

Dear Editor,
The analysis of hundreds of chronic lymphocytic leukemia (CLL) exomes has shed new light on the heterogeneous genomic background characterizing this disease1,2. At the same time, the increased availability of exome-sequencing data comes along with a big bottleneck in the interpretation of its results, which is related to the remarkable heterogeneity in mutation detection between different bioinformatic protocols. Differences in clonality, purity, sequencing coverage, and quality constitue difficulties for most variant callers. The methods with the highest sensitivity are frequently accompanied by lower precision, leading to remarkable differences in mutation detection3–5. Therefore, we hypothesize that numerous variants in large sequencing projects have passed unnoticed.
Here, we report the results of a complementary analysis performed on the International Cancer Genome Consortium (ICGC) CLL cohort6. The final analysis included 49 monoclonal B cell lymphocytosis and 390 treatment-naive CLL samples. Mutation detection was performed with two different methods: VarsCan2, which uses a heuristic/statistical method for variant detection; and Platypus, which implements a Bayesian approach and local realignment of reads for indel and complex mutation detection. Variant quality was recalibrated using a logistic model, and drivers were detected by integrating the results of methods based on mutation frequency (MuSiC2), functional impact (OncodriveFM), co-localization (OncodriveClust and Mutation3D), and pathogenicity prediction (VEST and CHASM) (Supplementary Methods). Cox regression was used for survival analysis. Assumption of proportional hazards was checked with Schoenfeld’s method. An unadjusted model was used to test the association of each mutated gene/pathway with time to treatment and overall survival. Similarly, we created an adjusted model which included variables associated with outcomes of interest at a nomial p-value < 0.2 (IGHV status, sex, and stage at diagnosis for time to treatment analysis; and IGHV status, age and stage at diagnosis for overall survival analysis). In the case of pathways analysis, the total number of mutations in genes belonging to each pathway were used as input. P-values were adjusted for multiple testing using the Benjamini–Hochberg (BH) method.
A total of 28,350 mutations were detected in 439 treatment-naive patient samples, of which 12,057 affected protein-coding regions (Supplementary Table 1). There were 8,965 non-silent and 3,095 silent mutations. The large majority of the non-silent mutations were missense (7,558 events). Point mutations were the most frequent (21,180), followed by short deletions (3,240) and insertions (2,041). There were 1,888 multi-nucleotide mutations (involving 2 or more consecutive nucleotides) (Supplementary Fig. 1).
Sixty-six genes were detected as putative drivers (Fig. 1, Supplementary Table 2, Supplementary Tables 3–8), of which thirty-two had been previously described by Puente et al.1 Among the novel ones, the most frequently mutated were DTX1, LPHN3, LRP1B, LTB, and WDFY3. LPHN2 and SI were mutated in six patients; BIRC6, DOCK1, MLL3, PCDH15, PTPN13, PTPRM, RELN, and TFEB were mutated in five patients and the remaining putative drivers were mutated in four different cases. Furthermore, WDFY3 harbored two additional silent mutations that are predicted to create new donor or acceptor cryptic sites. BIRC6, DOCK1, KMT2C/MLL3, PTPRB, and PTPRT were each affected by one silent mutation predicted to create a new cryptic splice site. Mutations in IGLL5 were frequent and located in hotspots, but they were accompanied by a high rate of silent mutations. Finally, we observed that FREM1 was targeted by four likely functional non-synonymous mutations and two additional silent mutations in the same position. Most of the new proposed drivers play well-defined roles in carcinogenesis, such as EPHA7;7 MYCBP2;8 PTPRM9. Other putative drivers have been linked to oncogenesis before, such as the autophagy regulator WDFY310, the Notch pathway gene DTX111, the latrophilin genes LPHN2 and LPHN312, as well as FREM1, which encodes the MYD88 and NFkB pathways related-protein TILRR13. Similarly, driver mutations in CARD11 and SI have been previously described in CLL2,14, and the genes BIRC6 and KMT2C/MLL3 are paralogs of the CLL drivers BIRC3 and KMT2D.
Fig. 1. Mutation plot representing the most frequent novel CLL drivers identified in this study.

Plot indicating the number and type of mutations in driver genes. Only drivers not described by Puente et al1. are represented
Low-frequency and likely pathogenic mutations in 60 genes (Supplementary Table 9) were detected. This type of mutations affected known cancer drivers (EGFR, ERBB4, MAP2K1, NF1, NFKB1, NOTCH3, and SRSF1), including multiple drivers of lymphoproliferation such as BAX, BCOR, BCR, BTG2, DIS3, IKZF3, KRAS, PPM1D, PTPN11, SETD1B, TLR2, and TRAF3. The list also includes regulators of lymphocyte pathways (CD19, CD36, ALCAM) and of relevant cancer pathways such as the Notch pathway (NOTCH3, DMXL2, and SBNO1), WNT/β-catenin pathway (DACT1); DNA polymerization (POLE) and epigenetic regulation (KDM5A, HIST1H1D, PHF1 and single mutations at HIST1H2BC and HIST1H2BG). Moreover, isolated missense mutations in relevant oncogenes and tumor suppressor genes such as EP300, KIT, MELK, and PTEN were among the most significant events.
Non-synonymous mutations in 16 genes were significantly associated with time to first treatment (q-value < 0.1, Supplementary Table 10). The list included known CLL drivers such as ATM, SF3B1, BRAF, NOTCH1, BIRC3, IRF4, and ZMYM3, as well as other putative novel drivers such as EPHA7 and SI. Mutations in IGLV3-21, DOCK1, and EPHA7 were associated with time to treatment after covariate adjustment (q-value < 0.1, Supplementary Table 11). In order to assess the potential effect of silent mutations on time to treatment, we included them in the regression, revealing new significant associations in IGHV1-69, IGKJ5, IGHV2-70, and FAT1. Furthermore, silent mutations in IGLV3-21 reduced the association p-value further (Supplementary Figure 2).Only two IGLV3-21 mutated cases co-expressed IGHV3-21, indicating an independent role of the IGHV3-21/IGLV3-21 stereotyped B cell receptor. This is in concordance with a recent report about the adverse prognosis of IGLV3-21 expression in CLL15. Finally, mutations in ASXL1, ATM, IGHV1-69, SPEN, SF3F1, PLCH1, and POT1 were associated with overall survival (q-value < 10%, Supplementary Table 12), but none of these was significant after covariate adjustment (q-value < 0.1; Supplementary Table 13).
The genes IGLL5, LTB, ZFP36L1, LRP1B, and PCDH15 were significantly enriched in intronic mutations (q-value < 0.1; Supplementary Table 14, Supplementary Table 15). Mutations in ZFP36L1 and DAPK1 were independently associated with time to first treatment (adjusted q-value < 0.1), whereas those in IGHV3-49 were independently associated with overall survival (adjusted q-value < 0.1; Supplementary Tables 16–19).
A pathway-level inquiry detected 62 terms enriched in mutations (Bonferroni p-value < 0.1) (Supplementary Table 20). The most significantly mutated pathways were “RB pathway”, “TP53 pathway”, “ATM pathway”, “Apoptotic Signaling in Response to DNA Damage”, “TP53 Hypoxia pathway” and the “G1 pathway”. Most of the significant associations with clinical evolution were influenced by the presence of frequent driver mutations within the pathway. However, the following four significant pathways did not include any high-frequency CLL-driver gene: “CDK5 pathway”, “Apoptosis-induced DNA fragmentation”, “FRS2 mediated cascade”, and the “RAF MAP Kinase cascade”. We detected an interesting pattern in the TP53 downstream pathway, which affected ~10% of the patients. Mutations in this pathway were strongly and independently associated with shorter time to first treatment (p-value 3.80 × 10−5, Fig. 2a, b, Supplementary Table 21), and removing TP53 mutated cases from the analysis did not affect the association substantially (p-value 5.3 × 10−4). These mutations were also significantly associated with lower overall survival (p-value 2.81 × 10−4), but not independently of IGHV status (p-value 0.54). These results suggest that the disruption of the TP53 pathway plays an active role in CLL.
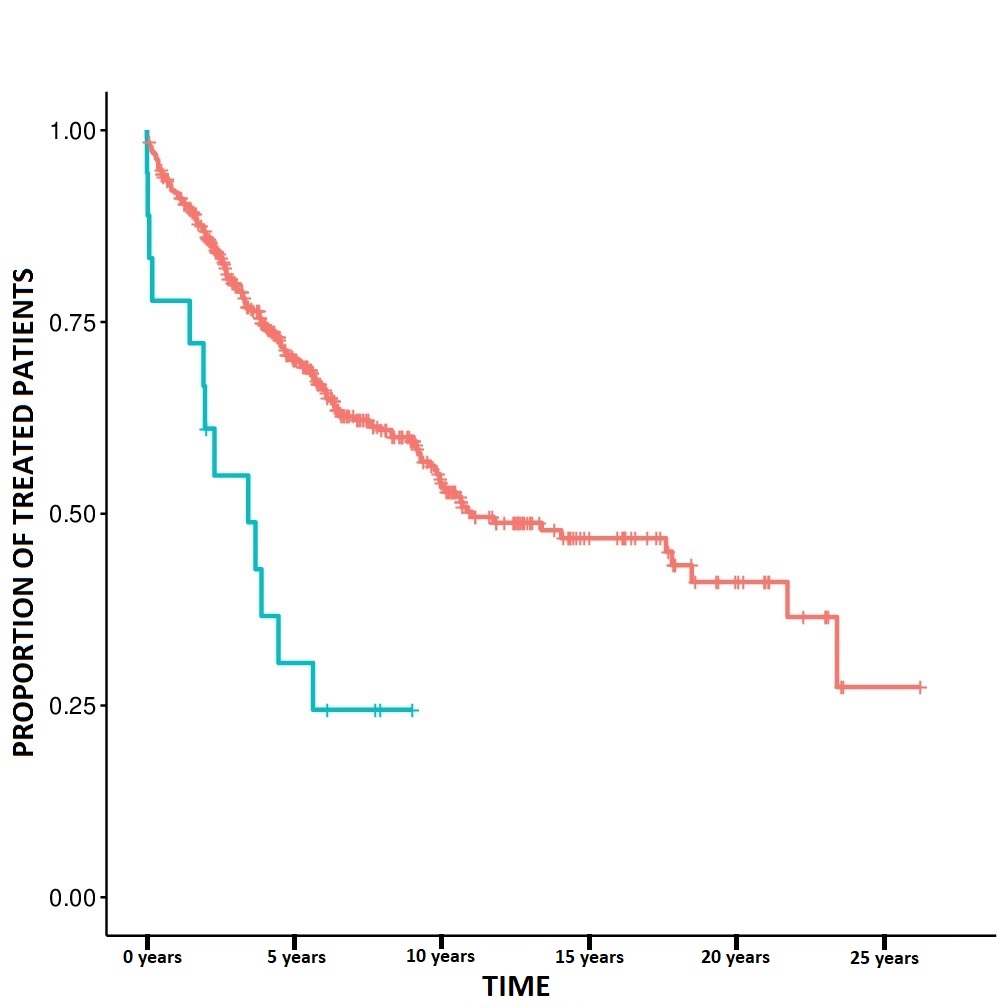
Fig. 2. New prognostic biomarkers of CLL clinical evolution.

a–b Kaplan–Meier plots indicating the association between mutations in the TP53 pathway and time to treatment with and without TP53 mutated cases (Fig. 2a, b, respectively). The red line indicates patients without mutation in this pathway, whereas the green and blue lines indicate patients with one or more than one mutation, respectively. c–d Kaplan–Meier plots indicating the association of mutations in the 3′ UTR and flanking region of IGKC with time to treatment. In Fig. 2c the red line indicates those patients without mutations in this region and the blue line indicate mutated cases. Similarly, in Fig. 2d the blue line indicates IGHV mutated cases with mutation in IGKC, the red line indicates IGHV mutated cases without IGKC mutation, the purple line indicates IGHV unmutated cases with IGKC mutation and the green line indicates those patients with both unmutated IGHV and IGKC
Finally, some analyzed non-coding regions located near immunoglobulin-related genes exhibited a remarkable mutation frequency. Mutations in the 3′ UTR of IGHV1-69 were independently associated with lower time to treatment (adjusted q-value < 0.1, 95% HR 1.09–4.33). Furthermore, hypermutation events occurred in a 1,543 base pair region located in the 5′ flank and UTR region of IGKC (172 patients, 40% of the total population, Supplementary Tables 22–23). These mutations were strongly associated with longer time to first treatment (p-value 7.23 × 10−11, HR 0.21–0.44; Fig. 2c) and were independent of IGHV status, sex, and clinical stage at diagnosis (p-value 6.3 × 10−3, q-value 3.7 × 10−2, HR 0.39–0.86; Fig. 2d). Similarly, an association with longer overall survival was detected (p-value 2.81 × 10−4), but not independently of other covariates (p-value 0.54). This region includes protein-coding sequences of some immunoglobulin genes (namely IGKJ1, IGKJ2, IGKJ3, IGKJ4, and IGJK5). Although these genes were mutated in 126 cases, most of them (96%) had concurrent mutations in the surrounding non-coding region.
For mutation validation, we matched whole genome sequencing data available in a subset of 88 samples which was used. We could validate 94.38, 100, and 97.75% mutations located in new putative exonic and intronic drivers, as well as in 5′UTR region of IGKC, respectively (Supplementary Table 24). Importantly, all non-confirmed mutations were subclonal.
Some of our results need further clarification in future approaches. Particularly, the frequency, functional and clinical implications of the new putative drivers needs to be replicated in independent cohorts. Nevertheless, the novelty and relevance of some of our results anticipate important implications in the biological comprehension and prognostic stratification of CLL.
Supplementary information
{kind=link}
{kind=link}
Acknowledgements
The authors gratefully thank CESGA (Supercomputing Center of Galicia) for providing the necessary resources for the development of this project, as well as the International Cancer Research Consortium and the European Bioinformatics Institute for supplying access to the data. We also thank Roche Pharmaceuticals for the financial support in order to pay the Article Processing Fee. The content of this paper is part of the doctoral thesis of Adrián Mosquera Orgueira to obtain a PhD in the Department of Medicine, University of Santiago de Compostela.
Authors’ contributions
A.M.O. conceived the study and performed the analysis. A.M.O., B.A.R. and J.A.D.A. analyzed the data. A.M.O., B.A.R., J.A.D.A. and J.L.B.L. wrote the paper.
Conflict of interest
The article processing fee of this paper has been partially funded by Roche Pharmaceuticals. Notwithstandingly, this company did not have any influence on the study design, data analysis, result interpretation, or article writing.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at (10.1038/s41408-019-0243-3).
References
- 1.Puente XS, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015;526:519–524. doi: 10.1038/nature14666. [DOI] [PubMed] [Google Scholar]
- 2.Landau DA, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526:525–530. doi: 10.1038/nature15395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sandmann S, et al. Evaluating variant calling tools for non-matched next-generation sequencing data. Sci. Rep. 2017;7:43169. doi: 10.1038/srep43169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hofmann AL, et al. Detailed simulation of cancer exome sequencing data reveals differences and common limitations of variant callers. BMC Bioinformatics. 2017;18:8. doi: 10.1186/s12859-016-1417-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cai L, Yuan W, Zhang Z, He L, Chou KC. In-depth comparison of somatic point mutation callers based on different tumor next-generation sequencing depth data. Sci. Rep. 2016;6:36540. doi: 10.1038/srep36540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramsay AJ, et al. Next-generation sequencing reveals the secrets of the chronic lymphocytic leukemia genome. Clin. Transl. Oncol. 2013;15:3–8. doi: 10.1007/s12094-012-0922-z. [DOI] [PubMed] [Google Scholar]
- 7.Oricchio E, et al. The Eph-receptor A7 is a soluble tumor suppressor for follicular lymphoma. Cell. 2011;147:554–564. doi: 10.1016/j.cell.2011.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ge Z, et al. Clinical significance of high c-MYC and low MYCBP2 expression and their association with Ikaros dysfunction in adult acute lymphoblastic leukemia. Oncotarget. 2015;6:42300–42311. doi: 10.18632/oncotarget.5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun PH, Ye L, Mason MD, Jiang WG. Protein tyrosine phosphatase µ (PTP µ or PTPRM), a negative regulator of proliferation and invasion of breast cancer cells, is associated with disease prognosis. PLoS One. 2012;7:e50183. doi: 10.1371/journal.pone.0050183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park HY, et al. Whole-exome and transcriptome sequencing of refractory diffuse large B-cell lymphoma. Oncotarget. 2016;7:86433–86445. doi: 10.18632/oncotarget.13239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meriranta L, et al. Deltex-1 mutations predict poor survival in diffuse large B-cell lymphoma. Haematologica. 2017;102:e195–e198. doi: 10.3324/haematol.2016.157495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kan Z, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–873. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- 13.Zhang X, Pino GM, Shephard F, Kiss-Toth E, Qwarnstrom EE. Distinct control of MyD88 adapter-dependent and Akt kinase-regulated responses by the interleukin (IL)-1RI co-receptor, TILRR. J. Biol. Chem. 2012;287:12348–12352. doi: 10.1074/jbc.C111.321711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodríguez D, et al. Functional analysis of sucrase-isomaltase mutations from chronic lymphocytic leukemia patients. Hum. Mol. Genet. 2013;22:2273–2282. doi: 10.1093/hmg/ddt078. [DOI] [PubMed] [Google Scholar]
- 15.Stamatopoulos, B. et al. The light chain IgLV3-21 defines a new poor prognostic subgroup in chronic lymphocytic leukemia: results of a multicenter study. Clin. Cancer Res. 10.1158/1078-0432.CCR-18-0133. (2018). [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.