

Abstract
The extinct marsupial Tasmanian tiger, or thylacine, and the eutherian gray wolf are among the most widely recognized examples of convergent evolution in mammals. Despite being distantly related, these large predators independently evolved extremely similar craniofacial morphologies, and evidence suggests that they filled similar ecological niches. Previous analyses revealed little evidence of adaptive convergence between their protein-coding genes. Thus, the genetic basis of their convergence is still unclear. Here, we identified candidate craniofacial cis-regulatory elements across vertebrates and compared their evolutionary rates in the thylacine and wolf, revealing abundant signatures of convergent positive selection. Craniofacial thylacine–wolf accelerated regions were enriched near genes involved in TGF beta (TGFB) and BMP signaling, both of which are key morphological signaling pathways with critical roles in establishing the identities and boundaries between craniofacial tissues. Similarly, enhancers of genes involved in craniofacial nerve development showed convergent selection and involvement in these pathways. Taken together, these results suggest that adaptation in cis-regulators of TGF beta and BMP signaling may provide a mechanism to explain the coevolution of developmentally and functionally integrated craniofacial structures in these species. We also found that despite major structural differences in marsupial and eutherian brains, accelerated regions in both species were common near genes with roles in brain development. Our findings support the hypothesis that, relative to protein-coding genes, positive selection on cis-regulatory elements is likely to be an essential driver of adaptive convergent evolution and may underpin thylacine–wolf phenotypic similarities.
The phenotypic resemblance between the extinct marsupial Tasmanian tiger, or thylacine (Thylacinus cynocephalus), and eutherian gray wolf (Canis lupus) is a widely recognized example of convergent evolution in mammals. Despite their large divergence time (∼160 Myr) (Bininda-Emonds et al. 2007), these species independently derived highly similar morphologies (Fig. 1A,B). Their skulls in particular were nearly identical in shape (Fig. 1C,D), and this similarity is strongly associated with their carnivorous feeding ecologies (Wroe and Milne 2007; Feigin et al. 2018). The thylacine occupied a similar niche in Australia to that filled by canids elsewhere in the world, that of an apex predator specialized in killing vertebrate prey (Jones and Stoddart 1998). Indeed, direct competition with dingoes may have contributed to their extinction on the mainland (Letnic et al. 2012). Previous morphometric analyses showed that much of the cranial morphospace between the thylacine and wolf was closed by convergent evolution (Feigin et al. 2018). However, the genomic changes underlying this convergence are still unclear. Recent comparative analyses of thylacine and canid protein-coding genes did not support adaptive amino acid homoplasy as a major contributor to their craniofacial similarities (Feigin et al. 2018). Therefore, it is necessary to interrogate other regions of the thylacine and wolf genomes to identify the molecular basis of their remarkable convergence.
Figure 1.

Comparison of thylacine (A) and gray wolf (B) gross morphology. Lateral view of thylacine (C) and gray wolf (D) skulls. (Image A courtesy of Kathryn Medlock of the Tasmanian Museum and Art Gallery; image B courtesy of photographer MacNeil Lyons.)
Cis-regulatory elements (CREs) such as promoters and enhancers are regions of noncoding DNA containing transcription factor binding sites (Latchman 1997). The combinatorial effect of interactions between multiple CREs with target gene promoters directs spatiotemporal patterns of gene expression (Spitz and Furlong 2012; Moorthy et al. 2017). Because many genes have pleiotropic functions, coding mutations that provide a fitness advantage in one trait can cause deleterious collateral effects in others (Stern and Orgogozo 2008). Thus, pleiotropy is thought to impose constraints on protein-coding genes that may limit their contribution to phenotypic adaptation. In contrast, many CREs have tissue- and stage-specific functions, reducing pleiotropy compared with coding genes. For these reasons, they have been proposed as an important source of adaptively relevant genetic variation (Carroll 2005).
Analysis of evolutionary rates has emerged as a powerful approach to study patterns of selection on CREs. It has been used to identify positive selection on forelimb-specific enhancers in bats (Booker et al. 2016) and across numerous traits in recent radiations of anole lizards (Tollis et al. 2018). This approach has also proven valuable for studying convergent evolution, revealing selection on enhancers of ocular genes in independent groups of subterranean mammals (Partha et al. 2017). Here, we performed comparative genomic analyses to investigate patterns of natural selection in the thylacine and wolf genomes in order to assess the potential for cis-regulatory evolution to underlie their adaptive morphological similarities.
Results
Identification of putative craniofacial CREs
Although the thylacine's axial skeleton was superficially similar the wolf, morphological convergence between these species has only been shown quantitatively in their skulls (Wroe and Milne 2007; Feigin et al. 2018). Therefore, we focused our study on CREs active during craniofacial development. We analyzed whole-genome alignments of 60 vertebrate species against the mouse genome (mm10) (Supplemental Table S1) and identified vertebrate conserved, noncoding regions overlapping epigenetic marks of cis-regulatory activity in mouse embryonic craniofacial tissues (Supplemental Table S2; Siepel et al. 2005; The ENCODE Project Consortium 2012). This yielded 57,227 regions with high potential for cis-regulatory activity in the face, which were termed putative craniofacial CREs (pcfCREs; Supplemental Table S3; Supplemental Figs. S1, S2). We next intersected the chromosomal coordinates of pcfCREs with those of 1135 experimentally validated human and mouse enhancers that show activity in the head (Supplemental Table S4; Visel et al. 2007). Despite the small fraction of the genome captured in these data sets, more than half (∼55%) of VISTA enhancers with cranial expression patterns overlapped with at least one pcfCRE (Supplemental Table S3), indicating that our approach for predicting craniofacial CREs is highly effective.
To investigate the potential contributions of cis-regulatory evolution to thylacine and wolf craniofacial adaptation, we tested thylacine and wolf craniofacial-active elements for signatures of positive selection. Analysis of evolutionary rates identified 10,910 thylacine accelerated regions (TARs) and 1923 wolf accelerated regions (WARs; FDR = 0.1) (Supplemental Tables S5, S6; Pollard et al. 2010). Three hundred thirty-nine of these elements showed accelerated evolution in both species, which we termed thylacine–wolf accelerated regions (TWARs) (Supplemental Table S7).
Patterns of GC-biased gene conversion differ between the thylacine and wolf
GC-biased gene conversion (gBGC) can confound evolutionary rates tests for positive selection by increasing the fixation rate of GC-rich alleles via biochemical bias during recombination. As much as 19% of human accelerated regions may be accounted for by gBGC (Kostka et al. 2012). Therefore, to exclude nonadaptive accelerated elements driven by gBGC, we performed two further likelihood ratio tests on TARs and WARs: one comparing a model with a parameter representing gBGC against the neutral model, and another comparing a model with parameters for both gBGC and selection (gBGC + Sel) against the model of gBGC alone (FDR = 0.1; for categorization rules, see Methods) (Hubisz et al. 2011; Kostka et al. 2012): 1018 WARs and 10,902 TARs, respectively (including 173 TWARs) (Supplemental Table S7), were retained as evolving adaptively (i.e., driven by selection alone or by selection and gBGC together).
Although accelerated evolution driven by gBGC was prevalent in the wolf, it was exceedingly rare in the thylacine (Supplemental Tables S5, S6). This finding is consistent with previously identified patterns of genome evolution in marsupial and canid genomes. The dog, a subspecies of gray wolf, has a similar overall GC content to humans (∼41%) (Lindblad-Toh et al. 2005). Unlike humans however, up to 40% of recombination hotspots in the dog genome contain significant peaks in GC content. This distribution of GC peaks is thought to be driven in part by a canid-specific loss of the recombination-directing transcription factor PRDM9, the loss of which corresponds to a long-term trend of strong and self-reinforcing gBGC in the dog lineage (Axelsson et al. 2012). In contrast, marsupials typically have far lower genomic GC content than eutherians (Johnson et al. 2018; Kasai et al. 2018). Indeed, the thylacine and its close living relative the Tasmanian devil have among the lowest GC contents observed in mammals (∼36%) (Murchison et al. 2012; Feigin et al. 2018). It has been suggested that the low GC content of marsupial genomes reduces their rate of gBGC (Rofe and Hayman 1985; Kasai et al. 2018).
Among elements showing signatures of gBGC, WARs were dominated by elements categorized as evolving under gBGC alone, whereas TARs tended to show signatures of both selection and gBGC (Supplemental Tables S5, S6). This difference may reflect difficulties in detecting weak selection pressure in the presence of extreme gBGC on the canid branch. Thus, although we have conservatively excluded elements explained solely by gBGC in our tests, it is possible that this underestimates the number of adaptively evolving WARs.
Regulatory evolution of TGF beta/BMP signaling may drive thylacine–wolf craniofacial convergence
Morphological convergence between thylacine and wolf skulls is the product of shape changes in homologous bones, and the strongest similarities in both form and function occur in those derived from the embryonic frontonasal and maxillary processes. Furthermore, the skull shapes of both species are adaptations to a carnivorous feeding ecology (Wroe et al. 2007; Feigin et al. 2018). Thus, we hypothesized that their craniofacial convergence should be reflected by selection pressure on many of the same loci and pathways. We therefore used the Genomic Regions Enrichment of Annotations Tool (GREAT) (McLean et al. 2010) to functionally annotate adaptively convergent, craniofacial TWARs based on protein-coding genes in close proximity (significance of enrichment tests are reported in Supplemental Tables). Two background sets were used in GREAT analyses. The first, corresponding to the whole genome, was used to ensure sufficient sensitivity to detect small enrichments of relevant ontology terms. The second, composed of pcfCREs with representative thylacine and wolf sequences, was used to ensure that top enriched terms identified against the whole-genome set did not solely reflect biases of the parent pcfCRE region set. This was performed because TWARs are a subset of pcfCREs, which are inherently skewed toward craniofacial functions.
Although few terms passed multiple testing correction (FDR = 0.1) against the pcfCRE background, we found that the same pathways identified using the whole-genome background were also among the top enriched terms based on raw P-value (P < 0.05) and fold enrichment (Supplemental Table S8). This indicates that although TWARs do reflect some innate craniofacial bias of the pcfCRE data set, the same molecular signaling pathways that distinguish TWARs from the genomic background are also the most divergent compared with pcfCREs in general. Thus, signaling pathways and related phenotypes identified by comparing TWARs against the whole-genome background are high-priority candidates to explain thylacine–wolf craniofacial convergence.
Genes associated with craniofacial TWARs were significantly enriched for actin binding and transcription factor activity (Supplemental Table S9). Many of these genes are involved in mouse phenotypes of the snout, jaw, and teeth (Blake et al. 2017). TWAR-associated genes were significantly enriched for involvement in transforming growth factor beta (TGF beta) and bone morphogenic protein (BMP) signaling and for genes whose expression is influenced by these pathways (Fig. 2A,B; Supplemental Tables S9–S11). Signaling through members of the TGF beta superfamily, which includes BMP, GDF, activin, and inhibin ligands, plays a central role in the differentiation and migration of cranial neural crest cells. These cells populate the frontonasal and maxillary processes (Dudas and Kaartinen 2005). TGF beta and BMP signaling also regulates the differentiation of bone, cartilage, tendons/ligaments, and skeletal muscle in the craniofacial region. These pathways help to establish connections and boundaries between these tissues (Baffi et al. 2006; Pryce et al. 2009; Elkasrawy and Hamrick 2010; Sartori and Sandri 2015; Wu et al. 2016). Thus, convergent changes in the regulation of TGF beta/BMP pathway genes are a plausible mechanism to facilitate the coevolution of developmentally and functionally integrated craniofacial structures. Consistent with this, we noted that TWAR-associated genes represent several components of these pathways, including receptors, ligands, modifiers of ligand availability, and mediators of downstream signaling (Fig. 2A,B; Supplemental Table S7).
Figure 2.

Genes involved in TGF beta signaling are enriched among TWAR-associated genes. (A) STRING functional protein association network of TWAR-associated, TGF beta/BMP signaling, and responsive genes. Edges represent predicted interactions based on data contained with the STRING database for Mus musculus proteins. Node colors represent cluster membership based on MCL clustering. The purple (“violet blue”) network represents genes belonging to the TGF beta and BMP signaling pathways. In this cluster, ligands are shown as diamonds, receptors as rectangles, inhibitors as octagons, and proteins affecting ECM bioavailability of ligands as hexagons. Other clusters are composed of genes whose expression is known to be regulated by TGF-beta/BMP signaling; for example the small green-yellow (“highball”) colored cluster contains WNT signaling genes, whereas the dull yellow (“sandwisp”) cluster is composed of genes related to signal transduction by RAS superfamily GTPases. (B) Diagram outlining major components of TGF beta/BMP signaling pathways and TWAR-associated genes in each class. TWAR-associated genes of each type are given, with distances between TWARs and the transcription start site (TSS) of each gene given in parentheses. Minus indicates distance upstream (5′); plus, downstream (3′).
Within the GREAT-defined distal gene region of growth differentiation factor 6 (Gdf6), we identified one adaptively evolving TWAR (TWAR2.Chr5). GDF6 has an inhibitory effect on osteoblast cell fate (Clendenning and Mortlock 2012) but enhances the differentiation of tendons (Mikic et al. 2010) and chondrocytes (Nochi et al. 2004). GDF6 also has a prominent role in craniofacial patterning and is required for formation of the coronal suture (Settle et al. 2003). Adaptation in other GDF6 CREs underpins variation in bony armor plates between freshwater and marine sticklebacks, with higher GDF6 expression in freshwater fish causing reduced armor plates (Indjeian et al. 2016). Another element (TWAR5.Chr1) was identified with the first intron of myostatin (Mstn) (Dankbar et al. 2015). Myostatin is a critical regulator of muscle development, best known for its involvement in the double-muscled phenotype observed in Piedmontese cattle, characterized by extreme muscular hypertrophy (Kambadur et al. 1997). Myostatin also contributes to skeletal development directly through signaling to osteoblasts and osteoclasts and indirectly by affecting muscle mass and consequent stress loads on bone (Hamrick 2003; Dankbar et al. 2015; Qin et al. 2017). Consistent with this, myostatin-deficient CD-1 mice, which show large increases in masseter muscle weight (∼83%), also show small but significant changes in facial length and shape (Vecchione et al. 2007).
Three TWARs (TWAR4.Chr1, TWAR5.Chr1, and TWAR6.Chr1) were identified within the GREAT-defined gene regulatory region of activin receptor IIA (Acvr2a). Acvr2a encodes a receptor for several TGF beta family ligands, including GDF6/7, myostatin, and BMP6/7, and is expressed during craniofacial development (Fig 2A; Dudas et al. 2006). The ACVR2A receptor mediates some of myostatin's effects on morphological development, including suppression of muscle growth and in associated bones (Fig. 2A; Elkasrawy and Hamrick 2010). Acvr2a-deficient osteoblasts show enhanced differentiation and up-regulation of key osteogenic genes, and mice lacking Acvr2a show increased bone volume, an effect similar to that of its ligand GDF6 (Clendenning and Mortlock 2012; Goh et al. 2017). Mutations in Acvr2a are also associated with craniofacial defects, including mandibular hypoplasia and cleft palate, as part of a complex of symptoms similar to Pierre Robin sequence (Matzuk et al. 1995). TWAR5.Chr2 and TWAR6.Chr2 (located ∼1.5 kb downstream from the Acvr2a transcription start site [TSS]) correspond to Ensembl regulatory labels of regional function active in both human osteoblasts and skeletal muscle myoblasts (Zerbino et al. 2015). The mouse homologous region overlaps with EP300 ChIP-seq peaks in craniofacial tissues of e11.5 and e14.5 fetuses, providing strong evidence of the craniofacial enhancer activity of these elements (Speir et al. 2016). A third element (TWAR4.Chr2) overlapping a separate Ensembl-annotated regulatory element was found ∼83 kb upstream of the Acvr2a TSS, corresponding to an annotated segment of open chromatin in the human genome (hg19). An osteoblast-active TWAR (TWAR25.Chr3) was also found in proximity of Bmpr1b, which encodes a type 1 receptor. Together with the partially redundant receptor BMPR1A, BMPR1B contributes to chondrogenesis (Yoon et al. 2005), which lays down a cartilaginous template during endochondral ossification. Like ACVR2A, BMPR1B binds to GDF6, and mutations in this receptor have been attributed to Pierre Robin sequence in humans (Erlacher et al. 1998; Yang et al. 2017).
TGF beta/BMP signals are mediated by SMAD proteins. Regulatory SMADs (R-SMADs) activate transcription, whereas inhibitory SMADs (I-SMADs) block downstream TGF beta/BMP signals through multiple mechanisms (Fig. 2B; Song et al. 2009). Two elements (TWAR15.Chr18 and TWAR16.Chr18) were found in the gene region of the I-SMAD Smad7, which has an inhibitory effect on osteoblast differentiation and bone mineralization (Yano et al. 2012). Upstream, TGF beta/BMP signaling can also be modulated by bioavailability of ligand modules in the extracellular matrix (ECM) (Robertson and Rifkin 2016). One TWAR (TWAR10.Chr17) was located near Ltbp1, which encodes a latent TGF beta binding protein. LTBP1 complexes with TGF beta precursors in the endoplasmic reticulum and is shuttled out of the cell in which it associates with ECM molecules such as fibrillins, influencing the extracellular availability of TGF-beta ligands (Dallas et al. 2000; Robertson et al. 2015). Correspondingly, disruption of TGF beta signaling in Ltbp1-null mice results in reduced snout length and a more compact head shape (Drews et al. 2008). Bioavailability of TGF beta ligands is further influenced by other fibrillin-interacting proteins, such as fibulins (Harikrishnan et al. 2015; Radice et al. 2015; Tian et al. 2015). Genes encoding two fibulins (Fbln2 and Fbln5) were also associated with TWARs (TWAR15.Chr6 and TWAR8.Chr12, respectively). FBLN5 enhances TGF beta–induced epithelial-to-mesenchymal transitions in mammary epithelial cells (Lee et al. 2008), and deficiency is known to cause craniofacial phenotypes, including premaxillary bone defects. However, it is unclear whether this is mediated by its effects on TGF beta signaling (Noda et al. 2015).
Next, we performed a more detailed functional annotation of TWAR-associated, TGF beta/BMP-related genes by compiling all relevant terms identified by GREAT, independent of statistical enrichment. In total, we cataloged 61 genes involved in or regulated by these pathways (Fig. 2A; Supplemental Table S12). Phenotype enrichment analyses revealed that TWAR-associated, TGF beta/BMP-signaling and responsive genes were significantly enriched for involvement in craniofacial morphology (short maxilla, premaxilla, nasal and basisphenoid bones, abnormal presphenoid morphology, and palatial shelf fusion) and other phenotypes related to body size, growth, and bone development (Supplemental Table S13; Kuleshov et al. 2016). Many of these phenotypes can be attributed to changes in postnatal life and as juveniles approach weaning, particularly those related to ossification and facial length. This is consistent with our understanding of the thylacine and wolf's developmental ontogeny. Like all marsupials, the thylacine was born at a stage equivalent to a eutherian embryo and had precocial development of the forelimb and jaw to facilitate the crawl to the pouch and attachment to the nipple (Newton et al. 2018). This marsupial mode of reproduction is a major driver of early developmental timing and morphology to such an extent that it constrains adult morphology (Sears and Janis 2004; Cooper and Steppan 2010). Thus, adaptation in late developmental processes may be preferred among marsupials, as these modifications are unlikely to impact required perinatal adaptations. Consistent with this, the thylacine's neonatal morphology closely resembled that of other marsupials, and the majority of species-specific morphological differentiation among marsupials arise after birth, while in the pouch. Indeed, a developmental series of the thylacine shows that as joeys approached weaning, their snout underwent considerable elongation (Newton et al. 2018). Thus, the thylacine began to acquire its canid-like morphology as it approached a developmental stage closer to that of a neonatal canid.
Taken together, the strong enrichment of positive selection around TGF beta and BMP signaling and response genes and the established roles for these loci in craniofacial development suggest that these pathways may represent critical genomic targets for adaptive morphological evolution in both the thylacine and wolf. Because TGF beta/BMP signaling is essential for tissue differentiation in the craniofacial region and for mediating communication between adjacent tissues, it is an attractive candidate to underlie thylacine-canid convergence. Morphological evolution involves modifications to structural elements, such as the skeleton, but also compensatory changes in musculature and the connective tissues that integrate them into functional modules, such as joints. Thus, mechanisms that can allow coevolution of integrated structures, such as TGF beta signaling, should be favored by natural selection.
Convergent coevolution in cis-regulators of craniofacial nerve development
It has long been established that innervation, particularly by branches of the cranial nerves, is critical for normal development of craniofacial tissues (Pagella et al. 2014). Extensive signaling exists between developing morphological structures and the peripheral nervous system in vertebrates (Adameyko and Fried 2016). For instance, BMP signaling from maxillomandibular tissues influences the phenotype of the trigeminal nerve, which is responsible for sensation and motor control in the jaw (Hodge et al. 2007). Conversely, sonic hedgehog signaling from the trigeminal nerve appears to influence the development of dental mesenchymal stem cells (Zhao et al. 2014). As morphological traits evolve, the peripheral nervous system may accommodate these changes, preserving normal motor control, sensory reception, and transmission.
Consistent with this, segments of several VISTA enhancers active in head and neck nerves were homologous to TARs and WARs. The human-origin enhancer hs1433, for instance, was tiled by nonoverlapping TARs and WARs (TAR Chr3.18214 and WAR Chr3.18205). This enhancer drives reporter gene expression in the trigeminal nerve of e11.5 embryonic mice (Fig. 3A; Visel et al. 2007). The nearest gene to this enhancer was the MDS1 and EVI1 complex locus (Mecom) (Speir et al. 2016), which in mouse embryos shows a nearly identical expression domain to that driven by hs1433 at a similar developmental stage (e10.5) (Fig. 3B; Gray et al. 2004; Finger et al. 2017), strongly suggesting that this enhancer is a direct regulator of Mecom expression. Mecom-mutant mice have a small trigeminal ganglion and show malformations of the superior vagal and glossopharyngeal ganglia, showing that this gene is critical for their development (Hoyt et al. 1997).
Figure 3.
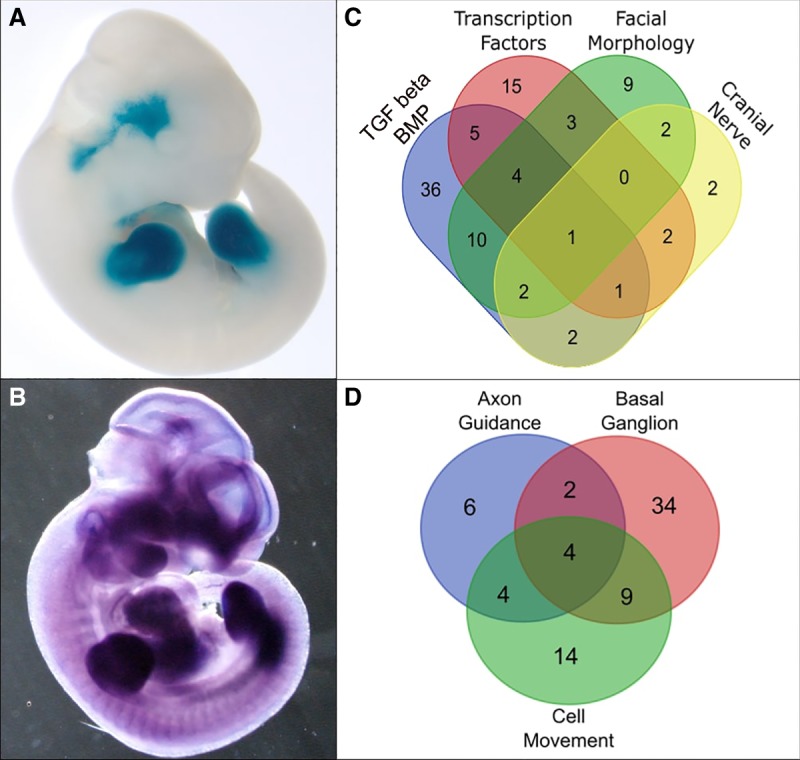
TWAR-associated genes are enriched for functions in cranial nerve and brain development. (A) Transgenic mouse fetus (e11.5) showing LacZ reporter gene expression in the trigeminal ganglion, driven by hs1433, an enhancer tiled by TARs and WARs (Gray et al. 2004; Visel et al. 2007). (B) In situ hybridization showing the mRNA localization of Mecom in the trigeminal ganglion at e10.5. Mecom is the nearest coding gene to enhancer hs1433. (From Gray et al. 2004. Reprinted with permission from the American Association for the Advancement of Science [AAAS].) (C) Unscaled Venn diagram showing overlap between TWAR-associated genes with roles in cranial nerve phenotypes, TGF beta/BMP signaling and responsive genes, transcription factors, and genes with roles in mouse facial morphology. (D) Unscaled Venn diagram showing overlap between TWAR-associated genes expressed in the basal ganglion with genes related to axon guidance and cell migration.
Because enhancer hs1433 is tiled by adjacent, but nonoverlapping accelerated regions, it was not considered a TWAR, and consequently, Mecom was not included in enrichment analyses. Despite this, TWAR-associated genes were significantly enriched for association with abnormal cranial nerve morphology in mice (12 genes) (Supplemental Table S14). Thus, it appears that cranial nerve enhancers were targeted by selection in both the thylacine and wolf, commensurate with their craniofacial convergence. Half of the cranial nerve genes identified were also functionally annotated as participating in or being responsive to TGF beta/BMP signaling (Fig. 3C), including Gdf6; Hes1, a key Notch signaling gene down-regulated by TGF beta inhibitors; and Phox2b, which encodes a TGF beta–regulated transcription factor that acts as a “switch” between sensory and visceral neuron identities and influences their physical position within cranial sensory pathways (D'Autreaux et al. 2011; Dias et al. 2014; Wang et al. 2017).
Convergence in thylacine and wolf brain enhancers
GREAT analysis of TWARs also revealed significant enrichment of nearby brain-expressed genes (Supplemental Table S15), and several nervous system genes had multiple TWARs within their GREAT-defined gene regulatory domains (Supplemental Table S7). Additionally, several adaptively evolving TARs and WARs (131 and seven, respectively) overlapped with VISTA brain enhancers, including one element (mm1660) tiled by nonoverlapping TARs and WARs (TAR Chr13.64577 and WAR Chr13.64567) (Supplemental Tables S16–S18).
Recent cortical map reconstructions of preserved thylacine brains revealed differences compared with their close living relative, the Tasmanian devil, a scavenger (Berns and Ashwell 2017). They observed that the basal ganglion was more highly modularized in the thylacine compared with the Tasmanian devil (Berns and Ashwell 2017). The caudate nucleus and the putamen projections, both subdivisions of the basal ganglion, are also larger in the thylacine. Although the putamen is involved primarily in motor control and stimulus-response (habit) learning, the caudate nucleus is associated with action–outcome learning, contributing to goal-directed action (Grahn et al. 2008). These evolved differences in the thylacine are suggested as adaptations to a predatory lifestyle, particularly decision-making behaviors (Berns and Ashwell 2017). We observed that TWARs were significantly enriched near genes expressed in the basal ganglion and ganglionic eminences (Supplemental Table S19). The basal ganglion contains projection neurons and interneurons that arise principally from the medial and lateral ganglionic eminences. During embryonic development, the progenitors of these neurons migrate to the basal ganglion via radial and tangential migration, respectively, contributing to both the putamen and caudate nucleus (Marín and Rubenstein 2001). This suggests that the mechanism underlying the relative increase in thylacine basal ganglion size may be increased neuron migration from the ganglionic eminence. Consistent with this, terms related to cell migration and axon guidance were enriched among TWAR-associated genes (Supplemental Tables S10, S20) and overlapped partially with basal ganglion genes (Fig. 3D; Supplemental Tables S19, S21, S22).
Discussion
Much of our knowledge of thylacine adaptations has been inferred through morphometric and biomechanical analyses, both of which reveal strong similarities between the structure and function of thylacine and wolf skulls. However, little was previously known about the genomic basis of their exquisite craniofacial convergence. Previous analyses of the thylacine and wolf genomes showed little evidence of adaptive amino acid homoplasy, suggesting that protein-coding genes are not primary drivers of their convergent morphologies. Here we have showed that in contrast, candidate craniofacial CREs show widespread evidence of convergent selection between the thylacine and wolf. We identified numerous convergently accelerated regions in proximity to key morphological transcription factors and members of conserved developmental pathways. Although the significance of many ontology terms enriched among these TWAR-associated genes was strongly influenced by background selection, we observed broad consistency in the top enriched signaling pathways identified when comparing TWARs to the whole-genomic background and to the set of orthologous pcfCREs. In particular, putative enhancers of TGF beta signaling genes show extensive signatures of adaptive convergence in these species. The craniofacial region is a complex assemblage of tissues, many of which are developmentally and functionally integrated. Establishing connections and boundaries of these tissues is critical in determining adult craniofacial morphology (Gross and Hanken 2008). Thus, as a craniofacial structure evolves in response to selection pressures, other developmentally and functionally integrated structures must coevolve in response. Because of the essential role that TGF beta/BMP signaling genes play in orchestrating the growth of adjacent facial tissues such as bone, cartilage, muscle, and peripheral nerves, changes in their regulation represent an attractive mechanism to explain the coevolution of craniofacial structures and to contribute to thylacine–wolf convergence.
There is strong evidence of niche overlap between the thylacine and canids, based on their historical competition with dingoes on mainland Australia (Jones and Stoddart 1998). The brains of the thylacine and wolf most closely resemble those of their closest marsupial and placental relatives, respectively, both in size and structure; thus, it should not be taken for granted that selection acts on many of the same regulators of brain development, even during the evolution of similar predatory behaviors. Given this, we were surprised to discover convergent selection pressure on TWARs near genes important in brain development. Little is known about the thylacine’s hunting and social behaviors, and the lack of clear convergence in brain structure makes the biological significance of convergent selection difficult to interpret. It is therefore not feasible at present to link genomic changes in the thylacine and wolf to similarities in behaviors. However, a living relative of the thylacine, the fat-tailed dunnart, has been developed as nontraditional model species, particularly for studies of neurogenomics and brain development (Suárez et al. 2017; Paolino et al. 2018). Recently developed experimental techniques now permit genetic manipulation of the forebrains of neonatal dunnarts, which are developmentally equivalent to a mouse fetus (Paolino et al. 2018). This opens the door to future functional studies of thylacine brain CREs in a biologically relevant context, yielding insights into their behavioral and adaptive significance.
Presently, the extent of thylacine–wolf convergence beyond their craniofacial morphology is poorly understood. Thus, we restricted the present study to enhancers active in this region. Because of this, it is likely that many adaptively evolving CREs have been excluded. Future comparative anatomical studies between these species would provide valuable phenotypic context for a more comprehensive assessment of CREs active in other body regions. A further limitation of our study is the exclusion of recently evolved, lineage-specific enhancers. The relative importance of conserved and novel enhancers remains an open question that is of significant interest to the field. It has been suggested that recently evolved enhancers have a somewhat smaller effect on gene expression than shared/conserved elements and have a slight association with genes showing evolutionarily stable expression levels (Berthelot et al. 2018). It also remains unclear the degree to which selection pressures have driven convergent, functional sequence changes such as in transcription factor binding sites in TWARs. Future studies examining the evolution of functional sequence changes will be of great interest.
Our study also supports distinct patterns of gBGC in the thylacine and wolf genomes. Numerous WARs were best explained by gBGC, whereas thylacine elements either lacked these signatures or were best explained by a model including both gBGC and selection. The canid lineage shows a distinct distribution of GC-rich regions compared with other eutherians with similar overall GC content, likely owing to lineage-specific loss of PRDM9 (Axelsson et al. 2012). Differing patterns of genome evolution and base composition between these species are likely to account for the large differences in the number of adaptively evolving elements detected in each species, highlighting the importance of accounting for lineage-specific genome properties in comparative genomic studies.
Our analyses show that noncoding regulatory regions of the thylacine and wolf genomes are rich in signatures of adaptation, suggesting that these regions are key drivers of their phenotypic convergence. The convergent CREs identified here are valuable targets for future studies to interrogate how sequence changes driven by selection affect the function of critical developmental pathways. Importantly, our findings are consistent with a more central role for CREs in phenotypic adaptation compared with protein-coding genes.
Methods
Data preparation
Precomputed whole-genome alignments of 59 vertebrates against mouse (60-way alignment) in MAF format were downloaded from the UCSC Genome Browser (http://hgdownload.cse.ucsc.edu/goldenPath/mm10/multiz60way/maf/) along with the corresponding neutral tree model (http://hgdownload.cse.ucsc.edu/goldenpath/mm10/phyloP60way/). The mouse reference genome (mm10/GRCm38) represents the most current major version of the mouse genome assembly. The human assembly included in the precomputed whole-genome alignments is hg19/GRCh37. This assembly has been superseded by hg38/GRCh38 and differs chiefly in its inclusion of centromeres and additional filled gap sequences. Minor assembly corrections are unlikely to include regions captured in the pcfCRE data set (described below) or to influence the prediction of vertebrate-conserved elements or subsequent analyses. Thylacine and wolf reference-based assemblies were generated previously (Feigin et al. 2018) by mapping read data against the Tasmanian devil (Murchison et al. 2012) and domestic dog (Hoeppner et al. 2014) genomes, respectively. Read data used to produce reference-based assemblies are available at the Sequence Read Archive (SRA) under the following accession numbers: thylacine at SRR5055303, SRR5055304, SRR5055305, and SRR5055306; wolf at SRR2149879, SRR2149880, and SRR2149881. Assemblies used in whole-genome alignments are provided in Supplemental Table S1. Thylacine and wolf reference-based assemblies have been deposited at the NCBI under accessions GCA_007646695.1 and GCA_007922845.1, respectively. The dog genome used to generate the wolf reference-based assembly is CanFam3.1 (Hoeppner et al. 2014), an improved assembly compared with the original dog genome (Lindblad-Toh et al. 2005). The thylacine and wolf reference-based assemblies share the coordinate system of their respective reference genomes. Positional information (start, length, and strand) in the MAF alignment headers of Tasmanian devil and dog sequences present in the 60-way alignments was used to identify orthologous regions of the thylacine and wolf genomes. These regions were extracted from the thylacine and wolf assemblies using SAMtools faidx (Li et al. 2009) and then projected into the appropriate subalignment using MAFFT (with –add and –keep length flags) (Katoh et al. 2002). Because the domestic dog has likely experienced different selective pressures compared with wild canids such as the wolf, it was stripped from all MAF alignments. This yielded a new 61-way alignment. Because the high-quality mouse genome served as the reference for whole-genome alignments, assembly errors affecting the structure of other aligned genomes are unlikely to have a large impact on alignment accuracy. Nonreference genomes are aligned against the reference and are therefore arranged based on homology with the reference genome.
For each large subalignment (i.e., MAF alignment block), sequences were removed if they contained a >50% gap or ambiguous characters. Subalignments with fewer than 30 species were also removed. However, small stretches within larger subalignments do contain gaps. For this reason, pcfCRE subalignments, which represent small regions typically in the 100s of base pairs, may lack data from one or more species (leading to different numbers of thylacine and wolf pcfCREs). Alignments corresponding to mouse Chromosomes X and Y were excluded from our neutral model and analyses because their evolutionary rate is expected to differ compared with the autosomes (Vicoso and Charlesworth 2006).
Annotations of mouse CDS sequences were downloaded from Ensembl (version 87) (Flicek et al. 2013) and were filtered to the longest complete CDS for each gene. Sufficient statistic (.ss) files were generated from whole-genome alignments using third codon positions (as a proxy for fourfold degenerate sites) and aggregated with msa_view (Hubisz et al. 2011). PhyloFit (Siepel and Haussler 2004) was then used to recompute the neutral tree model. Although third codon positions are not an exact representation of neutral evolution (as is usually assumed of fourfold degenerate sites), we found that our 61-way tree model was highly consistent with the original 60-way model provided by the UCSC Genome Browser for equivalent branches (see below for further discussion) (Supplemental Fig. S3, Supplemental Table S23). Scripts used for filtering data can be found in the Supplemental Materials under the Supplemental Code and online at https://github.com/charlesfeigin/TWARs_scripts.
Identification of vertebrate-conserved elements
Before scanning for vertebrate-conserved regions, thylacine and wolf sequences were temporarily removed from the modified 61-way alignments. This was performed to avoid accelerated evolution in these species from hampering detection of vertebrate conservation. Vertebrate-conserved regions were identified with phastCons (Siepel et al. 2005), which was run using the following parameters: --expected-length 200 –target-coverage 0.15, with rho being estimated directly from the data. Enhancers predicted in other studies (such as those bound by EP300) have an average length of 750–800 bp but are not contiguous stretches of high conservation. Instead, they are typically composed of shorter conserved segments of 10–100 bp (Visel et al. 2009; Lee et al. 2011). In contrast, the majority of exons are <200 bp in length but are often highly conserved across their length (Sakharkar et al. 2004; Zhu et al. 2009). Thus, we selected 200 bp as the expected length of conserved elements, as exons should be almost entirely contiguous stretches of conserved sequence. A target coverage of 0.15 was selected, as this represents higher estimates of the percentage of the genome that is evolutionarily constrained (Ponting and Hardison 2011). To identify elements with craniofacial activity, vertebrate-conserved regions were intersected with publicly available mouse craniofacial ChIP data for H3K9ac and H3K27ac (Supplemental Table S2; The ENCODE Project Consortium 2012). Consistent with enhancer conservation patterns, small gaps of nonconserved bases were observed to punctuate longer stretches of high conservation. We thus merged adjacent conserved regions such that the average percentage of all merged regions that was composed of nonconserved stretches did not exceed 10% and such that known protein-coding exons from our CDS annotation were covered by less than two contiguous, conserved segments on average. Finally, conserved elements overlapping exons by >25% of their length were removed to exclude coding genes. The resulting set of noncoding elements were considered pcfCREs (Supplemental Table S3). In total, 40,547 thylacine and 56,278 wolf orthologs of pcfCREs were represented.
Identification of TARs and WARs
To identify adaptively evolving thylacine and wolf pcfCREs, we performed a likelihood ratio test for acceleration implemented in phyloP (Pollard et al. 2010). First, thylacine and wolf sequences were added back into subalignments corresponding to pcfCREs, and phyloP was run with following parameters: --mode ACC --method LRT, with each species selected as the foreground branch. This test returned 12,960 alignments with significant P-values (P < 0.05) for acceleration in the thylacine and 3644 elements in the wolf. After multiple testing correction using the Benjamini–Hochberg (FDR = 0.1), 10,910 thylacine and 1923 wolf elements (terms TARs and WARs, respectively) were retained after FDR correction (Supplemental Tables S5, S6; Benjamini and Hochberg 1995). We next intersected the set of TARs and WARs, yielding a subset of 339 convergently accelerated regions elements termed TWARs (Supplemental Table S7). TWARs are named based on the reference mouse chromosome for the subalignment in which they were found and the order in which they occurred along the reference chromosome (e.g., TWAR6.Chr3 was identified in a subalignment corresponding to mouse Chromosome 3 and was the sixth TWAR to be identified along the chromosome). The abbreviations “TARs,” “WARs,” and “TWARs” are based on conventions used in the literature describing human and bat accelerated regions (HARs and BARs, respectively) (Pollard et al. 2006; Eckalbar et al. 2016).
We found that the number of craniofacial TARs considerably exceeds the number of WARs. We thus attempted to identify explanations other than different rates of positive selection (which were previously observed in coding genes) (Feigin et al. 2018) or differing rates of gBGC in the thylacine and wolf. First, we tested for acceleration in several other mammal species. Although the thylacine showed the most accelerated sequences, we observed considerable variation among species (Supplemental Fig. S4; Supplemental Table S24). We next validated our 61-way phyloP tree model by comparing pairwise distances (summed branch lengths) to all equivalent branches in the original UCSC 60-way model. Branch lengths in both models were nearly identical (Supplemental Fig. S3; Supplemental Table S23). Sequencing errors and contamination in thylacine data are unlikely, as the data were quality filtered with high stringency, and contaminant sequences were removed. Damage patterns characteristic of ancient DNA that can create false SNPs (e.g., cytosine deamination) are absent from the thylacine data (Feigin et al. 2018). The average identity, calculated using BioPerl (Stajich et al. 2002), was high for both thylacine–Tasmanian devil and wolf–panda alignments (∼94.68% and 93.36%, respectively), indicating that TAR and WAR evolutionary rates are not driven by poor sequence quality. Given this, we concluded that the differing number of TARs and WARs is unlikely to be the result of artifacts.
Detection of gBGC in accelerated regions
To assess rates of gBGC in the thylacine and wolf, we used the R package rphast (Hubisz et al. 2011). We implemented a reduced classification system compared with that implemented by Kostka et al. (2012), using three likelihood ratio tests aimed at simply excluding elements that are unlikely to be evolving adaptively at all. Accelerated regions in each species were tested using two additional likelihood ratio tests: (1) comparing a model with a parameter for gBGC on the foreground branch against the neutral model, and (2) comparing a model with parameters for both gBGC and selection on the foreground branch (gBGC + Sel) against the model of foreground gBGC alone. In both cases, we fixed the strength of the gBGC parameter at three (as used during the construction of the UCSC Genome Browser gBGC predictions track) (Speir et al. 2016). This parameter value was chosen, as a low value for B has been shown to improve sensitivity while minimizing false positives (Capra et al. 2013). Elements with a significant P-value (FDR = 0.1) (Benjamini and Hochberg 1995) for gBGC alone versus null were initially removed from the TAR/WAR data sets, but elements that were better explained by the gBGC + Sel model were “rescued” and considered to be evolving adaptively. Significance was determined using the asymptotic null (chi-squared distribution, accounting for the differing numbers of degrees of freedom in each test) (Hubisz et al. 2011).
Characterization of TWARs by proximity to protein-coding genes
TWARs were analyzed using GREAT (McLean et al. 2010; http://bejerano.stanford.edu/great/public/html/index.php). GREAT analysis was performed using default parameters (gene regulatory domain definition: basal plus extension; proximal: 5.0 kb upstream, 1.0 kb downstream; plus distal: up to 1000.0 kb; background regions: whole genome mm10). For GREAT ontology supplemental tables (Supplemental Tables S8-S11, S14, S15, S19, S20), the top 20 significant terms (binomial P-value over-regions test, P < 0.05 after Benjamini–Hochberg adjustment) with a fold enrichment of two or more (GREAT default setting) are given. When fewer than 20 terms passed filtering, only those terms are shown. GREAT analysis of TWARs was repeated using an alternate background set composed of the 38,993 pcfCREs with representative and thylacine and wolf sequences (up to top 20 terms exported based on raw P-value, P < 0.05) (Supplemental Table S24).
We also examined the “bracketing genes” (the genes immediately upstream and downstream in the UCSC Genome Browser) of VISTA enhancers overlapping accelerated regions. Known phenotypes associated with TWAR-associated genes were accessed from the Mouse Genome Informatics (MGI) database (Blake et al. 2017). Gene expression patterns were visualized where possible by referring to the Gene Expression Database (GXD) (Finger et al. 2017). Mammalian phenotype ontology analyses of TWAR-associated, TGF beta, and BMP signaling and responsive genes (Supplemental Table S13) were performed using the Enrichr web server provided by the Ma'ayan laboratory (Kuleshov et al. 2016; https://amp.pharm.mssm.edu/Enrichr/). Enrichment was inferred with a Fisher's exact test (FDR q-value <0.05) using Gene Ontology annotations from Mus musculus. Although many high-ranking Enrichr Mammalian phenotype ontology terms were directly relevant to craniofacial development, numerous other significant and biologically relevant terms were identified at lower rank. Thus, we exported all significant results with an adjusted P-value <0.05 (Benjamini and Hochberg 1995; Kuleshov et al. 2016).
Characterization by closest gene has important caveats. Because the 61-way alignment is referenced against the mouse (mm10) genome, positions of loci presented are derived from mouse coordinate system. It is known that many mammalian species have undergone extensive karyotypic rearrangements over time, so the nearest genes and their respective distances from predicted elements in mouse may not perfectly reflect those in thylacine and wolf. However, large syntenic blocks spanning many megabases are readily identifiable between even distantly related mammalian species (Ferguson-Smith and Trifonov 2007), whereas the vast majority of pcfCREs are <100 kb from their putative target genes (Supplemental Fig. S2; Supplemental Table S3). Synteny of most pcfCREs with nearby genes is therefore unlikely to be disrupted by larger-scale genome rearrangements. The potential impact of small-scale rearrangements is more difficult to assess. However, the frequency of enhancer–promoter interactions decreases with linear distance along a chromosome (Chepelev et al. 2012). Closest genes are thus the most likely regulatory targets of pcfCREs, so we expect rearrangements disrupting interactions between enhancers and target genes to be limited by purifying selection.
Annotation of TGF beta/BMP-related genes
GREAT analysis revealed enriched terms related to TGF beta/BMP signaling and responsiveness to signaling by these pathways among TWAR-associated genes (Supplemental Tables S9–S11). However, the genes that fell within these enriched terms did not reflect all TWAR-associated genes related to these pathways. Therefore, to comprehensively annotate all TGF beta/BMP-related, TWAR-associated genes, we compiled all GREAT-identified terms related to these pathways regardless of enrichment, yielding a set of 61 genes (Supplemental Table S12). These genes were then tested for enriched mammalian phenotype enrichment using the Enrichr web server (Kuleshov et al. 2016).
Network analysis and TGF beta/BMP pathway diagram
The gene network graph in Figure 2A was generated using the STRING network analysis tool (https://string-db.org/) and Cytoscape (Shannon et al. 2003; Szklarczyk et al. 2017). The network includes TWAR-associated genes involved in TGF beta/BMP signaling and response identified by GREAT (Supplemental Table S12). The gene list was first uploaded to the STRING web interface for network prediction (Szklarczyk et al. 2017). Gene names were mapped against the M. musculus gene set. Edges were calculated using an interaction score of 0.2, including evidence from all sources in the STRING database excluding gene fusions. Disconnected nodes were removed from the network, and MCL clustering was performed. An inflation parameter of two was chosen because it yielded a coherent module containing TGF beta/BMP signaling genes. STRING combined score and MCL clusters were exported to tabular files (Supplemental Tables S25, S26), and MCL cluster assignments were assigned hexadecimal color codes and imported into Cytoscape to produce a network graph (Shannon et al. 2003). Edge thickness represents STRING combined score. Node spacing was calculated using the prefuse force-directed layout algorithm (also based on combined score), with small manual adjustments to avoid hidden node labels. The diagram illustrating TGF beta/BMP signaling pathways was created using BioRender (https://biorender.com/).
Venn diagrams
Venn diagrams were produced using the Venn diagram web tool hosted by the University of Gent Bioinformatics and Systems Biology group (http://bioinformatics.psb.ugent.be/webtools/Venn/). In Figure 3C, the gene list for each category was obtained by functional annotation using GREAT. The TGF beta/BMP set is contained in Supplemental Table S12. Facial morphology is the list of genes within the MGI mouse phenotype ontology terms “abnormal craniofacial morphology” and “abnormal facial morphology” returned by GREAT analysis. Abnormal cranial nerve is the list of genes associated with abnormal cranial nerve morphology (Supplemental Table S14). Transcription factors contain all genes identified as having DNA-binding activity (Supplemental Table S9). In Figure 3D, the axon guidance set is the list of GREAT-identified genes with roles in axon guidance (MSigDB ontology). Cell movement is the combined lists of genes involved in cell migration, cell motility, and locomotion (Supplemental Table S21). Relevant ontology terms that are subsets of other terms are not mentioned individually.
Overlap with experimentally validated craniofacial enhancers
To identify TWARs, TARs, and WARs overlapping experimentally validated craniofacial enhancers, the coordinates of 1135 (mm9) mouse and human enhancers were downloaded from the VISTA Enhancer Browser (Supplemental Table S4; Visel et al. 2007; enhancer.lbl.gov). The coordinates of these elements were converted to mm10 using the UCSC Genome Browser liftOver tool (https://genome.ucsc.edu/cgi-bin/hgliftOver). For human enhancers with no homolog present in mm9, the human coordinates (hg19) were converted directly to mm10 using liftOver. Enhancers that failed to convert and enhancers located on mouse sex chromosomes were excluded. These included elements active in the ear, branchial arch, eye, nose, cranial nerve, facial mesenchyme, hindbrain, mesenchyme derived from neural crest, trigeminal V (ganglion, cranial), forebrain, and midbrain (shown by LacZ reporter). Images of enhancers with annotated expression in dorsal root ganglia or as “other” were visually inspected for cranial expression.
Software availability
The custom Perl and R scripts used for data parsing and filtering are contained in Supplemental Materials under “Supplemental Code.” Code is also hosted at https://github.com/charlesfeigin/TWARs_scripts.
Supplementary Material
Acknowledgments
We thank Katie Pollard of the Department of Epidemiology and Biostatistics at UCSF for providing advice into evolutionary analyses performed in this paper; Ricardo Mallarino of the Department of Molecular Biology at Princeton University for editorial advice; Bernard Meade from the National eResearch Collaboration Tools and Resources project for providing computational resources; MacNeil Lyons for the use of his wolf photograph (Fig. 1B); and Elise Ireland for proofreading. A.J.P. was supported by an Australian Research Council future fellowship (FT140100964).
Author contributions: C.Y.F. and A.J.P. conceived the study and wrote the manuscript. C.Y.F. performed comparative genomic analyses. A.H.N. assisted with annotation of craniofacial enhancers and edited the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.244251.118.
References
- Adameyko I, Fried K. 2016. The nervous system orchestrates and integrates craniofacial development: a review. Front Physiol 7: 49 10.3389/fphys.2016.00049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsson E, Webster MT, Ratnakumar A, LUPA Consortium, Ponting CP, Lindblad-Toh K. 2012. Death of PRDM9 coincides with stabilization of the recombination landscape in the dog genome. Genome Res 22: 51–63. 10.1101/gr.124123.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baffi MO, Moran MA, Serra R. 2006. Tgfbr2 regulates the maintenance of boundaries in the axial skeleton. Dev Biol 296: 363–374. 10.1016/j.ydbio.2006.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 57: 289–300. 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- Berns GS, Ashwell KWS. 2017. Reconstruction of the cortical maps of the Tasmanian tiger and comparison to the Tasmanian devil. PLoS One 12: e0168993 10.1371/journal.pone.0168993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthelot C, Villar D, Horvath JE, Odom DT, Flicek P. 2018. Complexity and conservation of regulatory landscapes underlie evolutionary resilience of mammalian gene expression. Nat Ecol Evol 2: 152–163. 10.1038/s41559-017-0377-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bininda-Emonds OR, Cardillo M, Jones KE, MacPhee RD, Beck RM, Grenyer R, Price SA, Vos RA, Gittleman JL, Purvis A. 2007. The delayed rise of present-day mammals. Nature 446: 507–512. 10.1038/nature05634 [DOI] [PubMed] [Google Scholar]
- Blake JA, Eppig JT, Kadin JA, Richardson JE, Smith CL, Bult CJ. 2017. Mouse Genome Database (MGD)-2017: community knowledge resource for the laboratory mouse. Nucleic Acids Res 45: D723–D729. 10.1093/nar/gkw1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker BM, Friedrich T, Mason MK, VanderMeer JE, Zhao J, Eckalbar WL, Logan M, Illing N, Pollard KS, Ahituv N. 2016. Bat accelerated regions identify a bat forelimb specific enhancer in the HoxD locus. PLoS Genet 12: e1005738 10.1371/journal.pgen.1005738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capra JA, Hubisz MJ, Kostka D, Pollard KS, Siepel A. 2013. A model-based analysis of GC-biased gene conversion in the human and chimpanzee genomes. PLoS Genet 9: e1003684 10.1371/journal.pgen.1003684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SB. 2005. Evolution at two levels: on genes and form. PLoS Biol 3: e245 10.1371/journal.pbio.0030245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chepelev I, Wei G, Wangsa D, Tang Q, Zhao K. 2012. Characterization of genome-wide enhancer-promoter interactions reveals co-expression of interacting genes and modes of higher order chromatin organization. Cell Res 22: 490–503. 10.1038/cr.2012.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clendenning DE, Mortlock DP. 2012. The BMP ligand Gdf6 prevents differentiation of coronal suture mesenchyme in early cranial development. PLoS One 7: e36789 10.1371/journal.pone.0036789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JW, Steppan SJ. 2010. Developmental constraint on the evolution of marsupial forelimb morphology. Aust J Zool 58: 1–15. 10.1071/ZO09102 [DOI] [Google Scholar]
- Dallas SL, Keene DR, Bruder SP, Saharinen J, Sakai LY, Mundy GR, Bonewald LF. 2000. Role of the latent transforming growth factor β binding protein 1 in fibrillin-containing microfibrils in bone cells in vitro and in vivo. J Bone Miner Res 15: 68–81. 10.1359/jbmr.2000.15.1.68 [DOI] [PubMed] [Google Scholar]
- Dankbar B, Fennen M, Brunert D, Hayer S, Frank S, Wehmeyer C, Beckmann D, Paruzel P, Bertrand J, Redlich K, et al. 2015. Myostatin is a direct regulator of osteoclast differentiation and its inhibition reduces inflammatory joint destruction in mice. Nat Med 21: 1085–1090. 10.1038/nm.3917 [DOI] [PubMed] [Google Scholar]
- D'Autreaux F, Coppola E, Hirsch MR, Birchmeier C, Brunet JF. 2011. Homeoprotein Phox2b commands a somatic-to-visceral switch in cranial sensory pathways. Proc Natl Acad Sci 108: 20018–20023. 10.1073/pnas.1110416108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias JM, Alekseenko Z, Applequist JM, Ericson J. 2014. Tgfβ signaling regulates temporal neurogenesis and potency of neural stem cells in the CNS. Neuron 84: 927–939. 10.1016/j.neuron.2014.10.033 [DOI] [PubMed] [Google Scholar]
- Drews F, Knöbel S, Moser M, Muhlack KG, Mohren S, Stoll C, Bosio A, Gressner AM, Weiskirchen R. 2008. Disruption of the latent transforming growth factor-β binding protein-1 gene causes alteration in facial structure and influences TGF-β bioavailability. Biochim Biophys Acta 1783: 34–48. 10.1016/j.bbamcr.2007.08.004 [DOI] [PubMed] [Google Scholar]
- Dudas M, Kaartinen V. 2005. TGF-β superfamily and mouse craniofacial development: interplay of morphogenetic proteins and receptor signaling controls normal formation of the face. Curr Top Dev Biol 66: 65–133. 10.1016/S0070-2153(05)66003-6 [DOI] [PubMed] [Google Scholar]
- Dudas M, Kim J, Li WY, Nagy A, Larsson J, Karlsson S, Chai Y, Kaartinen V. 2006. Epithelial and ectomesenchymal role of the type I TGF-β receptor ALK5 during facial morphogenesis and palatal fusion. Dev Biol 296: 298–314. 10.1016/j.ydbio.2006.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckalbar WL, Schlebusch SA, Mason MK, Gill Z, Parker AV, Booker BM, Nishizaki S, Muswamba-Nday C, Terhune E, Nevonen KA, et al. 2016. Transcriptomic and epigenomic characterization of the developing bat wing. Nat Genet 48: 528–536. 10.1038/ng.3537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkasrawy MN, Hamrick MW. 2010. Myostatin (GDF-8) as a key factor linking muscle mass and bone structure. J Musculoskelet Neuronal Interact 10: 56–63. [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlacher L, McCartney J, Piek E, ten Dijke P, Yanagishita M, Oppermann H, Luyten FP. 1998. Cartilage-derived morphogenetic proteins and osteogenic protein-1 differentially regulate osteogenesis. J Bone Miner Res 13: 383–392. 10.1359/jbmr.1998.13.3.383 [DOI] [PubMed] [Google Scholar]
- Feigin CY, Newton AH, Doronina L, Schmitz J, Hipsley CA, Mitchell KJ, Gower G, Llamas B, Soubrier J, Heider TN, et al. 2018. Genome of the Tasmanian tiger provides insights into the evolution and demography of an extinct marsupial carnivore. Nat Ecol Evol 2: 182–192. 10.1038/s41559-017-0417-y [DOI] [PubMed] [Google Scholar]
- Ferguson-Smith MA, Trifonov V. 2007. Mammalian karyotype evolution. Nat Rev Genet 8: 950–962. 10.1038/nrg2199 [DOI] [PubMed] [Google Scholar]
- Finger JH, Smith CM, Hayamizu TF, McCright IJ, Xu J, Law M, Shaw DR, Baldarelli RM, Beal JS, Blodgett O, et al. 2017. The mouse Gene Expression Database (GXD): 2017 update. Nucleic Acids Res 45: D730–D736. 10.1093/nar/gkw1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, et al. 2013. Ensembl 2013. Nucleic Acids Res 41: D48–D55. 10.1093/nar/gks1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh BC, Singhal V, Herrera AJ, Tomlinson RE, Kim S, Faugere MC, Germain-Lee EL, Clemens TL, Lee SJ, DiGirolamo DJ. 2017. Activin receptor type 2A (ACVR2A) functions directly in osteoblasts as a negative regulator of bone mass. J Biol Chem 292: 13809–13822. 10.1074/jbc.M117.782128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grahn JA, Parkinson JA, Owen AM. 2008. The cognitive functions of the caudate nucleus. Prog Neurobiol 86: 141–155. 10.1016/j.pneurobio.2008.09.004 [DOI] [PubMed] [Google Scholar]
- Gray PA, Fu H, Luo P, Zhao Q, Yu J, Ferrari A, Tenzen T, Yuk D-I, Tsung EF, Cai Z, et al. 2004. Mouse brain organization revealed through direct genome-scale TF expression analysis. Science 306: 2255–2257. 10.1126/science.1104935 [DOI] [PubMed] [Google Scholar]
- Gross JB, Hanken J. 2008. Review of fate-mapping studies of osteogenic cranial neural crest in vertebrates. Dev Biol 317: 389–400. 10.1016/j.ydbio.2008.02.046 [DOI] [PubMed] [Google Scholar]
- Hamrick MW. 2003. Increased bone mineral density in the femora of GDF8 knockout mice. Anat Rec A Discov Mol Cell Evol Biol 272A: 388–391. 10.1002/ar.a.10044 [DOI] [PubMed] [Google Scholar]
- Harikrishnan K, Cooley MA, Sugi Y, Barth JL, Rasmussen LM, Kern CB, Argraves KM, Argraves WS. 2015. Fibulin-1 suppresses endothelial to mesenchymal transition in the proximal outflow tract. Mech Dev 136: 123–132. 10.1016/j.mod.2014.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge LK, Klassen MP, Han BX, Yiu G, Hurrell J, Howell A, Rousseau G, Lemaigre F, Tessier-Lavigne M, Wang F. 2007. Retrograde BMP signaling regulates trigeminal sensory neuron identities and the formation of precise face maps. Neuron 55: 572–586. 10.1016/j.neuron.2007.07.010 [DOI] [PubMed] [Google Scholar]
- Hoeppner MP, Lundquist A, Pirun M, Meadows JRS, Zamani N, Johnson J, Sundström G, Cook A, FitzGerald MG, Swofford R, et al. 2014. An improved canine genome and a comprehensive catalogue of coding genes and non-coding transcripts. PLoS One 9: e91172 10.1371/journal.pone.0091172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt PR, Bartholomew C, Davis AJ, Yutzey K, Gamer LW, Potter SS, Ihle JN, Mucenski ML. 1997. The Evil proto-oncogene is required at midgestation for neural, heart, and paraxial mesenchyme development. Mech Dev 65: 55–70. 10.1016/S0925-4773(97)00057-9 [DOI] [PubMed] [Google Scholar]
- Hubisz MJ, Pollard KS, Siepel A. 2011. PHAST and RPHAST: phylogenetic analysis with space/time models. Brief Bioinform 12: 41–51. 10.1093/bib/bbq072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indjeian VB, Kingman GA, Jones FC, Guenther CA, Grimwood J, Schmutz J, Myers RM, Kingsley DM. 2016. Evolving new skeletal traits by cis-regulatory changes in bone morphogenetic proteins. Cell 164: 45–56. 10.1016/j.cell.2015.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RN, O'Meally D, Chen Z, Etherington GJ, Ho SYW, Nash WJ, Grueber CE, Cheng Y, Whittington CM, Dennison S, et al. 2018. Adaptation and conservation insights from the koala genome. Nat Genet 50: 1102–1111. 10.1038/s41588-018-0153-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones ME, Stoddart DM. 1998. Reconstruction of the predatory behaviour of the extinct marsupial thylacine (Thylacinus cynocephalus). J Zool 246: 239–246. 10.1111/j.1469-7998.1998.tb00152.x [DOI] [Google Scholar]
- Kambadur R, Sharma M, Smith TP, Bass JJ. 1997. Mutations in myostatin (GDF8) in double-muscled Belgian blue and Piedmontese cattle. Genome Res 7: 910–915. 10.1101/gr.7.9.910 [DOI] [PubMed] [Google Scholar]
- Kasai F, O'Brien PCM, Pereira JC, Ferguson-Smith MA. 2018. Marsupial chromosome DNA content and genome size assessed from flow karyotypes: invariable low autosomal GC content. R Soc Open Sci 5: 171539 10.1098/rsos.171539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K-i, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30: 3059–3066. 10.1093/nar/gkf436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostka D, Hubisz MJ, Siepel A, Pollard KS. 2012. The role of GC-biased gene conversion in shaping the fastest evolving regions of the human genome. Mol Biol Evol 29: 1047–1057. 10.1093/molbev/msr279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. 2016. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44: W90–W97. 10.1093/nar/gkw377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latchman DS. 1997. Transcription factors: an overview. Int J Biochem Cell Biol 29: 1305–1312. 10.1016/S1357-2725(97)00085-X [DOI] [PubMed] [Google Scholar]
- Lee YH, Albig AR, Regner M, Schiemann BJ, Schiemann WP. 2008. Fibulin-5 initiates epithelial–mesenchymal transition (EMT) and enhances EMT induced by TGF-β in mammary epithelial cells via a MMP-dependent mechanism. Carcinogenesis 29: 2243–2251. 10.1093/carcin/bgn199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Karchin R, Beer MA. 2011. Discriminative prediction of mammalian enhancers from DNA sequence. Genome Res 21: 2167–2180. 10.1101/gr.121905.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letnic M, Fillios M, Crowther MS. 2012. Could direct killing by larger dingoes have caused the extinction of the thylacine from mainland Australia? PLoS One 7: e34877 10.1371/journal.pone.0034877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, Kulbokas EJ, Zody MC 3rd, et al. 2005. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438: 803–819. 10.1038/nature04338 [DOI] [PubMed] [Google Scholar]
- Marín O, Rubenstein JL. 2001. A long, remarkable journey: tangential migration in the telencephalon. Nat Rev Neurosci 2: 780–790. 10.1038/35097509 [DOI] [PubMed] [Google Scholar]
- Matzuk MM, Kumar TR, Bradley A. 1995. Different phenotypes for mice deficient in either activins or activin receptor type II. Nature 374: 356–360. 10.1038/374356a0 [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. 2010. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28: 495–501. 10.1038/nbt.1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikic B, Rossmeier K, Bierwert L. 2010. Identification of a tendon phenotype in GDF6 deficient mice. Anat Rec 292: 396–400. 10.1002/ar.20852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorthy SD, Davidson S, Shchuka VM, Singh G, Malek-Gilani N, Langroudi L, Martchenko A, So V, Macpherson NN, Mitchell JA. 2017. Enhancers and super-enhancers have an equivalent regulatory role in embryonic stem cells through regulation of single or multiple genes. Genome Res 27: 246–258. 10.1101/gr.210930.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murchison EP, Schulz-Trieglaff OB, Ning Z, Alexandrov LB, Bauer MJ, Fu B, Hims M, Ding Z, Ivakhno S, Stewart C, et al. 2012. Genome sequencing and analysis of the Tasmanian devil and its transmissible cancer. Cell 148: 780–791. 10.1016/j.cell.2011.11.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton AH, Spoutil F, Prochazka J, Black JR, Medlock K, Paddle RN, Knitlova M, Hipsley CA, Pask AJ. 2018. Letting the ‘cat’ out of the bag: pouch young development of the extinct Tasmanian tiger revealed by X-ray computed tomography. R Soc Open Sci 5: 171914 10.1098/rsos.171914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nochi H, Sung JH, Lou J, Adkisson HD, Maloney WJ, Hruska KA. 2004. Adenovirus mediated BMP-13 gene transfer induces chondrogenic differentiation of murine mesenchymal progenitor cells. J Bone Miner Res 19: 111–122. 10.1359/jbmr.2004.19.1.111 [DOI] [PubMed] [Google Scholar]
- Noda K, Nakamura T, Komatsu Y. 2015. Fibulin-5 deficiency causes developmental defect of premaxillary bone in mice. Biochem Biophys Res Commun 466: 585–591. 10.1016/j.bbrc.2015.09.089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagella P, Jiménez-Rojo L, Mitsiadis TA. 2014. Roles of innervation in developing and regenerating orofacial tissues. Cell Mol Life Sci 71: 2241–2251. 10.1007/s00018-013-1549-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolino A, Fenlon LR, Kozulin P, Richards LJ, Suárez R. 2018. Multiple events of gene manipulation via in pouch electroporation in a marsupial model of mammalian forebrain development. J Neurosci Methods 293: 45–52. 10.1016/j.jneumeth.2017.09.004 [DOI] [PubMed] [Google Scholar]
- Partha R, Chauhan BK, Ferreira Z, Robinson JD, Lathrop K, Nischal KK, Chikina M, Clark NL. 2017. Subterranean mammals show convergent regression in ocular genes and enhancers, along with adaptation to tunneling. eLife 6: e25884 10.7554/eLife.25884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Salama SR, Lambert N, Lambot MA, Coppens S, Pedersen JS, Katzman S, King B, Onodera C, Siepel A, et al. 2006. An RNA gene expressed during cortical development evolved rapidly in humans. Nature 443: 167–172. 10.1038/nature05113 [DOI] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. 2010. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 20: 110–121. 10.1101/gr.097857.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponting CP, Hardison RC. 2011. What fraction of the human genome is functional? Genome Res 21: 1769–1776. 10.1101/gr.116814.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryce BA, Watson SS, Murchison ND, Staverosky JA, Dunker N, Schweitzer R. 2009. Recruitment and maintenance of tendon progenitors by TGFβ signaling are essential for tendon formation. Development 136: 1351–1361. 10.1242/dev.027342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Peng Y, Zhao W, Pan J, Ksiezak-Reding H, Cardozo C, Wu Y, Divieti Pajevic P, Bonewald LF, Bauman WA, et al. 2017. Myostatin inhibits osteoblastic differentiation by suppressing osteocyte-derived exosomal microRNA-218: a novel mechanism in muscle-bone communication. J Biol Chem 292: 11021–11033. 10.1074/jbc.M116.770941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radice PD, Mathieu P, Leal MC, Farías MI, Ferrari C, Puntel M, Salibe M, Chernomoretz A, Pitossi FJ. 2015. Fibulin-2 is a key mediator of the pro-neurogenic effect of TGF-β1 on adult neural stem cells. Mol Cell Neurosci 67: 75–83. 10.1016/j.mcn.2015.06.004 [DOI] [PubMed] [Google Scholar]
- Robertson IB, Rifkin DB. 2016. Regulation of the bioavailability of TGF-β and TGF-β-related proteins. Cold Spring Harb Perspect Biol 8: a021907 10.1101/cshperspect.a021907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson IB, Horiguchi M, Zilberberg L, Dabovic B, Hadjiolova K, Rifkin DB. 2015. Latent TGF-β-binding proteins. Matrix Biol 47: 44–53. 10.1016/j.matbio.2015.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rofe R, Hayman D. 1985. G-banding evidence for a conserved complement in the Marsupialia. Cytogenet Cell Genet 39: 40–50. 10.1159/000132101 [DOI] [PubMed] [Google Scholar]
- Sakharkar MK, Chow VT, Kangueane P. 2004. Distributions of exons and introns in the human genome. In Silico Biol 4: 387–393. [PubMed] [Google Scholar]
- Sartori R, Sandri M. 2015. BMPs and the muscle-bone connection. Bone 80: 37–42. 10.1016/j.bone.2015.05.023 [DOI] [PubMed] [Google Scholar]
- Sears KE, Janis C. 2004. Constraints on the morphological evolution of marsupial shoulder girdles. Evolution 58: 2353–2370. [DOI] [PubMed] [Google Scholar]
- Settle SH Jr, Rountree RB, Sinha A, Thacker A, Higgins K, Kingsley DM. 2003. Multiple joint and skeletal patterning defects caused by single and double mutations in the mouse Gdf6 and Gdf5 genes. Dev Biol 254: 116–130. 10.1016/S0012-1606(02)00022-2 [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504. 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Haussler D. 2004. Phylogenetic estimation of context-dependent substitution rates by maximum likelihood. Mol Biol Evol 21: 468–488. 10.1093/molbev/msh039 [DOI] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15: 1034–1050. 10.1101/gr.3715005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B, Estrada KD, Lyons KM. 2009. Smad signaling in skeletal development and regeneration. Cytokine Growth Factor Rev 20: 379–388. 10.1016/j.cytogfr.2009.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speir ML, Zweig AS, Rosenbloom KR, Raney BJ, Paten B, Nejad P, Lee BT, Learned K, Karolchik D, Hinrichs AS, et al. 2016. The UCSC Genome Browser database: 2016 update. Nucleic Acids Res 44: D717–D725. 10.1093/nar/gkv1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz F, Furlong EEM. 2012. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet 13: 613––626.. 10.1038/nrg3207 [DOI] [PubMed] [Google Scholar]
- Stajich JE, Block D, Boulez K, Brenner SE, Chervitz SA, Dagdigian C, Fuellen G, Gilbert JG, Korf I, Lapp H, et al. 2002. The Bioperl toolkit: Perl modules for the life sciences. Genome Res 12: 1611–1618. 10.1101/gr.361602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern DL, Orgogozo V. 2008. The loci of evolution: How predictable is genetic evolution? Evolution 62: 2155–2177. 10.1111/j.1558-5646.2008.00450.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suárez R, Paolino A, Kozulin P, Fenlon LR, Morcom LR, Englebright R, O'Hara PJ, Murray PJ, Richards LJ. 2017. Development of body, head and brain features in the Australian fat-tailed dunnart (Sminthopsis crassicaudata; Marsupialia: Dasyuridae): a postnatal model of forebrain formation. PLoS One 12: e0184450 10.1371/journal.pone.0184450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al. 2017. The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res 45: D362–D368. 10.1093/nar/gkw937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian H, Liu J, Chen J, Gatza ML, Blobe GC. 2015. Fibulin-3 is a novel TGF-β pathway inhibitor in the breast cancer microenvironment. Oncogene 34: 5635–5647. 10.1038/onc.2015.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollis M, Hutchins ED, Stapley J, Rupp SM, Eckalbar WL, Maayan I, Lasku E, Infante CR, Dennis SR, Robertson JA, et al. 2018. Comparative genomics reveals accelerated evolution in conserved pathways during the diversification of anole lizards. Genome Biol Evol 10: 489–506. 10.1093/gbe/evy013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecchione L, Byron C, Cooper GM, Barbano T, Hamrick MW, Sciote JJ, Mooney MP. 2007. Craniofacial morphology in myostatin-deficient mice. J Dent Res 86: 1068–1072. 10.1177/154405910708601109 [DOI] [PubMed] [Google Scholar]
- Vicoso B, Charlesworth B. 2006. Evolution on the X chromosome: unusual patterns and processes. Nat Rev Genet 7: 645–653. 10.1038/nrg1914 [DOI] [PubMed] [Google Scholar]
- Visel A, Minovitsky S, Dubchak I, Pennacchio LA. 2007. VISTA Enhancer Browser: a database of tissue-specific human enhancers. Nucleic Acids Res 35: D88–D92. 10.1093/nar/gkl822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. 2009. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457: 854–858. 10.1038/nature07730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Shen R-W, Han B, Li Z, Xiong L, Zhang F-Y, Cong B-B, Zhang B. 2017. Notch signaling mediated by TGF-β/Smad pathway in concanavalin A-induced liver fibrosis in rats. World J Gastroenterol 23: 2330–2336. 10.3748/wjg.v23.i13.2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wroe S, Milne N. 2007. Convergence and remarkably consistent constraint in the evolution of carnivore skull shape. Evolution 61: 1251–1260. 10.1111/j.1558-5646.2007.00101.x [DOI] [PubMed] [Google Scholar]
- Wroe S, Clausen P, McHenry C, Moreno K, Cunningham E. 2007. Computer simulation of feeding behaviour in the thylacine and dingo as a novel test for convergence and niche overlap. Proc Biol Sci 274: 2819–2828. 10.1098/rspb.2007.0906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Chen G, Li Y-P. 2016. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res 4: 16009 10.1038/boneres.2016.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Yuan J, Yao X, Zhang R, Yang H, Zhao R, Guo J, Jin K, Mei H, Luo Y, et al. 2017. BMPR1B mutation causes Pierre Robin sequence. Oncotarget 8: 25864–25871. 10.18632/oncotarget.16531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano M, Inoue Y, Tobimatsu T, Hendy G, Canaff L, Sugimoto T, Seino S, Kaji H. 2012. Smad7 inhibits differentiation and mineralization of mouse osteoblastic cells. Endocr J 59: 653–662. 10.1507/endocrj.EJ12-0022 [DOI] [PubMed] [Google Scholar]
- Yoon BS, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR, Lyons KM. 2005. Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc Natl Acad Sci 102: 5062–5067. 10.1073/pnas.0500031102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Wilder SP, Johnson N, Juettemann T, Flicek PR. 2015. The Ensembl Regulatory Build. Genome Biol 16: 56 10.1186/s13059-015-0621-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Feng J, Seidel K, Shi S, Klein O, Sharpe P, Chai Y. 2014. Secretion of Shh by a neurovascular bundle niche supports mesenchymal stem cell homeostasis in the adult mouse incisor. Cell Stem Cell 14: 160–173. 10.1016/j.stem.2013.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Zhang Y, Zhang W, Yang S, Chen JQ, Tian D. 2009. Patterns of exon-intron architecture variation of genes in eukaryotic genomes. BMC Genomics 10: 47 10.1186/1471-2164-10-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.