serum albumin (BSA) in PBST and incubated with the following primary antibodies in PBST + 5% BSA overnight at 4°C: rabbit anti‐NCAPH2 (1:200) and mouse anti‐TRF1 (ab10579, 1:200, Abcam). Coverslips were washed three times in PBST and incubated with secondary antibodies conjugated with Cy3 or Alexa488 (1:500, Jackson Laboratories, West Grove, PA). Nuclei were counterstained with 0.1 μg/μl 4′, 6‐diamidino‐2‐phenylindole (DAPI) and coverslips were mounted in Vectashield Mounting medium (Vector Laboratories, Burlingame, CA). Slides were imaged using a Nikon A1RSi confocal microscope with Plan Apo 60X and 100X oil immersion objectives and the Nikon Elements 4.0 software package (Nikon, Melville, NY). Fluorescence intensity was measured on a single z‐stack slice using ImageJ and plots were generated in Microsoft Excel.
2.7. IF‐FISH and TIF assay
RPE‐1 cells were treated with siRNAs and nucleofected as described above. Cells were grown on coverslips in 12‐well plates for 48 hr after which they were fixed with 4% formaldehyde for 10 min, permeabilized in 0.5% Triton X‐100 in PBS, and washed three times in PBS + 0.1% Triton X‐100 (PBST). For RPA staining, pre‐extraction with 0.2% Triton X‐100 in PBS was performed for 1 min on ice before fixation. Immunostaining was performed as above using the following primary and secondary antibodies: rabbit anti‐53BP1 (ab21083, 1:500, Abcam), mouse anti‐γH2AX (clone JBW301, 1:500, Millipore, Burlington, MA), mouse anti‐RPA32/RPA2 (ab2175, 1:200, Abcam), antimouse, or rabbit Alexa488 (1:500, Jackson Laboratories). Coverslips were washed with PBST, post fixed with 4% formaldehyde in 1X PBS for 10 min, and dehydrated in a series of 70%, 90%, and 100% ethanol for 2 min each at room temperature. Samples were hybridized with a TelC‐Cy3 probe (F1002, 1:800, PNA Bio, Thousand Oaks, CA) in hybridization buffer containing 10 mM Tris‐HCl pH 7.5, 70% formamide, and 1X blocking reagent (Roche). Cells and probe were first denatured at 80°C for 5 min then incubated for 4 hr at room temperature. After hybridization, coverslips were washed twice in PNA wash A (70% formamide and 10 mM Tris‐HCl, pH 7.5) and two times in PNA wash B (50 mM Tris‐HCl pH 7.5, 50 mM NaCl, and 0.08% Tween‐20). Nuclei were counterstained by adding 0.1 μg/μl DAPI to the final wash and coverslips were mounted on slides using Vectashield Mounting medium (Vector Laboratories, West Grove, PA). Slides were imaged as above. The telomere dysfunction‐induced foci (TIF) analysis was performed on a single z‐stack slice using Nikon Elements Software and graphs were generated in Microsoft Excel.
2.8. Telomere FISH on metaphase spreads
RPE‐1 cells were incubated for 2 hr with 0.1 µg/ml colchicine, harvested, and swollen in 0.075 KCl at 37°C for 10 min. Cells were fixed in 3:1 methanol:acetic acid and dropped onto glass slides which were then aged overnight at room temperature. Slides were washed with 1X PBS for 10 min, and dehydrated in a series of 70%, 90%, and 100% ethanol for 2 min each at room temperature. Slides were hybridized with a TelC‐Cy3 probe (F1002, 1:800, PNA Bio) in hybridization buffer containing 10 mM Tris‐HCl pH 7.5, 70% formamide, and 1X blocking reagent (Roche). Cells and probe were first denatured at 80°C for 5 min then incubated for 4 hr at room temperature. After hybridization, coverslips were washed two times in PNA wash A (70% formamide, 10 mM Tris‐HCl pH 7.5) and two times in PNA wash B (50 mM Tris‐HCl pH 7.5, 50 mM NaCl, and 0.08% Tween‐20). Nuclei were counterstained by adding 0.1 μg/μl DAPI to the final wash and coverslips were mounted on slides using Vectashield Mounting medium (Vector Laboratories). Slides were imaged as above.
2.9. Proximity ligation assay (PLA)
In situ proximity ligation assays (PLAs) were performed using the Duolink In Situ Red Starter Kit (Sigma‐Aldrich, Natick, MA). RPE‐1 cells were fixed in 4% formaldehyde in 1X PBS for 10 min, permeabilized in 0.5% Triton X‐100 in PBS, and washed three times in PBS + 0.1% Triton X‐100 (PBST). Cells were blocked in 5% BSA in PBST and incubated with the following primary antibodies in PBST + 5% BSA overnight at 4°c: rabbit anti‐NCAPH2 (1:200), mouse anti‐TRF1 (ab10579, 1:200, Abcam), and rabbit anti‐PML (sc‐5621, 1:100, Santa Cruz). PLA probe incubation, ligation, amplification, and detection were carried out according to the manufacturer's instructions and slides were imaged as above.
3. RESULTS
3.1. NCAPH2 contributes to telomere stability and prevents DNA damage signaling
Depletion of condensins in mouse neural stem cells and RPE‐1 cells indicates that loss of condensin II leads to DNA damage, p53 nuclear accumulation, and p21 upregulation (Nishide & Hirano, 2014). Furthermore, depletion of condensin via siRNA targeting of SMC2 has been shown to result in γH2AX enrichment at repetitive regions in dividing HeLa cells, including centromeres, subtelomeric regions, and rDNA repeats (Samoshkin, Dulev, Loukinov, Rosenfeld, & Strunnikov, 2012). We set out to further investigate whether condensin II specifically contributes to telomere stability by preventing DNA damage at telomeric repeat regions. Telomere dysfunction is caused by either short telomeres or loss of shelterin proteins, resulting in activation of DNA damage response at telomeres. DNA damage foci at telomeres are referred to as TIFs, and are measured by immunofluorescence with antibodies against 53BP1 and γH2AX coupled with fluorescence in situ hybridization (FISH) using PNA probes which hybridize to telomeric TTAGGG repeats (de Lange, 2005; Takai, Smogorzewska, & de Lange, 2003). To determine if loss of condensin proteins leads to telomere dysfunction, cells were treated with siRNAs against NCAPH2, NCAPD3, or SMC2. Efficacy of siRNA‐depletion was validated by western blot (Supplemental Figure S1 and Supplemental Figure S2 B). For NCAPH2, we generated an antibody against a C‐terminal fragment of human NCAPH2 as described previously (Ono et al., 2003). This antibody recognizes a 90 kDa band in immunoblots of RPE‐1 cell lysates and siRNA treatment results in efficient depletion of this band (Supplemental Figure S2 A and B). siRNA‐mediated depletion of TRF1 was used as a positive control for our TIF assay, as loss of TRF1 has been shown to result in accumulation of 53BP1 and γH2AX TIFs (Takai et al., 2003). We combined immunofluorescence with telomere PNA FISH and quantified TIFs by observing the colocalization of either γH2AX or 53BP1 with telomeric FISH signal. Cells containing four or more telomeric DNA damage foci were scored as positive, as described previously (Takai et al., 2003). As expected, TRF1‐depleted cells exhibited an increase in both 53BP1 (25.23%) and γH2AX (7.53%) TIFs relative to controls (0.37% and 0.35%, respectively). In NCAPH2‐depleted cells, the number of TIF‐positive cells was also significantly higher than controls, with 4.44% and 4.71% of cells scoring positive for telomeric 53BP1 and γH2AX, respectively (Figure 1A‐D). Consistent with previous work showing that RPE‐1 cells depleted for condensin II accumulate nuclear p53, p21, and γH2AX (Nishide & Hirano, 2014), we observed an additive increase in number of cells scoring positive for > 4 γH2AX or 53BP1 foci (telomeric + nontelomeric) in cells depleted for NCAPH2 (11.45% and 9.63%, respectively) relative to control (1.85% and 6.05%, respectively; Figure 1a,b,d,e). Unlike NCAPH2, cells depleted of NCAPD3 or SMC2 had no significant induction of the DNA damage response. Only 2.91% and 1.61% of NCAPD3 and SMC2 siRNA‐treated cells exhibited more than four foci per cell, respectively (Figure 1a,b,d,e). Depletion of NCAPD3 and SMC2 resulted in 0.48% of cells scoring positive for 53BP1 TIFs, whereas 2.35% of NCAPD3 and 0.81% of SMC2 siRNA‐treated cells scored as positive for γH2AX TIFs (Figure 1a,c,d,f). Thus, despite efficient depletion of the SMC2 core ATPase subunit and NCAPD3 (Supplemental Figure S1), we failed to detect significant induction of a DNA damage response.
Figure 1.
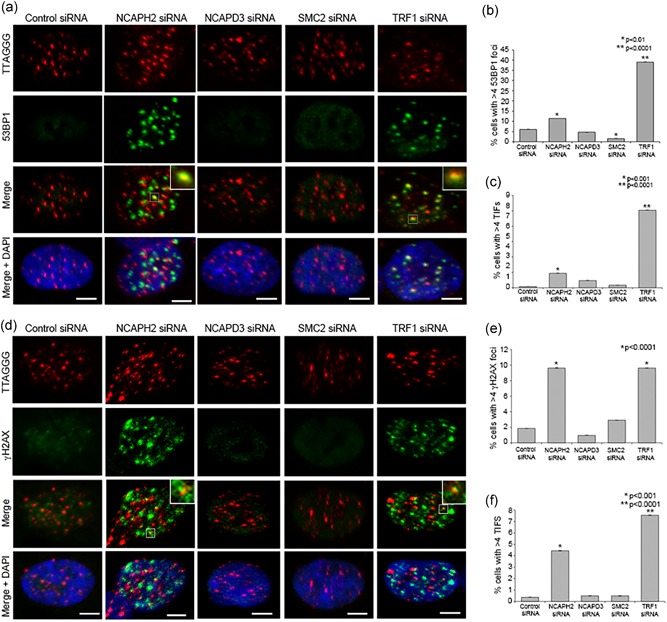
NCAPH2 depletion induces accumulation of telomeric DNA damage markers. (a) IF‐FISH in RPE‐1 cells treated with the indicated siRNA labeled with telomeric FISH probe (red) and immunostained for 53BP1 (green) and DNA (blue) as indicated. Insets show examples of telomeric 53BP1 signal (TIFs) scored in graph shown in (c). Scale bar, 5 μm. (b) Percentage of cells transfected as in (a) with > 4 53BP1 foci. At least 200 cells were counted for each category shown. *p < 0.01, **p < 0.001, ***p < 0.0001, two‐tailed Student's t test. Error bars, SEM; n.s. = nonsignificant. (c) Percentage of cells with > 4 TIFs. At least 200 cells were counted for each category shown. *p < 0.01, **p < 0.0001, two‐tailed Student's t test. Error bars, SEM; n.s. = nonsignificant. (d) IF‐FISH in RPE‐1 cells treated with the indicated siRNA labeled with telomeric FISH probe (red) and immunostained for γH2AX (green) and DNA (blue) as indicated. Insets show examples of telomeric γH2AX signal (TIFs) scored in graph shown in (f). Scale bar, 5 μm. (e) Percentage of cells transfected as in (a) with > 4 γH2AX foci. At least 200 cells were counted for each category shown. *p < 0.0001, two‐tailed Student's t test. Error bars, 95% confidence intervals. n.s. = nonsignificant. (f) Percentage of cells with > 4 TIFs. At 200 cells were counted for each category shown. *p < 0.01, **p < 0.0001, Fisher's exact test. Error bars, SEM; n.s. = nonsignificant. FISH, fluorescence in situ hybridization; TIF, telomere dysfunction‐induced foci [Color figure can be viewed at wileyonlinelibrary.com]
To confirm the NCAPH2 depletion results, an additional pool of siRNA was tested. The NCAPH2 siRNA pool and its individual components were tested for 53BP1 and γH2Ax foci as well as TIFs (Supplementary Figure S3). Individual NCAPH2 siRNA‐treated cells showed marginal increase in 53BP1 foci whereas the pool‐depleted cells showed significantly increased number (14.11%) of foci, comparable to the previously tested NCAPH2 siRNA SMARTpool (Supplementary Figure S3 A,C). All three individual siRNAs showed significant increase in γH2AX foci ranging from 7.10% to 7.72% and the pool with 8.48% positive cells (Supplementary Figure S3 B,E). The TIF analysis of 53BP1‐positive cells showed a significant increase only in the pooled NCAPH2 siRNA treatment compared with control (4.57% to 0.97%, respectively), although the single siRNAs show a slight increase (Supplementary Figure 3D). For γH2AX cells, the pooled treatment was significantly higher (5.65%) to control at 1.24% TIF‐positive cells (Supplementary Figure S3 F). The single NCAPH2.1 siRNA was also significantly increased with 4.34% of cells TIF positive (p = 0.044). Collectively, these data show loss of NCAPH2 consistently leads to an increase in both global and telomere specific DNA damage. Together, these data suggest that NCAPH2 acts to prevent telomeric DNA damage accumulation, but it remains unclear whether this NCAPH2 telomeric function is executed in the context of an active condensin II complex.
Figure 3.

NCAPH2 interacts with TRF1. (a) Schematic representation of the mammalian NCAPH2 protein. Numbers represent amino acid positions. Location of FMLEP sequence is indicated by black line. (b) Clustalw multiple sequence alignment of CapH2 proteins from the indicated vertebrates. Conserved FxLxP motif residues are highlighted in pink. (c,d) NCAPH2 and TRF1 coimmunoprecipitation. (c) RPE‐1 cells were cotransfected with plasmids expressing MYC‐NCAPH2 and either FLAG‐TRF1 or FLAG alone as a negative control. FLAG‐TRF1 immunoprecipitates (IP) and whole cell lysates (WCL) were analyzed by immunoblotting (WB) with indicated antibodies. (d) Reciprocal co‐IP. RPE‐1 cells were cotransfected with plasmids expressing FLAG‐TRF1 and either MYC‐NCAPH2 or MYC alone as a negative control. MYC‐NCAPH2 immunoprecipitates (IP) and WCL (WCL) were analyzed by immunoblotting (WB) with indicated antibodies. (e) In situ proximity ligation assay in RPE‐1 cells. Single antibody incubations for anti‐NCAPH2 or anti‐TRF1 were used as negative controls, whereas interaction of TRF1 and PML was used as a positive control. Nuclei were counterstained with DAPI and PLA interactions are represented by red fluorescent signal. Scale bar, 5 μm. (f) Average number of spots per cell nucleus counted using Duolink Image Tool software. Error bars represent SEM of three biological replicates. *p < 0.001, **p < 0.0001, Student's t‐test. DAPI, 4′, 6‐diamidino‐2‐phenylindole; PLA, proximity ligation assay [Color figure can be viewed at wileyonlinelibrary.com]
3.2. NCAPH2 associates with telomeric repeats
Although it was possible that siRNA depletion of SMC2 and NCAPD3 was not sufficient for TIF formation, the observations above raised the possibility that NCAPH2 may function at telomeres in a condensin II independent manner. We therefore asked whether NCAPH2 could have a more direct role in telomere replication and/or protection. To further study the function of NCAPH2 in telomere stability, we assessed its localization pattern in cultured cell lines. Antibody validation experiments show that our NCAPH2 antibody exhibits the expected pattern of localization on metaphase and interphase chromosomes, and this localization is specific as we observed loss of immunofluorescence signal in siRNA‐treated cells (Nishide & Hirano, 2014; Ono et al., 2004; Supplemental Figure S2 C,D). In cells fixed with methanol, which precipitates soluble proteins, immunofluorescence staining with anti‐NCAPH2 reveals a more punctate localization pattern in RPE‐1 and HeLa cells (Figure 2a). Costaining with an anti‐TRF1 antibody shows overlap of these two proteins at numerous sites throughout the nucleus. Although NCAPH2 is distributed broadly throughout the nucleus, fluorescence intensity plots show that many discrete TRF1 foci overlap with NCAPH2 signals (Figure 2a,b) suggesting that a pool of NCAPH2 localizes to telomeric regions. We performed chromatin immunoprecipitation (ChIP) in RPE‐1 cells followed by DNA dot blots hybridized to a TTAGGG repeat probe to determine if NCAPH2 binds to these repeat sequences (Figure 2c). NCAPH2 ChIP showed significant enrichment at TTAGGG repeats (0.25% input) when compared with preimmune sera (IgG; 0.05% input). Alu sequences that are vastly more abundant than TTAGGG repeats, used as a negative control, showed no enrichment of NCAPH2 above background levels (Figure 2c,d). These findings provide evidence that NCAPH2 can associate with TTAGGG repeat DNA.
Figure 2.
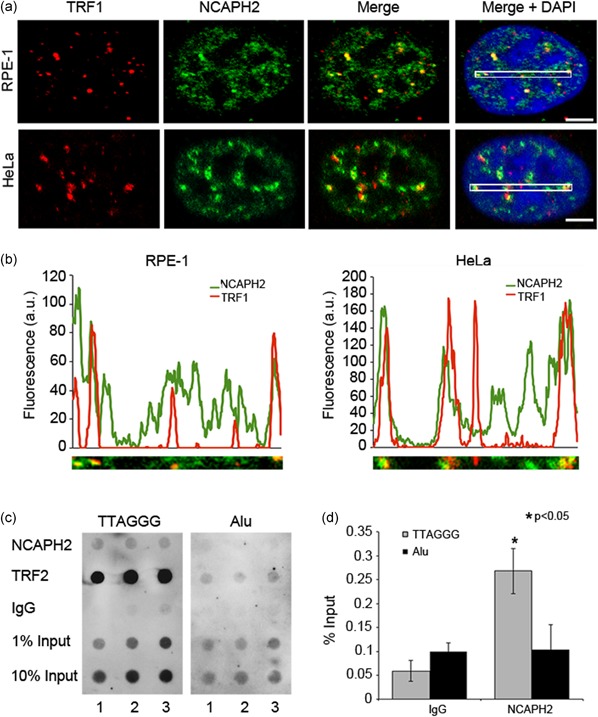
NCAPH2 associates with telomeric DNA. (a) RPE‐1 and HeLa cells immunostained for anti‐TRF1 (red), anti‐NCAPH2 (green), and DNA (blue). White boxes in Merge + DAPI panels indicate regions analyzed in (b). (b) Fluorescence intensity plots of regions outlined in (a) showing regions of overlap of NCAPH2 and TRF1. (c) ChIP assay in RPE‐1 cells with anti‐NCAPH2, anti‐TRF1, or IgG control. Dot blots were hybridized with a TTAGGG repeat probe (left panel) or Alu repeat probe (right panel). (d) Densitometric analysis of ChIP data in (c) expressed as % input ± SEM of three biological replicates. *p < 0.05, Student's t‐test. DAPI, 4′, 6‐diamidino‐2‐phenylindole [Color figure can be viewed at wileyonlinelibrary.com]
A number of shelterin accessory proteins have been identified contribute to maintenance and protection of chromosome ends, and the majority of these are recruited by shelterin (Bhanot & Smith, 2012; Palm & de Lange, 2008). Proteins are typically recruited and bind to the TRF homology (TRFH) domain of either TRF1 or TRF2 which recognizes a consensus FxLxP (TRF1) or YxLxP (TRF2) motif on the target protein (Chen et al., 2008). A search for these motifs revealed the presence of the FxLxP motif within the sequence of NCAPH2 (Figure 3a), this motif appears to be evolutionarily conserved among many CapH2 orthologs (Figure 3b). The presence of the FxLxP motif in NCAPH2 suggested potential interaction with TRF1. To determine if NCAPH2 interacts with TRF1 we performed coimmunoprecipitations using protein extracts from cells transiently coexpressing N‐terminally FLAG‐tagged TRF1 (Smogorzewska & de Lange, 2002) and MYC‐NCAPH2. In RPE‐1 cells, the MYC‐tagged NCAPH2 fusion protein exhibits the expected pattern of localization throughout the nucleus with some discrete foci, similar to the pattern observed with our anti‐NCAPH2 antibody (Supplemental Figure S4). MYC‐NCAPH2 coimmunoprecipitated with FLAG‐TRF1, but was not observed in immunoprecipitates from cells expressing FLAG alone as a control (Figure 3c). In the reciprocal co‐IP, we detected FLAG‐TRF1 in the immunoprecipitates from anti‐MYC IPs in cells coexpressing MYC‐NCAPH2 but not in cells expressing MYC alone (Figure 3d). To test whether this interaction is dependent on the FxLxP motif, site‐directed mutagenesis was used to mutate the F192, L194, and P196 residues to alanine. FLAG‐TRF1 was also detected in coimmunoprecipitations from cells transiently coexpressing MYC‐NCAPH2‐AxAxA, suggesting that the FxLxP motif is dispensable for the interaction with TRF1 (Supplemental Figure S5 A). The cells used for these experiments, however, still express endogenous NCAPH2 and we cannot eliminate the possibility that the mutated MYC‐NCAPH2 associates with NCAPH2/TRF1 complexes, allowing for its detection in co‐IPs. In support of this possibility, we have found that in cells cotransfected with MYC‐NCAPH2‐AxAxA and a GFP‐tagged wild type NCAPH2, GFP‐NCAPH2 co‐immunoprecipitates with both MYC‐NCAPH2 and MYC‐NCAPH2‐AxAxA (Supplemental Figure S5 B).
In addition, we validated the interaction between endogenous TRF1 and NCAPH2 with in situ proximity ligation assays (PLA) using antibodies against TRF1 and NCAPH2 and species‐specific oligonucleotide‐labeled secondary antibodies (Figure 3e,f). If the proteins of interest are within 40 nm in proximity, these sequences can be ligated upon addition of oligonucleotides, amplified and detected by hybridization of fluorescently labeled oligonucleotides (Soderberg et al., 2006; Weibrecht et al., 2010). Signals are visible as fluorescent spots that can be analyzed by fluorescence microscopy. Single antibody incubations were used as negative controls to determine background and as a positive control interaction between TRF1 and PML was similarly probed, as previously described (Brouwer et al., 2009). We detected 4.38 ± 0.23 (mean ± SEM) nuclear signals for TRF1 and NCAPH2, which was comparable with the number of signals detected for TRF1 and PML (4.59 ± 0.24). PLA signals for TRF1 and NCAPH2 were significantly higher than in either anti‐NCAPH2 (0.59 ± 0.08) or anti‐TRF1(3.27 ± 0.19) single antibody controls (Figure 3e,f). Similarly, NCAPH2 was also detected in close proximity to TRF2, with a PLA signal of 6.3 ± 0.06 spots per nucleus (Supplemental Figure S6). Taken together results, the co‐IP and PLA results confirm the association between TRF1 and NCAPH2 and is consistent with their colocalization at telomeres.
3.3. Loss of NCAPH2 leads to accumulation of ATR‐dependent DNA damage and RPA foci
Given the interaction with TRF1 and its known role in telomere replication (Sfeir et al., 2009; Zimmermann et al., 2014), we next asked whether this accumulation of telomeric DNA damage signals was dependent on ATM or ATR kinase phosphorylation of H2AX and 53BP1. ATM and ATR have distinct DNA‐damage specificities, with ATM responding primarily to double‐strand breaks, whereas ATR responds to replication stress (Awasthi, Foiani, & Kumar, 2015). RPE‐1 cells were treated with NCAPH2 siRNA in combination with either ATM or ATR siRNA and TIFs were scored as above. Western blot confirmed siRNA codepletion of NCAPH2 and ATM or ATR (Supplemental Figure S7). Although we observed no change in γH2AX or 53BP1 TIF accumulation upon codepletion of NCAPH2 and ATM (7.63% and 5.70% of cells scoring TIF positive, respectively) relative to NCAPH2 alone (6.15% and 6.67%, respectively; Figure 4a,c,d,f), codepletion of NCAPH2 and ATR suppressed the accumulation of telomeric γH2AX and 53BP1 (1.27% and 1.33%, respectively). This effect was not limited to telomeric regions. As with telomeric regions, codepletion of NCAPH2 and ATR suppressed the accumulation of whole cell levels of γH2AX or 53BP1 (Figure 4a,d,b,e). This finding suggests the accumulation of γH2AX and 53BP1 foci in cells depleted for NCAPH2 requires ATR kinase signaling and suggests that NCAPH2 normally acts to help suppress ATR‐mediated DNA damage signaling.
Figure 4.
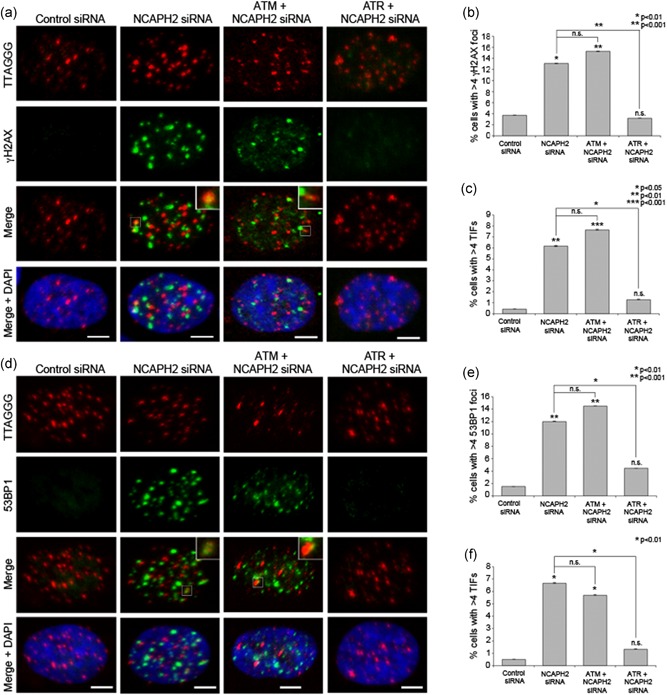
Accumulation of telomeric DNA damage in NCAPH2‐depleted cells is mediated by ATR kinase. (a) IF‐FISH in RPE‐1 cells treated with the indicated siRNA labeled with telomeric FISH probe (red) and immunostained for 53BP1 (green) and DNA (blue) as indicated. Insets show examples of telomeric 53BP1 signal (TIFs) scored in graph shown in (c). Scale bar, 5 μm. (b) Percentage of cells transfected as in (a) with > 4 53BP1 foci. At least 150 cells were counted for each category shown. *p < 0.01, **p < 0.001, two‐tailed Student's t test. Error bars, SEM, n.s. = nonsignificant. (c) Percentage of cells with > 4 TIFs. At least 150 cells were counted for each category shown. *p < 0.05, **p < 0.01, **p < 0.001, two‐tailed Student's t test. Error bars, SEM; n.s. = nonsignificant. (d) IF‐FISH in RPE‐1 cells treated with the indicated siRNA labeled with telomeric FISH probe (red) and immunostained for γH2AX (green) and DNA (blue) as indicated. Insets show examples of telomeric γH2AX signal (TIFs) scored in graph shown in (f). Scale bar, 5 μm. (e) Percentage of cells transfected as in (a) with > 4 γH2AX foci. At least 100 cells were counted for each category shown. *p < 0.01, *p < 0.001, two‐tailed Student's t test. Error bars, SEM; n.s. = nonsignificant. (f) Percentage of cells with > 4 TIFs. At least 100 cells were counted for each category shown. *p < 0.01, two‐tailed Student's t test. Error bars, SEM; n.s. = nonsignificant. FISH, fluorescence in situ hybridization; siRNA, small interfering RNA; TIF, telomere dysfunction‐induced foci [Color figure can be viewed at wileyonlinelibrary.com]
TRF1 is known to facilitate telomere replication, and TIF formation in the absence of TRF1 relies on ATR kinase and results in phosphorylation of downstream effector Chk1 (Sfeir et al., 2009). ATR recruitment to sites of DNA damage and downstream Chk1 activation requires recruitment and binding of replication protein A (RPA) to single‐stranded DNA (Zou & Elledge, 2003). To determine if depletion of NCAPH2 results in accumulation of telomeric RPA, we performed IF‐FISH with an anti‐RPA32 antibody and a telomere PNA FISH probe (Figure 5a). The number of cells with > 4 telomeric RPA foci showed a similar increase in NCAPH2 siRNA‐treated RPE cells as in TRF1 siRNA‐treated cells (4.1% and 4.3%, vs. 0.7% in control; Figure 5a,c). Previous studies in mouse embryonic fibroblasts showed that depleting 53BP1 in TRF1 null cells strengthened the activation of Chk1 and accumulation of RPA foci at telomeres (Martinez, Flores, & Blasco, 2012). Consistent with these findings, we observed a similar trend upon codepletion of 53BP1 and TRF1 (Figure 5a,c Supplemental Figure S8 A). We also observed an increase in % of RPA‐positive cells similar to that observed in hydroxyurea (HU)‐treated positive control cells, when 53BP1 was codepleted with NCAPH2 (Figure 5a,c). This RPA accumulation was detected at both telomeric and nontelomeric regions (Figure 5b), suggesting that the replication stress caused by depletion of NCAPH2 is not limited to telomeres. Unlike cells treated with HU, we were unable to detect any noticeable increase in either phosphorylated Chk1 or phosphorylated RPA relative to controls when cells were depleted for NCAPH2 or TRF1 alone or in combination with 53BP1 (Supplemental Figure S8 B).
Figure 5.
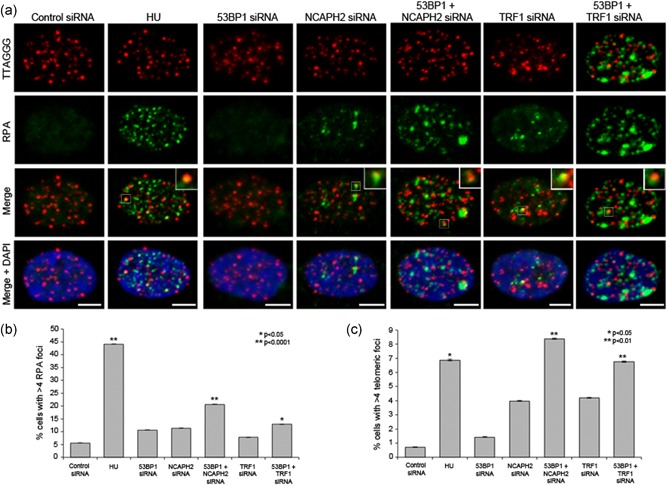
Codepletion of NCAPH2 and 53BP1 leads to increased telomeric RPA foci. (a) IF‐FISH in RPE‐1 cells treated with the indicated siRNA labeled with telomeric FISH probe (red) and immunostained for RPA (green) and DNA (blue) as indicated. Insets show examples of telomeric RPA signal scored in graph shown in (c). Scale bar, 5 μm. (b) Percentage of cells transfected as in (a) with > 4 RPA foci. At least 100 cells were counted for each category shown. *p < 0.05, **p < 0.001, two‐tailed Student's t test. Error bars, SEM; n.s. = nonsignificant. (c) Percentage of cells with > 4 telomeric RPA foci. At least 100 cells were counted for each category shown. *p < 0.05, **p < 0.01, two‐tailed Student's t test. Error bars, SEM; n.s. = nonsignificant. FISH, fluorescence in situ hybridization; RPA, replication protein A; siRNA, small interfering RNA [Color figure can be viewed at wileyonlinelibrary.com]
Telomere dysfunction can lead to chromosome structural abnormalities, and the strongest phenotype associated with TRF1 deletion in mouse cells is metaphase chromosome abnormalities (Sfeir et al., 2009). Cells lacking TRF1 exhibit sister telomere associations, as well as a fragile telomere phenotype, characterized by the appearance of multiple telomeric FISH signals at a proportion of the chromatids (Sfeir et al., 2009). We treated RPE‐1 cells with either siRNA against TRF1 or 0.2 µM of the replication inhibitor aphidicolin, which also induces fragile sites (Glover, Berger, Coyle, & Echo, 1984), and observed levels of fragile telomeres that were consistent with previous findings (Sfeir et al., 2009; Figure 6a). For example, aphidicolin induced abnormal telomeric FISH signals consistent with fragile telomeres in 8.42% of the chromosomes (n = 387), and TRF1 siRNA induced this phenotype at a similar level of 8.22% (n = 560; Figure 6a). In cells treated with NCAPH2 siRNA, we observed a significantly increased level of fragile telomeres (5.27%, n = 521) when compared with control siRNA‐treated cells (3.05%, 564; Figure 6a). Similarly, as expected association of sister telomeres was observed more frequently in aphidicolin (7.07%) and TRF1 siRNA (6.38%) controls than in control siRNA‐treated cells (2.67%). In NCAPH2‐depleted cells, sister telomere association also followed this trend, though the proportion (4.88%) was not significant (p = 0.1107, Fisher's exact test).
Figure 6.
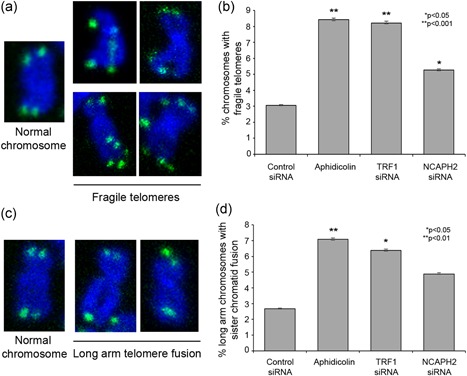
N‐CapH2 prevents formation of fragile telomeres/sister chromosome associations. (a) Examples of fragile telomeres scored in (b). (b) Quantification of fragile telomeres and sister telomere association. *p < 0.05, **p < 0.0001, Fisher's exact test. Error bars, 95% confidence intervals. (c). Examples of sister telomere associations scored in (d). Quantification of fragile telomeres and sister telomere association. *p < 0.05, **p < 0.01, Fisher's exact test. Error bars, 95% confidence intervals [Color figure can be viewed at wileyonlinelibrary.com]
Taken together results, our data suggest that NCAPH2 siRNA‐treated cells show phenotypes consistent with defects in DNA replication and that, like TRF1, NCAPH2 is required to maintain the stability of telomeres by promoting replication and preventing accumulation of DNA damage marks.
4. DISCUSSION
Here, we present evidence that NCAPH2 is a novel shelterin accessory factor that interacts with TRF1 to contribute to proper replication of telomeres in human cells. Similar to TRF1 (Sfeir et al., 2009), cells depleted of NCAPH2 showed an increase in 53BP1 and γH2AX foci at telomeres, as well as at nontelomeric regions (Figures 1, 4), resulting from activation of an ATR‐mediated DNA damage response (Figure 4). It is interesting to note that this effect was observed only when NCAPH2 was knocked down; neither siRNA depletion of NCAPD3 or SMC2 induced accumulation of DNA damage at telomeres. This finding raises the possibility that NCAPH2 is acting in a specialized manner independently of the rest of the condensin II complex components to suppress activation of DNA damage pathways. It has been suggested that individual components of condensins may associate with different protein complexes to perform specific functions (Hagstrom & Meyer, 2003). This idea of condensin subunit‐specific functions is supported by studies of silencing at the Saccharomyces cerevisiae mating‐type locus which found that YCS4 (CapD2) but not SMC2, contributes to repression of this locus (Bhalla, Biggins, & Murray, 2002). Furthermore, Caenorhabditis elegans MIX‐1(SMC2) is a member of a specialized dosage compensation complex that regulates genes on the X chromosome. We note that although siRNA depletion of SMC2 or NCAPD3 does not induce damage signaling, these treatments do lead to reduction of NCAPH2 protein (Supplemental Figure S2 B). This observation is consistent with previous reports where siRNA depletion of one condensin II subunit leads to reduction of other condensin subunits (George, Bozler, Nguyen, & Bosco, 2014; Hagstrom & Meyer, 2003), presumably through targeted proteolysis of subunits not bound into a condensin II complex. In this case either the level of NCAPH2 reduction is insufficient to induce DNA damage signaling or a pool of condensin II‐complexed NCAPH2 protein is specifically depleted by SMC2 and NCAPD3 siRNA.
Arguing against a specialized role for NCAPH2 in suppression of DNA damage signaling previous studies have associated loss of condensin II subunit CAPG2 in mouse neural stem cells (NSCs) as well as RPE‐1 cells with accumulation of γH2AX and 53BP1, presumably due to alterations in nuclear architecture and defects in mitotic chromosome segregation (Nishide & Hirano, 2014). The authors also observed a strong increase in γH2AX positive cells upon siRNA knockdown of SMC2 in RPE‐1 cells, a result which we were unable to recapitulate in our assay (Figure 1). This discrepancy could be explained by differences in scoring stringency, with our assay scoring for stronger effect by considering only those nuclei with more than four foci as positive, as has been described previously for the TIF analysis (Takai et al., 2003). However, conflicting data exists for SMC2 in U2OS cells, where siRNA depletion of SMC2 reduced DNA breakage as measured by the number of γH2AX foci and suppressed formation of 53BP1 nuclear bodies (Lukas et al., 2011). Our data are consistent with this finding, as we observe a reduction in SMC2 siRNA‐treated cells with 53BP1 foci. Nevertheless, we cannot exclude the possibility that the condensin II complex as a whole contributes to suppression of DNA damage signaling, both at telomeres and throughout the nucleus. Our data suggests, however, that NCAPH2 is an important factor required for this process, as its depletion results in a stronger effect than that observed for knockdown of SMC2 and NCAPD3.
Shelterin‐associated proteins are recruited to telomeres by TRF1 or TRF2 through their TRF homology (TRFH) domains, and those recruited by TRF1 contain a conserved FxLxP binding motif (Chen et al., 2008). The presence of an FMLEP sequence matching the consensus motif (Figure 2) within NCAPH2 suggests that it may be targeted to telomeric TTAGGG repeats by a direct interaction with TRF1 mediated by the FxLxP motif. TRF1 facilitates telomere replication by recruiting interactors TPP1/POT1, which suppresses ATR signaling, and the BLM helicase, which promotes replication fork progression (Zimmermann et al., 2014). Our data are consistent with recruitment of NCAPH2 to telomeres where it interacts with TRF1 and contributes to replication of telomeric sequences, as evidenced by accumulated RPA foci and appearance of fragile telomeres upon siRNA knockdown of NCAPH2. We note that we could not observe any noticeable increase in phosphorylation of Chk1, even in our control TRF1 knockdowns. However, other studies that have reported increased phospho‐Chk1 upon loss of TRF1 generated TRF1 null MEFs via Cre‐mediated recombination (Martinez et al., 2012; Sfeir et al., 2009), and therefore it is possible that incomplete siRNA‐depletion coupled with duration of the siRNA treatment (48 hr) was not sufficient to produce detectable accumulation of this downstream effector of the ATR pathway.
It is unclear how NCAPH2 might cooperate with TRF1 to facilitate replication of the telomeric repeats. Condensins have been linked to replication stress in budding yeast, where condensin proteins localize to hydroxyurea‐induced stalled replication forks (D'Ambrosio et al., 2008) and mutants of fission yeast non‐SMC subunit Cnd2 fail to activate the replication checkpoint after the hydroxyurea treatment (Aono, Sutani, Tomonaga, Mochida, & Yanagida, 2002). Interestingly, a mutation in the hinge domain of fission yeast Cut14(SMC2) protein resulted in DNA damage repair defects and accumulation of RPA. Condensin was found to antagonize RPA function and promote removal of RPA through its DNA reannealing. Exclusion of RPA from binding telomeric DNA is required to prevent activation of ATR at telomere ends, and it is thought that TPP1/POT1 may prevent replication‐dependent ATR activation through this same mechanism (Zimmermann et al., 2014). POT1/TPP1 occur in low abundance (approximately 1,000‐fold less than RPA), thus it has been suggested that POT/TPP1 alone may be insufficient to prevent RPA binding. The shelterin protein TIN2 has been shown to stabilize POT1/TPP1 and contribute to RPA exclusion (Takai, Kibe, Donigian, Frescas, & de Lange, 2011). We speculate that NCAPH2/condensin II could act in conjunction with TIN2 bound POT1/TPP1 to contribute to RPA removal from DNA similar to Cut14 in yeast. Thus, it will be important to determine whether NCAPH2 interacts with POT1/TPP1 or whether loss of NCAPH2 function interferes with recruitment of POT1/TPP1 to telomeres or their binding to TIN2.
It is possible that the chromatin compacting activity of NCAPH2/condensin II contributes to its suppression of telomeric ATR signaling. A recent study used super‐resolution microscopy to show that hypercondensation of human telomeres is mediated by TRF1 and TRF2 independent of histone deacetylation or methylation, and that loss of either shelterin component led to decondensation and TIF accumulation (Bandaria, Qin, Berk, Chu, & Yildiz, 2016). These observations suggest that shelterin components may prevent DNA damage signaling by compacting telomeric chromatin, thus reducing the accessibility of telomere to DNA damage signaling. Conflicting data from two studies has recently been presented, however, demonstrating a limited contribution of TRF1 and TRF2 to telomere compaction and a DNA damage response that remains intact at telomeres upon depletion or conditional knockout of these shelterin components in human and mouse cells, respectively (Timashev, Babcock, Zhuang, & de Lange, 2017; Vancevska, Douglass, Pfeiffer, Manley, & Lingner, 2017). These findings suggest that shelterin does not primarily repress DNA damage signaling at telomeres through chromatin compaction. Still, numerous studies support the idea that chromatin state affects DNA damage signaling (Burgess, Burman, Kruhlak, & Misteli, 2014; Kim, Kruhlak, Dotiwala, Nussenzweig, & Haber, 2007; Murga et al., 2007), and it remains possible that chromatin compaction at telomeres might act to diminish or slow the DNA damage response. Interestingly, condensin II has been shown to be recruited by the bromodomain protein BRD4 to sites of ionizing radiation‐induced chromosomal breaks where NCAPH2 recruitment mediates compaction of chromatin, resulting in attenuation of DNA damage response signaling (Floyd et al., 2013). Furthermore, the multitelomeric FISH signals observed in fragile telomeres upon loss of TRF1 have been proposed to result from altered chromatin organization that may arise as the result of incomplete replication and large areas of single‐stranded DNA (Sfeir et al., 2009). Our finding that NCAPH2 depletion, like loss of TRF1, leads to formation of fragile telomeres (Figure 6) is compatible with this hypothesis. Therefore, it seems plausible to predict that NCAPH2/condensin II may contribute to the structural organization of telomeric chromatin to promote efficient replication and suppress DNA damage signaling.
FUNDING
This study was supported by funds from the Geisel School of Medicine at Dartmouth.
CONFLICT OF INTERESTS
Authors declare that they have no conflict of interests.
AUTHOR CONTRIBUTIONS
HAW, VR, HQN, and GB conceived and designed experiments; HAW, VR, and HQN performed experiments and analyzed data. HAW and VR co‐wrote the manuscript with guidance from GB.
Supporting information
Supporting information
Supporting information
ACKNOWLEDGEMENTS
We thank members of the Bosco laboratory for helpful comments and suggestions.
Wallace HA, Rana V, Nguyen HQ, Bosco G. Condensin II subunit NCAPH2 associates with shelterin protein TRF1 and is required for telomere stability. J Cell Physiol. 2019;234:20755–20768. 10.1002/jcp.28681
References
REFERENCES
- Aono, N. , Sutani, T. , Tomonaga, T. , Mochida, S. , & Yanagida, M. (2002). Cnd2 has dual roles in mitotic condensation and interphase. Nature, 417, 197–202. [DOI] [PubMed] [Google Scholar]
- Awasthi, P. , Foiani, M. , & Kumar, A. (2015). ATM and ATR signaling at a glance. Journal of Cell Science, 128, 4255–4262. [DOI] [PubMed] [Google Scholar]
- Bandaria, J. N. , Qin, P. , Berk, V. , Chu, S. , & Yildiz, A. (2016). Shelterin protects chromosome ends by compacting telomeric chromatin. Cell, 164, 735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman, J. R. , Larschan, E. , D'Souza, R. , Marshall, L. S. , Dempsey, K. E. , Johnson, J. E. , Mellone, B. G. , & Kuroda, M. I. (2012). A genome‐wide screen identifies genes that affect somatic homolog pairing in Drosophila. G3 (Bethesda), 2, 731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer, C. R. , Hartl, T. A. , & Bosco, G. (2012). Condensin II promotes the formation of chromosome territories by inducing axial compaction of polyploid interphase chromosomes. PLoS Genetics, 8, e1002873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla, N. , Biggins, S. , & Murray, A. W. (2002). Mutation of YCS4, a budding yeast condensin subunit, affects mitotic and nonmitotic chromosome behavior. Molecular Biology of the Cell, 13, 632–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhanot, M. , & Smith, S. (2012). TIN2 stability is regulated by the E3 ligase Siah2. Molecular and Cellular Biology, 32, 376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi, A. , Smith, S. , Chong, L. , Elias, P. , & de Lange, T. (1997). TRF1 is a dimer and bends telomeric DNA. EMBO Journal, 16, 1785–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broccoli, D. , Smogorzewska, A. , Chong, L. , & de Lange, T. (1997). Human telomeres contain two distinct Myb‐related proteins, TRF1 and TRF2. Nature Genetics, 17, 231–235. [DOI] [PubMed] [Google Scholar]
- Brouwer, A. K. , Schimmel, J. , Wiegant, J. C. , Vertegaal, A. C. , Tanke, H. J. , & Dirks, R. W. (2009). Telomeric DNA mediates de novo PML body formation. Molecular Biology of the Cell, 20, 4804–4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess, R. C. , Burman, B. , Kruhlak, M. J. , & Misteli, T. (2014). Activation of DNA damage response signaling by condensed chromatin. Cell Reports, 9, 1703–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buster, D. W. , Daniel, S. G. , Nguyen, H. Q. , Windler, S. L. , Skwarek, L. C. , Peterson, M. , Roberts, M. , Meserve, J. H. , Hartl, T. , & Klebba, J. E. , et al. (2013). SCFSlimb ubiquitin ligase suppresses condensin II‐mediated nuclear reorganization by degrading Cap‐H2. Journal of Cell Biology, 201, 49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenci, G. , Ciapponi, L. , & Gatti, M. (2005). The mechanism of telomere protection: A comparison between Drosophila and humans. Chromosoma (Berlin), 114, 135–145. [DOI] [PubMed] [Google Scholar]
- Chen, Y. , Yang, Y. , van Overbeek, M. , Donigian, J. R. , Baciu, P. , de Lange, T. , & Lei, M. (2008). A shared docking motif in TRF1 and TRF2 used for differential recruitment of telomeric proteins. Science, 319, 1092–1096. [DOI] [PubMed] [Google Scholar]
- D'Ambrosio, C. , Schmidt, C. K. , Katou, Y. , Kelly, G. , Itoh, T. , Shirahige, K. , & Uhlmann, F. (2008). Identification of cis‐acting sites for condensin loading onto budding yeast chromosomes. Genes and Development, 22, 2215–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd, S. R. , Pacold, M. E. , Huang, Q. , Clarke, S. M. , Lam, F. C. , Cannell, I. G. , Bryson, B. D. , Rameseder, J. , Lee, M. J. , & Blake, E. J. , et al. (2013). The bromodomain protein Brd4 insulates chromatin from DNA damage signalling. Nature, 498, 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George, C. M. , Bozler, J. , Nguyen, H. Q. , & Bosco, G. (2014). Condensins are required for maintenance of nuclear architecture. Cells, 3, 865–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover, T. W. , Berger, C. , Coyle, J. , & Echo, B. (1984). DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Human Genetics, 67, 136–142. [DOI] [PubMed] [Google Scholar]
- Green, L. C. , Kalitsis, P. , Chang, T. M. , Cipetic, M. , Kim, J. H. , Marshall, O. , Turnbull, L. , Whitchurch, C. B. , Vagnarelli, P. , & Samejima, K. , et al. (2012). Contrasting roles of condensin I and condensin II in mitotic chromosome formation. Journal of Cell Science, 125, 1591–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagstrom, K. A. , & Meyer, B. J. (2003). Condensin and cohesin: More than chromosome compactor and glue. Nature Reviews Genetics, 4, 520–534. [DOI] [PubMed] [Google Scholar]
- Harrison, J. C. , & Haber, J. E. (2006). Surviving the breakup: The DNA damage checkpoint. Annual Review of Genetics, 40, 209–235. [DOI] [PubMed] [Google Scholar]
- Hartl, T. A. , Smith, H. F. , & Bosco, G. (2008). Chromosome alignment and transvection are antagonized by condensin II. Science, 322, 1384–1387. [DOI] [PubMed] [Google Scholar]
- Hirano, T. , Kobayashi, R. , & Hirano, M. (1997). Condensins, chromosome condensation protein complexes containing XCAP‐C, XCAP‐E and a Xenopus homolog of the Drosophila Barren protein. Cell, 89, 511–521. [DOI] [PubMed] [Google Scholar]
- Hirota, T. , Gerlich, D. , Koch, B. , Ellenberg, J. , & Peters, J. M. (2004). Distinct functions of condensin I and II in mitotic chromosome assembly. Journal of Cell Science, 117, 6435–6445. [DOI] [PubMed] [Google Scholar]
- Hug, N. , & Lingner, J. (2006). Telomere length homeostasis. Chromosoma (Berlin), 115, 413–425. [DOI] [PubMed] [Google Scholar]
- Jain, D. , & Cooper, J. P. (2010). Telomeric strategies: Means to an end. Annual Review of Genetics, 44, 243–269. [DOI] [PubMed] [Google Scholar]
- Joyce, E. F. , Williams, B. R. , Xie, T. , & Wu, C. T. (2012). Identification of genes that promote or antagonize somatic homolog pairing using a high‐throughput FISH‐based screen. PLoS Genetics, 8, e1002667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlseder, J. , Hoke, K. , Mirzoeva, O. K. , Bakkenist, C. , Kastan, M. B. , Petrini, J. H. , & de Lange, T. (2004). The telomeric protein TRF2 binds the ATM kinase and can inhibit the ATM‐dependent DNA damage response. PLoS Biology, 2, E240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. A. , Kruhlak, M. , Dotiwala, F. , Nussenzweig, A. , & Haber, J. E. (2007). Heterochromatin is refractory to gamma‐H2AX modification in yeast and mammals. Journal of Cell Biology, 178, 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. H. , Zhang, T. , Wong, N. C. , Davidson, N. , Maksimovic, J. , Oshlack, A. , Earnshaw, W. C. , Kalitsis, P. , & Hudson, D. F. (2013). Condensin I associates with structural and gene regulatory regions in vertebrate chromosomes. Nature Communications, 4, 2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange, T. (2009). How telomeres solve the end‐protection problem. Science, 326, 948–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange, T. (2005). Shelterin: The protein complex that shapes and safeguards human telomeres. Genes and Development, 19, 2100–2110. [DOI] [PubMed] [Google Scholar]
- Lingner, J. , & Cech, T. R. (1998). Telomerase and chromosome end maintenance. Current Opinion in Genetics & Development, 8, 226–232. [DOI] [PubMed] [Google Scholar]
- Lukas, C. , Savic, V. , Bekker‐Jensen, S. , Doil, C. , Neumann, B. , Pedersen, R. S. , Grofte, M. , Chan, K. L. , Hickson, I. D. , & Bartek, J. , et al. (2011). 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nature Cell Biology, 13, 243–253. [DOI] [PubMed] [Google Scholar]
- Lydall, D. (2009). Taming the tiger by the tail: Modulation of DNA damage responses by telomeres. EMBO Journal, 28, 2174–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez, P. , Flores, J. M. , & Blasco, M. A. (2012). 53BP1 deficiency combined with telomere dysfunction activates ATR‐dependent DNA damage response. Journal of Cell Biology, 197, 283–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez, J. , & Stillman, B. (2000). Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: Assembly of prereplication complexes in late mitosis. Molecular and Cellular Biology, 20, 8602–8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motwani, T. , Doris, R. , Holmes, S. G. , & Flory, M. R. (2010). Ccq1p and the condensin proteins Cut3p and Cut14p prevent telomere entanglements in the fission yeast Schizosaccharomyces pombe . Eukaryotic Cell, 9, 1612–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murga, M. , Jaco, I. , Fan, Y. , Soria, R. , Martinez‐Pastor, B. , Cuadrado, M. , Yang, S. M. , Blasco, M. A. , Skoultchi, A. I. , & Fernandez‐Capetillo, O. (2007). Global chromatin compaction limits the strength of the DNA damage response. Journal of Cell Biology, 178, 1101–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuwald, A. F. , & Hirano, T. (2000). HEAT repeats associated with condensins, cohesins, and other complexes involved in chromosome‐related functions. Genome Research, 10, 1445–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishide, K. , & Hirano, T. (2014). Overlapping and non‐overlapping functions of condensins I and II in neural stem cell divisions. PLoS Genetics, 10, e1004847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono, T. , Fang, Y. , Spector, D. L. , & Hirano, T. (2004). Spatial and temporal regulation of Condensins I and II in mitotic chromosome assembly in human cells. Molecular Biology of the Cell, 15, 3296–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono, T. , Losada, A. , Hirano, M. , Myers, M. P. , Neuwald, A. F. , & Hirano, T. (2003). Differential contributions of condensin I and condensin II to mitotic chromosome architecture in vertebrate cells. Cell, 115, 109–121. [DOI] [PubMed] [Google Scholar]
- Palm, W. , & de Lange, T. (2008). How shelterin protects mammalian telomeres. Annual Review of Genetics, 42, 301–334. [DOI] [PubMed] [Google Scholar]
- Rong, Y. S. (2008). Telomere capping in Drosophila: Dealing with chromosome ends that most resemble DNA breaks. Chromosoma (Berlin), 117, 235–242. [DOI] [PubMed] [Google Scholar]
- Samoshkin, A. , Dulev, S. , Loukinov, D. , Rosenfeld, J. A. , & Strunnikov, A. V. (2012). Condensin dysfunction in human cells induces nonrandom chromosomal breaks in anaphase, with distinct patterns for both unique and repeated genomic regions. Chromosoma (Berlin), 121, 191–199. [DOI] [PubMed] [Google Scholar]
- Sfeir, A. , Kosiyatrakul, S. T. , Hockemeyer, D. , MacRae, S. L. , Karlseder, J. , Schildkraut, C. L. , & de Lange, T. (2009). Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell, 138, 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir, A. , & de Lange, T. (2012). Removal of shelterin reveals the telomere end‐protection problem. Science, 336, 593–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintomi, K. , & Hirano, T. (2011). The relative ratio of condensin I to II determines chromosome shapes. Genes and Development, 25, 1464–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska, A. , & de Lange, T. (2002). Different telomere damage signaling pathways in human and mouse cells. EMBO Journal, 21, 4338–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska, A. , van Steensel, B. , Bianchi, A. , Oelmann, S. , Schaefer, M. R. , Schnapp, G. , & de Lange, T. (2000). Control of human telomere length by TRF1 and TRF2. Molecular and Cellular Biology, 20, 1659–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderberg, O. , Gullberg, M. , Jarvius, M. , Ridderstrale, K. , Leuchowius, K. J. , Jarvius, J. , Wester, K. , Hydbring, P. , Bahram, F. , & Larsson, L. G. , et al. (2006). Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nature Methods, 3, 995–1000. [DOI] [PubMed] [Google Scholar]
- van Steensel, B. , & de Lange, T. (1997). Control of telomere length by the human telomeric protein TRF1. Nature, 385, 740–743. [DOI] [PubMed] [Google Scholar]
- Takai, H. , Smogorzewska, A. , & de Lange, T. (2003). DNA damage foci at dysfunctional telomeres. Current Biology, 13, 1549–1556. [DOI] [PubMed] [Google Scholar]
- Takai, K. K. , Kibe, T. , Donigian, J. R. , Frescas, D. , & de Lange, T. (2011). Telomere protection by TPP1/POT1 requires tethering to TIN2. Molecular Cell, 44, 647–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timashev, L. A. , Babcock, H. , Zhuang, X. , & de Lange, T. (2017). The DDR at telomeres lacking intact shelterin does not require substantial chromatin decompaction. Genes and Development, 31, 578–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vancevska, A. , Douglass, K. M. , Pfeiffer, V. , Manley, S. , & Lingner, J. (2017). The telomeric DNA damage response occurs in the absence of chromatin decompaction. Genes and Development, 31, 567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viera, A. , Gomez, R. , Parra, M. T. , Schmiesing, J. A. , Yokomori, K. , Rufas, J. S. , & Suja, J. A. (2007). Condensin I reveals new insights on mouse meiotic chromosome structure and dynamics. PLoS One, 2, e783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace, H. A. , & Bosco, G. (2013). Condensins and 3D Organization of the Interphase Nucleus. Current Genetic Medicine Reports, 1, 219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson, J. D. (1972). Origin of concatemeric T7 DNA. Nature: New biology, 239, 197–201. [DOI] [PubMed] [Google Scholar]
- Weibrecht, I. , Leuchowius, K. J. , Clausson, C. M. , Conze, T. , Jarvius, M. , Howell, W. M. , Kamali‐Moghaddam, M. , & Soderberg, O. (2010). Proximity ligation assays: A recent addition to the proteomics toolbox. Expert Review of Proteomics, 7, 401–409. [DOI] [PubMed] [Google Scholar]
- Zimmermann, M. , Kibe, T. , Kabir, S. , & de Lange, T. (2014). TRF1 negotiates TTAGGG repeat‐associated replication problems by recruiting the BLM helicase and the TPP1/POT1 repressor of ATR signaling. Genes and Development, 28, 2477–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, L. , & Elledge, S. J. (2003). Sensing DNA damage through ATRIP recognition of RPA‐ssDNA complexes. Science, 300, 1542–1548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information