

Abstract
Malignant brain tumors, including glioblastoma, represent some of the most difficult to treat of solid tumors. Nevertheless, recent progress in immunotherapy, across a broad range of tumor types, provides hope that immunological approaches will have the potential to improve outcomes for patients with brain tumors. Chimeric antigen receptors (CAR) T cells, a promising immunotherapeutic modality, utilizes the tumor targeting specificity of any antibody or receptor ligand to redirect the cytolytic potency of T cells. The remarkable clinical response rates of CD19‐targeted CAR T cells and early clinical experiences in glioblastoma demonstrating safety and evidence for disease modifying activity support the potential of further advancements ultimately providing clinical benefit for patients. The brain, however, is an immune specialized organ presenting unique and specific challenges to immune‐based therapies. Remaining barriers to be overcome for achieving effective CAR T cell therapy in the central nervous system (CNS) include tumor antigenic heterogeneity, an immune‐suppressive microenvironment, unique properties of the CNS that limit T cell entry, and risks of immune‐based toxicities in this highly sensitive organ. This review will summarize preclinical and clinical data for CAR T cell immunotherapy in glioblastoma and other malignant brain tumors, including present obstacles to advancement.
Keywords: brain tumors, chimeric antigen receptors, glioblastoma, T cells
1. INTRODUCTION
Survival rates for many cancers have improved significantly over the past several decades due to progress in early detection, the advent of precision medicine, and advances in treatments.1 By contrast, brain tumors continue to be an exception to this trend with long‐term survivorship remaining unacceptably low. Glioblastoma (GBM) is the most common primary brain tumor in adults and is almost uniformly lethal with less than 10% of patients living beyond 5 years, and a median survival of 16‐20 months.2, 3 In children and young adults, primary brain tumors continue to be the leading cause of cancer‐related deaths.4 Patients who develop brain metastases also have an extremely poor prognosis with an average survival of typically less than 6 months after diagnosis.5 Current management options for local control of brain cancer, both primary and metastatic, include maximal safe resection, radiotherapy, and chemotherapy.6, 7 Such treatments are rarely curative and have a host of toxicities due to the sensitivity of this vital organ. The refractory nature of brain tumors to standard therapies provides compelling motivation for developing novel treatment interventions.
The tremendous progress in T cell immunotherapy across a broad range of tumor types provides hope that the immune system can be augmented to improve outcomes for patients with brain tumors. The brain, however, is an immune‐specialized organ presenting unique and specific challenges to the application of immunotherapy. Immune system access to the brain is tightly controlled and the immunosuppressive nature of the central nervous system (CNS) has evolved to protect against immunologic attack.8 Toxicity risks are also significant and potentially life‐threatening, including off‐tumor targeting of normal brain tissue and CNS inflammatory reactions.9 Although these challenges are considerable, emerging data supports the promise of the T cell immunotherapy for brain tumors.
Approaches to stimulate or enhance endogenous T cell immune responses to treat brain tumors are showing evidence of bioactivity, including clinical studies of tumor neoantigen vaccines,10 oncolytic viruses,11 and immune checkpoint inhibitors (ICIs).12, 13 The engagement of checkpoint receptors such as programmed cell death protein‐1 (PD‐1) and cytotoxic T‐lymphocyte‐associated antigen 4 (CTLA4) dampen anti‐tumor immune responses, and these checkpoint pathways have emerged as critical drivers of immunosuppression in solid cancers.14 In melanoma patients with CNS metastases, a phase II study evaluating nivolumab (anti‐PD‐1) combined with ipilimumab (anti‐CTLA‐4), reported that ICIs mediate intracranial clinical benefit in 57% of the patients with a 26% complete response rate.15 Remarkably, ICI clinical response rates against melanoma CNS metastases were durable and on par with systemic anti‐tumor responses. High tumor mutational burden (TMB; >20 mutations/mb) has previously been identified as an independent prognostic variable to response with ICI,16 and melanoma has some of the highest TMB of all malignancies, possibly contributing to the encouraging clinical results. Similar observations have also been reported in a subset of GBMs with high mutational burden due to mismatch repair deficiencies, and in these cases ICIs have also been shown to achieve complete and durable responses.17, 18 Most brain tumors, however, including GBM and pediatric brain tumors, have low TMB and have been less responsive to ICIs.19 Encouragingly, neoadjuvant ICI prior to surgical resection has been shown to mediate a survival benefit in recurrent GBM, possibly by augmenting endogenous T cell responses.20 These studies are important as they establish that brain tumors are not impervious to immunological recognition and attack, and point to immunotherapy making inroads in brain tumor treatment.
For most patients, endogenous immune responses are not potent or abundant enough to mount sufficient anti‐tumor responses even with ICI treatment, and thus the engineering of new T cell immunity using chimeric antigen receptors (CARs) is a rapidly advancing approach for the treatment of cancer.21 CAR T cells can be efficiently expanded ex vivo and delivered in large quantities to elicit tumor destruction. Tumor targeting by CARs is independent of major histocompatibility complex (MHC) antigen presentation and, therefore, may provide new options for tumors with low TMB or defects in antigen presentation. Early results with CAR T therapy against brain tumors have shown promise while at the same time illustrating multiple challenges. These include addressing tumor antigen heterogeneity, overcoming an immune‐suppressive tumor microenvironment (TME), ensuring sufficient T cell trafficking to the tumor, enhancing CAR T persistence, and avoiding CNS toxicity. In this review, we will present the most recent progress and challenges for CAR T cell immunotherapy in the treatment of brain tumors.
2. ENGINEERING TUMOR IMMUNITY WITH CARs
2.1. The design of CARs
Chimeric antigen receptors are synthetic immune receptors that redirect T cells to eradicate tumors through specific recognition of surface proteins expressed on tumor cells. CARs are fusion proteins comprised of three main components: the extracellular domain responsible for antigen recognition, the intracellular domain responsible for signal transmission, and region that links these two components for flexibility, stability, and dimerization potential composed of the extracellular spacer and transmembrane domain (Figure 1).21, 22 The intracellular domain contains the T cell co‐receptor CD3ζ, and second‐generation CARs include one co‐stimulatory molecule, such as CD137 (4‐1BB) or CD28, to enhance expansion and persistence.23 Third‐generation CARs contain two costimulatory domains in series with CD3ζ. One of the advantages of CARs is their modular design, which provides the flexibility to adjust the antigen recognition and signaling domains based on the targeted cancer types. Further, CAR T cells can be used as delivery vehicles when paired with constitutively or inducibly expressed chemokine such as IL‐12, antibody fragments, or other biomolecules.24, 25 Optimal CAR design is still an empirical process and highly dependent on antigen‐ and tumor‐specific properties. Innovations in CAR design and its application to the advancement of CAR T cell therapy have been previously reviewed.26 For brain tumor therapy, we will discuss how CAR design is evolving to address the challenges of tumor antigen escape, the suppressive TME and tumor trafficking.
Figure 1.
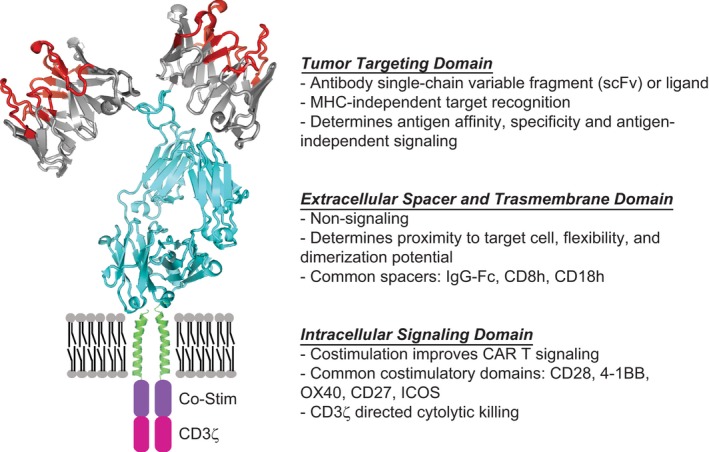
Chimeric antigen receptor (CAR) design. CARs are modular synthetic immunoreceptors that consist of a tumor targeting domain fused to an intracellular T cell signaling domain via the extracellular spacer and transmembrane (TM) domains. The tumor targeting domain has been designed and tested against multiple brain tumor antigens including IL13Rα2, HER2, and EGFRvIII (Table 2). The TM domain and extracellular spacer influence effector‐target cell interaction by providing flexibility, allowing dimerization to occur, and influencing stability. The cytoplasmic intracellular signaling domain is composed of a CD3ζ activation domain, and is most often paired with cognate T‐cell co‐stimulatory signaling domains (CD28, 4‐1BB, OX40, CD27, and ICOS), which improves CAR T‐cell proliferation, survival, and recursive killing
2.2. CAR T cell manufacture
Chimeric antigen receptors T cell manufacturing processes vary widely across therapeutic platforms, and the final product phenotype represents a critical variable impacting the potency of the therapy.27 The general workflow for autologous cell transfer begins with collection of patient T cells through leukapheresis followed by, in some protocols, the enrichment of bulk T cells or selection of specific T cell subsets. T cells are then stimulated by engagement of their TCR and costimulatory receptors, typically using anti‐CD3 and anti‐CD28 reagents. Variations on the T cell stimulation step include use of coated beads, plate‐bound antibody, soluble CD3 antibody only, and inclusion of other costimulatory or adhesion molecules (ie, CD2).27, 28, 29 Following activation, T cells are genetically modified to express the CAR, most commonly through lentiviral or retroviral transduction. The engineered CAR T cells are then ex vivo expanded in media containing common gamma chain (γc)‐cytokine cocktails, most commonly IL‐2 (eg, combinations of IL‐2, IL‐7, IL‐15, or IL‐21) to support T cell expansion. The choice of γc‐cytokine and concentration greatly impact the final phenotype of the expanded cells. Most manufacturing platforms utilize IL‐2, but there is evidence for advantages of using other cytokines, including IL‐15, IL‐7, and IL‐21.30, 31, 32, 33, 34, 35 A generalizable principle is that manufacturing processes yielding a less‐differentiated memory phenotype with greater mitochondrial fitness generates superior CAR T cell products.36, 37, 38 Reducing the time of ex vivo culture is one strategy that has been shown to yield more potent CAR T cell products, and most patient‐specific products are produced within two weeks or less.39 Ultimately this process is capable of generating millions to billions of engineered therapeutic CAR T cells ready to be infused back to the patient. A detailed summary of CAR T manufacturing has been reviewed previously.40, 41 In clinical manufacturing setting of brain tumors, there are many outstanding issues to be addressed, including the following: how intrinsic T cell product variability impacts potency; the effect of concomitant dexamethasone on the quality or quantity of manufactured product; whether certain T cell subsets traffic to the CNS more efficiently; and whether off‐the‐shelf manufacturing platforms will improve the consistency, timing, and availability of CAR T therapy.
3. ADOPTIVE T CELL THERAPY: FROM BLOOD TO BRAIN
Clinical success with CD19‐CAR T therapies and adoptive cellular therapy (ACT) studies in melanoma, while not initially intended to treat brain tumors, have nevertheless raised optimism for the potential of ACT for the treatment of CNS disease. In trials of ACT for metastatic melanoma, Hong and colleagues reported on a subset of patients (26 of 264) who were retrospectively identified to have had untreated melanoma brain metastases as well as extracranial disease.42 Remarkably, after treatment with ACT using either autologous tumor infiltrating lymphocytes or lymphocytes transduced to express a T cell receptor targeting a melanocyte‐specific antigen, nine patients (35%) achieved a complete response in the brain and seven patients achieved an overall partial response.42 Clinical experience with CD19‐CAR T therapy also serves as a roadmap for CAR targeting of brain tumors. CD19‐CARs have demonstrated remarkable clinical outcomes for patients with CD19‐positive B cell malignancies, which led to FDA approval in 2017 of the first two CAR T cell therapies: tisagenlecleucel (Kymriah) for relapsed/refractory (r/r) acute lymphoblastic leukemia (ALL),43 and axicabtagene ciloleucel (Yescarta) for r/r large B cell lymphoma (BCL).44 Early phase 1 testing of CD19‐CAR T cells excluded patients with CNS involvement, but evaluation of cerebrospinal fluid (CSF) post‐therapy revealed that the systemically administered CAR T cells often trafficked to the CNS. In a clinical trial of pediatric patients with r/r ALL, Lee and colleagues were the first to report that CD19‐CAR T cells were both detected in the CSF in the majority of patients evaluated, and also capable of eliminating CNS leukemia.45 Another case report described a patient with refractory BCL that had metastasized to the brain, and this patient had a complete remission of an intraparenchymal brain metastasis after receiving CD19‐CAR T cell therapy.46 While ACT for brain tumors is still in early stages of investigation and clinical responses are often ineffective, these experiences illustrate the potential of T cell therapy to provide clinical benefit for patients with CNS disease, and have generated enthusiasm for furthering the application of this therapeutic approach to brain tumors. Indeed, there are currently more clinical trials evaluating CAR T cell therapy for brain tumors than any other solid tumor indication (Figure 2).
Figure 2.
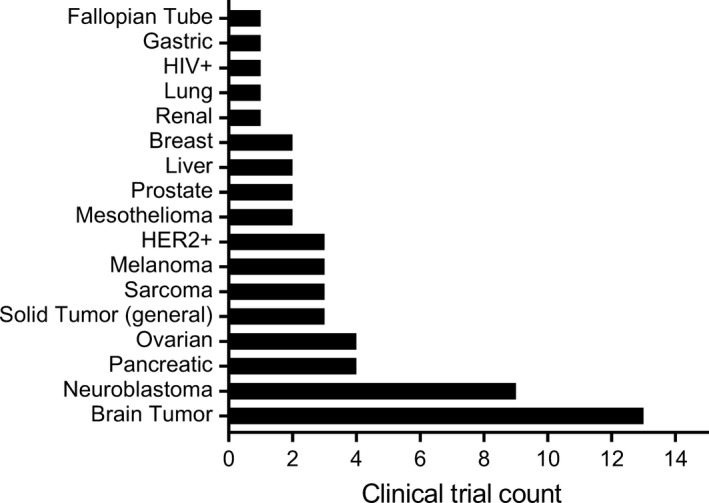
Chimeric antigen receptor (CAR) T cell trials for brain tumors vs other solid tumors. Clinical trial count in the United States evaluating CAR T cell therapy for solid tumors as of April 2019. As graphically represented, the largest number of CAR T cell trials for solid tumors is for brain tumors, which include trials for glioblastoma and other malignant gliomas (n = 12), as well as brain metastases (n = 1). Listed trials include those that are pending activation, enrolling patients, and completed
CD19‐CAR T clinical experiences have also provided points of caution for neurotoxicity that can be associated with CAR T cells. The incidence of high‐grade neurotoxicity occurs in approximately 12%‐32% of patients treated with CD19‐CARs across indications, and includes symptoms of delirium, confusion, and seizures.47, 48, 49, 50 Lethal cerebral edema has also been reported in a handful of patients treated with CD19‐CAR T cells in B‐ALL and non‐Hodgkin's lymphoma,51, 52 highlighting the life‐threatening risks of immune inflammatory reactions in the CNS. The gamut of these neurologic toxicities are referred to as CAR T‐related encephalopathy syndrome (CRES). Several studies have focused on the etiology of CD19‐CAR CRES. Gust et al evaluated 133 patients with B‐cell malignancies treated with CD19‐CARs by interrogating pre‐ and post‐therapy cytokine and metabolite levels in CSF and serum, and MR imaging changes, as well as post‐mortem brain for correlatives of CRES.48 This study revealed that patients with severe neurotoxicity demonstrated evidence of endothelial cell activation, including disseminated intravascular coagulation, capillary leakage, and increased blood‐brain barrier permeability. The permeable BBB failed to protect the CSF from high concentrations of systemic cytokines, which resulted in brain vascular pericyte stress and secretion of endothelium‐activated cytokines. This is supported by other studies demonstrating that CAR‐related neurotoxicity is associated with disease burden, high CAR T dose, and cytokine release syndrome (CRS).53 Norelli and colleagues simulated CAR T cell‐mediated CRES in a humanized mouse model with high leukemia burden.54 Human monocytes were the major source of IL‐6 and IL‐1 during CRS and CRES, and both toxicities were prevented by monocyte depletion. IL‐1‐receptor blockade inhibited both CRS and neurotoxicity in mice, whereas blocking IL‐6 decreased CRS‐like symptoms without reducing the incidence of lethal neurotoxicity. Non‐human primate models also have demonstrated an association with CD19‐CAR‐mediated neurotoxicity, proinflammatory CSF cytokines, BBB disruption, and pan T cell encephalitis.55 The clinical trial experience in CD19‐CAR will help inform future trials related to CARs for brain tumors. Important clinical trial correlates should include pro‐inflammatory CSF cytokines measurements, including IL‐1 and IL‐6, magnetic resolution tomography (MRI) evaluation for BBB disruption, and neuro‐monitoring. Whether CAR therapy applied to CNS tumors will cause similar problems requires further investigation, but has not been observed to date in initial clinical studies with CAR T cell therapy for GBM.
4. INITIAL CLINICAL EXPERIENCE WITH CAR T CELL THERAPY FOR GBM
Initial clinical experiences with CAR T cells for brain tumors have focused on treating recurrent or refractory GBM in both adult and pediatric patients and targeting a limited set of antigens: IL13Rα2, EGFR variant III (EGFRvIII), and HER2 (Table 1). These lead therapeutic candidates were selected based on evidence for negligible expression on normal brain and documented expression by GBM. For brain tumors, restricted expression is particularly critical because off‐tumor targeting of normal brain can result in lethal toxicity, as has been unfortunately observed with MAGE‐A3 TCR‐engineered T cells. The therapeutic T cells recognized an off‐target epitope of MAGE‐A12 expressed in the brain, resulting in lethal neuronal cell destruction and inflammatory responses.56 Given the risks associated with targeting normal brain tissue and neuroinflammation, caution is required regarding many factors influencing adoptive cell activity, such as cell dose and preparative regimens, as well as the identification of tumor‐specific targets. First‐in‐human clinical trials targeting IL13Rα2, EGFRvIII, and HER2 have elucidated important insights regarding safety and bioactivity that are guiding future translation of CARs for brain tumors.
Table 1.
First‐in‐human clinical experience with CAR‐T therapy for glioblastoma
| Target antigen | Trial | Aim | CAR T product | Route of delivery | Antigen loss | TME | Toxicity | Patient outcome |
|---|---|---|---|---|---|---|---|---|
| IL13Rα2 |
NCT00730613 City of Hope66, 68 |
First to evaluate intracranial delivery of IL13Rα2 CAR in rGBM |
IL13(E13Y)‐CD3ζ CD8+ CTL clones |
ICT | YES: IL13Rα2 negative/low (1 patient tested) | Transient inflammatory response/necrosis at tumor site by MRI | No DLTs |
3 pts evaluated Median OS 10.9 mo Increased necrotic volume at treatment site (1 pt) Decreased IL13Rα2 expression (1 pt) |
| IL13Rα2 |
NCT01082926 City of Hope67 |
First off‐the‐shelf allogeneic CAR T cells for rGBM. Evaluated feasibility of [18F]FHBG gene reporter imaging to monitor T‐cell distribution in rGBM |
IL13(E13Y)‐CD3ζ [18F]FHBG HSV1‐tk Glucocorticoid‐receptor‐depleted allogeneic CD8+ CTL clone |
ICT | Not reported | Not reported | No DLTs |
6 pts evaluated [18F]FHBG gene reporter allowed longitudinal imaging of ICT CAR‐T Clinical outcomes not reported |
| IL13Rα2 |
NCT02208362 City of Hope69 |
Evaluate safety of ICV and dual ICT‐ICV CAR delivery in rGBM |
IL13(E13Y)‐41BBζ Memory‐derived T cells |
ICT, ICV and dual ICT‐ICV | YES: IL13Rα2 negative/low tumors (1 patient reported) | Increased CD3+ CD14+ and CD15+ immune cells and inflammatory cytokines | No DLTs | Case study demonstrated CAR‐Ts mediate complete response that was durable for 7.5 mo |
| EGFRvIII |
NCT02209376 UPenn80 |
To determine safety and efficacy of single‐dose IV EGFRvIII CAR‐T in rGBM |
EGFRVIII‐41BBζ Bulk T cells |
IV | YES: EGFRvIII decreased in 5/7 patients | Increased IDO, FOXP3, IL‐10, PD‐L1 and TGFβ | No DLTs |
10 pts evaluated Median OS 8.3 mo 1/10 extended SD, alive @ 18 mo Decreased EGFRvIII expression (5 of 7 pts) |
| EGFRvIII |
NCT01454596 NCI81 |
To determine MTD and DLT of IV EGFRvIII CAR‐T and IL‐2 following lymphodepletion in rGBM |
EGFRvIII‐CD28‐41BBζ Bulk T cells |
IV | Not evaluated | Not evaluated |
2 DLTs 1 grade 5 at highest dose |
18 pts evaluated Median PFS 1.3 mo Median OS 6.9 mo 1/18 pts median PFS of 12.5 mo and alive at 59 mo |
| HER2 |
NCT01109095 Baylor, TX260 |
To determine safety and activity of HER2 CAR‐Tin adult and pediatric rGBM |
HER2‐CD28ζ VST: (EBV‐CMV‐AD) |
IV | Not evaluated | Not evaluated | No DLTs |
16 pts evaluated Median OS 11.1 mo 1/16 PR 7/16 SD (3 extended SD) |
Abbreviations: CAR, chimeric antigen receptors; DLT, dose limiting toxicity; ICT, intra‐cranial tumoral; ICV, intra‐cranial ventricular; IV, intravenous; rGBM, recurrent glioblastoma; VST, virus specific T cells.
4.1. CAR T cells targeting IL13Rα2
The first CAR T cells developed and optimized for brain tumors targeted IL13Rα2, a high affinity IL‐13 receptor first discovered by Debinski and colleagues to be overexpressed by the majority of GBMs.57 IL13Rα2 expression has been shown to increase with malignancy grade, to be a prognostic indicator of poor patient survival, and to be associated with gene signatures defining the mesenchymal subclass of GBM.58, 59 IL13Rα2 is expressed by both glioma stem‐like cells (GSCs) and differentiated tumor populations,60 rendering both malignant subpopulations susceptible to CAR T cell cytotoxicity. Importantly, IL13Rα2 is not expressed at significant levels on normal brain tissue.59, 61
Our group generated fully human IL13Rα2‐CARs with the human IL‐13 cytokine for tumor recognition, which represents a distinct class of CARs utilizing receptor ligands vs antibody scFvs for tumor targeting. Drawing from work on cytotoxin‐conjugated IL‐13 that defined mutations which increased the specificity of IL‐13 for IL13Rα2 over the more ubiquitously expressed IL13Rα1/IL‐4Rα complex, an E13Y site‐directed mutation was introduced in IL‐13.62, 63 In vitro studies showed that CAR T cells were specific to glioma cells, and consistent with the different affinities of the IL‐13(E13Y) mutein for IL‐13 receptor forms, the engineered T cell activity was observed only against cell lines expressing IL13Rα2.63, 64 First‐generation IL13Rα2‐targeted CARs, termed IL13‐zetakine, demonstrated anti‐glioma activity in vitro and in vivo in mice,63 and second‐generation CAR designs with the inclusion of a 4‐1BB costimulatory domain and an optimized spacer domain improved anti‐tumor potency more than 10‐fold against human GBM xenografts.64, 65
IL13Rα2‐targeted CAR T cells were the first to be clinically translated for the treatment of malignant glioma in trials at City of Hope. First‐generation IL13‐zetakine CD8+ T cell clones were evaluated in two FDA‐authorized clinical trials employing autologous (NCT00730613, 3 patients) and allogeneic (NCT01082926, 6 patients) engineered T cells for resectable and non‐resectable recurrent GBM, respectively.66, 67, 68 These initial clinical experiences demonstrated the feasibility and safety of repetitive locoregional intratumoral delivery (intracranial tumoral: ICT) of ≥1 × 108 CAR T cells through a reservoir/catheter system. For all 9 patients, no dose‐limiting toxicities (DLTs) were observed, and a subset of patients presented clinical evidence of transient anti‐glioma activity.66 This included increased tumor necrotic volume by MRI and PET, significant reduction in IL13Rα2+ tumor cells, and detection of transferred T cells at tumor microfoci distal from the site of injection.66, 67, 68 However, first‐generation IL‐13‐zetakine CAR T cell therapy yielded limited T cell persistence,66, 67, 68 and improvements in CAR design and manufacturing were initiated with the goal of improving therapeutic potency.64, 69
In 2015, our program at City of Hope initiated a phase I clinical trial evaluating second‐generation IL13Rα2‐targeted, 4‐1BB‐costimulatory CAR T cells for patients with r/r IL13Rα2+ malignant glioma (NCT02208362). Preclinical studies (reviewed below) led to our hypothesis that the CAR T cell route of administration is a key parameter for maximizing the benefit of therapy. The trial was expanded to evaluate not only CAR T cells administered ICT, but also by two additional routes of local delivery: intraventricular (intracranial ventricular: ICV) and dual intratumoral/intraventricular (ICT/ICV). Interim findings to date demonstrate that locoregional delivery of second‐generation IL13Rα2‐CAR T cells is safe and well‐tolerated, with no observed DLTs. With the study still ongoing, we described in a New England Journal of Medicine case report, a patient who responded strongly to CAR T cell therapy.69
This patient had stable disease while undergoing ICT delivery of CAR T cells, and later demonstrated a complete radiographic response of multifocal lesions after undergoing ICV delivery of CAR T cells.69 During the response that lasted approximately 7.5 months, the patient experienced dramatic improvements in his quality of life, including the discontinuation of systemic glucocorticoids and a return to normal life activities. Correlative studies of samples from this patient indicate a potential role of the endogenous immune system in anti‐tumor responses. Observed increases in endogenous immune cells and inflammatory cytokines after each intraventricular infusion of CAR T cells may reflect recruitment and stimulation of the host immune system. This may explain how a complete response was achieved despite non‐uniform expression of IL13Rα2 on the responding tumors. Unfortunately, the patient developed tumor recurrence at non‐adjacent areas within the brain. Recurrent tumors exhibited lower expression of IL13Rα2, thus evading targeted killing by IL13Rα2‐CAR T cells. These correlative results highlight the potential interplay between host and CAR‐mediated immune responses, as well as the challenge of antigen escape as a pathway of therapeutic resistance. Based on the safety thus far established for IL13Rα2‐CAR T cells, as well as the striking result of a patient who had a radiographic complete response,69 we aim to perform more robust clinical investigations of this CAR design. Other groups are also developing IL13Rα2‐CARs for clinical translation, using both ligand‐based and antibody‐based antigen targeting domains.70, 71, 72, 73, 74
4.2. CAR T cells targeting EGFRvIII
Epidermal growth factor receptor (EGFR) is a tyrosine kinase receptor that is genetically amplified and/or mutated in approximately 50% of adult primary GBM, making it an attractive candidate for targeted therapy.75 EGFR variant III (EGFRvIII), the genetic mutation most commonly found in glioma, has a truncated extracellular domain that leads to constitutive signaling activation and is associated with GBM development and progression.76 This truncated variant presents a novel, tumor‐specific immunogenic epitope for generating scFv‐based CAR‐targeting domains, and because of its tumor‐restricted expression, EGFRvIII is one of the most actively investigated CAR targets for GBM.77, 78, 79, 80
The NCI published results of a dose escalation study of EGFRvIII CAR T cells in recurrent EGFRvIII+ GBM utilizing a third‐generation human scFv‐based EGFRvIII CAR with CD28 and 4‐1BB costimulation (NCT01454596).78, 81 CAR T cells were delivered intravenously after lymphodepleting chemotherapy, followed by systemic IL‐2 support. At initial dose levels (107−109 cells), EGFRvIII‐CAR T cells were well‐tolerated, with no evidence of off‐tumor targeting of EGFR. At the highest dose levels of ≥1010 T cells, pulmonary toxicities were observed, including one treatment‐related mortality after IV infusion of 6 × 1010 cells, resulting in respiratory symptoms within hours of cell infusion. Post‐mortem analysis demonstrated significant pulmonary edema.81 In the majority of patients (14 of 18), low levels of CAR T cell persistence could be detected one month after infusion. For the 18 patients treated, the median survival was 6.9 months and no objective responses were observed, although one patient was reported to have progression free survival (PFS) of 12.5 months, and was still alive at 59 months.81 This study highlights challenges associated with treating brain tumors with targeted therapy, including the need to better understand the mechanism of action of the therapeutic cells at the tumor site.
Researchers at the University of Pennsylvania have also developed a humanized EGFRvIII‐specific CAR and showed that it efficiently targeted orthotopically implanted EGFRvIII+ GBM in preclinical models. The scFv was optimized for specificity against EGFRvIII over wild‐type EGFR, and showed minimal reactivity to primary human tissue in vitro and to human skin grafts in vivo.82 This CAR construct was employed in an EGFRvIII‐CAR T phase I clinical trial in which 10 patients with EGFRvIII+ GBM received a single intravenous infusion prior to surgical resection with the goal of understanding safety and CAR activity (NCT02209376).80 Analysis of the resected tumor post CAR T cell infusion showed that the IV‐infused CAR T cells trafficked to the brain and also revealed downregulation of EGFRvIII expression in the recurrent tumor suggesting antigen‐specific activity. This study further highlights the important challenges to CAR T therapy, including tumor heterogeneity and the suppressive TME. Baseline EGFRvIII expression in patient GBMs was heterogeneous so the CARs targeted only a fraction of cancerous cells, and relapse tumors emerged that had decreased EGFRvIII expression levels. Further, the immunosuppressive TME intensified upon CAR T cell administration, including upregulation of IDO1, PD‐L1, and IL‐10. Non‐CAR polyclonal T cells were observed to increase the TME, and phenotypic analysis indicated the cells were mostly immunosuppressive regulatory T cells based on their expression of CD4, CD25, and FoxP3. This immunosuppressive response to CAR T treatment suggests that countermeasures such as immune checkpoint blockade might work synergistically with EGFRvIII‐CAR T therapy, and a combination trial of EGFRvIII‐CAR T cells with the anti‐PD‐1 antibody pembrolizumab is currently underway (NCT03726515).
Although the tumor‐restricted expression profile of EGFRvIII render this receptor an attractive target, its expression is reported to be unstable throughout the course of disease,83 increasing the likelihood of antigen‐negative escape variants under targeted therapy. For this reason, wild‐type EGFR may be a more attractive target for CAR T targeting, given that it is over‐expressed in more than 60% of GBM.84 However, because EGFR is expressed on many normal tissues, including skin, bladder, and liver, there is significant risk of on‐target off‐tumor toxicity. To address this concern, antibodies have been generated to target activated and over‐expressed EGFR and EGFRvIII. One such selective antibody, mAb806, has been shown to selectively bind EGFRvIII and amplified EGFR.85 A clinical study of the biodistribution and tumor localization of 111Indium‐EGFR806 antibody in seven patients demonstrated no evidence of normal tissue uptake nor any significant toxicity.86 An EGFR806‐CAR is currently being evaluated in a phase I trial in pediatric solid tumors at Seattle Children's Hospital and will provide important safety information regarding on‐target off‐tumor toxicity. Other groups have affinity‐tuned antibodies to amplified EGFR and EGFRvIII, demonstrating that lower affinity toward endogenous EGFR can improve the specificity of CAR targeting.87, 88, 89
4.3. CAR T cells targeting HER2
Human epidermal growth factor receptor 2 (HER2) is overexpressed in many cancers and well‐studied in breast cancer.90 The majority of GBMs are reported to express HER2, but the receptor is not observed in normal brain tissue (www.proteinatlas.org).91 Safety considerations regarding targeting HER2, however, are important to note due to the death of the first patient treated with HER2‐CAR T cell therapy. The patient received lymphodepleting chemotherapy prior to intravenous (IV) infusion of 1010 HER2‐CAR T cells, a comparatively high dose of transferred cells.92 The death was speculated to be the result of off‐tumor targeting of normal lung epithelial tissue, which triggered cytokine release resulting in respiratory distress and pulmonary edema. This finding raised questions about CAR design and therapeutic safety, particularly of the tumor‐targeting scFv, which was based on the high‐affinity trastuzumab antibody. In addition, the CAR costimulatory signaling was produced by both CD28 and 4‐1BB in a third‐generation design. This clinical experience has prompted the preclinical development of more selective CAR designs, including lower affinity mutants of the trastuzumab scFv,89 optimization of the trastuzumab‐CAR costimulatory domain for lower cytokine production and improved tumor selectivity,93 and the use of the lower affinity HER2‐monoclonal antibody FRP5 for the tumor‐targeting domain.94, 95 An understanding of how HER2‐CAR designs impact both safety and anti‐tumor potency in the clinic remains under investigation.
To date, the most extensive HER2‐CAR T cell clinical experience has been from the Baylor group evaluating the FRP5 HER2‐CAR in a second‐generation CD28 costimulatory format.94, 95 HER2‐CAR T cells specifically killed HER2‐positive GBM cells and CD133‐positive GSCs, which have been shown to be resistant to radio‐ and chemo‐therapies. Preclinical studies also showed that HER2‐CAR T cells do not target HER2‐negative cells, and primary endothelial and epithelial cells did not activate HER2‐CAR T cells.95 An initial safety evaluation of HER2‐CAR T cells (up to 1 × 108/m2) without lymphodepletion in patients with sarcoma demonstrated safety and indications of anti‐tumor activity despite limited persistence of the CAR T cells.96 With the goal of further improving persistence of adoptively transferred T cells, the team engineered CARs into virus‐specific T cells97 where costimulation results from T cell engagement with latent virus antigens on antigen‐presenting cells. A phase 1 dose‐escalation study established the safety of autologous HER2‐CAR virus‐specific T cells in 17 patients with progressive GBM (NCT01109095). The CAR T cells did not expand, but they were detectable in the peripheral blood for up to 12 months. Of 8 patients, one had a partial response and seven had stable disease. The median overall survival (OS) was 11.1 months. This HER2‐CAR T cell clinical experience provides initial evidence of safety, but also illustrates the need for improved expansion, function, and persistence of the HER2‐CAR T cells.
In studies of the HER2‐CAR design, our preclinical work demonstrated decreased cytokine production and improved anti‐tumor efficacy of 4‐1BB vs CD28 costimulation.93 Moreover, ICV‐delivered CAR T cells were shown to eradicate tumors implanted in both hemispheres. City of Hope has recently initiated two phase I trials using an optimized HER2‐CAR for patients with HER2+ malignant glioma and for patients with breast metastases to the brain (NCT03389230 and NCT03696030, respectively). Given our clinical experience with IL13Rα2‐CAR in glioma and preclinical studies evaluating route of delivery,64, 69, 93 we surmised that locoregionally delivered HER2‐CAR may be effective for both primary brain tumors as well as brain metastases. These clinical studies are evaluating locoregional delivery of HER2‐CAR T cells to shed light on whether locoregional delivery methods may reduce the potential for systemic toxicity. One important distinction between these two trials is the relatively lower expression of HER2 in glioma compared with HER2‐amplified breast cancer, as well as the distinctly different genetic backgrounds and TME. Only with clinical testing and multi‐parameter molecular correlative data analysis will we determine how these vastly different tumors respond to the same CAR T treatment.
4.4. Lessons learned from initial clinical experience with CAR T cells for GBM
The clinical trial experience of IL13Rα2‐, EGFRvIII‐, and HER2‐CAR T cells against GBM provide initial evidence of the safety and anti‐tumor activity of CAR T cell immunotherapy in patients with malignant brain tumors. Clinical results published from studies support the safety of targeting these GBM‐associated antigens in the CNS. Also encouraging, a subset of patients appeared to show benefit from treatment. After treatment with IL13Rα2‐CAR T, one patient had a CR lasting about 7.5 months, which was remarkable given the aggressive multifocal nature of his recurrent disease. After treatment with EGFRvIII‐CAR T cells on the clinical trial NCT02209376, one of 10 patients had extended stable disease and remained alive more than 18 months,80 and on the NCT01454596 trial, one patient also had extended PFS of 12.5 months.81 Finally, after HER2‐CAR T treatment, 1 of 17 patients had a partial response and seven of 17 patients had stable disease (three extended stable disease). More investigation is needed to determine whether these patients were more responsive to therapy due to underlying differences in tumor biology and immune landscape.
Analysis of tumor tissue after IL13Rα2‐ or EGFRvIII‐CAR T cell infusions provide evidence for antigen loss as a pathway of therapeutic escape. Recurrent tumors in two patients treated with IL13Ra2‐CARs (NCT02208362 and NCT00730613) showed lower levels of the IL13Rα2, and in patients tested on NCT02209376, five of seven had lower levels of EGFRvIII and two of seven patients had undetectable antigen. Although these results may indicate tumor cell killing by CAR T cells, they also illustrated that antigen loss must be overcome in developing next‐generation CAR therapies (discussed in detail below). These initial clinical experiences also highlight the potential interplay between the host immune system and CAR T cells in the CNS. In response to IL13Rα2‐CAR T therapy, the TME had increased CD3+ CD14+ CD15+ immune cells in addition to inflammatory cytokines after each loco‐regional CAR infusion. In response to EGFRvIII‐CAR T therapy, the TME had increased IDO, FOXP3, IL‐10, PD‐L1 markers, and TGFβ. Understanding the determinants of response will help us understand who will benefit most from treatment, and importantly, may help us to modify adoptive immunotherapies to overcome obstacles presented by the tumor and TME.
5. OVERCOMING TUMOR HETEROGENEITY
5.1. Understanding the heterogeneity of brain tumors
To expand the repertoire of antigens for CAR T cells and identify optimal targets to limit antigen escape of brain tumors, an understanding of the complexity of tumor heterogeneity is essential. Tumor cell plasticity has been rigorously investigated as a major factor complicating cancer diagnosis and treatment, as well as mediating therapeutic resistance.98 Molecular and cellular heterogeneity manifests as one of the most notable characteristics of brain tumors, especially GBMs.99 The past two decades have seen the evolution of GBM genetic characterization, leading to the identification of subtypes based on mutations in key oncogenic pathways and differences in gene expression profiles.100 The existence of GBM subtypes helps explain the different responses to therapies, and some of the genetic signatures (eg, IDH1/2, MGMT, H3K27) have been used to guide diagnosis and treatment.3 The switch between subsets has also been shown to associate with tumor relapse.101 However, the complexity of brain tumor heterogeneity appears to go beyond the differential expression of certain genes and molecular subtypes.
Classifying GBMs into subtypes has focused on genetic signature at the whole‐tumor level, thereby simplifying the substantial intratumoral heterogeneity of GBMs. Independent studies have elucidated the differential gene expression patterns in distinct anatomic regions within a tumor.102, 103 More specifically, the leading‐edge of GBMs was shown to display a proneural‐like signature, while the tumor core appeared more mesenchymal‐like.103 The intratumoral heterogeneity of GBMs was further illustrated by cellular and genomic analysis at the single‐cell level. Meyer and colleagues profiled individual tumorigenic clones from patient‐derived GBM cells, identifying pre‐existing clones harboring the potential to resist anti‐tumor treatments such as TMZ.104 The results indicated that pre‐treatment tumor heterogeneity might be responsible for clonal selection toward recurrence. In addition to isolating and characterizing subclones from GBM tumors, other studies used an approach to directly sequence primary tumor cells with advanced single‐cell RNA‐sequencing (scRNAseq) technologies.105, 106 All of these studies used a subset of brain tumors categorized by well‐established markers such as IDH1/2 or H3K27, but a significant level of intratumoral cell‐to‐cell heterogeneity was still discovered. Variation was observed in a diverse array of transcriptional programs regulating oncogenic signaling, proliferation, complement/immune response, and hypoxia. Furthermore, the scRNAseq data were compared to the original classification scheme established by The Cancer Genome Atlas (TCGA) to distinguish four GBM subtypes: proneural, neural, classical, and mesenchymal. Importantly, the TCGA subtypes were established from bulk tumor profiles, and the scRNAseq phenocopied the four subtypes when similarly compared as a bulk tumor. However, on a single‐cell RNAseq analysis, all five evaluated tumors had individual cells with proneural subtype regardless of the dominant subtype of the tumor.105 Such molecular and cellular dynamics better explained the complexity of the disease in addition to the population‐level classification.
Intriguingly, scRNAseq has also revealed a nonautonomous regulatory mechanism of brain tumor heterogeneity. In a comparison between the single‐cell transcripts of IDH‐mutant astrocytomas and oligodendroglioma, no significant difference in glial lineage composition was found, suggesting a common malignant developmental re‐programming; instead, the TME signatures were distinct between the two types of tumors.106 Further, higher grade astrocytomas and oligodendrogliomas were found to associate with the enrichment of macrophage over microglia.106 In GBMs, immunological signatures were found to differ between subtypes, with the mesenchymal‐like tumors harboring an enrichment of immune‐related genes.107 Moreover, compared with primary GBMs, recurrent GBMs demonstrated an altered composition of infiltrating immune cells, particularly a noted decrease in monocytes.101 Together, these findings illustrate the heterogeneity in brain tumors at multiple levels including inter‐patient, intratumoral, and within the TME.
Tumor heterogeneity has complicated CAR T cell therapy for brain, as well as other tumors. Indeed, even with the success of CD19‐CAR T cell therapy against B cell malignancies, patients with complete responses often relapsed following therapy with undetectable CD19 antigen.108 Further, the loss and/or decrease in target antigen expression has also been observed in CAR T cell clinical studies of GBM.66, 69, 80 We have reported examples of IL13Rα2 loss/decrease in relapsed/recurrent tumors following IL13Rα2‐CAR T therapy.69 Likewise, in a study using EGFRvIII‐CAR T cells, 4 of 6 of resected tumors post CAR T cell infusion displayed significant down‐regulation of EGFRvIII.80 Therefore, advancing CAR therapy against brain tumors requires strategies to minimize antigen escape caused by tumor heterogeneity.
5.2. Discovering new targets
Tumor antigen heterogeneity in brain tumors leads to resistance against CAR T cells, pointing to the need for an expanded repertoire of targeted antigens. The success of CD19‐CAR T cells highlight the criteria that an ideal CAR target should be widely expressed across different tumors and intratumoral cellular subsets. However, finding such antigens in brain tumors has been challenging since brain tumor cells express many markers which are shared by regions of the normal brain (eg, CD133, CD44, Nestin, GFAP), and the off‐tumor targeting consequences in the CNS are far less tolerable than most other parts of the human body. Therefore, the discovery of CAR T cell targets in brain tumors requires consideration of the breadth and specificity of tumor‐antigen expression.
In addition to targets mentioned in the sections above (IL13Rα2, EGFRvIII and HER2), other antigens are under investigation for CAR therapy in brain tumors (Table 2). Erythropoietin‐producing hepatocellular carcinoma A2 (EphA2) was found to be over‐expressed in GBMs, and to enhance tumorigenesis and migration.109 An EphA2 inhibitor showed anti‐tumor effects in mouse models of pancreatic ductal adenocarcinoma.110 EphA2‐targeted CAR T cells with second‐ or third‐generation CAR designs were able to eradicate GBMs in preclinical studies, yet with limited persistence.111 EphA2‐CAR T cells are currently being evaluated in GBM patients, but no clinical results have been reported (NCT02575261).
Table 2.
Brain TAA targeted by CAR
| Antigen | Expression on brain tumors | Expression on normal tissues | Preclinical investigation of CAR targeting the brain TAA |
|---|---|---|---|
| B7‐H3 | Highly expressed in high‐grade gliomas and other brain tumors | Liver, lung, bladder, testis, prostate, breast, placenta, and lymphoid organs | 118 |
| CD133 | Glioma tumor‐initiating cancer stem cells | Hematopoetic stem cells, endothelial progenitor cells, neuronal stem cells | 123 |
| CSPG4 | Uniform in GBMs (67% high expression) | Chondroblasts, pericytes, cardiomyocytes | 119 |
| EGFRvIII | Most common EGFR mutation in GBM; approximately 30% of GBMs | Restricted | 77, 82 |
| EphA2 | Uniform in high‐grade glioma with various levels | Epithelial tissue | 111, 261 |
| GD2 | Uniform in DIPGs; low in high‐grade gliomas | Central nervous system, peripheral nerves, and skin melanocytes | 112 |
| HER2 | Moderate expression on GBM; highly expressed on other solid tumors that metastasize to the brain | Epithelial tissue, skin and muscle | 93, 95 |
| IL13Rα2 | Majority of GBM and other high‐grade gliomas | Testis | 60, 70, 262 |
Abbreviations: CAR, chimeric antigen receptors; GBM, glioblastoma; TAA, tumor‐associated antigens.
Another antigen that has been intensively investigated as a CAR target is the disialoganglioside GD2. GD2 was found to be expressed in a class of pediatric brain tumors called diffuse midline gliomas (DMGs) that bear mutations in the histone H3K27. GD2‐CAR T cells effectively controlled tumor growth in preclinical models of DMGs, even with tumors diffused into the spinal cord.112 GD2‐CAR T cells were well tolerated and exhibited some clinical activity in neuroblastoma patients,97, 113 but their clinical anti‐tumor efficacy and safety against DMGs remain to be addressed.
B7‐H3 (CD276) is an immune‐checkpoint molecule, which negatively regulates T cell activation,114, 115 and is also associated with tumor migration and invasion.116 An analysis on the TCGA database showed that B7‐H3 expression is up‐regulated particularly in high‐grade gliomas.117 Majzner and colleagues developed a CAR‐targeting B7‐H3, showing preclinical anti‐tumor activity against multiple types of pediatric tumors including medulloblastoma,118 indicating potential clinical application of B7‐H3‐CAR T cells against certain types of brain tumors.
Another study screened a panel of primary GBM samples representing various molecular subtypes, and identified Chondroitin sulfate proteoglycan 4 (CSPG4, also known as neuron‐glial antigen 2, NG2) to be widely expressed across these tumors.119 Moreover, CSPG4 expression can be induced by the immune‐stimulatory cytokine TNFα, which is produced during CAR T cell anti‐tumor responses thereby making tumor antigen escape less likely.119
Glioblastoma tumors contain specific subsets with the characteristics of self‐renewal and tumor regeneration, and studies have shown these GBM stem‐like cells (GSCs) to mediate resistance against radiotherapy and chemotherapy.120 The ability to eliminate GSCs, therefore, is essential for GBM‐targeting therapies to minimize tumor recurrence. On the one hand, using the strategy to enrich and expand tumor spheres from resected GBMs,121 different studies have proven the CAR T cells targeting IL13Rα2, HER2 and EGFRvIII are able to eliminate differentiated GBM cells as well as GSCs,60, 78, 95 indicating that CAR‐mediated cytotoxicity is not dependent on the “stemness” of GBM cells. On the other hand, CAR T cells have been developed against CD133 which is one of the surface markers of cancer stem cells including GSCs.122 CD133‐CAR T cells have shown preclinical cytotoxicity against patient‐derived GSCs,123 as well as anti‐tumor response in patients with tumors in liver, pancreas, and colon.124 However, CD133 is also expressed in neural stem cells,125, 126 thus raising the safety concerns of applying CD133‐CARs to GBM patients.
As an alternative to antibody‐based CAR designs, tumor‐binding peptides can also be exploited as the tumor‐targeting domain of a CAR. The utility of tumor‐binding peptides has already been demonstrated clinically for both diagnosis and treatment of cancers.127, 128 Recently, we have developed a GBM‐targeting CAR bearing the scorpion‐derived peptide chlorotoxin (CLTX).129 Studies have confirmed the specificity of CTLX binding to GBM, demonstrating its exquisite ability to distinguish between tumor and normal brain tissues.130 A fluorescently labeled CLTX agent is currently under clinical investigation as a tumor imaging agent to enable more precise surgical resection.131, 132 When incorporated into CARs, CLTX redirects T cells for specific tumor recognition with negligible off‐target effects on normal cells or tissues.129 The development of a CLTX‐CAR has shown the potential for peptide binding to redirect CAR T cell cytotoxicity.
Further, the pool of brain tumor antigens amenable to CAR T cell targeting is expected to expand beyond membrane‐associated proteins. Chheda and colleagues discovered a shared neoantigen across DIPG patients derived from a mutation in the H3.3K27.133 Stimulating HLA‐A2+, CD8+ T cells with the mutated peptide generated a TCR clone which, when expressed on T cells, mediated cytoxicity against tumor cells harboring the same mutation.133 Moreover, some recent studies have developed CARs against soluble proteins,134, 135 indicating the potential to target brain tumor‐specific secreted factors.
These efforts to expand the identification of tumor targets for GBM and other brain tumors provides new options for CAR T cell therapeutic development. Of course, the critical benchmark will be to establish safety for these new antigens in well‐designed clinical trials (see below). Once safety is established, tremendous opportunities become available to utilize CAR T cells targeting one or several of these antigens, against a wide‐range of brain tumors as well as a broader range of intratumoral cell subsets. Meanwhile, the capability of CAR T therapy against GSCs may further reduce tumor recurrence.
5.3. Addressing antigen escape by advancing CAR designs
Despite the emergence of new targets to direct CAR T cell therapy against brain tumors, recurrent tumors will likely still be able to bypass monovalent CAR T cells through downregulation of the targeted antigen or emergence of antigen negative clones. Consequently, one strategy against antigen escape is to extend the conventional single‐targeted CAR design to encompass bispecific targeting domains, rendering the recognition of either of two antigens sufficient to trigger T cell activation.136 The design of these “OR‐gated” CARs may increase safety concerns for the potential of non‐tumor targeting. Therefore, the targets for bispecific CARs are generally selected from the pool of well‐characterized tumor‐specific antigens or after safety of monovalent CAR‐targeting has been established. One of these examples is CD22, another cell surface marker for normal and malignant B cells in addition to CD19.137 With CD22 single‐targeted CAR T cells well tolerated in patients with B‐ALL,138 CD19/CD22 bispecific CAR T cells are currently under investigation in multiple clinical trials (NCT03448393, NCT03233854, NCT03241940). Additionally, CD20, another B cell surface marker, has been combined with CD19 to generate a bispecific CAR which eradicates lymphomas expressing either antigen in preclinical models.139
Likewise, designs of bispecific CARs against GBMs have focused on the antigens well characterized in preclinical and clinical studies, including IL13Rα2, HER2, and EphA2. Hedge and colleagues developed a CAR with bispecific targeting of IL13Rα2 and HER2.140 Using an orthotopic GBM xenograft model expressing both antigens, the authors showed that bispecific CARs were able to control the tumors for approximately 1 month, while single‐targeted CAR treatment did not lead to tumor regression due to antigen escape.140 This approach was further extended with a third targeting domain against EphA2.70 However, tumor relapse was found in all preclinical studies of these CARs, with the loss of all targeted antigens.70, 140 These studies raise questions as to the minimal number of targets and optimal combinations to “box‐in” tumor antigen escape.
The optimal structural design of bi‐ or tri‐specific CARs is still an open area of investigation and highly dependent on the antigens being targeted and structural consideration for the antibody or ligand binding domains. Currently two different structural designs have been tested for “OR‐gate” targeting strategy. The “tandem” design puts the heavy and light chains of each single‐chain variable fragment (scFv) in a sequential order,139 while the “loop” design resembles the structure of bivalent antibodies.141 The comparison between different “OR‐gate” designs was performed in the context of CD19/CD22 bispecific CARs, showing that the “loop” CAR outperformed “tandem” CAR in mediating anti‐tumor responses.142 No similar studies have been performed on bispecific CARs targeting other antigens, but the results from CD19/CD22 CARs indicated that different components of the CAR molecule (bivalent designs, linker lengths, and scFv orders) are critical to the potency of CAR products. Moreover, little is known about the potential alteration of downstream molecular events following dual‐antigen recognition, and the difference of effector potency between single‐ and dual‐targeted CARs. Selection of the optimized CAR design requires extensive functional evaluation recognizing single or dual targets by the engineered T cells and/or computational simulation of the thermodynamic CAR‐antigen interactions.140
Multi‐antigen targeting can also be achieved by expressing two CAR molecules on the same T cell, or mixing different single‐targeted CAR T cells.136 Both approaches have been investigated in preclinical models of relapsed B‐ALL by co‐targeting CD19 and CD123.143 Clinical experience with sequential infusion of CD22‐ and CD19‐CAR T cells into B‐ALL patients provide evidence that combining different CAR products is a feasible strategy.144 Notably, the safety and toxicity might be easier to monitor when using mixed CAR T cells given the experience with individual single‐targeted CAR T products. Other studies, however, suggest that bispecific CAR T cells are more potent than pooling single‐targeted CAR T cells both in vitro and in vivo, possibly due to local competition effects.139, 140 The development of “universal CARs” is also expected to significantly benefit the challenge of tumor heterogeneity. These designs use adapters to connect CAR with the targeted antigens, thus allowing for antigen switch without re‐engineering T cells.145, 146
Tremendous progress has been made in the identification of tumor‐specific antigens and the development of novel CAR designs against brain tumors. While combinatorial targeting has shown promise in addressing tumor heterogeneity, it remains under investigation with regard to optimizing the number and combination of targeted antigens. Most of the uncertainty comes from the lack of understanding on the dynamic changes of tumors following immunological attack. Therefore, uncovering the evolution of brain tumors following CAR treatment, especially the alterations in intratumoral subpopulations at single‐cell resolution, will provide valuable information to guide CAR T cell therapy and overcome tumor heterogeneity.
6. CAR T CELLS AND THE SUPPRESSIVE MICROENVIRONMENT OF BRAIN TUMORS
6.1. Unique aspects of TME of GBM
In addition to recognizing antigen‐positive tumor cells, effective immune responses mediated by CAR T cells require the therapeutic cells to persist and retain effective effector function in the TME. The unique anatomical and phenotypic features of GBM render their microenvironment exceptionally immunosuppressive. Several factors have been implicated in the glioma suppressive microenvironment: (a) CNS‐specific anatomical characteristics, (b) genetic composition of glioma tumor cells, (c) metabolic competition and hypoxia, (d) upregulation of immune inhibitory molecules (ie, immune checkpoints), (e) the presence of soluble factors, such as cytokines and growth factors, and (f) tumor‐associated immunosuppressive cells, such as tumor‐associated myeloid cells.
While the notion that the CNS is immunologically “privileged” has been recently challenged, the unique anatomical features of the CNS pose challenges that may impede the ability of T cells to recognize and respond to antigens within the brain. The molecular events required for immune recognition of brain tumors are still under investigation; however, as evident in the settings of autoimmune diseases such as multiple sclerosis, T cell immunosurveillance occurs in the CNS. In fact, several studies have identified chemokines and adhesion molecules that may be critical for T cell trafficking into the brain.147 Identifying mechanisms that induce T cell surveillance and trafficking and implementing these into CAR T design or as adjuvant therapy could improve the systemic anti‐tumor response to brain tumors (see below).
In addition to the anatomical features that limit T cell infiltration, glioma intrinsic factors based on the mutational profile and gene expression patterns also contribute to the suppressive TME. Gliomas exhibit a complex and unique mutational signature that can contribute to the immunosuppressive landscape. A study by Kohanbash and colleagues has shown that mutations in isocitrate dehydrogenase genes IDH1 and IDH2 in glioma cells suppress STAT1 expression, leading to reduced accumulation of CD8 T cells, type 1‐associated effector molecules, and chemokines such as CXCL10, thereby shaping the tumor immune environment.148 In line with these findings, Berghoff and colleagues demonstrated that IDH‐mutant gliomas exhibited significantly lower rate of T cell infiltration compared to IDH‐wildtype.149 Tumor‐intrinsic mechanisms can dictate the TME landscape; therefore, therapeutic interventions are needed to convert gliomas into an immunologically responsive microenvironment.
The glioma TME is characterized by low nutrients and hypoxic regions. The lack of nutrients, especially essential amino acids such as tryptophan, lysine, and arginine, is responsible for autophagic processes and stress responses that negatively impact T cell function.150 Enzymes such as indoleamine 2,3 dioxygenase (IDO) and arginase (Arg1) catabolize essential amino acids tryptophan and arginine, respectively. These enzymes are highly expressed by tumor cells and/or myeloid cells within the TME and can cause T cell suppression. In fact, kynurenine—a metabolite of L‐tryptophan—has been shown to reduce memory CD4 T cell survival.151 Studies by our group and others have shown that Arg1‐expressing tumor‐associated myeloid cells exhibit suppressive activity against T cells.152, 153 Lactic acid, a by‐product of tumor metabolism, has been found to suppress T cell proliferation and production of cytokines.154 Furthermore, immunosuppressive factors such as prostaglandin E2 (PGE2) and adenosine, released in large quantities by tumor cells and macrophages in hypoxic conditions, can inhibit T lymphocyte proliferation by activating protein kinase A (PKA). A study by Newick and colleagues demonstrated that inhibiting PKA enhanced trafficking and efficacy of CAR T cells.155 Increased hypoxia‐inducible factor‐1 alpha (HIF‐1α) activity and hypoxia in tumor tissues have been correlated with poor prognosis of cancer patients.156 Hypoxia has been shown to upregulate PD‐L1 expression by tumor cells and to promote tumor proliferation.157 while increasing the suppressive activity of tumor‐associated myeloid cells,158 resulting in impaired CD8+ TIL‐functioning. Together, these data show that hypoxia and metabolic pathways may contribute to reduced immune responses. Therefore, targeting and altering metabolic components in the TME could enhance CAR T therapy.
Tumor‐associated myeloid cells represent the dominant immune population in the glioma TME. Tumor‐ associated myeloid cells are frequently polarized toward a pro‐tumoral phenotype, and in combination with regulatory T cells, produce immunosuppressive cytokines/ligands including TGFβ, IL‐4, IL‐10, Arg1, IDO and PD‐L1.159 Strategies to limit myeloid recruitment or reprogram the myeloid populations have been proven beneficial.160 In preclinical studies, blockade of colony stimulating factor receptor (CSFR; a receptor exclusively expressed by myeloid cells) on glioma xenografts enhanced anti‐tumor response to radiotherapy by reducing the recruitment of bone marrow‐derived macrophages.161 Additionally, inhibiting STAT3, a key regulator in pro‐tumoral macrophages, significantly reduced macrophage polarization in patients with malignant glioma.162 Furthermore, treatment with tyrosine kinase inhibitors such as sunitinib that inhibit STAT3 signaling pathways, induced cancer cell apoptosis and reversed immunosuppressive cytokine profile.163 These studies suggest that selective targeting of immunosuppressive myeloid cells in the TME may synergize with CAR T therapy.
In addition to suppressive immune cells in the glioma TME, soluble factors secreted by both tumor and tumor‐associated immune cells can inhibit immune‐mediated cytolytic responses. Specifically, TGFβ has been found to inhibit T cell cytotoxic activity and promote regulatory T cell generation. TGFβ has been implicated in resistance to PD‐L1 therapy by contributing to T cell exclusion in the tumor bed.164 Targeting the TGFβ pathway has been shown to improve anti‐tumor activity in several tumor models including gliomas.165 Glioma‐associated IL‐10, a potent anti‐inflammatory cytokine secreted by myeloid cells and a subset of CD4+ T cells,166 has been shown to induce STAT3 in macrophage and dendritic cells,167 down‐regulate MHC class II expression on monocytes and inhibit IFN‐γ and TNF‐α production by immune cells.168 Another immunosuppressive cytokine that synergizes with IL‐10 and TGFβ is IL‐4. IL‐4 promotes generation of Th2 cells and polarization of suppressive macrophages. In fact, neutralizing IL‐4 during radiotherapy resulted in significant improvement in anti‐tumor immunity with a decrease in immunosuppressive macrophages.169 Changing the tumor milieu by reprogramming suppressive cells and neutralizing the suppressive soluble factors could enhance CAR T cell function and persistence in the tumor.
The suppressive properties of the GBM TME known to limit anti‐tumor immune responses are generalizable to the majority of brain tumors, both primary and metastatic. The key is to deconstruct the minimal set of pathways required to unleash effective immunological attack by CAR T cells. This can be accomplished by engineering into the T cell themselves resistance to suppression or by combining with other agents that promote CAR and/or endogenous T cell anti‐tumor activity.
6.2. Approaches to improve CAR T cell activity
6.2.1. Engineering resistance into CAR T cells
Various approaches have been implemented to overcome the suppressive effects of cytokines in the TME. In the context of CAR T therapy, several studies have investigated “the reverse approach,” which consists of inhibiting immunosuppressive cytokines or converting their signals to pro‐inflammatory. Several groups have designed CAR T cells to co‐express dominant‐negative TGFβRII (dnTGFβRII), which blocks TGFβ signaling within the engineered T cells. In a murine prostate cancer model, prostate‐specific membrane antigen (PSMA)‐targeted CAR T cells that co‐express dnTGFβRII exhibited enhanced proliferation and persistence, reduced exhaustion, and superior anti‐tumor activity.170 These preclinical studies led to a phase 1 clinical trial of PSMA‐CAR T cell co‐expressing dnTGFβRII for patients with metastatic prostate cancer (NCT03089203). Similarly, Epstein‐Barr virus‐specific cytotoxic T lymphocytes (CTLs) transduced with dnTGFβRII continued to produce cytokines and maintain cytolytic activity in response to antigenic stimulus in the presence of TGFβ.171 To protect adoptively transferred T cells from the immunosuppressive tumor‐milieu, T cells can also be engineered to recognize soluble ligands and potentially convert the immunosuppressive cytokine signal to an immunostimulatory signal. Chang and colleagues developed a CAR consisting of scFv TGFβ neutralizing antibodies incorporated into a second‐generation CAR containing a CD28 co‐stimulatory domain. Binding to TGFβ, an immunosuppressive factor, resulted in stimulation and activation of CAR T cells.134 Along the same line, Mohammed and colleagues, developed CAR T cells that express the IL‐4 receptor ectodomain fused to the IL‐7 receptor endodomain. When the CAR T cells bound to IL‐4, typically an immunosuppressive cytokine, the IL‐4/IL‐7 chimera promoted cell proliferation while maintaining the anti‐tumor efficiency in vivo.172
6.2.2. Engineering cytokine support into CAR T cells
Chimeric antigen receptors T cell persistence and survival is of utmost importance especially in solid tumors. Longer persistence of CAR T cells posttreatment has been associated with better clinical response.113 Our team has modified the manufacturing to select for T cells with a less differentiated phenotype.69 Our group and others have evaluated reduced ex vivo culture duration and addition of cytokines such as IL‐7 and/or IL‐15 to culture conditions.30, 31 Certain cytokines support survival and expansion of T cells, and this is crucial especially when they encounter hostile conditions of solid tumors. CAR T cells have been designed to secrete pro‐inflammatory cytokines to support function and proliferation and to shield themselves from immunosuppressive cytokines. IL‐12− and IL‐18−secreting CAR T cells have thus been shown to persist longer and lead to enhanced tumor responses in preclinical models of solid cancers.173, 174 Other investigators have described improved anti‐tumor efficiencies of CAR T cells equipped with constitutive IL‐7 and IL‐15 signaling, as well as by inducible delivery of an IL‐15 super‐agonist complex by T cells upon encounter with the cognate antigen.175, 176, 177 Approaches involving secretion of pro‐inflammatory molecules may have additional paracrine effects, eg, remodeling the TME and activating by‐stander immune cells such as tumor‐associated macrophages. An example of this effect was demonstrated by the co‐expression of the single‐chain IL‐12 by CAR T cells, which resulted in tumor regression through repolarization of myeloid cells.178 Another IL‐12 secreting CAR T cell design has advanced to the clinic in a phase I trial of IL‐12─secreting CAR T cells targeting MUC‐16ecto for the treatment of recurrent ovarian cancer (NCT02498912).179 The use of engineered CAR T cells to deliver a range of cytokines, and their potential to support not only the T cell persistence and function, but also to remodel the TME to be anti‐tumorigenic is of importance, especially in the context of solid tumor therapies.
6.3. Combination therapies to augment CAR T cell function
Combination therapies designed to overcome the hostile glioma environment while improving CAR T cell persistence and function may be a promising strategy. Targeting immune inhibitory molecules such as PD‐1, CTLA‐4 and PD‐L1 has been the focus of many studies as a potential therapy that could enhance CAR T cell efficacy.180 Combining CAR T cell therapy with checkpoint blocking agents could overcome impediments to T cell infiltration and functionality. Several preclinical studies have shown benefit in blocking PD‐1181 or CTLA‐4182 in murine glioma models. Importantly, two recent clinical reports have shown benefit for neoadjuvant anti‐PD‐1 immunotherapy in promoting survival20 and modifying the TME183 in glioma patients. These promising reports suggest that blocking PD‐1 changes the TME and could synergize with CAR T therapy in improving the survival of glioma patients. Genetic removal of PD‐1 from CAR T cell products, or engineering CAR T cells to produce a blocking antibody against PD‐1/PD‐L1 have been considered as alternative strategies. A study by Ren and colleagues demonstrated that PSCA‐CAR T cells that had PD‐1 genetically removed exhibit enhanced anti‐tumor efficacy both in vitro and in vivo in a murine prostate cancer model.184 Currently, clinical studies are underway to assess combination of CAR T cell therapy with checkpoint inhibitors in GBM (EGFRvII‐CAR T + Pembrolizumab; NCT03726515) and (IL13Rα2‐CAR T + Nivolumab). Although, the clinical impact of CAR‐T cells combined with checkpoint inhibitors in GBM is still unknown, results from these trials will provide important information regarding safety, feasibility, and potential anti‐tumor activity.
Oncolytic viruses are also promising agents for the treatment of solid tumors such as gliomas. Oncolytic viruses can specifically target cancer cells, while sparing normal cells, and the resulting tumor lysis can release danger signals and stimulate immune system.185 Furthermore, oncolytic viruses can be genetically modified to express therapeutic transgenes to target a suppressive TME, potentially synergizing with CAR T therapy. Indeed, studies have reported enhanced CAR T cell efficacy by combining oncolytic viruses expressing either cytokines, chemokines, or an anti‐PD‐L1 minibody against solid tumors in pre‐clinical mouse models.186 Additional promising combinatorial approaches include agonistic antibodies specific for the 4‐1BB costimulatory receptor,187 which can directly activate CAR T cells. Vaccines in a form of glioma‐associated antigens or dendritic cell loaded with mRNA or tumor lysate have been used for primary brain tumors,188 and could also synergize with CAR T therapy to overcome tumor heterogeneity and induce an endogenous immune response.
6.4. Combining standard‐of‐care radiation therapy plus CAR T cells
Radiation therapy has long been an important component of standard‐of‐care management of brain tumors. Evidence for potential synergy of CAR T and radiation therapy lies in the retrospective clinical series and preclinical models of ICI and stereotactic radio‐surgery (SRS). Retrospective meta‐analysis of patients with melanoma and non‐small cell lung cancer brain metastases has demonstrated improved outcomes of combined vs sequential ICI and SRS.189 Preclinical orthotopic glioma models also have demonstrated efficacy of combination ICI and SRS.190 Mechanisms by which radiation promotes immunologic memory are under investigation. Preclinical studies have demonstrated that radiation alters the TME to potentiate an adaptive immune response. Tumor irradiation functions as an in situ vaccine because it results in the release of tumor‐associated antigens, which activate antigen presenting cells to migrate to draining lymph nodes where they prime cytotoxic CD8+ T cells to generate an adaptive immune response,191 including recruitment of endogenous TILs and enhanced TCR expansion.190, 192, 193 Specifically, high dose tumor irradiation increases T cell priming due to cross‐presentation of tumor peptides via MHC class I pathway. Recent studies have shown hypofractionated radiation (8 Gy × 3 fractions) results in a more robust systemic tumor rejection compared to high dose single fraction (20 Gy × 1 fraction) in the context of immune checkpoint blockade.194 Mechanistically, radiation doses above 12‐18 Gy induce DNA exonuclease Trex1, which enzymatically attenuates their immunogenicity by degrading DNA that accumulates in the cytosol upon radiation. Cytosolic double‐stranded DNA is a potent stimulator of the cGAS/STING/IFN‐beta pathway to recruit Batf3+ dendritic cells that can activate CD8+ T cell‐mediated systemic immune responses.195
Given radiation's role in STING‐dependent recruitment of the adaptive immune response, radiation may also help to address shortcomings of CAR T cells in solid tumors, namely T cell trafficking and persistence. Indeed, in a preclinical pancreatic adenocarcinoma model, a small‐molecule STING agonist co‐delivered with CAR‐T cells resulted in a host immune response to eliminate tumor cells not recognized by the adoptively transferred lymphocytes.196 Going forward, radiation and concomitant STING activation may also address post‐CAR T antigen escape, as TCR expansion and dendritic cell activation may result in “epitope spreading” and immunologic memory against multiple tumor antigens.
7. GETTING CARs TO GO: CAR T CELL TRAFFICKING TO BRAIN TUMORS
7.1. Anatomical considerations for T cell trafficking beyond the blood‐brain barrier (241)
Brain tumors pose unique obstacles for T cell homing due to the selective properties of the blood‐brain barrier (BBB) and the blood‐CSF barrier (BCSFB), which strictly regulate immune cell entry to the brain. Much of what is known about immune cell infiltration into the brain is derived from models of infection or experimental autoimmune encephalomyelitis (EAE).197 These barriers regulate extravasation through postcapillary vesicles and limit immune entry in the brain or CSF due to the presence of endothelial cell layers with tight junctions and astrocyte foot process known as glia limitans.198, 199 From circulation, T cells must first adhere to vascular endothelium via a multitude of integrins, adhesion molecules, and chemokines.198, 199 Activated T cells, but not naive or resting memory T cells,200 can be recruited beyond the BBB in the absence of inflammation although CD8 T cells require the presentation of MHC class I antigens on luminal endothelium.201 Understanding how to best ensure optimal trafficking of CAR T cells to brain tumors remains an active area of investigation, and we will summarize some of the considerations below.
7.2. Route of delivery of CAR T cells for brain tumor therapy
One key question for the advancement of CAR T cell therapy for brain tumors is the choice of delivery route and whether systemic or locoregional delivery is more advantageous, particularly because brain tumors are unique in that they are regionally localized and primary brain tumors such as GBM rarely metastasize outside the CNS.202 Systemic delivery, whereby CAR T cells are given intravenously (IV), is the most common delivery approach for hematological and solid cancers. For IV delivery, cryopreserved cells can be thawed at bedside and infused directly without the need for reformulation or a delivery device. As reviewed above, melanoma‐targeted and CD19‐CAR T cells delivered systemically have been shown to traffic to the brain and eliminate malignant disease.42, 45, 46 However, in both of these clinical settings, the adoptively transferred cells were activated in the periphery due to the presence of disease. Since activated T cells more readily traffic to the CNS, this could significantly influence CNS trafficking efficiency.
Given the complexity of immune cell homing, locoregional delivery strategies can be used to bypass some of the anatomical barriers involved in trafficking from circulation. Locoregional delivery methods include intratumoral (ICT) administration whereby CAR T cells are delivered into the tumor bed or resection cavity, and intraventricular (ICV) administration whereby CAR T cells are delivered into the CSF via the ventricular system. ICV delivery bypasses all but the glia limitans in delivery to the brain parenchyma. Treating patients with locoregional delivery of CAR T cells has been reported in GBM66, 68, 69, 79 as well as other malignant diseases including ovarian, lung, and breast cancers.198, 203 For brain tumors, locoregional delivery requires implantation of a reservoir/catheter delivery device that is typically placed during surgical resection or biopsy, with the reservoir accessible under the scalp and the catheter placed to drain into the tumor bed/cavity or CSF. Such devices have been used routinely for CNS delivery of chemotherapies or biologics.204 These devices should be monitored closely due to the risk of infection, but they are generally well‐tolerated by patients in our experience.
Studies comparing routes of delivery for both preclinical models of GBM and breast cancer brain metastases have shown that local delivery (ICT or ICV) outperforms systemic delivery (IV) of CAR T cells. HER2‐CAR T cells (0.5 M dose) delivered ICV was more effective than a 10‐fold higher dose (5 M dose) delivered IV in orthotopic models of breast cancer brain metastases.93 Similarly, ICT administered IL13Rα2‐CAR T cells resulted in long‐term survival in orthotopic GBM models, whereas IV delivery provided no significant benefit over mock transduced T cells.64 When comparing locoregional delivery routes in a multifocal GBM model where tumors were implanted on both hemispheres, ICV exhibited improved targeting of multifocal disease.64 In fact, preclinical studies in which CAR T cells were labeled with the radionuclide 89Zr and followed by PET imaging in mice, we show that CAR T cells delivered into the brain parenchyma remain localized in the brain, whereas ICV‐delivered cells distribute throughout the CNS over 6 days of monitoring by PET (Figure 3).205 Indeed, direct infusion into the CSF via intraventricular delivery achieved a complete clinical response in a patient with multifocal disease, including a distal lesion in the spine,69 illustrating the surveillance of CAR T cells throughout the CNS.
Figure 3.
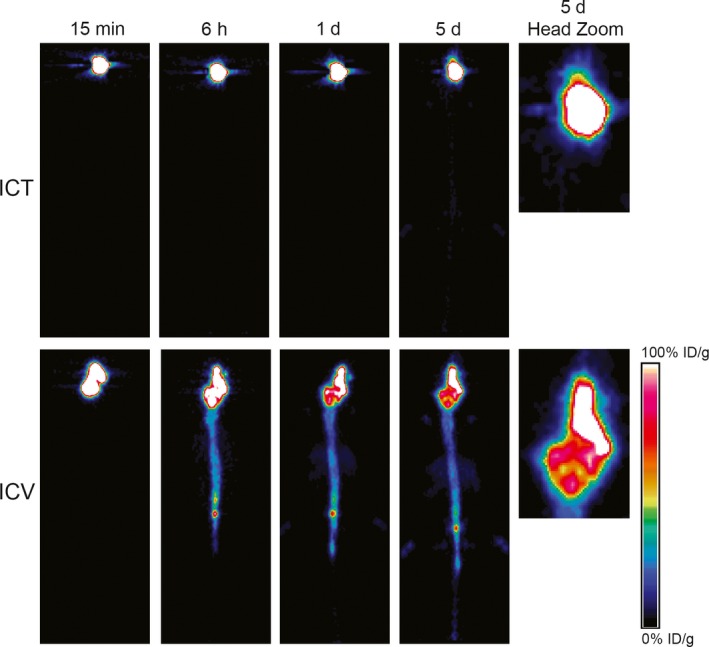
Chimeric antigen receptor (CAR) T cell distribution following locoregional delivery. Primary glioblastoma cells (1 × 105 PBT030‐2) were implanted intracranially (IC) in the left hemisphere (2.0 mm lateral, 0.5 mm anterior to the bregma, 2.15‐3.0 mm depth from dura) of NSG mice, and 8 d later 89Zr‐oxine labeled IL13Rα2‐CAR T cells (2 × 106) were administered either IC in the right hemisphere, or into the right ventricle (ICV; 0.9 mm lateral, 0.3 mm caudal to the bregma, 2.5 mm depth from dura). Representative PET images are depicted at 15 min, 6 h, 1 d or 5 d after ICT (top) or ICV (bottom) delivery of 89Zr‐oxine labeled IL13Rα2‐CAR T cells are depicted. Color scale indicates percentages of injected radioactive dose (ID) per gram weight of voxel that were calculated using Vivo‐Quant (Invicro) analysis of the PET images205
Locoregional CNS delivery of CAR T cells may also reduce the risk of systemic toxicities associated with the therapeutic cells. For instance, in clinical trials of EGFRvIII‐ and HER2‐CAR T cells,81, 92 high‐dose peripheral (IV) infusion increased the risk of serious pulmonary toxicities, and in the most severe cases resulted in death. Locoregional delivery is expected to limit intravenous first‐pass pulmonary toxicity as well as off‐tumor targeting of other systemic tissues. Together these studies suggest that locoregional delivery may improve trafficking, infiltration, and safety of the adoptively transferred cells for the treatment of brain tumors. However, further clinical testing is required to understand how delivery route impacts both safety and patient outcomes.
7.3. Engineering CAR T cells for improved tumor tropism
One strategy to improve T cell trafficking and infiltration is to engineer the cells to express proteins that facilitate tumor tropism. This requires an understanding of the mechanism of T cell extravasation into the brain parenchyma as well as characterization of vascular properties of the specific brain tumor. The interactions between chemokines and their corresponding chemokine receptors in T cells have been studied extensively,206 and research is underway to apply these fundamentals to CAR T cells.
Engineering T cells to express chemokine receptors have improved the tumor tropism of therapeutic lymphocytes by delivering more therapeutic cells to the site of the tumor. The most clinically advanced example to date is the expression of CCR4 in CD30‐CARs for treating Hodgkin's lymphoma. Di Stassi et al first recognized CD8+ T cells lacked CCR4 (2.5%) despite Hodgkin's lymphoma overexpressing the CCR4 ligands CCL17 and CCL22,207 and the forced expression of CCR4 enhance CAR T cell accumulation and therapeutic response in mice. This work is under further investigation in a phase I clinical trial (NCT03602157). Expressed on CD4+ but not on mature CD8+ T cells,207 CCR4 could be a versatile, albeit promiscuous, strategy as it recognizes the chemokines CCL2, CCL4, CCL5, CCL17, and CCL21, which have all been associated with glioma.208, 209, 210, 211 Despite this connection, engineered chemokine receptors have not specifically been evaluated in CAR T cells for the treatment of brain tumors. CCL2 is also produced by the glioma microenvironment to recruit both Treg and myeloid‐derived suppressor cells.212 Through manipulating tumor chemokine release, we have demonstrated the tumor tropism of adoptively transferred T cells to GBM lines expressing CCL2, which is a ligand for CCR4.213 Similar to CCR4, CCR2 is a receptor for CCL2, is poorly expressed on activated T cells (<7%), and its addition in CAR T cells has been shown to increase efficacy in neuroblastoma and melanoma models.214, 215 Other lymphocyte models have leverage chemokines to increase tumor accumulation with the introduction of receptors such as CXCR4,216 CXCR1,217 CXCR2,218, 219 CCR7,220 and CX3CR1.221 Additional targeting of endothelial adhesion molecules or vascular cytokines upregulated in brain tumors could also be a worthwhile approach to enhance CAR T cell accumulation at the tumor site.178, 222 To improve cell localization, chemokine receptor‐engineered T cells should be designed for the chemokine signaling unique to each tumor. Such strategies to improve tumor trafficking are important, as preclinical models suggest that trafficking can often be highly inefficient with only a minor fraction of the adoptively transferred cells infiltrating the tumor.205, 215
7.4. Imaging of T cell trafficking
While clinical sampling of blood and CSF or invasive biopsies provide general information about cells in circulation or in the biopsied tissue, respectively, incorporating imaging techniques to visualize the migration of CAR T cells in real‐time can provide important evidence for on‐ and off‐target localization, treatment efficiency, T cell persistence, and the influence of alternative delivery methods or combination interventions. Clinically available nuclear imaging modalities such as positron tomography (PET), which has a higher relative resolution in comparison to single photon emission computed tomography (SPECT),223 enable sensitive and non‐invasive detection of radionuclides when associated with cells or other biomolecules. When combined with computed tomography (CT) or MRI, the sensitivity of nuclear imaging is co‐registered with anatomical detail. MRI also enables additional diagnostic modes based on the pulse sequences, especially in the context of blood perfusion and diffusion in brain tumors.224 Researchers have used these imaging modalities for tracking cells with techniques for ex vivo labeling as well as in vivo detection of cells using reporter genes or radiolabeled antibodies.
The first study to image CAR T cells for brain tumors evaluated first‐generation IL‐13‐zetakine CAR T cells that were engineered the co‐express the herpes simplex virus type‐1 thymidine kinase reporter gene (HSV1‐tk) in a patient with GBM.67, 68 Cellular expression of HSV1‐tk enabled T cell detection based on uptake of the radiolabeled nucleoside analog 9‐[4‐[18F]fluoro‐3‐(hydroxymethyl)butyl]guanine (18F‐FHBG). HSV1‐tk and its variants have also been used preclinically for studying CAR T cells trafficking.225, 226, 227 However, the immunogenicity of HSV1‐tk has also been reported in the clinical setting.228, 229 To reduce the risk of immunological rejection, several reporter systems using human genes as non‐immunogenic alternatives are being evaluated preclinically to study the trafficking of T cells in mice.230 In direct comparisons of the reporter systems in T cells,231, 232, 233 human deoxycytidine kinase (dCK) mutants have shown high relative sensitivity and selectivity over native human dCK. This imaging strategy enables longitudinal imaging sessions yielding signal which correlates with the number of metabolically active cells present, but is limited by tissue permeability of the probe and background uptake of the nucleoside in surrounding tissue. Because these radioactive nucleoside probes have poor BBB penetration,234 they may not be useful for imaging dCK expressing cells beyond intact BBB as distribution at regions other than disrupted tumor vasculature will be limited.
As an alternative strategy, approaches to directly label cells with radionuclide ex vivo are being developed which enable cell detection with PET imaging after infusion. 111In‐oxine has specifically been used to image the tumor tropism of antigen‐specific T cells in preclinical models235 and patients.236, 237 This approach is the current clinical standard for imaging cells and platelets with SPECT, where lipophilic metal complexes passively diffuse across cell membranes in a rapid and efficient manner.238, 239 Recently adopted for PET,240 we imaged 89Zr‐oxine‐labeled CAR T cells in both subcutaneous prostate and intracranial glioma models.205 CAR T cells demonstrated tumor tropism and retained therapeutic potential after labeling. Since 89Zr (with antibodies241) and oxine (with 111In‐oxine) have already been used in patients, this new technique for imaging cell trafficking with PET may be clinically available in the near future. Ex vivo labeling approaches offer cell‐specific signal which correlates with the initial cell number. However, imaging is limited by radionuclide half‐life and is not proportional to the number of cells if proliferation occurs. The limitations of each technique should be carefully considered before proceeding with study design.
Other approaches for tracking cells use antibodies conjugated with metal chelators (ex. 1,4,7,10‐Tetraazacyclododecane‐1,4,7,10‐tetraacetic acid [DOTA]) which bind radionuclides (ex. 64Cu) for visualizing cell‐antigen‐specific targeting with PET. This approach has been most commonly used for cancer cell detection and has been more recently applied to the tracking of endogenous T cells242, 243 or cells with engineered antigen tags.244, 245 To reduce signal background and circumvent issues with BBB permeability, these antibodies could be used for ex vivo pre‐labeling of cells to study initial trafficking246 as long as the antibody stays cell‐associated and radionuclide half‐life is permissible. In comparison to passive labeling mechanisms like 89Zr‐oxine, prelabeling with antibody uses the antibody binding affinity while potentially reducing cell damage by localizing radionuclide to the cell surface prior to any receptor internalization. Another approach utilizes the metal binding properties of DOTA after the introduction of a membrane‐bound single‐chain fragment (scFv) into T cells, which binds to DOTA‐lanthanide complexes (DAbR1).247 More studies are required to determine which of these methods will be most effective in imaging CAR T cell therapy for brain tumors, given the limitations of antibodies permeating the BBB and the sensitivity required for tumor visualization. Taken together, prelabeling techniques should be utilized when sensitivity and initial trafficking is most valuable while infused antibodies and reporter genes are best for multiple imaging sessions and signal which correlates with live cell numbers. Future combinations of these techniques could yield more comprehensive information regarding T cell trafficking into the brain.
8. FROM BENCH TO BEDSIDE: CLINICAL TRIAL CONSIDERATIONS
This is an auspicious time for the field of immunotherapy, with opportunities to advance brain cancer treatment through comparing experiences and ideas across many adoptive cellular platforms. Clinicaltrials.gov lists more CAR T trials for brain tumors than any other solid tumor indication (Figure 2). The majority are for malignant glioma (12 of 13), but the extension to metastatic tumors in the brain has begun with the first trial targeting brain metastases (NCT03696030). Five of the 13 trials have been performed at City of Hope: two completed trials in recurrent GBM targeting IL13Rα2, and 3 trials currently enrolling for either IL13Rα2+ or HER2+ GBM, or HER2+ breast metastasis in the brain (Table 1). Our experience has shaped our trial designs, which are described here in the context of the ideas presented in this review.
8.1. CAR T cell trials at City of Hope
For CAR T cell trials at City of Hope, patients with malignant brain tumors (high‐grade malignant glioma or breast metastases) are enrolled following confirmation of target antigen expression (IL13Rα2 or HER2) by histology on prior tumor specimens (most recent resection or biopsy) and after medical assessment meeting the additional study‐specific eligibility criteria. Patients first undergo leukapheresis to collect peripheral blood mononuclear cells (PBMC) for manufacturing of autologous CAR T cell product. While the CAR T cells are being produced (~3‐4 weeks), patients may proceed with treatment for management of their cancer, but specified washout periods are required before beginning CAR T cell treatment. Prior to the start CAR T cell infusion, patients undergo surgery for placement of an intraventricular and/or intratumoral/intracavitary reservoir/catheter delivery device. During that surgical procedure, tumor resection or biopsy may be performed per the discretion of the neurosurgeon. Once the CAR T cell product has been released for clinical use, and there is evidence of progressive disease in the CNS, patients undergo baseline PET and MR imaging and then begin treatment with CAR T cells. Depending on the trial design, the treatment phase usually consists of 3 to 4 weekly cycles of CAR T cell infusions followed by PET and MR imaging of the brain to assess response. After the treatment phase, patients can continue to receive optional CAR T cell infusions as long as they are tolerating the treatment well and continue to meet eligibility criteria. During these cycles or until progression is established, study patients cannot be treated with any other anti‐cancer therapy.
Immunologic correlative studies are performed throughout the treatment time course to assess the persistence of the CAR T cells in the CSF (obtained through the delivery device) and peripheral blood, monitor for evidence of activation of the endogenous immune system, and describe changes in cytokine levels in the CSF and peripheral blood. If a patient undergoes tumor resection or biopsy while on study treatment or afterwards, then evaluation of CAR T cell persistence and changes in cytokine levels in the TME, as well as changes in antigen expression levels in tumor tissue pre‐ and post‐treatment is performed.
City of Hope's approach of locoregional delivery and repeat dosing is intended to prioritize for both patient safety and maximum therapeutic benefit. As discussed above, our preclinical studies suggest that locoregional delivery of CAR T cells outperform systemic IV delivery, so our trials have prioritized delivering CAR T cells directly into the tumor or resected tumor cavity (ICT) and/or into the CSF via the lateral ventricle (ICV). The dosing regimen splits the intended total cell dose over 3 to 4 weekly infusions. Splitting the intended total dose into smaller doses delivered weekly is anticipated to minimize safety risks compared with a single bolus intracranial dose, which may cause unintended inflammatory toxicities, including CRS. Multiple or split dosing regimens have been used previously to reduce risk of infusion‐related toxicities.248, 249 Repetitive dosing also affords a larger total dose of cells, and is intended to introduce functional CAR T cells over a longer timeframe, thereby extending the therapeutic window in the immunosuppressive TME.
8.2. Concomitant medicines
Concomitant medicines, such as temozolomide, dexamethasone, and bevacizumab, which are commonly used for brain tumor management and treatment, may impact CAR T cell activity either positively or negatively. While a detailed understanding of which concomitant medicines impact CAR T cell function are still unknown, their effects require careful monitoring. Future trials and analyses of correlative endpoints may shed light on how best to combine these agents with CAR T cells for both clinical disease management and therapeutic activity.
Temozolomide (Temodar [TMZ]) is a DNA alkylating chemotherapy that is routinely used for standard‐of‐care management of GBM.250 A common side‐effect of TMZ is bone marrow suppression and lymphopenia.251 In the context of CAR T therapy, preclinical models have demonstrated lymphodepleting pre‐conditioning with a dose‐intensified regimen of TMZ (TMZ‐DI), but not standard TMZ dose regimens. In a syngeneic mouse model of GBM, TMZ‐DI pretreatment prompted enhanced EGFRvIII‐CAR (IV‐administered) expansion and persistence in circulation, as well as tumor eradication.252 Based on these findings, an ongoing EGFRvIII‐CAR T trial at Duke University (NCT02664363) is utilizing TMZ‐DI as a preconditioning lymphodepleting regimen. The dual use of TMZ for the treatment of GBM and for lymphodepleting pre‐conditioning is an attractive strategy, as lymphodepletion prior to adoptive transfer of T cell‐based immunotherapies has been shown to be essential for effective TIL, TCR‐, and CAR‐engineered T cell anti‐tumor activity for systemic cancers when the therapeutic cells are delivered IV. While it is reasonable to assume that lymphodepletion will also augment CAR T cell clinical responses for brain tumors, particularly for IV delivery, its therapeutic benefit in the context of brain tumors requires further clinical validation, since most clinical studies to date have not included lymphodepleting regimens. Additionally, for the two reported EGFRvIII‐CAR clinical trials, the NCI study that included lymphodepletion pre‐conditioning (Fludarabine + Cyclophosphamide) and systemic IL‐2 cytokine support (NCT01454596)81 did not show significantly improved CAR T cell persistence, expansion, or patient outcomes compared with the University of Pennsylvania study that did not include lymphodepletion (NCT02209376).80 While this could be due to many factors, such as CAR design, target selection, and patient enrollment, it highlights the need for further clinical assessment of the role lymphodepletion in the application of CAR T cells for brain tumors, particularly for locoregional delivery.
Dexamethasone (Dex) is a brain‐penetrant steroidal anti‐inflammatory used often in the glioma setting to alleviate tumor‐associated brain swelling. Dex is also highly toxic to immune cells including T cells, which may limit the CAR T cell efficacy. In particular, Dex is known to inhibit priming of T cell immune response by dendritic cells, and was shown to be deleterious to vaccine trials for GBM at very low levels.10 In preclinical mouse models, intratumorally delivered CAR T cells were functional with low‐levels of Dex (1 and 0.2 mg/kg), but were strongly suppressed by high levels (5 mg/kg).64 Further, CD19‐CAR T cell trials report high response rates, even for patients who have been given steroids to manage CRS, although prolonged systemic corticosteroid use has been shown to disrupt CAR persistence and efficacy.248 Given the importance of Dex in glioma symptom management, successful CAR T cell trial design must identify allowable Dex concentrations. Most CAR T cell trials limit Dex levels to no more than 4‐6 mg/d, and at this concentration, we have successfully manufactured clinically sufficient levels of CAR T cells from patients, have detected increased inflammatory cytokines post CAR T cell infusion, and in one patient CAR T cells elicited a complete response.69 These findings provide initial evidence that low levels of Dex may be able to be used to manage tumor and/or immune inflammatory reactions without significant deleterious effects on CAR T cell function, but more clinical experience is required to fully delineate the impact on therapeutic activity and patient outcomes.
Bevacizumab (Avastin) is FDA‐approved for the treatment of recurrent GBM, and acts as a ligand‐sink against the vascular endothelial growth factor receptor (VEGF). Disrupting VEGF/VEGFR2 signaling inhibits angiogenesis, normalizes tumor vasculature and re‐establishes the blood‐brain barrier. The effects of bevacizumab on CAR T cell therapy are anticipated to be multifaceted. First, by normalizing the tumor vasculature, bevacizumab alters MR imaging features, resulting in decreased T1 contrast enhancement and FLAIR sequences.253 These imaging changes confound disease assessment when using bevacizumab in conjunction with CAR T cells therapy and therefore necessitate using OS vs PFS for a response endpoint. Second, bevacizumab can be an effective non‐steroid anti‐inflammatory for managing symptoms of cerebral edema, thereby providing an alternative to immunosuppressive corticosteroids such as dexamethasone.254 Additionally, bevacizumab may augment CAR T cell therapy by inhibiting the immune suppressive effects of VEGF, as well as promote tumor lymphocyte trafficking.255, 256 Preclinical models in neuroblastoma have demonstrated enhanced efficacy of combination bevacizumab and GD2‐CAR‐T cells.257 Combination treatment resulted in high infiltration of GD2‐CAR T cells which resulted in increased IFNγ production. Given bevacizumab's role in enhancing lymphocyte trafficking and blocking VEGF immunosuppressive effects, it may be a promising combination therapy with CARs for treatment of solid tumors. Whether there is enhanced clinical efficacy with combination CAR T cells and bevacizumab in GBM and other brain tumors still requires clinical validation.
8.3. Clinical and molecular endpoints and correlative studies
Clinical and molecular endpoints and correlative studies collected prior, during, and after therapy are designed to gain more understanding of the mechanisms of response and resistance to treatment and can inform future combination trials. An important molecular endpoint is tumor antigen expression after CAR infusion. The IL13Rα2‐ and EGFRvIII‐CAR studies both observed loss of antigen expression in tumor tissues post treatment. In addition, molecular correlative data may identify predictive biomarkers of response and resistance. O'Rourke and colleagues incorporated tumor TCR‐β sequencing to identify expansion of the T cell repertoire after EGFRvIII‐CAR delivery.80 This important molecular correlative can identify endogenous TCR clones responsible for therapeutic response and may be used to predict treatment response. Also, for studies that use a device for locoregional intracranial delivery (ICT of ICV), tumor fluid or CSF samples for this analysis can be collected relatively non‐invasively. CAR T cell expansion and persistence is also be an important molecular endpoint, as it has been associated with clinical response in CD19‐CAR T for ALL.45, 258 Additionally, persistence of CAR T cells of at least 1 month has been associated with overall response in patients with metastatic melanoma.259 Other clinically important correlates include an assessment of the tumor and TME, which require biopsy. Indeed, tumor PD‐L1 upregulation was found after EGFRvIII‐CAR delivery.80 PD‐L1 upregulation is clinically meaningful in the context of future combination trials with CAR T cells and immune‐checkpoint inhibitors. Well‐designed correlative studies will provide answers as well as raise new questions, such as what is the optimum frequency of CSF biopsy and tumor biopsy to evaluate for treatment response.
9. SUMMARY AND PERSPECTIVES
The success of immunotherapy is revolutionizing cancer therapy and improving outcomes for patients with a wide variety of cancers. While brain tumors are considered some of the most difficult to treat of solid tumors, progress in the immunotherapy is raising optimism for its potential to make inroads in their treatment. Evidence that adoptively transferred T cells, including CD19‐CARs and melanoma‐targeted TCR‐engineered or TILs, can traffic to the brain and eliminate disease supports the notion that CAR T cells may eventually prove effective against other primary and metastatic brain tumors. Advances in the understanding of neuroimmunology alongside advances in cancer immunotherapy have provided a platform to create more effective treatments for brain tumors. CAR T cell therapy for brain tumors faces major obstacles, such CAR T cell persistence and trafficking to the CNS, adaptive immune resistance and other immunosuppressive elements of the TME, and tumor heterogeneity. CAR T cell clinical studies have provided valuable insights that will help in shaping future trials, and initial indications from these trials point to the need for combination therapies that can address several obstacles simultaneously or in sequence. It is critical to ensure that future trial designs not only inform on safety, but also provide information on mechanisms underlying response and resistance, as this information will provide the path forward to overcoming the formidable obstacles to treatment.
CONFLICT OF INTEREST
Patents associated with CAR design, T cell manufacturing, and delivery have been licensed by Mustang Bio., Inc, for which CEB receives licensing and consulting payments.
ACKNOWLEDGEMENTS
We thank Dr. Julie R. Ostberg for manuscript and figure preparation, and Dr. Lawrence A. Stern for the CAR schematic. We also thank Drs. Michael E. Barish and Behnam Badie for scientific feedback. This work was supported by California Institute for Regenerative Medicine (CLIN2‐10248), IVY Foundation, NCI 1R01CA236500, Curing Kids Cancer, Gateway for Cancer Research, Norris Foundation, Meringoff Family Foundation, and Rising Tide. D.W. is supported by NCI fellowship 1F99CA234923‐01.
Akhavan D, Alizadeh D, Wang D, Weist MR, Shepphird JK, Brown CE. CAR T cells for brain tumors: Lessons learned and road ahead. Immunol Rev. 2019;290:60–84. 10.1111/imr.12773
This article is part of a series of reviews covering Bench to Bedside Successes in Immunology in Immune Cells appearing in Volume 290 of Immunological Reviews.
[The copyright line for this article was changed on 2nd August 2019 after original online publication]
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7‐30. [DOI] [PubMed] [Google Scholar]
- 2. Stupp R, Taillibert S, Kanner A, et al. Effect of tumor‐treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA. 2017;318(23):2306‐2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alexander BM, Cloughesy TF. Adult glioblastoma. J Clin Oncol. 2017;35(21):2402‐2409. [DOI] [PubMed] [Google Scholar]
- 4. Pollack IF, Agnihotri S, Broniscer A. Childhood brain tumors: current management, biological insights, and future directions. J Neurosurg Pediatr. 2019;23(3):261‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sperduto PW, Kased N, Roberge D, et al. Summary report on the graded prognostic assessment: an accurate and facile diagnosis‐specific tool to estimate survival for patients with brain metastases. J Clin Oncol. 2012;30(4):419‐425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kirkpatrick JP, Laack NN, Shih HA, Gondi V. Management of GBM: a problem of local recurrence. J Neurooncol. 2017;134(3):487‐493. [DOI] [PubMed] [Google Scholar]
- 7. Kotecha R, Gondi V, Ahluwalia MS, Brastianos PK, Mehta MP. Recent advances in managing brain metastasis. F1000Res. 2018;7:1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kipnis J. Multifaceted interactions between adaptive immunity and the central nervous system. Science. 2016;353(6301):766‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Titov A, Petukhov A, Staliarova A, et al. The biological basis and clinical symptoms of CAR‐T therapy‐associated toxicites. Cell Death Dis. 2018;9(9):897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Keskin DB, Anandappa AJ, Sun J, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565(7738):234‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Desjardins A, Gromeier M, Herndon JE, et al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med. 2018;379(2):150‐161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kamath SD, Kumthekar PU. Immune checkpoint inhibitors for the treatment of central nervous system (CNS) metastatic disease. Front Oncol. 2018;8:414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Simonelli M, Persico P, Perrino M, et al. Checkpoint inhibitors as treatment for malignant gliomas: "a long way to the top". Cancer Treat Rev. 2018;69:121‐131. [DOI] [PubMed] [Google Scholar]
- 14. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tawbi HA, Forsyth PA, Algazi A, et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N Engl J Med. 2018;379(8):722‐730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goodman AM, Kato S, Bazhenova L, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16(11):2598‐2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. AlHarbi M, Ali Mobark N, AlMubarak L, et al. Durable response to nivolumab in a pediatric patient with refractory glioblastoma and constitutional biallelic mismatch repair deficiency. Oncologist. 2018;23(12):1401‐1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bouffet E, Larouche V, Campbell BB, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol. 2016;34(19):2206‐2211. [DOI] [PubMed] [Google Scholar]
- 19. Omuro A, Vlahovic G, Lim M, et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. 2018;20(5):674‐686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cloughesy TF, Mochizuki AY, Orpilla JR, et al. Neoadjuvant anti‐PD‐1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25(3):477‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Priceman SJ, Forman SJ, Brown CE. Smart CARs engineered for cancer immunotherapy. Curr Opin Oncol. 2015;27(6):466‐474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang C, Liu J, Zhong JF, Zhang X. Engineering CAR‐T cells. Biomark Res. 2017;5:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maus MV, June CH. Making better chimeric antigen receptors for adoptive T‐cell therapy. Clin Cancer Res. 2016;22(8):1875‐1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shum T, Kruse RL, Rooney CM. Strategies for enhancing adoptive T‐cell immunotherapy against solid tumors using engineered cytokine signaling and other modalities. Expert Opin Biol Ther. 2018;18(6):653‐664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yeku OO, Brentjens RJ. Armored CAR T‐cells: utilizing cytokines and pro‐inflammatory ligands to enhance CAR T‐cell anti‐tumour efficacy. Biochem Soc Trans. 2016;44(2):412‐418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guedan S, Calderon H, Posey AD Jr, Maus MV. Engineering and design of chimeric antigen receptors. Mol Ther Methods Clin Dev. 2019;12:145‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang X, Riviere I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther Oncolytics. 2016;3:16015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Levine BL, Miskin J, Wonnacott K, Keir C. Global manufacturing of CAR T cell therapy. Mol Ther Methods Clin Dev. 2017;4:92‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cheadle EJ, Rothwell DG, Bridgeman JS, Sheard VE, Hawkins RE, Gilham DE. Ligation of the CD2 co‐stimulatory receptor enhances IL‐2 production from first‐generation chimeric antigen receptor T cells. Gene Ther. 2012;19(11):1114‐1120. [DOI] [PubMed] [Google Scholar]
- 30. Alizadeh D, Wong RA, Yang X, et al. IL‐15‐mediated reduction of mTORC1 activity preserves the stem cell memory phenotype of CAR‐T cells and confers superior antitumor activity. Cancer Immunol Res. 2019;7:759‐772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cieri N, Camisa B, Cocchiarella F, et al. IL‐7 and IL‐15 instruct the generation of human memory stem T cells from naive precursors. Blood. 2013;121(4):573‐584. [DOI] [PubMed] [Google Scholar]
- 32. Gattinoni L, Finkelstein SE, Klebanoff CA, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor‐specific CD8+ T cells. J Exp Med. 2005;202(7):907‐912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by gamma(c) family cytokines. Nat Rev Immunol. 2009;9(7):480‐490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Santegoets SJ, Turksma AW, Suhoski MM, et al. IL‐21 promotes the expansion of CD27+ CD28+ tumor infiltrating lymphocytes with high cytotoxic potential and low collateral expansion of regulatory T cells. J Transl Med. 2013;11:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xu Y, Zhang M, Ramos CA, et al. Closely related T‐memory stem cells correlate with in vivo expansion of CAR.CD19‐T cells and are preserved by IL‐7 and IL‐15. Blood. 2014;123(24):3750‐3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Blaeschke F, Stenger D, Kaeuferle T, et al. Induction of a central memory and stem cell memory phenotype in functionally active CD4(+) and CD8(+) CAR T cells produced in an automated good manufacturing practice system for the treatment of CD19(+) acute lymphoblastic leukemia. Cancer Immunol Immunother. 2018;67(7):1053‐1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kawalekar OU, O’Connor RS, Fraietta JA, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44(2):380‐390. [DOI] [PubMed] [Google Scholar]
- 38. Fraietta JA, Lacey SF, Orlando EJ, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. 2018;24(5):563‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ghassemi S, Nunez‐Cruz S, O'Connor RS, et al. Reducing ex vivo culture improves the antileukemic activity of chimeric antigen receptor (CAR) T cells. Cancer Immunol Res. 2018;6(9):1100‐1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Piscopo NJ, Mueller KP, Das A, et al. Bioengineering solutions for manufacturing challenges in CAR T cells. Biotechnol J. 2018;13(2):1700095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Roddie C, O'Reilly M, Dias Alves Pinto J, Vispute K, Lowdell M. Manufacturing chimeric antigen receptor T cells: issues and challenges. Cytotherapy. 2019;21(3):327‐340. [DOI] [PubMed] [Google Scholar]
- 42. Hong JJ, Rosenberg SA, Dudley ME, et al. Successful treatment of melanoma brain metastases with adoptive cell therapy. Clin Cancer Res. 2010;16(19):4892‐4898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B‐cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439‐448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T‐cell therapy in refractory large B‐cell lymphoma. N Engl J Med. 2017;377(26):2531‐2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee DW, Kochenderfer JN, Stetler‐Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose‐escalation trial. Lancet. 2015;385(9967):517‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Abramson JS, McGree B, Noyes S, et al. Anti‐CD19 CAR T cells in CNS diffuse large‐B‐cell lymphoma. N Engl J Med. 2017;377(8):783‐784. [DOI] [PubMed] [Google Scholar]
- 47. Abramson JS, Gordon LI, Palomba ML, et al. Updated safety and long term clinical outcomes in TRANSCEND NHL 001, pivotal trial of lisocabtagene maraleucel (JCAR017) in R/R aggressive NHL. J Clin Oncol. 2018;36(15_suppl):7505‐7505. [Google Scholar]
- 48. Gust J, Hay KA, Hanafi LA, et al. Endothelial activation and blood‐brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR‐T cells. Cancer Discov. 2017;7(12):1404‐1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Locke FL, Ghobadi A, Jacobson CA, et al. Long‐term safety and activity of axicabtagene ciloleucel in refractory large B‐cell lymphoma (ZUMA‐1): a single‐arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019;20(1):31‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B‐cell lymphoma. N Engl J Med. 2019;380(1):45‐56. [DOI] [PubMed] [Google Scholar]
- 51. De Angelo DJ, Ghobadi A, Park JH, et al. Clinical Outcomes for the Phase 2, Single‐Arm, Multicenter Trial of JCAR015 in Adult B‐ALL (ROCKET Study). Oxon Hill, MD: Society for Immunotherapy of Cancer; 2017. [Google Scholar]
- 52. Harris J. Kite reports cerebral edema death in ZUMA‐1 CAR T‐cell trial. OncLive 2017.
- 53. Santomasso BD, Park JH, Salloum D, et al. Clinical and biological correlates of neurotoxicity associated with CAR T‐cell therapy in patients with B‐cell acute lymphoblastic leukemia. Cancer Discov. 2018;8(8):958‐971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Norelli M, Camisa B, Barbiera G, et al. Monocyte‐derived IL‐1 and IL‐6 are differentially required for cytokine‐release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24(6):739‐748. [DOI] [PubMed] [Google Scholar]
- 55. Taraseviciute A, Tkachev V, Ponce R, et al. Chimeric antigen receptor T cell‐mediated neurotoxicity in nonhuman primates. Cancer Discov. 2018;8(6):750‐763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Morgan RA, Chinnasamy N, Abate‐Daga D, et al. Cancer regression and neurological toxicity following anti‐MAGE‐A3 TCR gene therapy. J Immunother. 2013;36(2):133‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Debinski W, Obiri NI, Powers SK, Pastan I, Puri RK. Human glioma cells overexpress receptors for interleukin 13 and are extremely sensitive to a novel chimeric protein composed of interleukin 13 and pseudomonas exotoxin. Clin Cancer Res. 1995;1(11):1253‐1258. [PubMed] [Google Scholar]
- 58. Brown CE, Warden CD, Starr R, et al. Glioma IL13Ralpha2 is associated with mesenchymal signature gene expression and poor patient prognosis. PLoS ONE. 2013;8(10):e77769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jarboe JS, Johnson KR, Choi Y, Lonser RR, Park JK. Expression of interleukin‐13 receptor alpha2 in glioblastoma multiforme: implications for targeted therapies. Cancer Res. 2007;67(17):7983‐7986. [DOI] [PubMed] [Google Scholar]
- 60. Brown CE, Starr R, Aguilar B, et al. Stem‐like tumor‐initiating cells isolated from IL13Ralpha2 expressing gliomas are targeted and killed by IL13‐zetakine‐redirected T Cells. Clin Cancer Res. 2012;18(8):2199‐2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Debinski W, Gibo DM, Hulet SW, Connor JR, Gillespie GY. Receptor for interleukin 13 is a marker and therapeutic target for human high‐grade gliomas. Clin Cancer Res. 1999;5(5):985‐990. [PubMed] [Google Scholar]
- 62. Debinski W, Gibo DM, Obiri NI, Kealiher A, Puri RK. Novel anti‐brain tumor cytotoxins specific for cancer cells. Nat Biotechnol. 1998;16(5):449‐453. [DOI] [PubMed] [Google Scholar]
- 63. Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13‐zetakine redirected cytolytic T cells. Cancer Res. 2004;64(24):9160‐9166. [DOI] [PubMed] [Google Scholar]
- 64. Brown CE, Aguilar B, Starr R, et al. Optimization of IL13Ralpha2‐targeted chimeric antigen receptor T cells for improved anti‐tumor efficacy against glioblastoma. Mol Ther. 2018;26(1):31‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Jonnalagadda M, Mardiros A, Urak R, et al. Chimeric antigen receptors with mutated IgG4 Fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Mol Ther. 2015;23(4):757‐768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Brown CE, Badie B, Barish ME, et al. Bioactivity and safety of IL13Ralpha2‐redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. 2015;21(18):4062‐4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Keu KV, Witney TH, Yaghoubi S, et al. Reporter gene imaging of targeted T cell immunotherapy in recurrent glioma. Sci Transl Med. 2017;9(373):eaag2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yaghoubi SS, Jensen MC, Satyamurthy N, et al. Noninvasive detection of therapeutic cytolytic T cells with 18F‐FHBG PET in a patient with glioma. Nat Clin Pract Oncol. 2009;6(1):53‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Brown CE, Alizadeh D, Starr R, et al. Regression of glioblastoma after chimeric antigen receptor T‐cell therapy. N Engl J Med. 2016;375(26):2561‐2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bielamowicz K, Fousek K, Byrd TT, et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. 2018;20(4):506‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Krebs S, Chow KK, Yi Z, et al. T cells redirected to interleukin‐13Ralpha2 with interleukin‐13 mutein – chimeric antigen receptors have anti‐glioma activity but also recognize interleukin‐13Ralpha1. Cytotherapy. 2014;16(8):1121‐1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Krenciute G, Krebs S, Torres D, et al. Characterization and functional analysis of scFv‐based chimeric antigen receptors to redirect T cells to IL13Ralpha2‐positive glioma. Mol Ther. 2016;24(2):354‐363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yin Y, Boesteanu AC, Binder ZA, et al. Checkpoint blockade reverses anergy in IL‐13Ralpha2 humanized scFv‐based CAR T cells to treat murine and canine gliomas. Mol Ther Oncolytics. 2018;11:20‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kong S, Sengupta S, Tyler B, et al. Suppression of human glioma xenografts with second‐generation IL13R‐specific chimeric antigen receptor‐modified T cells. Clin Cancer Res. 2012;18(21):5949‐5960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Brennan C, Verhaak R, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Feldkamp MM, Feldkamp MM, Lala P, et al. Expression of activated epidermal growth factor receptors, Ras‐guanosine triphosphate, and mitogen‐activated protein kinase in human glioblastoma multiforme specimens. Neurosurgery. 1999;45(6):1442‐1453. [DOI] [PubMed] [Google Scholar]
- 77. Sampson JH, Choi BD, Sanchez‐Perez L, et al. EGFRvIII mCAR‐modified T‐cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor‐antigen loss. Clin Cancer Res. 2014;20(4):972‐984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Morgan RA, Johnson LA, Davis JL, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther. 2012;23(10):1043‐1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Choi BD, Suryadevara CM, Gedeon PC, et al. Intracerebral delivery of a third generation EGFRvIII‐specific chimeric antigen receptor is efficacious against human glioma. J Clin Neurosci. 2014;21(1):189‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. O’Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII‐directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399):eaaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Goff SL, Morgan RA, Yang JC, et al. Pilot trial of adoptive transfer of chimeric antigen receptor‐transduced T cells targeting EGFRvIII in patients with glioblastoma. J Immunother. 2019;42(4):126‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Johnson LA, Scholler J, Ohkuri T, et al. Rational development and characterization of humanized anti‐EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med. 2015;7(275):275ra22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. van den Bent MJ, Gao YA, Kerkhof M, et al. Changes in the EGFR amplification and EGFRvIII expression between paired primary and recurrent glioblastomas. Neuro Oncol. 2015;17(7):935‐941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hicks MJ, Chiuchiolo MJ, Ballon D, et al. Anti‐epidermal growth factor receptor gene therapy for glioblastoma. PLoS ONE. 2016;11(10):e0162978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Johns TG, Stockert E, Ritter G, et al. Novel monoclonal antibody specific for the de2‐7 epidermal growth factor receptor (EGFR) that also recognizes the EGFR expressed in cells containing amplification of the EGFR gene. Int J Cancer. 2002;98(3):398‐408. [DOI] [PubMed] [Google Scholar]
- 86. Scott AM, Lee F‐T, Tebbutt N, et al. A phase I clinical trial with monoclonal antibody ch806 targeting transitional state and mutant epidermal growth factor receptors. Proc Natl Acad Sci USA. 2007;104(10):4071‐4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jiang H, Gao H, Kong J, et al. Selective targeting of glioblastoma with EGFRvIII/EGFR bitargeted chimeric antigen receptor T cell. Cancer Immunol Res. 2018;6(11):1314‐1326. [DOI] [PubMed] [Google Scholar]
- 88. Caruso HG, Hurton LV, Najjar A, et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res. 2015;75(17):3505‐3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Liu X, Jiang S, Fang C, et al. Affinity‐tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. 2015;75(17):3596‐3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Pernas S, Tolaney SM. HER2‐positive breast cancer: new therapeutic frontiers and overcoming resistance. Ther Adv Med Oncol. 2019;11:1758835919833519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Liu R, Qu Y, Chen L, et al. Genomic copy number gains of ErbB family members predict poor clinical outcomes in glioma patients. Oncotarget. 2017;8(54):92275‐92288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843‐851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Priceman SJ, Tilakawardane D, Jeang B, et al. Regional delivery of chimeric antigen receptor‐engineered T cells effectively targets HER2+ breast cancer metastasis to the brain. Clin Cancer Res. 2018;24(1):95‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Moritz D, Wels W, Mattern J, Groner B. Cytotoxic T lymphocytes with a grafted recognition specificity for ERBB2‐expressing tumor cells. Proc Natl Acad Sci USA. 1994;91(10):4318‐4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Ahmed N, Salsman VS, Kew Y, et al. HER2‐specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res. 2010;16(2):474‐485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ahmed N, Brawley VS, Hegde M, et al. Human epidermal growth factor receptor 2 (HER2)‐specific chimeric antigen receptor‐modified T cells for the immunotherapy of HER2‐positive sarcoma. J Clin Oncol. 2015;33(15):1688‐1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Pule MA, Savoldo B, Myers GD, et al. Virus‐specific T cells engineered to coexpress tumor‐specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14(11):1264‐1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501(7467):328‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Parker NR, Khong P, Parkinson JF, Howell VM, Wheeler HR. Molecular heterogeneity in glioblastoma: potential clinical implications. Front Oncol. 2015;5:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Verhaak R, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Wang Q, Hu B, Hu X, et al. Tumor evolution of glioma‐intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell. 2017;32(1):42‐56.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Puchalski RB, Shah N, Miller J, et al. An anatomic transcriptional atlas of human glioblastoma. Science. 2018;360(6389):660‐663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Jin X, Kim L, Wu Q, et al. Targeting glioma stem cells through combined BMI1 and EZH2 inhibition. Nat Med. 2017;23(11):1352‐1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Meyer M, Reimand J, Lan X, et al. Single cell‐derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc Natl Acad Sci USA. 2015;112(3):851‐856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Patel AP, Tirosh I, Trombetta JJ, et al. Single‐cell RNA‐seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396‐1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Venteicher AS, Tirosh I, Hebert C, et al. Decoupling genetics, lineages, and microenvironment in IDH‐mutant gliomas by single‐cell RNA‐seq. Science. 2017;355(6332):eaai8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Doucette T, Rao G, Rao A, et al. Immune heterogeneity of glioblastoma subtypes: extrapolation from the cancer genome atlas. Cancer Immunol Res. 2013;1(2):112‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Brown CE, Mackall CL. CAR T cell therapy: inroads to response and resistance. Nat Rev Immunol. 2019;19(2):73‐74. [DOI] [PubMed] [Google Scholar]
- 109. Wykosky J, Gibo DM, Stanton C, Debinski W. EphA2 as a novel molecular marker and target in glioblastoma multiforme. Mol Cancer Res. 2005;3(10):541‐551. [DOI] [PubMed] [Google Scholar]
- 110. Dobrzanski P, Hunter K, Jones‐Bolin S, et al. Antiangiogenic and antitumor efficacy of EphA2 receptor antagonist. Cancer Res. 2004;64(3):910‐919. [DOI] [PubMed] [Google Scholar]
- 111. Chow K, Naik S, Kakarla S, et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol Ther. 2013;21(3):629‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Mount CW, Majzner RG, Sundaresh S, et al. Potent antitumor efficacy of anti‐GD2 CAR T cells in H3–K27M(+) diffuse midline gliomas. Nat Med. 2018;24(5):572‐579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Louis CU, Savoldo B, Dotti G, et al. Antitumor activity and long‐term fate of chimeric antigen receptor‐positive T cells in patients with neuroblastoma. Blood. 2011;118(23):6050‐6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Prasad D, Nguyen T, Li Z, et al. Murine B7–H3 is a negative regulator of T cells. J Immunol. 2004;173(4):2500‐2506. [DOI] [PubMed] [Google Scholar]
- 115. Suh W‐K, Gajewska BU, Okada H, et al. The B7 family member B7–H3 preferentially down‐regulates T helper type 1‐mediated immune responses. Nat Immunol. 2003;4(9):899‐906. [DOI] [PubMed] [Google Scholar]
- 116. Xie C, Liu D, Chen Q, Yang C, Wang B, Wu H. Soluble B7–H3 promotes the invasion and metastasis of pancreatic carcinoma cells through the TLR4/NF‐kappaB pathway. Sci Rep. 2016;6:27528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Wang Z, Wang Z, Zhang C, et al. Genetic and clinical characterization of B7–H3 (CD276) expression and epigenetic regulation in diffuse brain glioma. Cancer Sci. 2018;109(9):2697‐2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Majzner RG, Theruvath JL, Nellan A, et al. CAR T cells targeting B7‐H3, a pan‐cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clin Cancer Res. 2019;25(8):2560‐2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Pellegatta S, Savoldo B, Di Ianni N, et al. Constitutive and TNFalpha‐inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: Implications for CAR‐T cell therapy. Sci Transl Med. 2018;10(430):eaao2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Prager BC, Xie Q, Bao S, Rich JN. Cancer stem cells: the architects of the tumor ecosystem. Cell Stem Cell. 2019;24(1):41‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Hemmati HD, Nakano I, Lazareff JA, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci USA. 2003;100(25):15178‐15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63(18):5821‐5828. [PubMed] [Google Scholar]
- 123. Zhu X, Prasad S, Gaedicke S, Hettich M, Firat E, Niedermann G. Patient‐derived glioblastoma stem cells are killed by CD133‐specific CAR T cells but induce the T cell aging marker CD57. Oncotarget. 2015;6(1):171‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Wang Y, Chen M, Wu Z, et al. CD133‐directed CAR T cells for advanced metastasis malignancies: a phase I trial. Oncoimmunology. 2018;7(7):e1440169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Coskun V, Wu H, Blanchi B, et al. CD133+ neural stem cells in the ependyma of mammalian postnatal forebrain. Proc Natl Acad Sci USA. 2008;105(3):1026‐1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Sanai N, Alvarez‐Buylla A, Berger MS. Neural stem cells and the origin of gliomas. N Engl J Med. 2005;353(8):811‐822. [DOI] [PubMed] [Google Scholar]
- 127. Sun XL, Li YS, Liu T, Li ZJ, Zhang XZ, Chen XY. Peptide‐based imaging agents for cancer detection. Adv Drug Deliv Rev. 2017;110:38‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Warner RR, O'Dorisio TM. Radiolabeled peptides in diagnosis and tumor imaging: clinical overview. Semin Nucl Med. 2002;32(2):79‐83. [DOI] [PubMed] [Google Scholar]
- 129. Wang DR, Wright S, Chang WC, et al. Therapeutic potential of chlorotoxin‐redirected Car‐T cells against heterogeneous glioblastomas. Neuro Oncol. 2017;19:114‐114. [Google Scholar]
- 130. Veiseh M, Gabikian P, Bahrami SB, et al. Tumor paint: a chlorotoxin:Cy5.5 bioconjugate for intraoperative visualization of cancer foci. Cancer Res. 2007;67(14):6882‐6888. [DOI] [PubMed] [Google Scholar]
- 131. Fidel J, Kennedy KC, Dernell WS, et al. Preclinical validation of the utility of BLZ‐100 in providing fluorescence contrast for imaging spontaneous solid tumors. Cancer Res. 2015;75(20):4283‐4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Parrish‐Novak J, Byrnes‐Blake K, Lalayeva N, et al. Nonclinical profile of BLZ‐100, a tumor‐targeting fluorescent imaging agent. Int J Toxicol. 2017;36(2):104‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Chheda ZS, Kohanbash G, Okada K, et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J Exp Med. 2018;215(1):141‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Chang ZL, Lorenzini MH, Chen X, Tran U, Bangayan NJ, Chen YY. Rewiring T‐cell responses to soluble factors with chimeric antigen receptors. Nat Chem Biol. 2018;14(3):317‐324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Hou AJ, Chang ZL, Lorenzini MH, Zah E, Chen YY. TGF‐β‐responsive CAR‐T cells promote anti‐tumor immune function. Bioeng Transl Med. 2018;3(2):75‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. 2016;16(9):566‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Raponi S, Stefania De Propris M, Intoppa S, et al. Flow cytometric study of potential target antigens (CD19, CD20, CD22, CD33) for antibody‐based immunotherapy in acute lymphoblastic leukemia: analysis of 552 cases. Leuk Lymphoma. 2011;52(6):1098‐1107. [DOI] [PubMed] [Google Scholar]
- 138. Fry TJ, Shah NN, Orentas RJ, et al. CD22‐targeted CAR T cells induce remission in B‐ALL that is naive or resistant to CD19‐targeted CAR immunotherapy. Nat Med. 2018;24(1):20‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Zah E, Lin MY, Silva‐Benedict A, Jensen MC, Chen YY. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol Res. 2016;4(6):498‐508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Hegde M, Mukherjee M, Grada Z, et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J Clin Invest. 2016;126(8):3036‐3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Perisic O, Webb PA, Holliger P, Winter G, Williams RL. Crystal structure of a diabody, a bivalent antibody fragment. Structure. 1994;2(12):1217‐1226. [DOI] [PubMed] [Google Scholar]
- 142. Qin H, Ramakrishna S, Nguyen S, et al. Preclinical development of bivalent chimeric antigen receptors targeting both CD19 and CD22. Mol Ther Oncolytics. 2018;11:127‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Ruella M, Barrett DM, Kenderian SS, et al. Dual CD19 and CD123 targeting prevents antigen‐loss relapses after CD19‐directed immunotherapies. J Clin Invest. 2016;126(10):3814‐3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Huang L, Wang N, Li CR, et al. Sequential infusion of anti‐CD22 and anti‐CD19 chimeric antigen receptor T cells for adult patients with refractory/relapsed B‐cell acute lymphoblastic leukemia. Blood. 2017;130(Suppl 1):846. [Google Scholar]
- 145. Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell. 2018;173(6):1426‐1438.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Rodgers DT, Mazagova M, Hampton EN, et al. Switch‐mediated activation and retargeting of CAR‐T cells for B‐cell malignancies. Proc Natl Acad Sci USA. 2016;113(4):E459‐468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Calzascia T, Di Berardino‐Besson W, Wilmotte R, et al. Cutting edge: cross‐presentation as a mechanism for efficient recruitment of tumor‐specific CTL to the brain. J Immunol. 2003;171(5):2187‐2191. [DOI] [PubMed] [Google Scholar]
- 148. Kohanbash G, Carrera DA, Shrivastav S, et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J Clin Invest. 2017;127(4):1425‐1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Berghoff AS, Kiesel B, Widhalm G, et al. Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro Oncol. 2017;19(11):1460‐1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Howie D, Waldmann H, Cobbold S. Nutrient sensing via mTOR in T cells maintains a tolerogenic microenvironment. Front Immunol. 2014;5:409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Dagenais‐Lussier X, Aounallah M, Mehraj V, et al. Kynurenine reduces memory CD4 T‐cell survival by interfering with interleukin‐2 signaling early during HIV‐1 infection. J Virol. 2016;90(17):7967‐7979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Alizadeh D, Trad M, Hanke NT, et al. Doxorubicin eliminates myeloid‐derived suppressor cells and enhances the efficacy of adoptive T‐cell transfer in breast cancer. Cancer Res. 2014;74(1):104‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Arlauckas SP, Garren SB, Garris CS, et al. Arg1 expression defines immunosuppressive subsets of tumor‐associated macrophages. Theranostics. 2018;8(21):5842‐5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Fischer K, Hoffmann P, Voelkl S, et al. Inhibitory effect of tumor cell‐derived lactic acid on human T cells. Blood. 2007;109(9):3812‐3819. [DOI] [PubMed] [Google Scholar]
- 155. Newick K, O'Brien S, Sun J, et al. Augmentation of CAR T‐cell trafficking and antitumor efficacy by blocking protein kinase A localization. Cancer Immunol Res. 2016;4(6):541‐551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Bos R, Zhong H, Hanrahan CF, et al. Levels of hypoxia‐inducible factor‐1 alpha during breast carcinogenesis. J Natl Cancer Inst. 2001;93(4):309‐314. [DOI] [PubMed] [Google Scholar]
- 157. Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia‐mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014;74(3):665‐674. [DOI] [PubMed] [Google Scholar]
- 158. Fang H‐Y, Hughes R, Murdoch C, et al. Hypoxia‐inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood. 2009;114(4):844‐859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Newick K, Moon E, Albelda SM. Chimeric antigen receptor T‐cell therapy for solid tumors. Mol Ther Oncolytics. 2016;3:16006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Poh AR, Ernst M. Targeting macrophages in cancer: from bench to bedside. Front Oncol. 2018;8:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Stafford JH, Hirai T, Deng L, et al. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro Oncol. 2016;18(6):797‐806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Hussain SF, Kong L‐Y, Jordan J, et al. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 2007;67(20):9630‐9636. [DOI] [PubMed] [Google Scholar]
- 163. Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 2009;69(6):2506‐2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Mariathasan S, Turley SJ, Nickles D, et al. TGFbeta attenuates tumour response to PD‐L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Maier A, Peille AL, Vuaroqueaux V, Lahn M. Anti‐tumor activity of the TGF‐beta receptor kinase inhibitor galunisertib (LY2157299 monohydrate) in patient‐derived tumor xenografts. Cell Oncol (Dordr). 2015;38(2):131‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Sher A, Fiorentino D, Caspar P, Pearce E, Mosmann T. Production of IL‐10 by CD4+ T lymphocytes correlates with down‐regulation of Th1 cytokine synthesis in helminth infection. J Immunol. 1991;147(8):2713‐2716. [PubMed] [Google Scholar]
- 167. Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O'Garra A. IL‐10 inhibits cytokine production by activated macrophages. J Immunol. 1991;147(11):3815‐3822. [PubMed] [Google Scholar]
- 168. Hishii M, Nitta T, Ishida H, et al. Human glioma‐derived interleukin‐10 inhibits antitumor immune responses in vitro. Neurosurgery. 1995;37(6):1166‐1167; discussion 1166‐1167. [DOI] [PubMed] [Google Scholar]
- 169. Shiao SL, Ruffell B, DeNardo DG, Faddegon BA, Park CC, Coussens LM. TH2‐polarized CD4(+) T cells and macrophages limit efficacy of radiotherapy. Cancer Immunol Res. 2015;3(5):518‐525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Kloss CC, Lee J, Zhang A, et al. Dominant‐negative TGF‐beta receptor enhances PSMA‐targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. 2018;26(7):1855‐1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Bollard CM, Rossig C, Calonge MJ, et al. Adapting a transforming growth factor beta‐related tumor protection strategy to enhance antitumor immunity. Blood. 2002;99(9):3179‐3187. [DOI] [PubMed] [Google Scholar]
- 172. Mohammed S, Sukumaran S, Bajgain P, et al. Improving chimeric antigen receptor‐modified T cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer. Mol Ther. 2017;25(1):249‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL‐12 secreting tumor‐targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology. 2015;4(3):e994446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Hu B, Ren J, Luo Y, et al. Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL‐18. Cell Rep. 2017;20(13):3025‐3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Shum T, Omer B, Tashiro H, et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor‐redirected T cells. Cancer Discov. 2017;7(11):1238‐1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Hurton LV, Singh H, Najjar AM, et al. Tethered IL‐15 augments antitumor activity and promotes a stem‐cell memory subset in tumor‐specific T cells. Proc Natl Acad Sci USA. 2016;113(48):E7788‐E7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Tang L, Zheng Y, Melo MB, et al. Enhancing T cell therapy through TCR‐signaling‐responsive nanoparticle drug delivery. Nat Biotechnol. 2018;36(8):707‐716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Chinnasamy D, Yu Z, Kerkar SP, et al. Local delivery of interleukin‐12 using T cells targeting VEGF receptor‐2 eradicates multiple vascularized tumors in mice. Clin Cancer Res. 2012;18(6):1672‐1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Koneru M, O'Cearbhaill R, Pendharkar S, Spriggs DR, Brentjens RJ. A phase I clinical trial of adoptive T cell therapy using IL‐12 secreting MUC‐16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J Transl Med. 2015;13:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Chong EA, Melenhorst JJ, Lacey SF, et al. PD‐1 blockade modulates chimeric antigen receptor (CAR)‐modified T cells: refueling the CAR. Blood. 2017;129(8):1039‐1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Reardon DA, Gokhale PC, Klein SR, et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol Res. 2016;4(2):124‐135. [DOI] [PubMed] [Google Scholar]
- 182. Fecci PE, Ochiai H, Mitchell DA, et al. Systemic CTLA‐4 blockade ameliorates glioma‐induced changes to the CD4+ T cell compartment without affecting regulatory T‐cell function. Clin Cancer Res. 2007;13(7):2158‐2167. [DOI] [PubMed] [Google Scholar]
- 183. Schalper KA, Rodriguez‐Ruiz ME, Diez‐Valle R, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med. 2019;25(3):470‐476. [DOI] [PubMed] [Google Scholar]
- 184. Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. 2017;23(9):2255‐2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer. 2014;14(8):559‐567. [DOI] [PubMed] [Google Scholar]
- 186. Rosewell Shaw A, Porter CE, Watanabe N, et al. Adenovirotherapy delivering cytokine and checkpoint inhibitor augments CAR T cells against metastatic head and neck cancer. Mol Ther. 2017;25(11):2440‐2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Mardiana S, John LB, Henderson MA, et al. A multifunctional role for adjuvant anti‐4‐1BB therapy in augmenting antitumor response by chimeric antigen receptor T cells. Cancer Res. 2017;77(6):1296‐1309. [DOI] [PubMed] [Google Scholar]
- 188. Sampson JH, Maus MV, June CH. Immunotherapy for brain tumors. J Clin Oncol. 2017;35(21):2450‐2456. [DOI] [PubMed] [Google Scholar]
- 189. Lehrer EJ, Peterson J, Brown PD, et al. Treatment of brain metastases with stereotactic radiosurgery and immune checkpoint inhibitors: an international meta‐analysis of individual patient data. Radiother Oncol. 2019;130:104‐112. [DOI] [PubMed] [Google Scholar]
- 190. Kim JE, Patel MA, Mangraviti A, et al. Combination therapy with anti‐PD‐1, anti‐TIM‐3, and focal radiation results in regression of murine gliomas. Clin Cancer Res. 2017;23(1):124‐136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Lee Y, Auh SL, Wang Y, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood. 2009;114(3):589‐595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Rudqvist N‐P, Pilones KA, Lhuillier C, et al. Radiotherapy and CTLA‐4 blockade shape the TCR repertoire of tumor‐infiltrating T cells. Cancer Immunol Res. 2018;6(2):139‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Zeng J, See AP, Phallen J, et al. Anti‐PD‐1 blockade and stereotactic radiation produce long‐term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86(2):343‐349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Vanpouille‐Box C, Alard A, Aryankalayil MJ, et al. DNA exonuclease Trex1 regulates radiotherapy‐induced tumour immunogenicity. Nat Commun. 2017;8:15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Spranger S, Dai D, Horton B, Gajewski TF. Tumor‐residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. 2017;31(5):711‐723.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Smith TT, Moffett HF, Stephan SB, et al. Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J Clin Invest. 2017;127(6):2176‐2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Exp Immunol. 2010;162(1):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Chuntova P, Downey KM, Hegde B, Almeida ND, Okada H. Genetically engineered T‐cells for malignant glioma: overcoming the barriers to effective immunotherapy. Front Immunol. 2019;9:3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Ratnam NM, Gilbert MR, Giles AJ. Immunotherapy in CNS cancers: the role of immune cell trafficking. Neuro Oncol. 2019;21(1):37‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Lehmann PV. The fate of T cells in the brain: veni, vidi, vici and veni, mori. Am J Pathol. 1998;153(3):677‐680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Galea I, Bernardes‐Silva M, Forse PA, van Rooijen N, Liblau RS, Perry VH. An antigen‐specific pathway for CD8 T cells across the blood‐brain barrier. J Exp Med. 2007;204(9):2023‐2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Rosen J, Blau T, Grau SJ, Barbe MT, Fink GR, Galldiks N. Extracranial metastases of a cerebral glioblastoma: a case report and review of the literature. Case Rep Oncol. 2018;11(2):591‐600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Sridhar P, Petrocca F. Regional delivery of chimeric antigen receptor (CAR) T‐cells for cancer therapy. Cancers. 2017;9(12):92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Cohen‐Pfeffer JL, Gururangan S, Lester T, et al. Intracerebroventricular delivery as a safe, long‐term route of drug administration. Pediatr Neurol. 2017;67:23‐35. [DOI] [PubMed] [Google Scholar]
- 205. Weist MR, Starr R, Aguilar B, et al. PET of adoptively transferred chimeric antigen receptor T cells with (89)Zr‐oxine. J Nucl Med. 2018;59(10):1531‐1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Hughes CE, Nibbs R. A guide to chemokines and their receptors. FEBS J. 2018;285(16):2944‐2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Di Stasi A, De Angelis B, Rooney CM, et al. T lymphocytes coexpressing CCR207 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113(25):6392‐6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Wang Y, Liu T, Yang N, Xu S, Li X, Wang D. Hypoxia and macrophages promote glioblastoma invasion by the CCL4‐CCR208 axis. Oncol Rep. 2016;36(6):3522‐3528. [DOI] [PubMed] [Google Scholar]
- 209. Pan Y, Smithson LJ, Ma Y, Hambardzumyan D, Gutmann DH. Ccl5 establishes an autocrine high‐grade glioma growth regulatory circuit critical for mesenchymal glioblastoma survival. Oncotarget. 2017;8(20):32977‐32989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Nguyen T, Lagman C, Chung LK, et al. Insights into CCL21's roles in immunosurveillance and immunotherapy for gliomas. J Neuroimmunol. 2017;305:29‐34. [DOI] [PubMed] [Google Scholar]
- 211. Kimura T, Takeshima H, Nomiyama N, et al. Expression of lymphocyte‐specific chemokines in human malignant glioma: essential role of LARC in cellular immunity of malignant glioma. Int J Oncol. 2002;21(4):707‐715. [DOI] [PubMed] [Google Scholar]
- 212. Chang AL, Miska J, Wainwright DA, et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid‐derived suppressor cells. Cancer Res. 2016;76(19):5671‐5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Brown CE, Vishwanath RP, Aguilar B, et al. Tumor‐derived chemokine MCP‐1/CCL2 is sufficient for mediating tumor tropism of adoptively transferred T cells. J Immunol. 2007;179(5):3332‐3341. [DOI] [PubMed] [Google Scholar]
- 214. Craddock JA, Lu AN, Bear A, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR214b. J Immunother. 2010;33(8):780‐788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Moon EK, Carpenito C, Sun J, et al. Expression of a functional CCR215 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin‐specific chimeric antibody receptor. Clin Cancer Res. 2011;17(14):4719‐4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Muller N, Michen S, Tietze S, et al. Engineering NK cells modified with an EGFRvIII‐specific chimeric antigen receptor to overexpress CXCR216 improves immunotherapy of CXCL12/SDF‐1alpha‐secreting glioblastoma. J Immunother. 2015;38(5):197‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Sapoznik S, Ortenberg R, Galore‐Haskel G, et al. CXCR217 as a novel target for directing reactive T cells toward melanoma: implications for adoptive cell transfer immunotherapy. Cancer Immunol Immunother. 2012;61(10):1833‐1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Idorn M, Skadborg SK, Kellermann L, et al. Chemokine receptor engineering of T cells with CXCR218 improves homing towards subcutaneous human melanomas in xenograft mouse model. Oncoimmunology. 2018;7(8):e1450715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Peng W, Ye Y, Rabinovich BA, et al. Transduction of tumor‐specific T cells with CXCR219 chemokine receptor improves migration to tumor and antitumor immune responses. Clin Cancer Res. 2010;16(22):5458‐5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Somanchi SS, Somanchi A, Cooper L, Lee DA. Engineering lymph node homing of ex vivo‐expanded human natural killer cells via trogocytosis of the chemokine receptor CCR220. Blood. 2012;119(22):5164‐5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Siddiqui I, Erreni M, Van Brakel M, Debets R, Allavena P. Enhanced recruitment of genetically modified CX3CR221‐positive human T cells into Fractalkine/CX3CL1 expressing tumors: importance of the chemokine gradient. J Immunother Cancer. 2016;4(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Wallstabe L, Mades A, Frenz S, Einsele H, Rader C, Hudecek M. CAR T cells targeting αvβ3 integrin are effective against advanced cancer in preclinical models. Adv Cell Gene Ther. 2018;1(2):e11 10.1002/acg2.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Livieratos L. Technical pitfalls and limitations of SPECT/CT. Semin Nucl Med. 2015;45(6):530‐540. [DOI] [PubMed] [Google Scholar]
- 224. Salama GR, Heier LA, Patel P, Ramakrishna R, Magge R, Tsiouris AJ. Diffusion weighted/tensor imaging, functional MRI and perfusion weighted imaging in glioblastoma‐foundations and future. Front Neurol. 2017;8:660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Dobrenkov K, Olszewska M, Likar Y, et al. Monitoring the efficacy of adoptively transferred prostate cancer‐targeted human T lymphocytes with PET and bioluminescence imaging. J Nucl Med. 2008;49(7):1162‐1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Vatakis DN, Koya RC, Nixon CC, et al. Antitumor activity from antigen‐specific CD8 T cells generated in vivo from genetically engineered human hematopoietic stem cells. Proc Natl Acad Sci USA. 2011;108(51):E1408‐E1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Najjar AM, Manuri PR, Olivares S, et al. Imaging of sleeping beauty‐modified CD19‐specific T cells expressing HSV1‐thymidine kinase by positron emission tomography. Mol Imaging Biol. 2016;18(6):838‐848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Jensen MC, Popplewell L, Cooper LJ, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19‐specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16(9):1245‐1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Berger C, Flowers ME, Warren EH, Riddell SR. Analysis of transgene‐specific immune responses that limit the in vivo persistence of adoptively transferred HSV‐TK‐modified donor T cells after allogeneic hematopoietic cell transplantation. Blood. 2006;107(6):2294‐2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. McCracken MN, Tavare R, Witte ON, Wu AM. Advances in PET detection of the antitumor T cell response. Adv Immunol. 2016;131:187‐231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Moroz MA, Zhang H, Lee J, et al. Comparative analysis of T cell imaging with human nuclear reporter genes. J Nucl Med. 2015;56(7):1055‐1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. McCracken MN, Vatakis DN, Dixit D, McLaughlin J, Zack JA, Witte ON. Noninvasive detection of tumor‐infiltrating T cells by PET reporter imaging. J Clin Invest. 2015;125(5):1815‐1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Lee JT, Zhang H, Moroz MA, et al. Comparative analysis of human nucleoside kinase‐based reporter systems for PET imaging. Mol Imaging Biol. 2017;19(1):100‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Miyagawa T, Gogiberidze G, Serganova I, et al. Imaging of HSV‐tk reporter gene expression: comparison between [18F]FEAU, [18F]FFEAU, and other imaging probes. J Nucl Med. 2008;49(4):637‐648. [DOI] [PubMed] [Google Scholar]
- 235. Ritchie DS, Neeson PJ, Khot A, et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther. 2013;21(11):2122‐2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. van Schalkwyk MC, Papa SE, Jeannon JP, Guerrero Urbano T, Spicer JF, Maher J. Design of a phase I clinical trial to evaluate intratumoral delivery of ErbB‐targeted chimeric antigen receptor T‐cells in locally advanced or recurrent head and neck cancer. Hum Gene Ther Clin Dev. 2013;24(3):134‐142. [DOI] [PubMed] [Google Scholar]
- 237. Stanton SE, Eary JF, Marzbani EA, et al. Concurrent SPECT/PET‐CT imaging as a method for tracking adoptively transferred T‐cells in vivo. J Immunother Cancer. 2016;4:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Roca M, de Vries EF, Jamar F, Israel O, Signore A. Guidelines for the labelling of leucocytes with (111)In‐oxine. Inflammation/Infection Taskgroup of the European Association of Nuclear Medicine. Eur J Nucl Med Mol Imaging. 2010;37(4):835‐841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. de Vries EF, Roca M, Jamar F, Israel O, Signore A. Guidelines for the labelling of leucocytes with (99m)Tc‐HMPAO. Inflammation/Infection Taskgroup of the European Association of Nuclear Medicine. Eur J Nucl Med Mol Imaging. 2010;37(4):842‐848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Ferris TJ, Charoenphun P, Meszaros LK, Mullen GE, Blower PJ, Went MJ. Synthesis and characterisation of zirconium complexes for cell tracking with Zr‐89 by positron emission tomography. Dalton Trans. 2014;43(39):14851‐14857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Jauw Y, Menke‐van der Houven van Oordt CW, Hoekstra OS, et al. Emission tomography with zirconium‐89‐labeled monoclonal antibodies in oncology: what can we learn from initial. Clinical Trials? Front Pharmacol. 2016;7:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Seo JW, Tavare R, Mahakian LM, et al. CD8(+) T‐cell density imaging with (64)Cu‐labeled Cys‐diabody informs immunotherapy protocols. Clin Cancer Res. 2018;24(20):4976‐4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Freise AC, Zettlitz KA, Salazar FB, et al. Immuno‐PET in inflammatory bowel disease: imaging CD4‐positive T cells in a murine model of colitis. J Nucl Med. 2018;59(6):980‐985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Barat B, Kenanova VE, Olafsen T, Wu AM. Evaluation of two internalizing carcinoembryonic antigen reporter genes for molecular imaging. Mol Imaging Biol. 2011;13(3):526‐535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Kenanova V, Barat B, Olafsen T, et al. Recombinant carcinoembryonic antigen as a reporter gene for molecular imaging. Eur J Nucl Med Mol Imaging. 2009;36(1):104‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Griessinger CM, Maurer A, Kesenheimer C, et al. 64Cu antibody‐targeting of the T‐cell receptor and subsequent internalization enables in vivo tracking of lymphocytes by PET. Proc Natl Acad Sci USA. 2015;112(4):1161‐1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Krebs S, Ahad A, Carter LM, et al. Antibody with infinite affinity for in vivo tracking of genetically engineered lymphocytes. J Nucl Med. 2018;59(12):1894‐1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987‐996. [DOI] [PubMed] [Google Scholar]
- 251. Hart MG, Garside R, Rogers G, Stein K, Grant R. Temozolomide for high grade glioma. Cochrane Database Syst Rev. 2013;(4):CD007415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Suryadevara CM, Desai R, Abel ML, et al. Temozolomide lymphodepletion enhances CAR abundance and correlates with antitumor efficacy against established glioblastoma. Oncoimmunology. 2018;7(6):e1434464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Li Y, Ali S, Clarke J, Cha S. Bevacizumab in recurrent glioma: patterns of treatment failure and implications. Brain Tumor Res Treat. 2017;5(1):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Dietrich J, Rao K, Pastorino S, Kesari S. Corticosteroids in brain cancer patients: benefits and pitfalls. Expert Rev Clin Pharmacol. 2011;4(2):233‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Huang H, Langenkamp E, Georganaki M, et al. VEGF suppresses T‐lymphocyte infiltration in the tumor microenvironment through inhibition of NF‐κB‐induced endothelial activation. FASEB J. 2015;29(1):227‐238. [DOI] [PubMed] [Google Scholar]
- 256. Yang J, Yan J, Liu B. Targeting VEGF/VEGFR to modulate antitumor immunity. Front Immunol. 2018;9:978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Bocca P, Di Carlo E, Caruana I, et al. Bevacizumab‐mediated tumor vasculature remodelling improves tumor infiltration and antitumor efficacy of GD2‐CAR T cells in a human neuroblastoma preclinical model. Oncoimmunology. 2018;7(1):e1378843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Turtle CJ, Hanafi L‐A, Berger C, et al. CD19 CAR‐T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126(6):2123‐2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Johnson LA, Morgan RA, Dudley ME, et al. Gene therapy with human and mouse T‐cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114(3):535‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Ahmed N, Brawley V, Hegde M, et al. HER2‐specific chimeric antigen receptor‐modified virus‐specific T cells for progressive glioblastoma: a phase 1 dose‐escalation trial. JAMA Oncol. 2017;3(8):1094‐1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Yi Z, Prinzing BL, Cao F, Gottschalk S, Krenciute G. Optimizing EphA2‐CAR T cells for the adoptive immunotherapy of glioma. Mol Ther Methods Clin Dev. 2018;9:70‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Brown CE, Starr R, Martinez C, et al. Recognition and killing of brain tumor stem‐like initiating cells by CD8+ cytolytic T cells. Cancer Res. 2009;69(23):8886‐8893. [DOI] [PMC free article] [PubMed] [Google Scholar]