

Abstract
Sustained reliance on androgen receptor (AR) after failure of AR-targeting androgen deprivation therapy (ADT) prevents effective treatment of castration-recurrent (CR) prostate cancer (CaP). Interfering with the molecular machinery by which AR drives CaP progression may be an alternative therapeutic strategy but its feasibility remains to be tested. Here, we explore targeting the mechanism by which AR, via RhoA, conveys androgen-responsiveness to Serum Response Factor (SRF), which controls aggressive CaP behavior and is maintained in CR-CaP. Following a siRNA screen and candidate gene approach, RNA-Seq studies confirmed that the RhoA effector Protein Kinase N1 (PKN1) transduces androgen-responsiveness to SRF. Androgen treatment induced SRF-PKN1 interaction, and PKN1 knockdown or overexpression severely impaired or stimulated, respectively, androgen regulation of SRF target genes. PKN1 overexpression occurred during clinical CR-CaP progression, and hastened CaP growth and shortened CR-CaP survival in orthotopic CaP xenografts. PKN1’s effects on SRF relied on its kinase domain. The multikinase inhibitor lestaurtinib inhibited PKN1 action and preferentially affected androgen regulation of SRF over direct AR target genes. In a CR-CaP patient-derived xenograft, expression of SRF target genes was maintained while AR target gene expression declined and proliferative gene expression increased. PKN1 inhibition decreased viability of CaP cells before and after ADT. In patient-derived CaP explants, lestaurtinib increased AR target gene expression but did not significantly alter SRF target gene or proliferative gene expression. These results provide proof-of-principle for selective forms of ADT that preferentially target different fractions of AR’s transcriptional output to inhibit CaP growth.
Introduction
Failure of androgen deprivation therapy (ADT) contributes directly to >29,000 American prostate cancer (CaP) deaths annually (1). ADT targets ligand-activation of the androgen receptor (AR). Castration-recurrent (CR-)CaP that recurs during ADT, however, continues to rely on AR because of adaptive CaP responses that facilitate, for instance, intracrine metabolism of precursor androgens to bioactive androgens, conversion of ADT drugs into (partial) AR agonists, or expression of AR versions that are ligand-independent or have broader ligand sensitivity (2–5).
Therefore, alternative ADT forms that inhibit AR activity by means other than interference with its ligand activation would be useful clinically. Targeting the transcriptional output by which AR ultimately drives lethal CaP progression may be a viable and novel therapeutic approach. Significant diversity exists in the manner in which AR interacts with and signals to hundreds of coregulators and transcription factors, that results in preferential control over subsets of target genes (6, 7). Theoretically, inhibiting the molecular mechanism(s) by which AR preferentially controls CaP progression could lead to an effective CaP treatment. Testing this hypothesis has been difficult because selecting a suitable mechanism of AR action requires that several criteria be met. An appropriate mechanism should mediate aggressive CaP behavior and impart survival benefit to CaP, demonstrate CaP-selectivity, and be maintained after conventional ADT fails. In addition, such AR action should be druggable; thus a therapeutic agent is needed that has limited toxicity and can be moved into clinical practice. In an ideal scenario, this drug would not affect other cellular functions mediated by AR, and its therapeutic efficacy would be evaluable via an appropriate biomarker of response.
We identified a mechanism of AR action that fulfills several of these criteria; the ability of AR to activate Serum Response Factor (SRF)-regulated genes (8, 9). In this mechanism, AR activates the transcription factor SRF that is bound constitutively to a CArG box, its genomic binding motif. This mechanism differs substantially from the traditional model of AR action in which androgen exposure leads to AR recruitment to Androgen Response Elements (AREs) in target genes (10, 11). SRF, a MADS box transcription factor, controls expression of genes involved in the immediate early response and regulation of the actin cytoskeleton, and thus is relevant to cell proliferation and migration (12). SRF-dependent androgen-responsive genes represent only 5.5% of AR-regulated genes in CaP cells, but unlike ARE-driven genes, they are enriched in CaP compared to benign prostate (9). This gene signature distinguishes benign from malignant prostate samples, and is associated with aggressive CaP behavior and recurrence (9). RhoA conveys androgen regulation to the majority of androgen-responsive SRF target genes, and RhoA’s control over SRF is maintained in CaP that has failed ADT and expresses aberrantly activated AR (13). The RhoA signaling axis contains druggable targets, and inhibitors are already in clinic [(14), clinicaltrials.gov)], which suggests that the AR-RhoA-SRF transcriptional mechanism may be amenable to therapeutic intervention.
Herein, we examine the molecular mechanism by which RhoA transfers androgen-dependence to SRF and isolate the Rho effector Protein Kinase N1 (PKN1) as a key mediator. Inhibition of PKN1 using the kinase inhibitor, lestaurtinib, which is used already in clinic (15), inhibited CaP cell growth while preferentially blocking SRF target gene over AR target gene expression. Our findings provide proof-of-principle for more selective inhibition of AR action that drives CaP progression.
Results
The RhoA effector PKN1 conveys androgen-responsiveness to SRF
Androgen treatment of CaP cells induces RhoA activation and increases cellular RhoA-GTP levels (13). Rho GTPase activation causes selective interaction with Rho effectors, which initiates downstream signaling. Class I RhoA effectors contains the kinase Protein Kinase N1 (PKN1) and the scaffold proteins rhophilin (RHPN1) and rhotekin (RTKN). Class II harbors the Rho-associated protein kinases ROCK1 and ROCK2, and citron and its splice variant citron kinase (CIT) belong to class III (16) (Figure 1A). The ability of these RhoA effectors to convey androgen signaling to SRF target genes was determined using an siRNA screen and a candidate gene approach. Transfection with siRNAs that individually target each RhoA effector, RhoA, or non-targeting siRNA, was combined with treatment with the synthetic androgen, R1881, or vehicle. RNA was isolated and processed for real-time RT-PCR analysis using primers targeting FHL2 and CCL8, two SRF target genes that rely on RhoA to maintain androgen-responsiveness (13). Expression of the androgen-responsive but RhoA-independent SRF target gene SSR3 (13) served as control. Silencing of RhoA effectors was verified using real-time RT-PCR (Supplemental Figure 1A). Loss of PKN1 decreased androgen regulation of FHL2 and CCL8 but not SSR3, without increasing the absolute expression levels of FHL2 or CCL8. Such increased androgen-stimulated expression did occur for FHL2 after knock-down of RHPN1 (average fold androgen induction increased from 18.5 to 23) or for CCL8 after loss of ROCK1 (average fold induction increased from 7.5 to 20), for instance. The impact of PKN1 silencing was most consistent with effect of RhoA knock-down, which slightly increased rather than decreased androgen regulation of SSR3 expression. These results suggested that PKN1 is an important mediator of androgen regulation of SRF target genes.
Figure 1. RhoA effector PKN1 mediates androgen-regulation of SRF target genes.
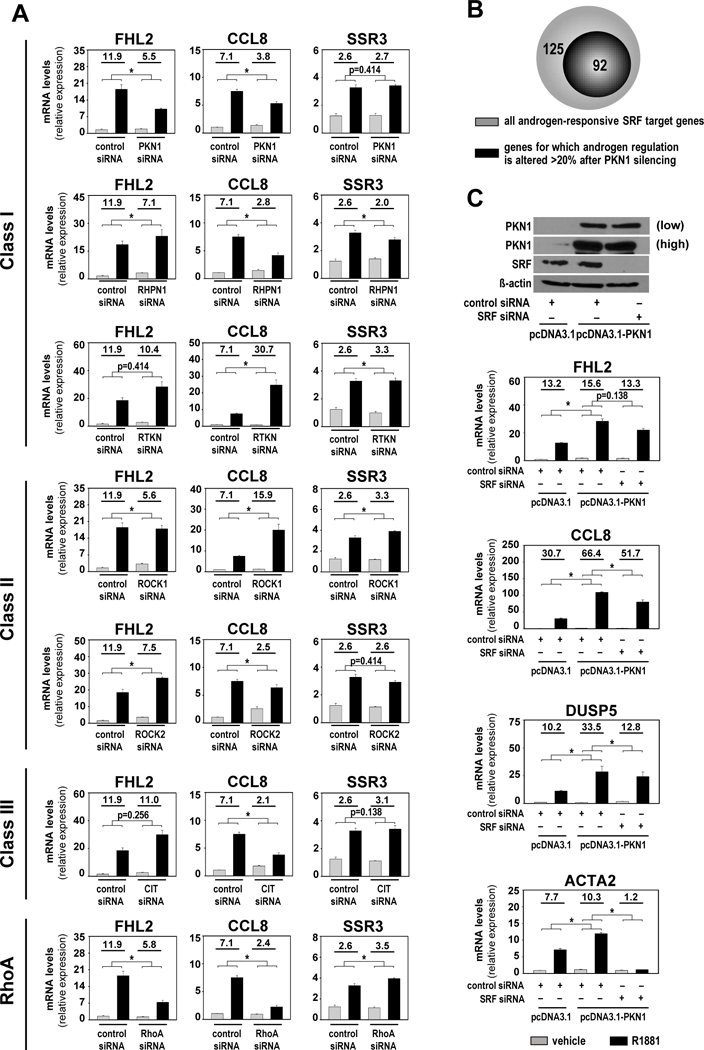
A. LNCaP cells were transfected with siRNA targeting RhoA or 6 RhoA effectors, or with non-specific control siRNA. At 42 hours after transfection, cells were treated with vehicle (ethanol) or 5nM of the synthetic androgen R1881. Cells were harvested 48 hours after treatment and androgen-induction of SRF target genes FHL2, CCL8 and SSR3 was evaluated via real-time RT-PCR. Target gene mRNA levels were normalized to GAPDH expression and are represented as relative expression using one of the values obtained from vehicle-treated control siRNA-transfected conditions as 1. Values obtained for specific siRNA transfections were evaluated against those from the same control siRNA group samples; all samples were from the same experiment. RhoA effectors Class I: PKN1, RHPN1, RTKN; Class II: ROCK1, ROCK2 and Class III: CIT. B. RNA-Seq was done on LNCaP cells after transfection with siRNAs targeting SRF or PKN1 or non-targeting siRNAs and treatment with 5nM R1881 or vehicle, using biological triplicates per treatment group as above. Genes that were ≥ 2-fold androgen-regulated (p adj <0.05) in control siRNA condition and for which androgen-responsiveness was decreased ≥1.5-fold upon loss of SRF were cross matched with gene sets that are bound directly by SRF. This approach isolated 217 androgen-responsive direct SRF target genes. Androgen regulation of 92 of these genes was affected >20% after PKN1 silencing. C. Bottom panel LNCaP cells were transfected with expression constructs encoding PKN1 (pcDNA3.1-PKN1) or empty vector (pcDNA3.1), in combination with control or SRF-targeting siRNA. At 42 hours after transfection, cells were treated with vehicle (ethanol) or 5nM R1881 and then harvested 48 hours after treatment. Androgen-induction of FHL2, CCL8, DUSP5 and ACTA2 was evaluated by real-time RT-PCR. Top panel Western blot analysis in a parallel experiment confirming the efficiency of PKN1 overexpression and siRNA-mediated knockdown of SRF. Blots were reprobed also for ß-actin as a loading control.
Columns, means of values obtained from independent biological triplicates; grey, vehicle treatment; black, R1881 treatment; bars, SEM values.*, p<0.05, all statistical analyses used Wilcoxon tests. Numeric values indicate the average fold change in androgen regulation. Low, low exposure, high, high exposure.
RNA-Seq studies were conducted to verify whether conclusions from candidate gene studies held true at a genome-wide level. LNCaP cells were transfected with siRNAs targeting SRF or PKN1 or non-targeting siRNAs and treated with R1881 or vehicle as above. RNA-Seq data were analyzed to identify genes that were ≥2-fold androgen-regulated (p adj < 0.05) in control siRNA condition and for which androgen-responsiveness was impacted ≥1.5-fold upon loss of SRF. The resulting gene list was crossmatched with gene sets that are bound directly by SRF (17–20) to identify direct SRF target genes. This approach isolated 217 androgen-responsive direct SRF target genes (Supplemental Table S1). RNA-Seq data obtained after PKN1 silencing showed that androgen regulation of 160, or 73.7%, of these genes was affected after PKN1 silencing, and for 92, or 42.4%, of SRF target genes the effect on fold androgen regulation was >20% (Figure 1B, Supplemental Table S1). Newly identified androgen-responsive PKN1-dependent SRF target genes included DUSP5 and ACTA2 (Supplemental Figure 1B and C) (12). Patterns of androgen regulation of representative target genes FHL2, CCL8, DUSP5 and ACTA2 was maintained in a second CaP cell line, VCaP, and when another PKN1 siRNA was used (Supplemental Figure 1D and E). SRF-dependence of PKN1 regulation was verified further by overexpressing PKN1 alone or in combination with SRF-targeting siRNA. For each SRF target gene studied (FHL2, CCL8, DUSP5 and ACTA2), overexpression of PKN1 significantly increased the level of androgen regulation and siRNA-mediated silencing of SRF markedly attenuated these stimulatory effects (Figure 1C). These data confirm an important role for PKN1 in androgen regulation of SRF target genes in CaP cells.
PKN1 overexpression during CaP progression provides growth advantage
Although PKN1 has been reported as overexpressed in localized treatment-naïve CaP compared to benign prostate (21), its expression status in clinical CR-CaP is unknown. To verify the evolution of PKN1 expression during CaP progression, immunohistochemistry for PKN1 was done on tissue microarrays constructed from benign prostate tissue (n=36), matching treatment-naïve localized CaP tissue (n=36), and CR-CaP (n=36) tissue. PKN1 expression tended to be higher in localized CaP compared to matching benign tissue, consistent with previous reports (21). PKN1 expression was significantly higher in CR-CaP than in benign or localized CaP (Figure 2A). Representative images of PKN1 expression in the three tissue types are shown in Figure 2B.
Figure 2. PKN1 overexpression provides CaP growth advantage.
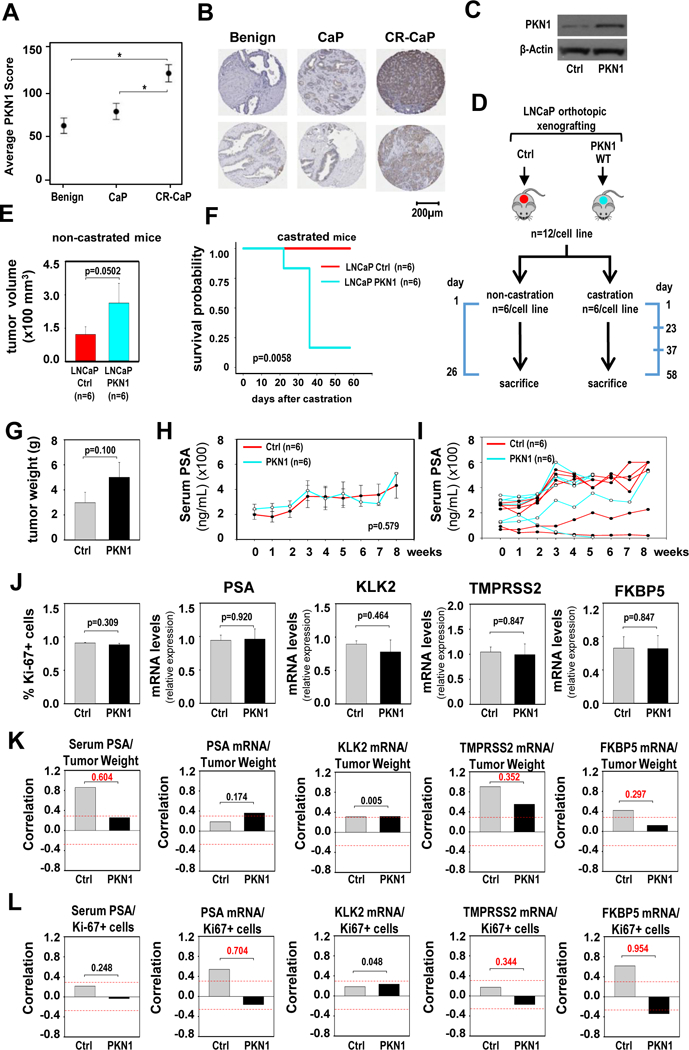
A. Average PKN1 immunostaining intensity score obtained from 36 benign, 36 CaP and 36 CR-CaP patient samples. Data are expressed as average intensity score +/− standard error. Statistical analysis was done using Tukey-Kramer tests. B. Two representative images of PKN1 immunostaining for each group of patient tissues. Matching benign prostate and localized CaP tissues from the same radical prostatectomy specimens are shown. Magnification: 40x. C. Western blot showing PKN1 expression levels in LNCaP cells that stably express pcDNA3.1 (Ctrl), or pcDNA3.1 vectors that encode (WT) wild-type PKN1. The blot was reprobed for ß-actin as a loading control. D. Schematic representation of the design and timeline for the xenograft experiment. Timeline marks key dates, and the scale is not proportional. Cells stably expressing pcDNA3.1 (Ctrl), or pcDNA3.1-PKN1-WT were orthotopically grafted in the anterior prostate of nude mice (n=12/cell line). After 2 months, mice were assigned into non-castration and castration groups (n=6/cell line). E. For each cell line, mice from the non-castration group were sacrificed 26 days later, and tumor volumes were measured. For the control group, values of 5 animals were recorded and shown. Columns, means of values obtained from each group; red, LNCaP-pcDNA3.1 (Ctrl); cyan, LNCaP-pcDNA3.1-PKN1-WT; bars, SEM values. Unit, mm3. Statistical analysis used Wilcoxon tests. F. Mice from the castration groups were scheduled to be sacrificed 2 months after castration. For some of these mice, this time point had to be moved up because their tumor burden exceeded that allowed by IACUC guidelines. A survival plot is shown for the animals from the castration group for each cell line. Red, LNCaP-pcDNA3.1 (Ctrl); cyan, LNCaP-pcDNA3.1-PKN1-WT. Statistical analyses were done using Mantel-Cox test (logrank test).
G. Tumor weights at time of harvest of castrated animals. Columns, means; bars, SEM values. Statistics used Wilcoxon tests. H. Kinetics of serum PSA levels in castrated animals, expressed in ng/ml. Weekly measurements are shown starting at time of castration (week 0). Columns, means; bars, SEM values. Statistics used ANOVA tests. I. Data from panel H, graphed for individual animals. J. Percentage of Ki67-positive cells (left panel), and mRNA expression levels for AR target genes PSA, KLK2, TMPRSS2 and FKBP5 (4 right panels) in PKN1-overexpressing tumors (PKN1) and control (Ctrl) tumors. mRNA expression for AR target genes was normalized to GAPDH expression and are represented as relative expression using one of the values from the control group as 1. Columns, means of values obtained from 6 independent xenografts; bars, SEM values. Statistical analyses was done using T-tests. K and L. Correlation between tumor weight (K) or percentage of Ki-67- positive tumor cells (L) and serum PSA levels and mRNA expression levels of PSA, KLK2, TMPRSS2 and FKBP5 at tumors harvested at sacrifice. Red dashes lines indicate Pearson correlation coefficient of 0.3. Numerical values indicate difference in correlation coefficient between PKN1-overexpressing and control group, in absolute values.
The impact of increased PKN1 expression on CaP progression was investigated in an orthotopic xenograft model system. LNCaP cells that stably express wild-type PKN1 and cells that express empty vector were grafted into the anterior prostate of nude mice. PKN1 expression levels in stable cell lines are shown in Figure 2C. Two months after grafting (at day 60), for each cell line CaP-bearing mice were assigned to either castration (n=6) or non-castration groups (n=6) based on serum PSA levels (Figure 2D). Non-castrated intact mice were sacrificed 86 days after grafting. Tumors with PKN1 overexpression had larger volumes than those expressing endogenous levels of PKN1 (Figure 2E), which indicated that PKN1 overexpression promotes growth under androgen-supplemented conditions. The 6 castrated animals of each cell line were followed for 2 months to explore whether this also occurs under ADT. Of the 6 animals with PKN1-overexpressing CaPs, 5 needed to be sacrificed before the expected time point because their tumor burden exceeded that allowable by animal welfare guidelines; sacrifice was required 23 days after castration for 1 animal and 37 days after castration for 4 other animals. None of the animals carrying tumors expressing endogenous PKN1 had to be sacrificed prior to the anticipated 2 month endpoint (Figure 2F), indicating significantly decreased survival under ADT when PKN1 is overexpressed. Tumor weights at sacrifice tended to be higher for PKN1-overexpressing tumors than control tumors also in the castrated group despite earlier harvests for 5 of these 6 animals (Figure 2G). Serum PSA levels, measured at weekly intervals for 8 weeks after castration, however were not different at any time point, and the kinetics of serum PSA levels were comparable between animals carrying tumors that overexpress PKN1 and those that express only endogenous PKN1 levels (Figure 2H, 2I). Percentage of tumor cells positive for the proliferation marker Ki67, and expression levels of 4 well-characterized direct AR target genes, PSA, KLK2, TMPRSS2 and FKBP5 (22–25), were also similar in tumors harvested at sacrifice for the 2 groups of animals (PKN1-overexpressing or control) (Figure 2J). These data suggested that impact of PKN1 overexpression on tumor progression does not depend on AR. We further calculated the correlation between tumor weight (Figure 2K), or the percentage of Ki67 positive tumor cells (Figure 2L), and the levels of the 5 markers of AR action (serum PSA and expression of 4 AR target genes). An absolute difference of 0.3 in correlation coefficient (CC) between tumors from the PKN1-overexpressing and the control group was considered relevant. In 7 of the 10 comparisons, there was a weaker correlation between tumor weight or percentage of Ki67-positive tumor cells and AR activity in PKN1-overexpressing tumors than in the control group, including 6 comparisons in which the CC difference was a magnitude of ≥ 0.3 (value 0.297 in Figure 2K considered as 0.3). On the other hand, for the other 3 comparisons where there was a stronger correlation between tumor weight or percentage of Ki67-positive tumor cells and AR activity in PKN1-overexpressing tumors than in control group, none of these had a CC difference of ≥0.2. Taken together, these data indicated that AR action is not a major determinant for CR-CaP progression by PKN1 overexpression.
PKN1’s control over androgen-responsive SRF target genes relies on its kinase domain
The relevance of the PKN1 kinase moiety for androgen-dependent SRF activity in CaP cells was examined because of its critical role in PKN1-mediated signal transduction. CaP cells that stably express PKN1 in which K is mutated to E at position 644 could not be established (data not shown). PKN1 K644E abrogates PKN1 kinase activity (26), indicating that CaP cells depend on PKN1 kinase action. These findings were consistent with more pronounced decreases in CaP cell viability with prolonged siRNA-mediated PKN1 silencing (Supplemental Figure 2A). Transient transfection of PKN1 K644E did not result in the overshoot in androgen-regulation of representative target genes encoding FHL2, CCL8, DUSP5 and ACTA2 that occurred with the construct encoding wild-type PKN1. Expression of the PKN1 K644E construct significantly decreased androgen regulation of these genes to levels below those observed in cells that express endogenous PKN1 only (Figure 3A), which demonstrated the importance of PKN1’s kinase function for androgen-regulation of SRF target genes. The possibility of kinase-dependent interaction between SRF and PKN was examined since PKN1 functions as coregulator for other transcription factors (27, 28). Co-IP studies in which IP was done using an antibody targeting SRF and immunoblot detected PKN1 indicated that SRF and PKN1 are part of the same immunocomplex in LNCaP and VCaP cells (Supplemental Figure 2B and C). This interaction was induced after stimulation with R1881 (Figure 3B), occurred also when IP was done for PKN1 and western blotting for SRF (Supplemental Figure 2D), and depended on an intact kinase domain since SRF-PKN1 immunocomplex formation did not occur when cells transiently overexpressed PKN1 K644E instead of wild-type PKN1 (Figure 3C).
Figure 3. PKN1 relies on its kinase activity for androgen regulation of SRF.
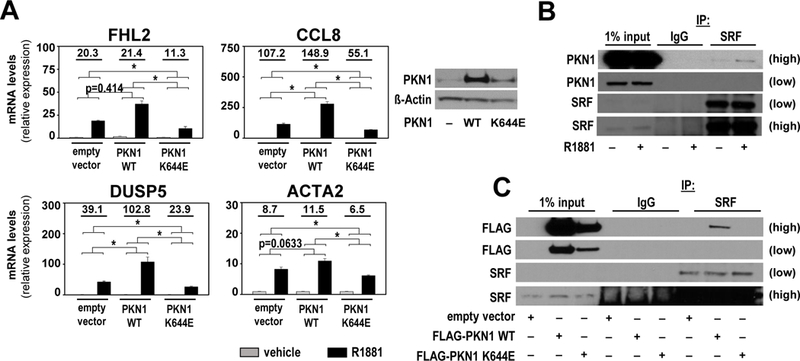
A. Left panel LNCaP cells were transfected with expression constructs encoding PKN1 WT, PKN1 K644E or empty vector. At 16 hours after transfection, cells were treated with vehicle (ethanol) or 5nM R1881 and then harvested 48 hours after treatment. Androgen-induction of FHL2, CCL8, DUSP5 and ACTA2 was evaluated by real-time RT-PCR. Target gene mRNA levels were normalized to GAPDH expression and are represented as relative expression values using one of the value obtained from vehicle-treated empty vector-transfected conditions as 1. Columns, means of values obtained from independent biological triplicates; grey, vehicle treatment; black, R1881 treatment; bars, SEM values.*, p<0.05, all statistical analyses used Wilcoxon tests. Numeric values indicate the average fold change in androgen regulation. Right panel Western blot analysis in a parallel experiment confirming overexpression of PKN1 forms. Blots were reprobed also for ß-actin as a loading control. B. Androgen-dependent interaction between SRF and PKN1. LNCaP cells were treated for 16 hours with 5nM R1881 or vehicle (ethanol). Co-immunoprecipitation assay was done in which immunoprecipation (IP) was done using non-targeting IgG antibody or an antibody targeting SRF. Western blotting was done using PKN1 antibody. Low, low exposure; high, high exposure. C. SRF-PKN1 interaction relies on PKN1’s kinase domain. LNCaP cells were transfected with expression constructs encoding FLAG-tagged PKN1 WT, FLAG-tagged PKN1 K644E or empty vector. At 72 hours after transfection, cells were harvested and co-immunoprecipitation performed as under B. Western blotting was done using FLAG antibody. Low, low exposure, high, high exposure.
Lestaurtinib inhibits androgen-regulated PKN1 function
Collectively, results shown in Figure 2 and 3 identify PKN1 an attractive target for CaP therapy. Literature was reviewed for a potential pharmacological inhibitor of PKN1 that could be moved forward clinically. The multikinase inhibitor lestaurtinib, aka CEP-701, inhibits PKN1 action, has been well-tolerated in phase I clinical trials for advanced carcinoma (29), and is studied for treatment of myelofibrosis and acute myeloid leukemia (15, 30). Lestaurtinib has been reported to blunt androgen-dependent gene expression in CaP cells at a dose of 5 μM (31). At that dose, we found that lestaurtinib killed all CaP cells (Supplemental Figure 2E). At 50nM, which is closer to its IC50 of 8.6±0.9 nM (32), lestaurtinib impeded androgen regulation of the representative SRF-driven genes FHL2, CCL8, DUSP5 and ACTA2 in LNCaP and VCaP cells (Figure 4A,Supplemental Figure 2G). However, androgen responsiveness of ACTA2 appeared diminished in the presence of DMSO in VCaP cells, which impedes assessment of effect of lestaurtinib on its androgen-regulation. In Co-IP studies, lestaurtinib also prevented SRF-PKN1 interactions (Figure 4B).
Figure 4. The multikinase inhibitor lestaurtinib inhibits PKN1 action.
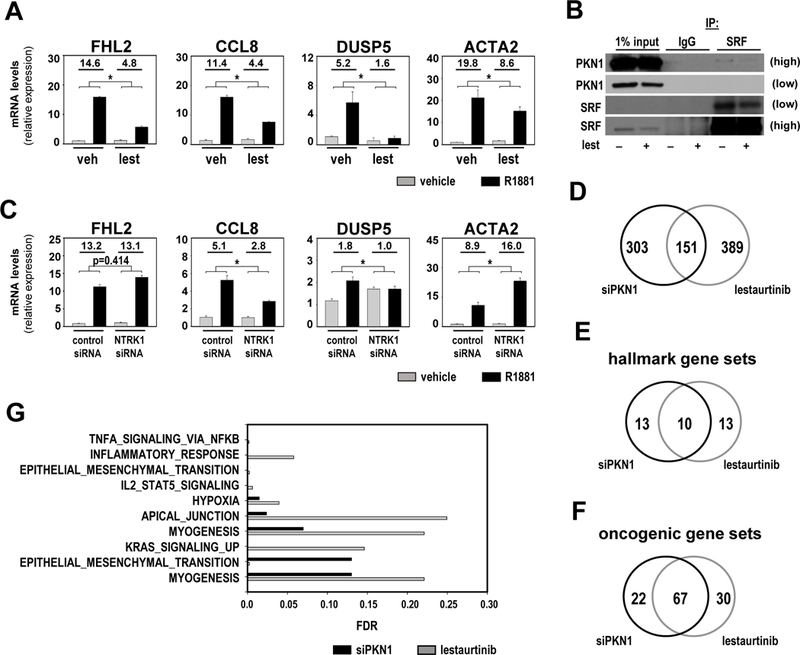
A. LNCaP cells were treated with either vehicle (veh, DMSO) or 50nM lestaurtinib (lest) in combination with vehicle (ethanol) or 5nM R1881. 48 hours later, cells were harvested and androgen-induction of FHL2, CCL8 DUSP5 and ACTA2 was evaluated by real-time RT-PCR and normalized to GAPDH expression. Target gene mRNA levels were normalized to GAPDH expression and are represented as relative expression values using one of the value obtained from vehicle-treated conditions as 1. Columns, means of values obtained from independent biological triplicates; grey, vehicle treatment; black, R1881 treatment; bars, SEM values.*, p<0.05, all statistical analyses used Wilcoxon tests. Numeric values in plots indicate the average fold change in androgen regulation. B. LNCaP cells were treated for 48 hours with vehicle (DMSO) or 50nM lestaurinib. Co-immunoprecipitation assay was done in which immunoprecipation (IP) was done using non-targeting IgG antibody or an antibody targeting SRF. Western blotting was done using PKN1 antibody. Low, low exposure; high, high exposure. C. LNCaP cells were transfected with siRNA targeting NTRK1 or nonspecific control siRNA. At 42 hours after transfection, cells were treated with vehicle (ethanol) or 5nM R1881. Cells were harvested 48 hours later. Real-time RT-PCR was done and normalized to GAPDH expression. Target gene mRNA levels were normalized to GAPDH expression and are represented as relative expression values using one of the value obtained from vehicle-treated control siRNA-transfected conditions as 1. D. LNCaP cells were transfected with siRNA targeting PKN1 (siPKN1) or non-specific control siRNA. 42 hours later, cells were treated with vehicle (ethanol) or 5nM R1881. After 48 hours of treatment, cells were harvested and RNA was isolated and processed for RNA-Seq analysis (siPKN1 experiment, left). LNCaP cells were treated with either vehicle (DMSO) or lestaurtinib (lest), in combination with vehicle (ethanol) or 5nM R1881. 48 hours later, cells were harvested and RNA was isolated and processed for RNA-Seq analysis (lestaurtinib experiment, right). Venn diagram shows the number of unique and common genes that fit the selection criteria between both experiments. E and F. Venn diagram showing unique and common MSigDB hallmark and oncogenic gene sets between the 2 experiments, respectively. G. Overview of hallmark gene sets that overlap between PKN1- and lestaurtinib-dependent gene sets at false discovery rate (FDR) < 0.25. Black, FDR for PKN1-dependent entry; grey, FDR for lestaurtinib-dependent entry.
Lestaurtinib had been demonstrated to also inhibit tyrosine kinases such as FLT3, JAK2, and Trks (30, 33, 34) before it was recognized as a potent inhibitor for PKN1. As initial verification of lestaurtinib’s selectivity for PKN1, its effect was compared to that of siRNA-mediated silencing of TrkA, which is known under the NCBI gene symbol NTRK1. Loss of NTRK1 decreased the level of androgen regulation of CCL8 and DUSP5, but increased that of ACTA2 and did not affect FHL2 (Figure 4C), which suggested that lestaurtinib more efficiently inhibits PKN1’s than NTRK1’s impact on expression of these genes. RNA-Seq profiling was performed on LNCaP cells that had been stimulated with R1881 combined with either PKN1 silencing or lestaurtinib (50nM) treatment to more fully determine lestaurtinib’s specificity to inhibit PKN1’s effects on CaP cell function. 454 and 540 genes that were ≥2-fold androgen-regulated (p adj <0.05) in the non-targeting siRNA or vehicle condition and for which androgen-responsiveness is altered ≥50% after PKN1 silencing or lestaurtinib inhibition, respectively, were isolated. 151 of these genes overlapped between the 2 experiments (Figure 4D). Closer examination of the common and unique genes show that several belong to a cluster of related genes that exert similar biological function, but did not all fulfill the cut-off criteria for change in androgen regulation to be retained. These observations suggested that the affected CaP biology may be more similar than the number of genes indicates. Molecular Signature Database (MSigDB) (35) analysis for hallmark gene sets that represent well-defined biological processes indicated considerable overlap (n=10) in gene sets affected after PKN1 silencing (n=23) or lestaurtinib treatment (n=23) (Figure 4E). More relevant to our studies, this overlap was larger for MSigDB oncogenic signature gene sets that reflect cellular pathways that are often dysregulated in cancer. 67 of the 89 gene sets isolated after PKN1 silencing overlapped with 108 gene sets derived after lestaurtinib treatment (Figure 4F).The specific pathways and processes that were enriched include several that relate to inflammatory signaling, EMT, and myogenesis in which PKN1 and SRF have been implicated in other cell and organ systems (e.g. (12)) (Figure 4G, Supplemental Figure 2H). This is consistent with an enrichment of PKN1-dependent SRF target genes in the lestautinib-dependent gene set above. Of 92 PKN1-dependent SRF genes (Figure 1B), 78 are detectable in the lestaurtinib-dependent RNA-Seq data, and for 38, or 50%, of these genes androgen regulation is changed >1.5 fold after lestaurtinib treatment (Supplemental Table 2). These results indicated that lestaurtinib has substantial specificity for PKN1 action in androgen-dependent CaP cell behavior.
Differential regulation of SRF and AR target gene expression
PKN1 has been reported as an AR-associated coregulator at ARE-driven genes such as PSA (36, 37), and secreted PSA was increased following lestaurtinib treatment (38). The impact of PKN1 silencing and lestaurtinib treatment on AR target genes was examined by verifying expression of 452 previously characterized bona fide direct AR target genes (39) in the RNA-Seq-derived gene sets described above. Only 42 (or 9.3%) and 48 (or 10.6%) of these 452 genes were present in the PKN1- or lestaurtinib-dependent gene set, respectively. These findings were consistent with our previous results using a custom AR target gene oligoarray, which showed that androgen regulation of only 12 of 452 (or 2.5%) AR target genes was altered significantly after PKN1 knock-down (39). Real-time RT-PCR confirmed no significant change in the fold androgen regulation of 2 representative AR target genes KLK2 and FKBP5 and significant increase in androgen regulation of 2 other AR target genes, PSA and TMPRSS2, after PKN1 knock-down in LNCaP cells. Lestaurtinib treatment significantly enhanced androgen response of these 4 genes (Figure 5A and B). However, lestaurtinib treatment of VCaP cells significantly increased androgen response of FKBP5 without affecting 3 other AR target genes, and PKN1 silencing did not impact androgen response for any of the AR target genes studied (Supplemental Figure 3A and B). These data suggested differences in PKN1-dependency of androgen-responsive SRF and AR target genes.
Figure 5. Differential regulation of SRF and AR target genes.
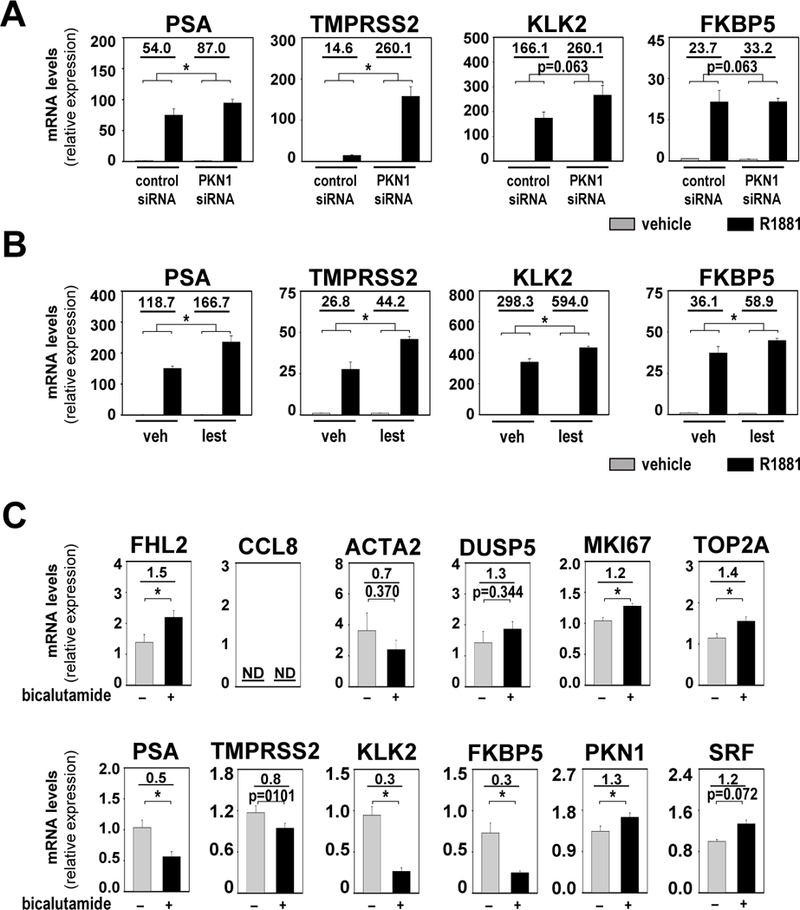
A and B. LNCaP cells were transfected with siRNA targeting PKN1 or non-specific control siRNA. At 42 hours after transfection, cells were treated with vehicle (ethanol) or 5nM of the synthetic androgen R1881 (A). LNCaP cells were treated with either vehicle (veh, DMSO) or 50nM lestaurtinib (lest) in combination with vehicle (ethanol) or 5nM R1881 (B). Cells were harvested 48 hours after treatment and androgen-induction of direct AR target genes PSA, KLK2, TMPRSS2 and FKBP5 was evaluated via real-time RT-PCR. Target gene mRNA levels were normalized to GAPDH expression and are represented as relative expression using one of the values obtained from vehicle-treated control siRNA-transfected conditions as 1. Columns, means of values obtained from independent biological triplicates; grey, vehicle treatment; black, R1881 treatment; bars, SEM values.*, p<0.05, all statistical analyses used Wilcoxon tests. Numeric values in plots indicate the average fold change in androgen regulation. Low, low exposure, high, high exposure. C. Analysis of RNA obtained from PCSD1 xenografts from mice that were treated with vehicle (0.9% benzyl alcohol, 1% Tween-80, 0.5% Methylcellulose) or 50mg/kg/day bicalutamide for 18 days. Effect of bicalutamide treatment on SRF target genes FHL2, CCL8, DUSP5 and ACTA2, AR target genes PSA, KLK2, TMPRSS2 and FKBP5, proliferation-associated genes MKI67 and TOP2A, and SRF and PKN1 was evaluated by real-time RT-PCR. Target gene mRNA levels were normalized to GAPDH expression and are represented as relative expression values using one of the value obtained from vehicle-treated control mice as 1. Columns, means of values obtained from 5 independent biological replicates; grey, vehicle treatment; black, bicalutamide treatment; bars, SEM values. *, p<0.05, all statistical analyses used T-tests tests. Numeric values in plots indicate the average fold change in mRNA expression. ND, not determined.
Differential regulation of SRF and AR target genes, even without PKN1 inhibition, is in line with earlier observations of enrichment of SRF target genes and gradual depletion of AR target genes during clinical CaP progression (9). Whether such dichotomy in regulation of SRF and AR target gene expression occurs in clinical CaP under ADT remains unknown. The patient-derived xenograft (PDX) PCSD1 (40), which models osteolytic/osteoblastic CR-CaP bone metastatis, was studied to address this therapeutically relevant question. Expression of representative SRF and AR target genes, and Ki67 and TOP2A, for which mRNA expression has been linked to CaP proliferation and aggressive progression (41, 42), was quantitated in PCSD1 tissues previously obtained after treatment with the anti-androgen bicalutamide (n=5) or vehicle (n=5). As reported before, bicalutamide treatment did not inhibit PCSD1 growth, as confirmed here by significantly increased TOP2A and Ki67 mRNA expression (Figure 5C) yet decreased PSA expression (40)(Figure 5C). Expression of KLK2 and FKBP5 was significantly decreased, while TMPRS22 expression tended to decrease. FHL2, DUSP5 and ACTA2 expression was not altered or slightly increased after bicalutamide treatment (Figure 5C). CCL8 expression was below the limit of detection. These data indicated discordance between SRF and AR target gene expression in growing CaP, and showed that SRF target gene expression is associated with CR-CaP growth.
Lestaurtinib decreases CaP cell viability
The effect of lestaurtinib on CaP cell viability was determined in several model systems that reflect different CaP stages. A dose of 50 nM, at which different effects on SRF and AR target gene expression have been observed, decreased the viability of LNCaP cells to an extent similar to that of siRNA-mediated PKN1 loss, which suggested that the drug may be suitable for CaP treatment (Figure 6A). This inhibitory effect of lestaurtinib was verified in VCaP cells (Figure 6B) and seen under androgen deprivation as well as supplementation (Supplemental Figure 2I). We investigated the effect of lestaurtinib in cells in which AR activation status mimics the AR status in CR-CaP because RhoA-SRF signaling occurs downstream of ligand-activated AR in ADT-naïve CaP and is maintained in CR-CaP cells. First, PKN1 inhibition using lestaurtinib decreased viability of CR LNCaP subline C4–2, as did knock-down of PKN1 (Figure 6C). Next, the effect of lestaurtinib on viability of LNCaP cells that were treated with 10 μM 5α-abiraterone was examined. Abiraterone is a CYP17A1 inhibitor used to treat CR-CaP patients. One of its metabolites, 5α-abiraterone, acts as a partial AR agonist that mediates CaP progression during abiraterone treatment (43). LNCaP cell viability that was induced by 5α-abiraterone was counteracted by lestaurtinib (Figure 6D). Treatment with lestaurtinib also decreased LAPC4 cell viability before and after development of resistance to the second-generation AR antagonist enzalutamide (Figure 6E), which causes compensatory upregulation of glucocorticoid receptor activity (44). Similar decreases in CaP cell viability were seen when CWR-R1 sublines engineered to express either constitutively active AR variant ARv567es or full-length AR (45) underwent lestaurtinib treatment (Figure 6F). These data confirmed the suitability of PKN1 as a therapeutic target and lestaurtinib as a drug to treat CaP even after failure of ADT.
Figure 6. Lestaurtinib decreases CaP cell viability.
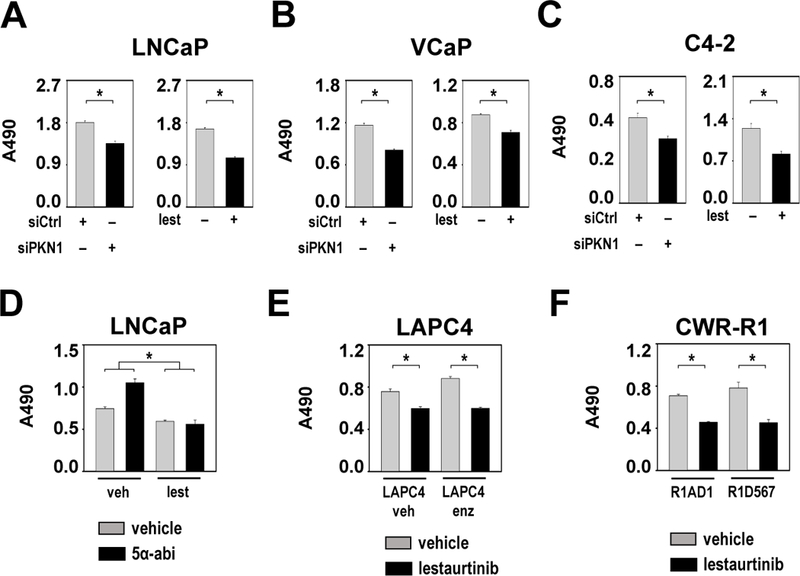
A. Left panel LNCaP cells were transfected with siRNA targeting PKN1 (siPKN1) or non-specific control siRNA (siCtrl). 96 hours later, cells were subjected to an MTS assay reading at 490 nm. Right panel LNCaP cells were treated with either vehicle (DMSO) or 50nM lestaurtinib (lest). 96 hours later, cells were subjected to an MTS assay read at 490 nm. B. VCaP cells were treated and transfected as in panel A. 96 hours later, cells were subjected to an MTS assay reading at 490 nm. Note the difference in Y axis scales between panels. C. C4–2 cells were treated and transfected as in panel A. 96 hours later, cells were subjected to an MTS assay reading at 490 nm. Note the difference in Y axis scales between panels. D. LNCaP cells were treated either with vehicle (veh, DMSO) or 50nM lestaurtinib (lest) in combination with 10μM 5α-abiraterone (5α-abi) or matching vehicle (ethanol). 6 days later, cells were subjected to an MTS assay reading at 490 nm. E. LAPC4 cells that are resistant to 1μM enzalutamide and matching vehicle (DMSO)-supplemented cell line (LAPC4 enz and LAPC4 veh) were treated with either vehicle (DMSO) or 50nM lestaurtinib. 96 hours later, cells were subjected to an MTS assay reading at 490 nm. F. CWR-R1 sublines R1AD1 and R1D567 cells were treated with either vehicle (DMSO) or 50nM lestaurtinib. 96 hours later, cells were subjected to an MTS assay reading at 490 nm. For all panels: Columns, means of values obtained from 4 independent biological replicates; bars, SEM values. *, p<0.05. statistical analyses were done using T-tests (A, B, C, E, F) or Wilcoxon tests (for fold-change, D).
In a final experiment, we assessed the effect of lestaurtinib on representative SRF and AR target gene expression and proliferation-associated genes in ex vivo explants of fresh CaP tissue obtained at radical prostatectomy (46). Tissue from patients who received neoadjuvant therapies was excluded. CaP cores from 10 patients, in which the presence of CaP was verified by an experienced genitourologic pathologist (CMG), were procured from radical prostatectomy specimens. Each core was divided in 2 parts. One half of was used to establish a CaP explant that was treated with 500 nM lestaurtinib. This concentration was used because it is about 2 times the concentration of lestaurtinib detected in CaP at radical prostatectomy in a neoadjuvant trial of lestaurtinib (38). The other half was used to establish an explant that was treated with vehicle (DMSO). Ten explants were established per treatment group. Tissues were collected 96 hours after treatment, and RNA was extracted and subjected to real-time RT-PCR. Expression of each of these genes was corrected for 5 individual housekeeping genes (GAPDH, 18SrRNA, TBP, HMBS, HPRT) that have been used for CaP patient specimen analysis (47, 48). Similar patterns of expression were obtained after correction for these different housekeeping genes. Results after correction for GAPDH expression are shown in Figures 7 and B; results corrected for HPRT are shown in Supplemental Figure 4. Expression of the AR target genes PSA, KLK2, TMPRSS2 and FKBP5 was induced significantly after lestaurtinib treatment. Expression of CCL8 significantly decreased after such treatment, and FHL2 and DUSP5 expression was unaffected. Reminiscent of observations in VCaP cells, expression of the SRF target gene ACTA2 was increased slightly under these conditions. Expression of proliferation-associated genes MKI67 and TOP2A did not change (Figure 7). These data indicate dichotomy in the effects of lestaurtinib on PSA and SRF target genes, and confirmed that expression patterns of SRF target genes associated with, that of while AR target genes opposed, the cytostatic effect of lestaurtinib on clinical CaP.
Figure 7. Differential regulation of SRF and AR target genes following lestaurtinib treatment of fresh clinical CaP tissue.
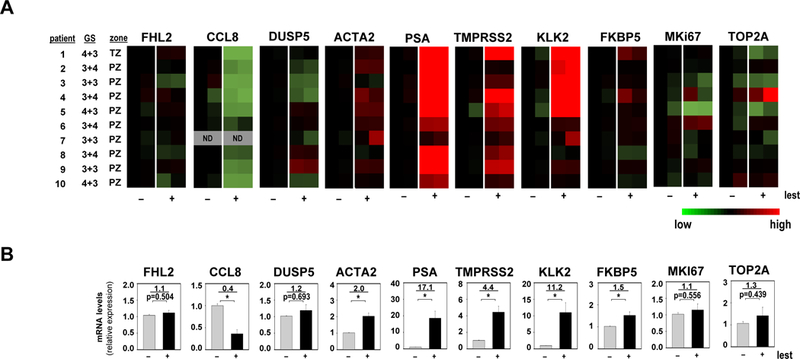
Ex vivo CaP explants were treated with either vehicle (DMSO) or 500nM lestaurtinib (lest). Tissues were harvested after 96 hours. Effect of lest on SRF and AR target genes and proliferation-associated genes was evaluated by real-time RT-PCR. Target gene mRNA levels were normalized to GAPDH expression and are represented as relative expression using one of the values obtained from vehicle-treated control conditions as 1. A. Heatmap representation of data obtained from 2 technical replicates per treatment group. GS, Gleason score; TZ, transition zone; PZ, peripheral zone; ND, not determined. B. Quantitation of data shown in panel A. Columns, means of values obtained from 2 technical replicates per explant; grey, vehicle treatment; black, lestaurtinib treatment; bars, SEM values. *, p<0.05. Statistical analyses were done using T-tests. Numeric values, average fold change in androgen regulation. Quantitation of CCL8 expression data was done using data from 9 patients for whom CCL8 mRNA expression was detectable.
Discussion
The persistence of active AR in the majority of CR-CaPs that no longer respond to ADT underscores the need for alternative strategies to interfere with AR action and improve patient survival (e.g. (49)). Our previous work delineated the AR-RhoA-SRF signaling axis as a signal transduction pathway downstream of activated AR that mediates aggressive CaP behavior (14). These findings and the relevance of SRF signaling in CaP have been validated by others (50–53). Here, we isolated the serine-threonine kinase, PKN1, as a drugable target and lestaurtinib, which is used already in other human malignancies (15), as a drug to preferentially silence SRF- and RhoA-dependent AR action in CaP.
PKN1 was identified as the mediator that signals between androgen-activated RhoA and SRF. Loss of this RhoA effector preferentially affected androgen regulation of SRF target genes with limited off-target effects on direct AR target genes. Moreover, PKN1 was relevant to clinical CaP progression, mediated aggressive CaP behavior, and pharmacological inhibition of PKN1 that inhibited CaP growth was achievable with a clinically tolerable inhibitor, lestaurtinib. Previous observation of decreased androgen regulation of a single SRF target gene, FHL2, after ROCK inhibitor treatment (13) was consistent with decreased fold androgen regulation of FHL2 mRNA expression after ROCK1 and ROCK2 silencing but did not extend to other SRF target mRNA gene expression.
Our identification of PKN1 as the functional link between androgen-activated RhoA and SRF was somewhat unexpected. PKN1 had previously been proposed as an AR-associated coactivator that controls androgen regulation of ARE-driven genes (36, 37). The latter conclusions for PKN1’s role in endogenous AR target gene expression in AR-positive CaP cells considered only 3 genes, PSA, KLK2, and TMPRSS2 (36, 37, 54). That PKN1 does not consistently and significantly alter androgen regulation of these genes in our studies but acts as a co-activator in previous reports may be related to cell culture conditions or data quantitation methods. For instance, increases in absolute PSA expression after PKN1 inhibition were confirmed here using various independent measurement methods (real-time RT-PCR, oligoarray, and RNA-Seq) in cell lines and CaP explants, and is consistent also with increased PSA secretion after lestaurtinib treatment that others have reported in multiple CaP cell lines and xenografts (38).
Our results indicate that PKN1 inhibition preferentially impacted androgen-dependent SRF action. Discordance between the molecular regulation of androgen-dependent expression of SRF and AR target genes was observed in CaP cell lines, and, more importantly, in PDXs and ex vivo CaP explants. Such dichotomy was reminiscent of our previous observations that SRF gene expression is maintained during clinical CaP progression, while direct AR target gene expression declined (9). A major novel finding was that (PKN1 inhibition of) SRF target gene expression is accompanied by (decreased) CaP growth. These results indicate that preferential targeting of SRF target gene expression may be feasible and tolerable clinically. Before lestaurtinib was recognized as a PKN1 inhibitor, it was demonstrated to inhibit growth of primary and metastatic CaP in both rodent and human models, without toxicity to the host or benign prostate tissue (38, 55, 56). In the Dunning H rat CaP model, lestaurtinib treatment increased median survival (56), and combined ADT and lestaurtinib led to durable regression of CaP without evidence of regrowth. ADT given before lestaurtinib sensitized CaP cells to lestaurtinib, which suggested clinical benefit of sequential therapy (55). To date, two phase II CR-CaP clinical trials of lestaurtinib have been performed in which patients were considered responders if they had > 50% decrease in serum PSA compared to baseline. No patients in either trial met these response criteria. Instead, serum PSA tended to increase during treatment and declined when treatment was stopped. The decision not to move lestaurtinib forward clinically for CaP treatment was made solely based on the failure to meet the inadequate endpoint of PSA response (38). Evaluating the therapeutic efficacy of PKN1-targeting therapies will require other endpoints. These could include determining clinical CR-CaP progression as outlined in Prostate Cancer Working Group 3 guidelines (57), monitoring PKN1- and lestaurtinib-dependent androgen-responsive gene signatures reported here in CR-CaP tumor biopsies, circulating tumor cells or cell-free RNA that are increasingly procured as part of routine disease monitoring (e.g.(58–60)), or examining expression or activity of representative target (genes) using other assays such as ELISA, which are commercially available already for, for instance, CCL8.
PKN1 did not impact on androgen regulation of all SRF target genes, which is consistent with our previous observations that RhoA controls the majority of but not all androgen-responsive SRF action (13). The stringent selection criteria used on biological triplicate samples may have prevented identification of all bona fide SRF target genes. For instance, CCL8 was previously identified as indirect SRF target gene (9), but FHL2 and SSR3 contain an SRF binding peak (8, 17–20) but SRF silencing did not cause the 1.5-fold change in their androgen response used as cut-off for inclusion in our studies (Supplemental Table 1). A more specific PKN1 inhibitor may identify the full spectrum of PKN1-dependent SRF target genes and improve on CaP treatment, but will likely be challenging in view of polypharmacology that impacts kinase inhibitors (61). Characterization of the newly identified PKN1-SRF interaction may facilitate such novel therapeutic options. Peptidomimetics targeting critical contact sites (62) could be pursued if interactions are direct. If signaling is indirect and involves kinase downstream of PKN1 (63), inhibition of the latter kinases and PKN1 could be explored alone or in combination.
In conclusion, these studies indicate for the first time that the diversity in molecular regulation of AR activity can be exploited for therapy. Such novel treatments are particularly relevant and needed in late-stage CR-CaP that is driven by activated AR yet resistant to conventional ADT that directly targets AR.
Materials and methods
Cell culture
LNCaP, C4–2 and VCaP cells were obtained and maintained as described (13). R1-AD1 and R1-D567 cells were obtained from the Dehm laboratory (45). LAPC4 cells that grow under enzalutamide selection and DMSO-treated control cells were received from the Sharifi laboratory (44). LNCaP sublines that stably express pcDNA3.1-PKN1 or empty vector were generated as before (13). Cell lines were cultured for ≤10 passages, Mycoplasma-tested using the LookOut® Mycoplasma PCR Detection Kit (Sigma-Aldrich, cat# MP0035), and authenticated via Short Tandem Repeat profiling by Genetica DNA Laboratories.
Reagents are described in Supplemental Information.
Cloning
The coding sequence for PKN1 was cloned into KpnI and NotI restriction enzyme sites of the pcDNA3.1+ vector (Life Technologies). A PKN1 mutation K644 were generated using a Quick Change II XL Site-Directed mutagenesis kit (Agilent). PKN1 coding sequence was cloned in frame in HindIII and KpnI sites of the pCMV-FLAG multiple cloning site (Sigma). Primers for cloning and site-directed mutagenesis are listed in Supplemental Information. Sequence integrity of constructs was verified via Sanger sequencing.
Transfections
siRNA and plasmid transfection of CaP cells has been described (8).
Western blotting
Cells were harvested in sample lysis buffer and analyzed as before (13).
Co-IP
Co-IP studies were performed as described (39).
Real-time RT-PCR
Real-time RT-PCR was done as before (9, 13). Novel primers are listed in Supplemental Information.
Immunohistochemistry
Detailed descriptions of patient population, PKN1 and Ki67 immunohistochemistry and quantitation are provided in Supplemental Information.
Cell viability assays
Cell viability assays were done as before (8).
Orthotopic CaP xenografts
A detailed description is provided in the Supplemental Information.
PCSD1 PDX
The PCSD1 PDX has been described before ((40), Supplemental Information).
RNA-Seq analysis
Descriptions of RNA isolation and RNA-Seq analyses are provided in Supplemental Information.
Statistical analyses
All experiments were performed at least twice, except for RNA-Seq, xenograft, and TMA studies. Cell lines studies analyzed in qRT-PCR, RNA-Seq, or cell viability assays, included 3, 3, and 4 biological replicates, respectively, per experiment. For comparison of fold change (e.g. in androgen regulation) wilcoxon test was used. For pair-wise comparisons that do not involve fold change, Student’s t-test was used. For survival analysis of orthotopic xenograft studies, logrank test was used. All statistical tests were 2-tailed unless otherwise specified. A p value less than 0.05 was considered significant.
Supplementary Material
Acknowledegements
The authors thank Dr. Cassandra Talerico for review of the manuscript and Heemers lab members for helpful discussions. RNA-Seq was supported by NCI grant P30CA016056 involving the use of Roswell Park Comprehensive Cancer Center’s Genomic Shared Resource.
Financial support: These studies were supported by DOD PCRP award W81XWH-16–1-0404 (HVH), NIH NCI grants CA166440 (HVH), CA174777 (SMD), CA077739 (JLM), CA232979 (SL) and CA016056 (JLM and SL).
Footnotes
Supplemental Information is available at Oncogene’s website.
Competing interests: The authors report no conflicts of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. [DOI] [PubMed] [Google Scholar]
- 2.Karantanos T, Evans CP, Tombal B, Thompson TC, Montironi R, Isaacs WB. Understanding the mechanisms of androgen deprivation resistance in prostate cancer at the molecular level. Eur Urol. 2015;67(3):470–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dai C, Heemers HV, Sharifi N. Androgen signalling in prostate cancer. Cold Spring HArb PErspect MEd. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luo J, Attard G, Balk SP, Bevan C, Burnstein K, Cato L, et al. Role of Androgen Receptor Variants in Prostate Cancer: Report from the 2017 Mission Androgen Receptor Variants Meeting. Eur Urol. 2018;73(5):715–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Viswanathan SR, Ha G, Hoff AM, Wala JA, Carrot-Zhang J, Whelan CW, et al. Structural Alterations Driving Castration-Resistant Prostate Cancer Revealed by Linked-Read Genome Sequencing. Cell. 2018;174(2):433–47 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mills IG. Maintaining and reprogramming genomic androgen receptor activity in prostate cancer. Nat Rev Cancer. 2014;14(3):187–98. [DOI] [PubMed] [Google Scholar]
- 7.Kumari S, Senapati D, Heemers H. Rationale for the development of alternative forms of androgen deprivation therapy. Endocr Relat Cancer. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heemers HV, Regan KM, Dehm SM, Tindall DJ. Androgen induction of the androgen receptor coactivator four and a half LIM domain protein-2: evidence for a role for serum response factor in prostate cancer. Cancer Res. 2007;67(21):10592–9. [DOI] [PubMed] [Google Scholar]
- 9.Heemers HV, Schmidt LJ, Sun Z, Regan KM, Anderson SK, Duncan K, et al. Identification of a clinically relevant androgen-dependent gene signature in prostate cancer. Cancer Res. 2011;71(5):1978–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heemers HV, Tindall DJ. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocr Rev. 2007;28(7):778–808. [DOI] [PubMed] [Google Scholar]
- 11.Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25(2):276–308. [DOI] [PubMed] [Google Scholar]
- 12.Sun Q, Chen G, Streb JW, Long X, Yang Y, Stoeckert CJ, Jr., et al. Defining the mammalian CArGome. Genome Res. 2006;16(2):197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmidt LJ, Duncan K, Yadav N, Regan KM, Verone AR, Lohse CM, et al. RhoA as a mediator of clinically relevant androgen action in prostate cancer cells. Mol Endocrinol. 2012;26(5):716–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heemers HV. Identification of a RhoA- and SRF-dependent mechanism of androgen action that is associated with prostate cancer progression. Curr Drug Targets. 2013;14(4):481–9. [DOI] [PubMed] [Google Scholar]
- 15.Shabbir M, Stuart R. Lestaurtinib, a multitargeted tyrosine kinase inhibitor: from bench to bedside. Expert opinion on investigational drugs. 2010;19(3):427–36. [DOI] [PubMed] [Google Scholar]
- 16.Treisman R, Alberts AS, Sahai E. Regulation of SRF activity by Rho family GTPases. Cold Spring Harb Symp Quant Biol. 1998;63:643–51. [DOI] [PubMed] [Google Scholar]
- 17.He A, Kong SW, Ma Q, Pu WT. Co-occupancy by multiple cardiac transcription factors identifies transcriptional enhancers active in heart. Proc Natl Acad Sci U S A. 2011;108(14):5632–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schlesinger J, Schueler M, Grunert M, Fischer JJ, Zhang Q, Krueger T, et al. The cardiac transcription network modulated by Gata4, Mef2a, Nkx2.5, Srf, histone modifications, and microRNAs. PLoS Genet. 2011;7(2):e1001313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Esnault C, Stewart A, Gualdrini F, East P, Horswell S, Matthews N, et al. Rho-actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts. Genes Dev. 2014;28(9):943–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gualdrini F, Esnault C, Horswell S, Stewart A, Matthews N, Treisman R. SRF Co-factors Control the Balance between Cell Proliferation and Contractility. Mol Cell. 2016;64(6):1048–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Metzger E, Muller JM, Ferrari S, Buettner R, Schule R. A novel inducible transactivation domain in the androgen receptor: implications for PRK in prostate cancer. EMBO J. 2003;22(2):270–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cleutjens KB, van Eekelen CC, van der Korput HA, Brinkmann AO, Trapman J. Two androgen response regions cooperate in steroid hormone regulated activity of the prostate-specific antigen promoter. J Biol Chem. 1996;271(11):6379–88. [DOI] [PubMed] [Google Scholar]
- 23.Mitchell SH, Murtha PE, Zhang S, Zhu W, Young CY. An androgen response element mediates LNCaP cell dependent androgen induction of the hK2 gene. Mol Cell Endocrinol. 2000;168(1–2):89–99. [DOI] [PubMed] [Google Scholar]
- 24.Wang Q, Li W, Liu XS, Carroll JS, Janne OA, Keeton EK, et al. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell. 2007;27(3):380–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Magee JA, Chang LW, Stormo GD, Milbrandt J. Direct, androgen receptor-mediated regulation of the FKBP5 gene via a distal enhancer element. Endocrinology. 2006;147(1):590–8. [DOI] [PubMed] [Google Scholar]
- 26.Taniguchi T, Kawamata T, Mukai H, Hasegawa H, Isagawa T, Yasuda M, et al. Phosphorylation of tau is regulated by PKN. J Biol Chem. 2001;276(13):10025–31. [DOI] [PubMed] [Google Scholar]
- 27.Takanaga H, Mukai H, Shibata H, Toshimori M, Ono Y. PKN interacts with a paraneoplastic cerebellar degeneration-associated antigen, which is a potential transcription factor. Exp Cell Res. 1998;241(2):363–72. [DOI] [PubMed] [Google Scholar]
- 28.Shibata H, Oda H, Mukai H, Oishi K, Misaki K, Ohkubo H, et al. Interaction of PKN with a neuron-specific basic helix-loop-helix transcription factor, NDRF/NeuroD2. Brain Res Mol Brain Res. 1999;74(1–2):126–34. [DOI] [PubMed] [Google Scholar]
- 29.Marshall JL, Kindler H, Deeken J, Bhargava P, Vogelzang NJ, Rizvi N, et al. Phase I trial of orally administered CEP-701, a novel neurotrophin receptor-linked tyrosine kinase inhibitor. Invest New Drugs. 2005;23(1):31–7. [DOI] [PubMed] [Google Scholar]
- 30.Knapper S, Burnett AK, Littlewood T, Kell WJ, Agrawal S, Chopra R, et al. A phase 2 trial of the FLT3 inhibitor lestaurtinib (CEP701) as first-line treatment for older patients with acute myeloid leukemia not considered fit for intensive chemotherapy. Blood. 2006;108(10):3262–70. [DOI] [PubMed] [Google Scholar]
- 31.Jilg CA, Ketscher A, Metzger E, Hummel B, Willmann D, Russeler V, et al. PRK1/PKN1 controls migration and metastasis of androgen-independent prostate cancer cells. Oncotarget. 2014;5(24):12646–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kohler J, Erlenkamp G, Eberlin A, Rumpf T, Slynko I, Metzger E, et al. Lestaurtinib inhibits histone phosphorylation and androgen-dependent gene expression in prostate cancer cells. PLoS One. 2012;7(4):e34973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hexner EO, Serdikoff C, Jan M, Swider CR, Robinson C, Yang S, et al. Lestaurtinib (CEP701) is a JAK2 inhibitor that suppresses JAK2/STAT5 signaling and the proliferation of primary erythroid cells from patients with myeloproliferative disorders. Blood. 2008;111(12):5663–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iyer R, Evans AE, Qi X, Ho R, Minturn JE, Zhao H, et al. Lestaurtinib enhances the antitumor efficacy of chemotherapy in murine xenograft models of neuroblastoma. Clin Cancer Res. 2010;16(5):1478–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27(12):1739–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim JY, Banerjee T, Vinckevicius A, Luo Q, Parker JB, Baker MR, et al. A role for WDR5 in integrating threonine 11 phosphorylation to lysine 4 methylation on histone H3 during androgen signaling and in prostate cancer. Mol Cell. 2014;54(4):613–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Metzger E, Yin N, Wissmann M, Kunowska N, Fischer K, Friedrichs N, et al. Phosphorylation of histone H3 at threonine 11 establishes a novel chromatin mark for transcriptional regulation. Nat Cell Biol. 2008;10(1):53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Collins C, Carducci MA, Eisenberger MA, Isaacs JT, Partin AW, Pili R, et al. Preclinical and clinical studies with the multi-kinase inhibitor CEP-701 as treatment for prostate cancer demonstrate the inadequacy of PSA response as a primary endpoint. Cancer Biol Ther. 2007;6(9):1360–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu S, Kumari S, Hu Q, Senapati D, Venkadakrishnan VB, Wang D, et al. A comprehensive analysis of coregulator recruitment, androgen receptor function and gene expression in prostate cancer. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Godebu E, Muldong M, Strasner A, Wu CN, Park SC, Woo JR, et al. PCSD1, a new patient-derived model of bone metastatic prostate cancer, is castrate-resistant in the bone-niche. J Transl Med. 2014;12:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Labbe DP, Sweeney CJ, Brown M, Galbo P, Rosario S, Wadosky KM, et al. TOP2A and EZH2 Provide Early Detection of an Aggressive Prostate Cancer Subgroup. Clin Cancer Res. 2017;23(22):7072–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamoah K, Johnson MH, Choeurng V, Faisal FA, Yousefi K, Haddad Z, et al. Novel Biomarker Signature That May Predict Aggressive Disease in African American Men With Prostate Cancer. J Clin Oncol. 2015;33(25):2789–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Z, Alyamani M, Li J, Rogacki K, Abazeed M, Upadhyay SK, et al. Redirecting abiraterone metabolism to fine-tune prostate cancer anti-androgen therapy. Nature. 2016;533(7604):547–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li J, Alyamani M, Zhang A, Chang KH, Berk M, Li Z, et al. Aberrant corticosteroid metabolism in tumor cells enables GR takeover in enzalutamide resistant prostate cancer. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nyquist MD, Li Y, Hwang TH, Manlove LS, Vessella RL, Silverstein KA, et al. TALEN-engineered AR gene rearrangements reveal endocrine uncoupling of androgen receptor in prostate cancer. Proc Natl Acad Sci U S A. 2013;110(43):17492–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Centenera MM, Raj GV, Knudsen KE, Tilley WD, Butler LM. Ex vivo culture of human prostate tissue and drug development. Nat Rev Urol. 2013;10(8):483–7. [DOI] [PubMed] [Google Scholar]
- 47.Nakagawa T, Kollmeyer TM, Morlan BW, Anderson SK, Bergstralh EJ, Davis BJ, et al. A tissue biomarker panel predicting systemic progression after PSA recurrence post-definitive prostate cancer therapy. PLoS One. 2008;3(5):e2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Itkonen HM, Brown M, Urbanucci A, Tredwell G, Ho Lau C, Barfeld S, et al. Lipid degradation promotes prostate cancer cell survival. Oncotarget. 2017;8(24):38264–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Attard G, Borre M, Gurney H, Loriot Y, Andresen-Daniil C, Kalleda R, et al. Abiraterone Alone or in Combination With Enzalutamide in Metastatic Castration-Resistant Prostate Cancer With Rising Prostate-Specific Antigen During Enzalutamide Treatment. J Clin Oncol. 2018:JCO2018779827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O’Hurley G, Prencipe M, Lundon D, O’Neill A, Boyce S, O’Grady A, et al. The analysis of serum response factor expression in bone and soft tissue prostate cancer metastases. Prostate. 2013. [DOI] [PubMed] [Google Scholar]
- 51.Prencipe M, Madden SF, O’Neill A, O’Hurley G, Culhane A, O’Connor D, et al. Identification of transcription factors associated with castration-resistance: Is the serum responsive factor a potential therapeutic target? Prostate. 2013. [DOI] [PubMed] [Google Scholar]
- 52.Prencipe M, O’Neill A, O’Hurley G, Nguyen LK, Fabre A, Bjartell A, et al. Relationship between serum response factor and androgen receptor in prostate cancer. Prostate. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu W, Feng S, Dakhova O, Creighton CJ, Cai Y, Wang J, et al. FGFR-4 Arg(3)(8)(8) enhances prostate cancer progression via extracellular signal-related kinase and serum response factor signaling. Clin Cancer Res. 2011;17(13):4355–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O’Sullivan AG, Mulvaney EP, Kinsella BT. Regulation of protein kinase C-related kinase (PRK) signalling by the TPalpha and TPbeta isoforms of the human thromboxane A2 receptor: Implications for thromboxane- and androgen- dependent neoplastic and epigenetic responses in prostate cancer. Biochim Biophys Acta. 2017;1863(4):838–56. [DOI] [PubMed] [Google Scholar]
- 55.George DJ, Dionne CA, Jani J, Angeles T, Murakata C, Lamb J, et al. Sustained in vivo regression of Dunning H rat prostate cancers treated with combinations of androgen ablation and Trk tyrosine kinase inhibitors, CEP-751 (KT-6587) or CEP-701 (KT-5555). Cancer Res. 1999;59(10):2395–401. [PubMed] [Google Scholar]
- 56.Weeraratna AT, Dalrymple SL, Lamb JC, Denmeade SR, Miknyoczki S, Dionne CA, et al. Pan-trk inhibition decreases metastasis and enhances host survival in experimental models as a result of its selective induction of apoptosis of prostate cancer cells. Clin Cancer Res. 2001;7(8):2237–45. [PubMed] [Google Scholar]
- 57.Scher HI, Morris MJ, Stadler WM, Higano C, Basch E, Fizazi K, et al. Trial Design and Objectives for Castration-Resistant Prostate Cancer: Updated Recommendations From the Prostate Cancer Clinical Trials Working Group 3. J Clin Oncol. 2016;34(12):1402–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Olmos D, Brewer D, Clark J, Danila DC, Parker C, Attard G, et al. Prognostic value of blood mRNA expression signatures in castration-resistant prostate cancer: a prospective, two-stage study. Lancet Oncol. 2012;13(11):1114–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miyamoto DT, Lee RJ, Kalinich M, LiCausi JA, Zheng Y, Chen T, et al. An RNA-Based Digital Circulating Tumor Cell Signature Is Predictive of Drug Response and Early Dissemination in Prostate Cancer. Cancer discovery. 2018;8(3):288–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miyamoto DT, Zheng Y, Wittner BS, Lee RJ, Zhu H, Broderick KT, et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science. 2015;349(6254):1351–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Klaeger S, Heinzlmeir S, Wilhelm M, Polzer H, Vick B, Koenig PA, et al. The target landscape of clinical kinase drugs. Science. 2017;358(6367). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ravindranathan P, Lee TK, Yang L, Centenera MM, Butler L, Tilley WD, et al. Peptidomimetic targeting of critical androgen receptor-coregulator interactions in prostate cancer. Nat Commun. 2013;4:1923. [DOI] [PubMed] [Google Scholar]
- 63.Kajimoto K, Shao D, Takagi H, Maceri G, Zablocki D, Mukai H, et al. Hypotonic swelling-induced activation of PKN1 mediates cell survival in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2011;300(1):H191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.