

Abstract
The alarming rise of antimicrobial resistance (AMR) imposes severe burdens on healthcare systems and the economy worldwide, urgently calling for the development of new antibiotics. Antimicrobial peptides could be ideal templates for next‐generation antibiotics, due to their low propensity to cause resistance. An especially promising branch of antimicrobial peptides target lipid II, the precursor of the bacterial peptidoglycan network. To develop these peptides into clinically applicable compounds, detailed information on their pharmacologically relevant modes of action is of critical importance. Here we review the binding modes of a selection of peptides that target lipid II and highlight shortcomings in our molecular understanding that, at least partly, relate to the widespread use of artificial membrane mimics for structural studies of membrane‐active antibiotics. In particular, with the example of the antimicrobial peptide nisin, we showcase how the native cellular membrane environment can be critical for understanding of the physiologically relevant binding mode.
Keywords: antibiotics, antimicrobial peptides, in-cell NMR spectroscopy, lipid II, membranes, solid-state NMR spectroscopy
1. An Urgent Need for New Antibiotics
Antibiotics are the bedrock of modern medicine, and the world is running out of these magic bullets at a dramatic pace. To date, antimicrobial resistance (AMR) has been observed against all clinically used antibiotics.1 The rapid and global emergence of AMR, which is forecast to cause a staggering 10 million human deaths annually by the year 2050,2 entails severe repercussions for public healthcare systems. At present, according to a recent extensive investigation, the healthcare burden of AMR in the European Economic Area (EEA) is already on a par with the combined effect of influenza, HIV, and tuberculosis, and about 33 000 people in the EEA die as a consequence of AMR every year.3 Likewise, AMR is estimated to cause costs of billions of euros for the European economy.4 To make things worse, the industry's drug‐discovery pipeline for antibiotics is almost empty. It is hence of pressing need to develop new antibiotics that target unexploited pathways and that are robust to the development of resistance mechanisms. This has led to an increased interest in unconventional agents such as naturally occurring antimicrobial peptides, against which bacteria have enormous difficulty in building up effective resistance mechanisms. Here we focus on a special class of powerful antimicrobial peptides that specifically target lipid II, the precursor of the bacterial peptidoglycan (PGN) network.5
2. Lipid II, the Achilles’ Heel of Bacteria
Every bacterium is surrounded by a PGN network, a protective hull that is essential for its survival. To ensure cell viability, growth, and division, PGN biosynthesis must be tightly regulated. The PGN network is composed of strands of alternating N‐acetylmuramic acid and N‐acetylglucosamine (MurNAc–GlcNAc) disaccharides linked through pentapeptide bridges. The PGN is synthesized from its plasma‐membrane‐anchored precursor lipid II, which carries one complete PGN subunit linked through a unique pyrophosphate group to a C55 polyisoprenoid moiety.
However, lipid II exists only in very low copy numbers, and it is constantly recycled through the so‐called lipid II cycle to ensure efficient PGN synthesis (Figure 1 A). This means that any drug that sequesters lipid II, and thereby stalls the PGN synthesis, is a potential antibiotic.5a, 6 In the case of Gram‐positive bacteria the plasma membrane is readily accessible from the outside, so certain peptides can exploit this weak spot and specifically bind lipid II at its pyrophosphate group.7 This is a unique mode of action that kills even highly resistant superbugs such as methicillin‐resistant Staphylococcus aureus (MRSA) at nanomolar concentrations, and against which resistance is difficult to acquire because the lipid II structure is highly conserved and the pyrophosphate group is even deemed irreplaceable.5a In addition, because human cells do not produce lipid II, it is a target with a low risk of unwanted toxicity. Peptides that bind lipid II could hence be promising templates for next‐generation antibiotics; however, detailed information on their modes of action is critically required in order to develop them into clinically applicable drugs.
Figure 1.
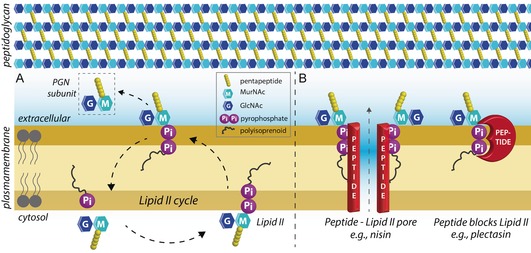
A) The cell envelope and the lipid II cycle. Lipid II is assembled in the cytosol and then transported across the plasma membrane to sustain PGN synthesis. B) Peptide antibiotics that bind lipid II kill bacteria by two known mechanisms: (left) targeted pore formation, and/or (right) blocking of the lipid II cycle and hence of PGN synthesis.
3. Peptide Antibiotics that Bind Lipid II
Below we discuss structural studies on a selection of representative peptides that bind lipid II, including plectasin,8 teixobactin,9 tridecaptin A1,10 and nisin.11 These peptides were chosen either because of their unique modes of action or because of their popularity. Unfortunately, due to the limited format of this minireview, we will not discuss the modes of action of other antimicrobials that bind lipid II, such as plusbacin A3,12 ramoplanin,13 nukacin ISK‐1,14 or vancomycin and its derivatives,15 most of which were recently reviewed elsewhere.6 Most of the antibiotics that bind lipid II kill bacteria by sequestering it, thereby blocking the lipid II cycle. Furthermore, some of the reviewed peptides such as teixobactin are also known to bind precursors of wall teichoic acids or other PGN precursors, whereas peptides such as the lantibiotic nisin11 additionally recruit lipid II to form pores through the plasma membrane (Figure 1 B). Such dual‐ or multi‐killing mechanisms render peptides that bind lipid II even less susceptible to causing AMR. In Section 4 we show that a physiologically relevant membrane environment can be indispensable for understanding of the modes of action of peptides that bind lipid II.
3.1. Plectasin
Defensins are a major class of host‐defense peptides, found in almost all eukaryotes. They are small (3–5 kDa) and contain three to five disulfide bonds. It was recently discovered that certain CSαβ defensins specifically target lipid II.16 These CSαβ defensins, defined by a common α‐helix‐β‐sheet fold, act by sequestering lipid II to block PGN synthesis. The best‐known example is plectasin, from the fungus Pseudoplectania nigrella (Figure 2 A).8 Plectasin tightly (K D=1.8×10−7 m) binds lipid II in a 1:1 stoichiometry.16 The activity spectrum of plectasin is broad and includes medically important Gram‐positive pathogens.8 In addition, plectasin is stable and can be produced in large quantities. Altogether, plectasin is an excellent lead for rational drug design, and the plectasin triple mutant NZ2114 (D9N, M13L, Q14R) is currently in clinical trials.17
Figure 2.
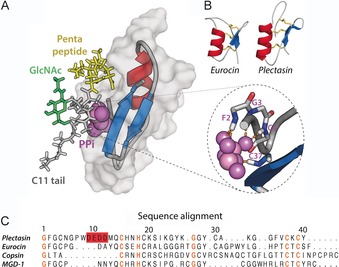
A) Docking model of the plectasin⋅lipid II complex, based on NMR data acquired in DPC micelles. Plectasin coordinates the pyrophosphate group (PPi) through backbone amide protons of the N terminus and the C‐terminal β‐strand (see zoom, right). Moreover, in this model the pentapeptide (in yellow) is involved in plectasin binding. Plectasin is shown in surface representation (in gray) and in cartoon representation. B) CSαβ defensins such as eurocin exhibit a fold similar to that of plectasin yet low sequence homology. C) Sequence alignment of several CSαβ defensins. Conserved residues are highlighted in orange whereas the anionic patch in plectasin is in red. The first ten residues of the copsin N terminus were not included in the alignment.
Our structural understanding of the binding mode of plectasin is based on a solution NMR study in dodecylphosphocholine (DPC) micelles with a truncated, water‐soluble lipid II construct. On titration with micelles containing lipid II, a small number of plectasin residues (F2, C4, A31, G33, C37, and K38) showed larger chemical shift perturbations (CSPs).16 A CSP‐based HADDOCK18 model suggests that the backbone amide protons of the N terminus and the C‐terminal β‐strand form hydrogen bonds with the lipid II pyrophosphate and the pentapeptide (Figure 2 A). That the pentapeptide is part of the complex interface agrees with the fact that the enzymatic amidation of the γ‐d‐Glu residue in the lipid II pentapeptide decreases the antimicrobial efficiency of plectasin.19
However, many aspects of plectasin's binding mode remain unclear. For example, the stark influence of the lipid II GlcNAc sugar on the binding affinity of plectasin16 cannot be explained by the micelle model, in which GlcNAc is far from the complex interface (Figure 2 A), whereas the conserved side chain of H18 (Figure 2 C), which is assumed to be critically involved in binding of lipid II, was not detectable in NMR experiments in micelles.16 Moreover, the model does not provide a structural explanation of why the triple mutant NZ2114 exhibits a favorable activity over WT plectasin. Furthermore, the 1:1 binding stoichiometry was determined with a water‐soluble version of lipid II, and this might not necessarily reflect the binding to membrane‐imbedded lipid II. In addition, the molecular function of the remarkable anionic stretch (9‐DEDD‐12) of plectasin remains unknown. Interestingly, although CSαβ defensins such as eurocin20 or copsin21 share a common fold with plectasin, they exhibit only low sequence homology and marked differences in the lengths of certain loops (Figure 2 B, C). It is unknown whether or not these structural differences modulate the binding mode to lipid II or if CSαβ defensins share a similar binding motif.
3.2. Teixobactin
Teixobactin (Figure 3) is an 11‐residue macrocyclic depsipeptide that is non‐ribosomally produced by the soil bacterium Eleftheria terrae. A highly promising compound for the development of antibiotics with excellent potency against multidrug‐resistant Gram‐positive bacteria, it exhibits favorable pharmacokinetics and activity in mouse models.9 Teixobactin binds lipid II and the cell‐wall teichoic acid precursor undecaprenyl‐PP‐GlcNAc (lipid III) by an as yet uncertain mechanism.22 Initial studies established that the pyrophosphate group is part of teixobactin's binding motif. Most notably, a recent solid‐state NMR (ssNMR) study in DPC micelles by the Lewandowski lab. provided more detailed structural insights into teixobactin's mode of action (Figure 3).23 That study suggests that teixobactin forms larger aggregates upon binding to lipid II; this was also supported by a recent X‐ray study in aqueous solution.24 Intriguingly, such higher‐order aggregates of peptide⋅ lipid II complexes could be of considerable importance under physiological conditions because lipid II is assumed to be predominately concentrated in specific cell membrane regions.5b, 6b
Figure 3.
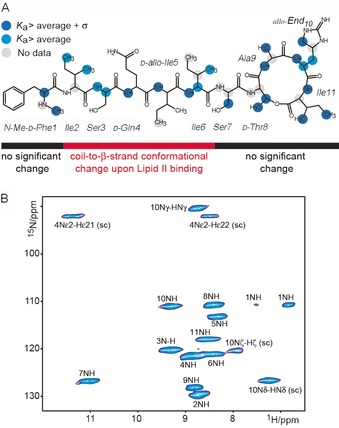
Characterization of the teixobactin⋅lipid II complex in DPC micelles by NMR. A) Results of solid‐state NMR titrations of teixobactin with Gram‐negative mDAP‐lipid II in DPC micelles. The blue dots relate to the binding affinity of teixobactin towards lipid II (see ref. 23 for details). B) 2D NH solid‐state NMR correlation spectrum of [13C,15N] teixobactin in complex with natural abundance mDAP‐lipid II. Figure adapted from ref. 23.
In the bound state, the C‐terminal macrocyclic part of teixobactin presumably complexes lipid II, whereas the N‐terminal residues, which are disordered in the unbound state,25 adopt a β‐strand structure involved in peptide aggregation. Such a binding mode is in line with recent long MD simulations of the teixobactin⋅lipid II complex in membranes.26 Interestingly, the solid‐state NMR study also strongly suggests that the pentapeptide or the sugar moieties of lipid II play a critical role for the binding mode and aggregation of teixobactin. Currently, extensive efforts are being undertaken to simplify the complex structure and to lower the financial constraints on the development of teixobactin‐based drugs. In the process, it was surprisingly revealed that the unusual enduracididine residue at position 10 can be replaced by branched l‐amino acids without any adverse effect on antimicrobial activity.22
3.3. Tridecaptin A1
Tridecaptins are linear, non‐ribosomally synthesized lipopeptides isolated from Bacillus and Paenbacillus species that bind lipid II and have the rare quality of showing activity against Gram‐negative bacterial strains including Escherichia coli, Klebsiella pneumoniae, and Acinetobacter baumannii. This activity profile is especially noteworthy because of the strong need for new antibiotics against Gram‐negative bacteria, as illustrated by the “Priority Pathogen List for Research and Development on Antibiotics” of the World Health Organization (WHO).27 The archetypical representative is tridecaptin A1 (Figure 4 A), which disrupts the proton‐motive force, presumably by forming small proton pores across the bacterial plasma membrane, as recently shown in an elegant study by the Vederas lab.10
Figure 4.
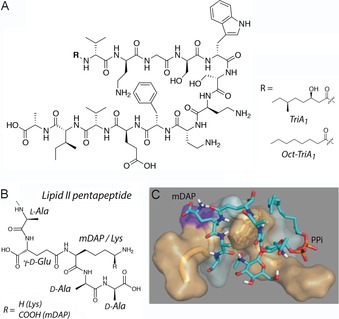
A) Chemical structures of the tridecaptin analogues TriA1 and Oct‐TriA1. B) Chemical structures of the pentapeptide of Gram‐negative and Gram‐positive lipid II. C) mDAP‐lipid II docked into TriA1. Hydrophobic residues interact with the lipid II terpene tail, and the pentapeptide occupies the binding pocket. Figures A) and C) reproduced from ref. 10. Copyright: 2015, the authors.
Tridecaptin A1 crosses the outer membrane of a Gram‐negative bacterium by specifically interacting with a chiral target of lipopolysaccharide (LPS). The N‐terminal lipid tail, however, is not necessary for crossing the outer membrane, although its presence is critical for the antimicrobial activity of tridecaptin A1. Notably, tridecaptin A1 displays selective targeting of the lipid II of Gram‐negative bacteria; this lipid II variant possesses a meso‐diaminopimelic acid (mDAP) residue—as opposed to a lysine residue—at the 3‐position in the pentapeptide (Figure 4 B, C).10 The mDAP residue is presumably directly involved in complex formation, given that the activity of tridecaptin A1 against Gram‐positive bacteria is strongly reduced. Indeed, solution NMR experiments in DPC micelles showed that tridecaptin A1 targets the lipid II pentapeptide, with no binding to the pyrophosphate group being observed.
Whereas unbound tridecaptin A1 adopts an elongated structure, in the bound state it wraps around lipid II in a process stabilized by a π‐stacking interaction between residues d‐Trp5 and l‐Ph9. The importance of this stacking interaction was recently corroborated by chemical engineering. Here, good antimicrobial activity was observed after covalent bridging of residues 5 and 9, forming a cyclic construct.28 Furthermore, the NMR structure suggests an interaction between the γ‐amino group of d‐Dab8, which is essential for antimicrobial activity of tridecaptins, and the ϵ‐carboxylate group of the mDAP residue in lipid II. However, because the small proton pores that tridecaptin A1 presumably forms could not be observed in DPC micelles, insights into the physiological mode of action most likely require a liposomal setting. This is a similar situation as for nisin, discussed in the next section.
4. Towards the Native Mode of Action
Until recently, quantitative structural data on peptide–lipid II interactions had exclusively been obtained in nonphysiological media such as DMSO or micelles. Such artificial media, as highlighted by recent publications,29 are strongly compromised in their capacity to mimic lipid membranes, can destabilize secondary structures, promote disorder, and generally perturb the native folding and topology of membrane‐embedded or associated biomolecules. As we elaborate with a few selected examples, many aspects of the modes of action of drugs that bind lipid II have thus far evaded deeper structural understanding. This lack of information might, at least partially, relate to the use of artificial membrane mimics. In the case of tridecaptin A1, for example, it seems highly likely that the small pores that represent the physiological binding mode cannot be observed in micelles. Using the lantibiotic nisin as a showcase, we have recently, for the first time, explored quantitative peptide–lipid II interactions at high resolution in lipid membranes and in native cellular bacterial membranes. Our study demonstrated that lipid membranes are essential for the physiologically relevant binding mode of nisin, and very likely also for the native binding modes of many other antimicrobials that target lipid II.
4.1. The membrane environment modulates the nisin pore
Lantibiotics are heavily modified peptides, characterized by thioether rings.6a The class A(I) lantibiotic nisin, produced by Lactococcus lactis, is the most widely studied peptide that binds lipid II (Figure 5 A). Nisin is globally used as a preservative in the food industry and has a recognized potential for clinical use,31 including against drug‐resistant bacterial strains and biofilms. As discovered in a pioneering study by Breukink et al., nisin kills bacteria at nanomolar concentrations through a unique mechanism called targeted pore formation,11 for which eight nisin and four lipid II molecules are assumed to form a lethal hole that spans the plasma membrane (Figure 5 B).6a
Figure 5.
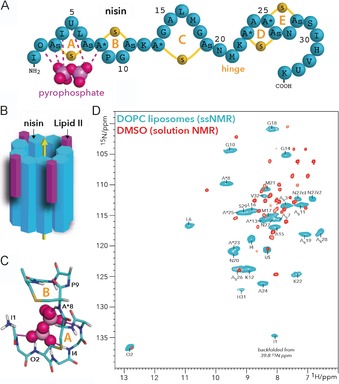
A) Nisin features five thioether rings, of which rings A and B are critical for binding lipid II. B) Nisin and lipid II form a defined pore that spans the bacterial plasma membrane. C) The pyrophosphate cage structure as determined in DMSO.7 The thioether rings A and B of nisin are annotated. D) Overlay of 2D NH spectra of the nisin⋅lipid II complex in DOPC (blue) and in DMSO (red). The ssNMR spectrum was measured at 950 MHz (1H frequency) and 60 kHz magic‐angle spinning. The solution NMR spectrum in DMSO was published previously.7 (A) and (D) reproduced from ref. 30. Copyright: 2018, the authors.
Although this mechanism was discovered >15 years ago, the pore structure is still unknown. The only structural information came from a solution NMR study in DMSO, which yielded the famous “pyrophosphate cage”, in which the amide backbone protons of the thioether rings A and B coordinate to the lipid II pyrophosphate group (Figure 5 C). However, by using an advanced solid‐state NMR approach in lipid membranes, we have recently shown that the nisin⋅lipid II complex as determined in DMSO corresponds to a non‐native state. We observed large and global chemical shift perturbations between nisin bound to lipid II in DMSO and in membranes; this demonstrates a major conformational change in lipid membranes, including in the pyrophosphate cage (Figure 5 D).30 Importantly, we performed this study with modern 1H‐detected solid‐state NMR,32 which was critical for direct investigation of the backbone amide protons of nisin that coordinate lipid II.
4.2. High‐resolution NMR in native bacterial cell membranes
Although reconstituted liposomes represent an important step towards physiologically relevant media, they fall short of the stunning complexity of cell membranes that host a plethora of different lipid and protein types. Furthermore, the variations in the chemical structure of lipid II that are present in different bacterial strains can decisively influence the binding modes of some antimicrobials.33 In principle, ssNMR enables structural studies of biomolecules directly in native cellular membranes. However, because of the heterogeneity of cellular membranes and the low native copy numbers of lipid II, such studies critically require the use of advanced highly sensitive ssNMR methods such as 1H‐detection32 and dynamic nuclear polarization (DNP).34
1H detection, driven by the advent of very high magic‐angle spinning frequencies, makes use of the direct detection of proton nuclei. Of all biological nuclei, protons have the highest gyromagnetic ratio, and the higher this physical constant the higher the NMR signal sensitivity. Moreover, 1H detection is compatible with physiological buffer conditions and temperatures, and enables direct access to amide protons, which generally seem immediately involved in the binding of lipid II. In DNP experiments, conducted at cryogenic temperatures, the system is doped with small radicals. The gyromagnetic ratio of an electron is more than 600 times higher than for a proton, and this can translate into enormous signal enhancements. Here, through the use of microwave irradiation, spin polarization is transferred from the radicals to the system of interest such as a peptide antibiotic, and this is then followed by a NMR experiment with improved sensitivity.
Using a combination of both NMR methods, we succeeded in investigating the nisin⋅lipid II pore complex directly in cellular membranes of Micrococcus flavus (Figure 6 A) with as little as 5–10 nmol of nisin. This study showed that 1,2‐dioleoyl‐sn‐glycero‐3‐phosphocholine (DOPC) and 1,2‐dioleoyl‐sn‐glycero‐3‐phosphoglycerol (DOPG) liposomes are very good mimics for the nisin⋅lipid II pore and enable physiologically relevant insights to be obtained. Intriguingly, this study, supported by 1H/2H exchange experiments,35 also revealed that the so‐called hinge region (Figure 5 A) and the linker between thio rings B and C are both plastic and enable nisin to adapt to the complexity of cellular membranes (Figure 6 C, D). This is an important finding because these linker regions are pharmaceutical hotspots, and mutations in these regions were indeed shown to modulate nisin's activity in a strain‐specific manner (Figure 6 E).36 These insights were only possible in native cellular membranes.
Figure 6.
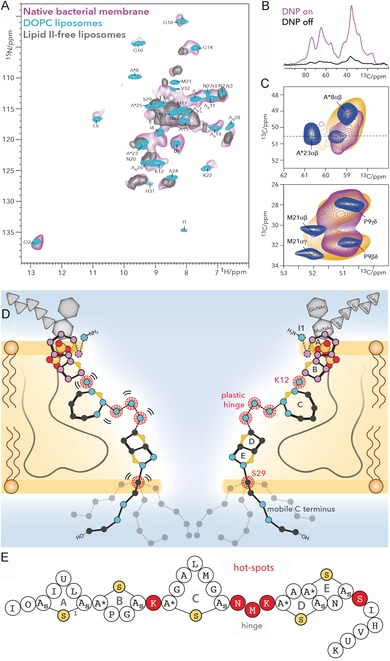
A) Comparison of 1H‐detected 2D NH spectra of nisin bound to lipid II in cellular M. flavus membranes (magenta) and in DOPC (cyan). The gray spectrum shows nisin nonspecifically bound to liposomes in the absence of lipid II. B) 13C spectra of nisin bound to lipid II in M. flavus membranes with (magenta) and without (black) DNP enhancement. C) The hinge domain is plastic and broadens out under DNP conditions with a 100 K sample temperature (orange). This is even more pronounced in cellular membranes (magenta). Upper panel: zoom into residue A*23, adjacent to the hinge. Lower panel: zoom into M21 of the hinge. D) Membrane arrangement of the nisin⋅lipid II topology from ssNMR. Plastic residues are highlighted with red circles. Residues that showed 1H/2H exchange are colored in blue and align the pore lumen. The C terminus is dynamically disordered. The A and B rings (in magenta) interact with the pyrophosphate group. E) Linker residues (in red) are pharmaceutical hotspots that enable improvement of nisin's activity upon mutation. These residues were all identified as important for nisin's cellular adaptability by cellular ssNMR.30 Figure reproduced from ref. 30. Copyright: 2018, the authors.
5. Summary and Outlook
Drugs that bind lipid II have aroused considerable interest, due to their potency and their robustness to the development of AMR. In order to use these promising compounds for the design of next‐generation antibiotics, a detailed understanding of the pharmacologically relevant binding modes is essential. In recent years a small number of structural studies have considerably advanced our comprehension of drugs that bind lipid II. However, these studies have predominantly been performed in membrane mimics such as DMSO and micelles, and the extent to which such artificial media enable native binding modes is uncertain. In order to understand the pharmacologically relevant binding modes, it is therefore essential to investigate membrane‐active drugs under physiological conditions. Here, ssNMR37 and the advent of highly sensitive signal enhancement and detection methods bode well for performance of structural studies directly in cellular membranes and, in the future, even with whole cells.38 Such cellular experiments could be of particular value for investigation of higher‐order complexes such as pores or aggregates, which seem to be more common among drugs that bind lipid II, and which are of special interest given that lipid II seems to be more concentrated in certain regions of the cell membrane.5b, 6b We can envision that such cellular structural studies could be used to understand and break bacterial drug‐resistance mechanisms, or to optimize antibiotics’ selectivity against specific bacterial strains. The latter is of particular importance in light of our growing understanding of the human microbiota.39 Altogether, such truly native studies of membrane‐active antibiotics should become an indispensable tool to sharpen our weapons against drug‐resistant bacteria.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
João Medeiros‐Silva obtained his Master's degree in biochemistry from NOVA University Lisbon, working on sugar–protein interactions and the synthesis of G‐quadruplex ligands. Currently he is a Ph.D. student in the lab. of Markus Weingarth at Utrecht University. His work involves the development of strategies to study complex systems such as membrane‐active antibiotics and ion channels in their native environment by solid‐state NMR.

Biographical Information
Shehrazade Jekhmane obtained her Master's degree in molecular life sciences from Wageningen University, working on the development of (m)RNA drug delivery systems for nucleic acid therapy purposes. She's currently doing a Ph.D. in the lab. of Markus Weingarth, where she studies membrane‐active antimicrobials in their native environment and at the atomic scale by solid‐state NMR.

Biographical Information
Eefjan Breukink did his Ph.D. at the University of Utrecht in the lab of Ben de Kruijff, working on the Sec‐dependent protein translocation pathway in E. coli. He is now an Associate Professor and his research focuses on the bacterial cell wall synthesis machinery in relation to antibiotics, the modes of action of antimicrobial peptides, and development of pathway‐specific antibiotic screening.

Biographical Information
Markus Weingarth did his Ph.D. at École normale supérieure (Paris) in the laboratory of Geoffrey Bodenhausen, working on solid‐state NMR methods, followed by a postdoc with Marc Baldus at Utrecht University, working on NMR methods for membrane proteins. Honors include a FEBS Distinguished Young Investigator Award and an NWO VIDI Award. He now is Assistant Professor, and his research deals with ion channels and bioactive peptides with a focus on antibiotics.

Acknowledgements
This work was funded by the Netherlands Science Organisation for Scientific Research (NWO, grants no. 723.014.003, 711.018.001 to M.W.).
J. Medeiros-Silva, S. Jekhmane, E. Breukink, M. Weingarth, ChemBioChem 2019, 20, 1731.
References
- 1. Ventola C. L., PT 2015, 40, 277–283. [PMC free article] [PubMed] [Google Scholar]
- 2.J. O′Neill, (Review on Antimicrobial Resistance, London) 2014 https://amr-review.org/.
- 3. Cassini A., Diaz Högberg L., Plachouras D., Quattrocchi A., Hoxha A., Skov Simonsen G., Colomb-Cotinat M., Kretzschmar M. E., Devleesschauwer B., Cecchini M., Ouakrim D. A., Oliveira T. C., Struelens M. J., Suetens C., Group) D. L. Monnet (Burden of AMR Collaborative, Lancet Infect. Dis. 2019, 19, 56–66.30409683 [Google Scholar]
- 4. Gandra S., Barter D. M., Laxminarayan R., Clin. Microbiol. Infect. 2014, 20, 973–980. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Breukink E., de Kruijff B., Nat. Rev. Drug Discovery 2006, 5, 321–332; [DOI] [PubMed] [Google Scholar]
- 5b. Scheffers D. J., Tol M. B., Plos Pathog. 2015, 11, e1005213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Oppedijk S. F., Martin N. I., Breukink E., Biochim. Biophys. Acta Biomembranes 2016, 1858, 947–957; [DOI] [PubMed] [Google Scholar]
- 6b. Müller A., Klockner A., Schneider T., Nat. Prod. Rep. 2017, 34, 909–932. [DOI] [PubMed] [Google Scholar]
- 7. Hsu S. T., Breukink E., Tischenko E., Lutters M., de Kruijff B., Kaptein R., Bonvin A., van Nuland N. A., Nat. Struct. Mol. Biol. 2004, 11, 963–967. [DOI] [PubMed] [Google Scholar]
- 8. Mygind P. H., Fischer R. L., Schnorr K. M., Hansen M. T., Sonksen C. P., Ludvigsen S., Raventos D., Buskov S., Christensen B., De Maria L., Taboureau O., Yaver D., Elvig-Jorgensen S. G., Sorensen M. V., Christensen B. E., Kjaerulff S., Frimodt-Moller N., Lehrer R. I., Zasloff M., Kristensen H. H., Nature 2005, 437, 975–980. [DOI] [PubMed] [Google Scholar]
- 9. Ling L. L., Schneider T., Peoples A. J., Spoering A. L., Engels I., Conlon B. P., Mueller A., Schaberle T. F., Hughes D. E., Epstein S., Jones M., Lazarides L., Steadman V. A., Cohen D. R., Felix C. R., Fetterman K. A., Millett W. P., Nitti A. G., Zullo A. M., Chen C., Lewis K., Nature 2015, 520, 388. [DOI] [PubMed] [Google Scholar]
- 10. Cochrane S. A., Findlay B., Bakhtiary A., Acedo J. Z., Rodriguez-Lopez E. M., Mercier P., Vederas J. C., Proc. Natl. Acad. Sci. USA 2016, 113, 11561–11566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Breukink E., Wiedemann I., van Kraaij C., Kuipers O. P., Sahl H. G., de Kruijff B., Science 1999, 286, 2361–2364. [DOI] [PubMed] [Google Scholar]
- 12. O'Connor R. D., Singh M., Chang J., Kim S. J., VanNieuwenhze M., Schaefer J., J. Phys. Chem. B 2017, 121, 1499–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fang X., Tiyanont K., Zhang Y., Wanner J., Boger D., Walker S., Mol. Biosyst. 2006, 2, 69–76. [DOI] [PubMed] [Google Scholar]
- 14. Fujinami D., Mahin A. A., Elsayed K. M., Islam M. R., Nagao J. I., Roy U., Momin S., Zendo T., Kohda D., Sonomoto K., Commun. Biol. 2018, 1, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Okano A., Isley N. A., Boger D. L., Proc. Natl. Acad. Sci. USA 2017, 114, E5052–E5061; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Antonoplis A., Zang X. Y., Huttner M. A., Chong K. K. L., Lee Y. B., Co J. Y., Amieva M. R., Kline K. A., Wender P. A., Cegelski L., J. Am. Chem. Soc. 2018, 140, 16140–16151; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15c. Kim S. J., Cegelski L., Stueber D., Singh M., Dietrich E., Tanaka K. S. E., Parr T. R., Far A. R., Schaefer J., J. Mol. Biol. 2008, 377, 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schneider T., Kruse T., Wimmer R., Wiedemann I., Sass V., Pag U., Jansen A., Nielsen A. K., Mygind P. H., Raventos D. S., Neve S., Ravn B., Bonvin A. M., De Maria L., Andersen A. S., Gammelgaard L. K., Sahl H. G., Kristensen H. H., Science 2010, 328, 1168–1172. [DOI] [PubMed] [Google Scholar]
- 17. Andes D., Craig W., Nielsen L. A., Kristensen H. H., Antimicrob. Agents Chemother. 2009, 53, 3003–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dominguez C., Boelens R., Bonvin A. M. J. J., J. Am. Chem. Soc. 2003, 125, 1731–1737. [DOI] [PubMed] [Google Scholar]
- 19. Münch D., Roemer T., Lee S. H., Engeser M., Sahl H. G., Schneider T., PLoS Pathog. 2012, 8, e1002509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Oeemig J. S., Lynggaard C., Knudsen D. H., Hansen F. T., Norgaard K. D., Schneider T., Vad B. S., Sandvang D. H., Nielsen L. A., Neve S., Kristensen H. H., Sahl H. G., Otzen D. E., Wimmer R., J. Biol. Chem. 2012, 287, 42361–42372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Essig A., Hofmann D., Munch D., Gayathri S., Kunzler M., Kallio P. T., Sahl H. G., Wider G., Schneider T., Aebi M., J. Biol. Chem. 2014, 289, 34953–34964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Parmar A., Iyer A., Prior S. H., Lloyd D. G., Goh E. T. L., Vincent C. S., Palmai-Pallag T., Bachrati C. Z., Breukink E., Madder A., Lakshminarayanan R., Taylor E. J., Singh I., Chem. Sci. 2017, 8, 8183–8192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Öster C., Walkowiak G. P., Hughes D. E., Spoering A. L., Peoples A. J., Catherwood A. C., Tod J. A., Lloyd A. J., Herrmann T., Lewis K., Dowson C. G., Lewandowski J. R., Chem. Sci. 2018, 9, 8850–8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang H., Wierzbicki M., Du Bois D. R., Nowick J. S., J. Am. Chem. Soc. 2018, 140, 14028–14032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Parmar A., Prior S. H., Iyer A., Vincent C. S., Van Lysebetten D., Breukink E., Madder A., Taylor E. J., Singh I., Chem. Commun. 2017, 53, 2016–2019. [DOI] [PubMed] [Google Scholar]
- 26. Wen P. C., Vanegas J. M., Rempe S. B., Tajkhorshid E., Chem. Sci. 2018, 9, 6997–7008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Antibacterial Agents in Clinical Development: An analysis of the antibacterial clinical development pipeline, including tuberculosis, WHO, 2017 https://apps.who.int/iris/bitstream/handle/10665/258965/WHO-EMP-IAU-2017.11-eng.pdf?sequence=1&isAllowed=y.
- 28. Ballantine R. D., Li Y. X., Qian P. Y., Cochrane S. A., Chem. Commun. 2018, 54, 10634–10637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.
- 29a. Kurauskas V., Hessel A., Ma P. X., Lunetti P., Weinhaupl K., Imbert L., Brutscher B., King M. S., Sounier R., Dolce V., Kunji E. R. S., Capobianco L., Chipot C., Dehez F., Bersch B., Schanda P., J. Phys. Chem. Lett. 2018, 9, 933–938; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29b. Chipot C., Dehez F., Schnell J. R., Zitzmann N., Pebay-Peyroula E., Catoire L. J., Miroux B., Kunji E. R. S., Veglia G., Cross T. A., Schanda P., Chem. Rev. 2018, 118, 3559–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Medeiros-Silva J., Jekhmane S., Paioni A. L., Gawarecka K., Baldus M., Swiezewska E., Breukink E., Weingarth M., Nat. Commun. 2018, 9, 3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shin J. M., Gwak J. W., Kamarajan P., Fenno J. C., Rickard A. H., Kapila Y. L., J. Appl. Microbiol. 2016, 120, 1449–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.
- 32a. Sinnige T., Daniels M., Baldus M., Weingarth M., J. Am. Chem. Soc. 2014, 136, 4452–4455; [DOI] [PubMed] [Google Scholar]
- 32b. Weingarth M., van der Cruijsen E. A., Ostmeyer J., Lievestro S., Roux B., Baldus M., J. Am. Chem. Soc. 2014, 136, 2000–2007; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32c. Mance D., Sinnige T., Kaplan M., Narasimhan S., Daniels M., Houben K., Baldus M., Weingarth M., Angew. Chem. Int. Ed. 2015, 54, 15799–15803; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 16025–16029; [Google Scholar]
- 32d. Medeiros-Silva J., Mance D., Daniels M., Jekhmane S., Houben K., Baldus M., Weingarth M., Angew. Chem. Int. Ed. 2016, 55, 13606–13610; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 13804–13808; [Google Scholar]
- 32e. Jekhmane S., Medeiros-Silva J., Li J., Kummerer F., Muller-Hermes C., Baldus M., Roux B., Weingarth M., Nat. Commun. 2019, 10, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Münch D., Engels I., Muller A., Reder-Christ K., Falkenstein-Paul H., Bierbaum G., Grein F., Bendas G., Sahl H. G., Schneider T., Antimicrob. Agents Chemother. 2015, 59, 772–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.
- 34a. Kaplan M., Narasimhan S., de Heus C., Mance D., van Doorn S., Houben K., Popov-Čeleketič D., Damman R., Katrukha E. A., Jain P., Geerts W. J. C., Heck A. J. R., Folkers G. E., Kapitein L. C., Lemeer S., Van Bergen En Henegouwen P. M. P., Baldus M., Cell 2016, 167, 1241–1251; [DOI] [PubMed] [Google Scholar]
- 34b. Salnikov E. S., Aisenbrey C., Aussenac F., Ouari O., Sarrouj H., Reiter C., Tordo P., Engelke F., Bechinger B., Sci. Rep. 2016, 6, 20895; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34c. Visscher K. M., Medeiros-Silva J., Mance D., Rodrigues J., Daniels M., Bonvin A., Baldus M., Weingarth M., Angew. Chem. Int. Ed. 2017, 56, 13222–13227; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 13404–13409. [Google Scholar]
- 35. Medeiros-Silva J., Jekhmane S., Baldus M., Weingarth M., Solid State Nucl. Magn. Reson. 2017, 87, 80–85. [DOI] [PubMed] [Google Scholar]
- 36. Zhou L., van Heel A. J., Kuipers O. P., Front. Microbiol. 2015, 6, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.
- 37a. Hong M., Su Y., Protein Sci. 2011, 20, 641–655; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37b. Bechinger B., Salnikov E. S., Chem. Phys. Lipids 2012, 165, 282–301; [DOI] [PubMed] [Google Scholar]
- 37c. Ramamoorthy A., Solid State Nucl. Magn. Reson. 2009, 35, 201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Romaniuk J. A., Cegelski L., Philos. Trans. R. Soc. London Ser. B 2015, 370, 20150024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.
- 39a. Blaser M., Nature 2011, 476, 393–394; [DOI] [PubMed] [Google Scholar]
- 39b. Blaser M. J., Cell 2018, 172, 1173–1177; [DOI] [PubMed] [Google Scholar]
- 39c. Tanoue T., Morita S., Plichta D. R., Skelly A. N., Suda W., Sugiura Y., Narushima S., Vlamakis H., Motoo I., Sugita K., Shiota A., Takeshita K., Yasuma-Mitobe K., Riethmacher D., Kaisho T., Norman J. M., Mucida D., Suematsu M., Yaguchi T., Bucci V., Inoue T., Kawakami Y., Olle B., Roberts B., Hattori M., Xavier R. J., Atarashi K., Honda K., Nature 2019, 565, 600–605. [DOI] [PubMed] [Google Scholar]