

Abstract
Fluorinated motifs have a venerable history in drug discovery, but as C(sp3)−F‐rich 3D scaffolds appear with increasing frequency, the effect of multiple bioisosteric changes on molecular recognition requires elucidation. Herein we demonstrate that installation of a 1,3,5‐stereotriad, in the substrate for a commonly used lipase from Pseudomonas fluorescens does not inhibit recognition, but inverts stereoselectivity. This provides facile access to optically active, stereochemically well‐defined organofluorine compounds (up to 98 % ee). Whilst orthogonal recognition is observed with fluorine, the trend does not hold for the corresponding chlorinated substrates or mixed halogens. This phenomenon can be placed on a structural basis by considering the stereoelectronic gauche effect inherent to F−C−C−X systems (σ→σ*). Docking reveals that this change in selectivity (H versus F) with a common lipase results from inversion in the orientation of the bound substrate being processed as a consequence of conformation. This contrasts with the stereochemical interpretation of the biogenetic isoprene rule, whereby product divergence from a common starting material is also a consequence of conformation, albeit enforced by two discrete enzymes.
Keywords: biocatalysis, conformation, fluorine, gauche effect, molecular recognition
Arigoni and Eschenmoser's stereochemical interpretation of the biogenetic isoprene rule is synonymous with placing enzyme function on a structural level.1 Predicated on efficient substrate pre‐organisation governed by a specific cyclase (Figure 1), this treatise rationalises the conversion of oxidosqualene to either lanosterol or β‐amyrin. Described as the “apotheosis of the isoprene rule” by Cornforth,2 this interpretation has proven to be expansive and continues to inform and inspire the design of cyclisation cascades for the rapid generation of structural complexity.3 This study, together with the contemporaneous work from Stork and Burgstahler,4 nucleates upon an acyclic conformational analysis of oxidosqualene.5 The manifest selectivity of these two, discrete enzymatic processes is a consequence of (i) minimisation of non‐bonding interactions, and (ii) further conformational refinement in the enzyme–substrate ensemble. Whilst the latter is pervasive in enzyme catalysis and ultimately responsible for structural divergence, achieving orthogonal reactivity encoded at the substrate conformation level is comparatively underexplored. This is a logical consequence of enzyme specificity and the need for effective bioisosteres that modulate conformation but do not inhibit recognition.6
Figure 1.

Stereochemical interpretation of the biogenetic isoprene rule.
Multiple hydrogen to fluorine substitution is ideally suited to the task, since this subtle steric modification induces a localised partial charge inversion (C‐Hδ+ → C‐Fδ−). This strategy enabling physicochemical modulation7 also provides a potentially expansive platform to modulate molecular recognition in a manner distinct from the role of fluorine in interrogating enzyme function.8 The strategic introduction of configurationally defined C(sp3)−F centres offers an additional degree of versatility in allowing the structural and electronic topology to be regulated by stereoelectronic and electrostatic effects.9
The stereoelectronic gauche effect10 intrinsic to the 1,2‐difluoro motif,11 together with electrostatic 1,3‐repulsion, validate the potential of synergistic fluorine effects and multiple bioisosterism in achieving acyclic conformational control. Consequently, stereochemically complex, fluorine‐rich architectures are suited to divert the intrinsic selectivity of a given enzyme, observed with the non‐fluorinated case, via two discrete substrate‐enzyme ensembles (Figure 2, top).12
Figure 2.

Conceptual framework of this study (EWG: electron withdrawing group).
As a model for this study, the desymmetrisation of a meso‐acetate (X=H) utilising lipase from Pseudomonas fluorescence was investigated (Figure 2, bottom).13, 14 This scaffold is a convenient platform to interrogate multiple bioisosterism (X=H, F and Cl) as a function of Van der Waals radii, and allows for conformational regulation by virtue of the fluorine gauche effect and 1,3‐repulsion. The desymmetrisation platform would also provide facile access to stereochemically complex, multiply fluorinated systems in an optically active form.15 Derivatives of the parent scaffold continue to be produced on an industrial scale for the production of various blockbuster pharmaceuticals such as Lipitor and related statins.16 Moreover, 1,3‐syn diols are ubiquitous in polyketides, and construction of the 1,3,5‐trifluoro motif is a highlight of Carreira's synthesis of a fluorosulfolipid (Figure 2, bottom).17
Initially, the notion of inverting the intrinsic sense of stereoselection by multiple H to F substitution was interrogated by docking studies using LeadIT.19 To that end, substrates 1 and 2
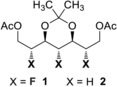 were docked into the active site of the Pseudomonas fluorescens HU380 lipase to establish if the bound topologies showed appreciable differences. This analysis revealed a striking difference in both the orientation and conformation of these closely similar molecules in the active site. The conformation of the bound, fluorinated meso‐substrate clearly manifested fluorine–ester gauche effects, and partial miminisation of 1,3‐repulsion (Figure 3 a,b. For a computational analysis of unbound 1 and 2, see the Supporting Information (SI)).
were docked into the active site of the Pseudomonas fluorescens HU380 lipase to establish if the bound topologies showed appreciable differences. This analysis revealed a striking difference in both the orientation and conformation of these closely similar molecules in the active site. The conformation of the bound, fluorinated meso‐substrate clearly manifested fluorine–ester gauche effects, and partial miminisation of 1,3‐repulsion (Figure 3 a,b. For a computational analysis of unbound 1 and 2, see the Supporting Information (SI)).
Figure 3.

Comparative orientations of the fluorinated and non‐fluorinated meso‐substrates docked in the active site of Pseudomonas fluorescens HU380 lipase. a) Fluorinated (1) and non‐fluorinated (2) meso‐acetates with protein surface and their interaction with amino acid residues in the binding pocket. b) Interaction of fluorinated and non‐fluorinated meso‐acetates with important amino acid residues. c) Superimposed fluorinated and non‐fluorinated meso‐acetates. Hydrogen bonds below 3.6 Å are shown as dashed, black lines. Colour code: protein surface: grey; protein skeleton: C: grey; fluorinated meso‐acetate: C: cyan; non‐fluorinated meso‐acetate: C: yellow; O: red; N: blue; F: green. This figure was generated using PyMOL (Schrödinger).18
Most importantly, the effect of fluorine bioisosterism is to reposition the internal carbonyl groups of 1 compared to the non‐fluorinated congener 2, without inhibiting recognition. Whilst the carbonyl groups diverge slightly in their spatial orientation (Figure 3 b), both are proximal to the nucleophilic serine residue (S374). This computational analysis thus provided confidence to proceed to experimental validation to establish that ground state changes achieved via molecular editing with fluorine can have significant implications for enzyme function (Figure 3 c).
Encouraged by the computational data, the tolerance of the lipase to the 1,3,5‐trifluoro motif was explored in the desymmetrisation of meso‐substrate 1 (Table 1). This required the identification of conditions that were compatible with both the trifluorinated substrate 1 and the non‐fluorinated control (2). Full details regarding the stereocontrolled synthesis of the substrates are provided in the Supporting Information. An optimisation process identified a solvent ratio of aq. buffer/DMF (3:1) and an enzyme loading of 0.1 mg μmol−1 to be highly effective, resulting in >95 % conversion to the product 3.
Table 1.
Optimisation of the hydrolytic enzymatic desymmetrisation of 1.[a]

| Entry | Solvent | Ratio [v/v] |
Enzyme loading [mg/μmol] |
Conversion [%][b] |
|---|---|---|---|---|
| 1 | aq. buffer | – | 0.3 | n.d. |
| 2 | aq. buffer/methanol | 1:1 | 0.3 | n.d. |
| 3 | aq. buffer/ethanol | 1:1 | 0.3 | 50 |
| 4 | aq. buffer/isopropanol | 1:1 | 0.3 | <5 |
| 5 | aq. buffer/acetonitrile | 1:1 | 0.3 | <5 |
| 6 | aq. buffer/THF | 1:1 | 0.3 | <5 |
| 7 | aq. buffer/chloroform | 1:1 | 0.3 | <5 |
| 8 | aq. buffer/DMF | 1:1 | 0.3 | >95 |
| 9[c] | aq. buffer/DMF | 3:1 | 0.1 | >95 |
[a] Standard reaction conditions: meso‐2,4,6‐trifluoro‐1,3,5,7‐tetrahydroxyheptane‐1,7‐diacetate (1) (17 mg, 50 μmol), aq. phosphate buffer (0.2 m)/ co‐solvent (1:1 v/v, 10 mL), lipase 15 mg (Pseudomonas fluorescens, ≥600 U/g immobilised on Immobead 150), ambient temperature, 18 h. [b] Conversion was monitored via GC analysis. [c] 1 (150 μmol), aq. phosphate buffer (0.2 m)/ DMF (3:1 v/v, 20 mL).
With these optimised conditions, the effect of fluorine introduction on the selectivity of the reaction with lipase from Pseudomonas fluorescens was investigated and compared directly to the non‐fluorinated analogue (Figure 4, top). Gratifyingly, after 18 h the all‐anti substrate (1) was cleanly processed to alcohol 3 in 98 % yield and with exquisite selectivity (99:1 e.r.). The key control experiment with the non‐fluorinated analogue (2) required doubling of the enzyme loading to 0.2 mg μmol−1 and stirring in neat aq. phosphate buffer (0.2 m, pH 7) for 112 h. Whilst the reaction proved to be efficient both in terms of yield (4, 98 %) and selectivity (3:97 d.r., determined by derivatisation to the Mosher ester, see SI), the effect of deleting the fluorine motif was to invert the intrinsic sense of stereoselection. To explore this phenomenon further, the C4 epimer 5 (anti,syn,syn,anti, see the SI) was investigated (Figure 4): Again, the sense of selectivity was in line with product 3 and furnished 6 in 95 % yield (94:6 d.r. determined by derivatisation to the Mosher ester, see SI). As an important set of control substrates, the tris‐chloro and mixed interhalogen examples were prepared (Figure 4, bottom). In all four cases, <5 % conversion was observed (7–10) further underscoring the ability of fluorinated arrays to effectively mimic hydrocarbon scaffolds. For completion, the desymmetrisation of the meso‐diol 11 was also explored and compared with the non‐fluorinated counterpart (12). Gratifyingly, this compound proved to be crystalline. Structural analysis by single‐crystal X‐ray diffraction revealed the gauche (σC‐H→σC‐F*) and 1,3‐repulsion effects that were part of the working hypothesis.
Figure 4.

Exploring the effect of F versus H on the selectivity of the transformation. [a] Enantiomeric ratio (e.r.) determined by chiral GC analysis. [b] Enantiomeric ratio could not be directly determined by GC or HPLC. Diastereomeric ratio (d.r.) determined by converting the alcohol to the Mosher ester and subsequent 19F NMR analysis (see Supporting Information).
Bonini and co‐workers have reported that the formation of ent‐4 arises from the lipase catalysed transesterification of the meso‐diol with vinyl acetate (Figure 5, top).13 However, upon repeating these conditions with the trifluorinated substrate 11 using 20 equiv. of vinyl acetate, only the diacetate (1) was isolated. By reducing the equivalents of vinyl acetate it was possible to demonstrate that the selectivity of his process using the fluorinated substrate was inverted, furnishing ent‐3 (7:93 e.r.).13 This key experiment thereby illustrates that selectivity is encoded at the substrate level thereby allowing product enantiomers to be generated depending on the starting material (diol versus bis‐acetate) (Figure 6).
Figure 5.

X‐ray structural analysis of the meso‐2,4,6‐trifluoro‐1,3,5,7‐tetrahydroxyheptanol 11. [a] Ref. 13: Meso‐diol 12 (100 μmol), Et2O (4 mL), Pseudomonas fluorescens Lipase (100 mg, 4.0 mass eq.), vinyl acetate (20.0 equiv.), ambient temperature, 18 h. [b] Meso diol 11 (100 μmol), Et2O (4 mL), Pseudomonas fluorescens Lipase (50 mg, 2.0 mass eq.), vinyl acetate (2.0 equiv.), ambient temperature, 4 h; 64 % recovered starting material 11.
Figure 6.

Demonstrating that 1,3,5‐trifluorination alters the intrinsic selectivity of catalysis in both directions (hydrolysis and esterification).
To demonstrate the synthetic utility of the enzymatic desymmetrisation in the generation of optically active, multiply fluorinated systems, the diol chain common to a series of blockbuster HMG‐CoA reductase inhibitors was prepared (Figure 7). Synthesis of the drug chemotype analogue common to Lipitor, Cerivastatin, Fluvastatin and Rosuvastatin was achieved by enzymatic desymmetrisation, and subsequent oxidation to the ester (13/14).20 Conversion to the azide (15/16) provides a valuable handle to install a multitude of aryl fragments as represented by the triazole (23/24). Reduction to the amine (17/18) and subsequent amidation furnished a crystalline derivative (19/20) thereby allowing the absolute and relative stereochemistry to be unequivocally established by X‐ray analysis (Figure 7, inset). These structural data are in line with the docking studies and support the gauche effect as being instrumental in inverting the selectivity of enzyme function.
Figure 7.

Generation of 1,3,5‐trifluoro modified chemotypes common to blockbuster HMG‐CoA reductase inhibitors such as Lipitor. a) TEMPO, NaOCl, CH2Cl2/H2O 6:1, 0 °C to rt, 1 h; b) NaClO2, tBuOH, 2‐methyl‐2‐butene, phosphate buffer, rt, pH 7, 3 h; c) MeI, KHCO3, DMF, rt, 16 h, S‐21 70 % (3 steps), S‐22 71 % (3 steps), ent ‐S‐23 86 % (3 steps), ent ‐S‐24 86 % (3 steps); d) NaOMe, MeOH/THF 1:1, 0 °C, 3 h, 13 90 % d.r. >12:1, 14 quant.; e) NEt3, DMAP, TsCl, CH2Cl2, rt, 18 h, S‐23 90 %, S‐24 65 %, S‐25 91 %, S‐26 94 %; f) NaN3, DMF, 80 °C, 18 h, 15 70 %, 16 94 %, ent ‐15 70 %, ent ‐16 75 %; g) Pd/C, H2, EtOAc, rt, 20 h, 17 quant., 18 quant., ent ‐17 quant., ent ‐18 quant.; h) RCOCl, Et3N, CH2Cl2, 16 h, 19 30 %, 20 45 %; i) CH2Cl2, TFA, H2O, 100:10:1, rt, 16 h, 21 68 %, 22 35 %, 23 85 %, 24 54 %; j) phenylacetylene, Na‐ascorbate, CuSO4⋅5 H2O, DMF, 50 °C, 16 h, S‐27 98 %, S‐28 96 %; k) KOH, MeOH, rt, 2 h, 25 80 %, 26 quant. Please note that “S” refers to substrates described in the Supporting Information.
In conclusion, this study reveals that a seemingly subtle 1,3,5‐trifluoro bioisostere motif inverts enzyme selectivity relative to the non‐fluorinated substrate. Curiously, this change does not inhibit activity, in contrast to the chlorinated analogues. This behaviour has been demonstrated for both the hydrolysis and transesterification to generate all possible stereoisomers. From a translational perspective, and as drug discovery expands into 3D chemical space,21 clarifying the effect of multiple C(sp3)−H to C(sp3)−F substitutions on enzyme function will become more urgent. Whereas single‐point alterations may well be tolerated, such as in Streptomyces cattleya,22 delineating the bioisosteric nature of larger fluorinated arrays requires clarification.7c The study also demonstrates the value of simple lipases in accessing optically active, stereochemically complex fluorinated species to modulate the physicochemical properties of bioactive small molecules.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge financial support from the WWU Münster, the Deutsche Forschungsgemeinschaft (SFB 858, P.B.), and the Helmholtz‐Association's Initiative and Networking Fund. The European Commission is acknowledged for an Intra‐European Marie Skłodowska‐Curie actions fellowship under Horizon‐2020 (796089‐NovInDXS, R.P.J.) and an ERC Consolidator Grant (R.G., 818949 RECON ERC‐2018‐CoG).
P. Bentler, K. Bergander, C. G. Daniliuc, C. Mück-Lichtenfeld, R. P. Jumde, A. K. H. Hirsch, R. Gilmour, Angew. Chem. Int. Ed. 2019, 58, 10990.
Dedicated to Professor François Diederich on the occasion of his retirement
Contributor Information
Dr. Patrick Bentler, http://www.uni‐muenster.de/Chemie.oc/gilmour/
Prof. Dr. Ryan Gilmour, Email: ryan.gilmour@uni-muenster.de.
References
- 1.
- 1a. Eschenmoser A., Ruzicka L., Jeger O., Arigoni D., Helv. Chim. Acta 1955, 38, 1890–1904; [Google Scholar]
- 1b. Eschenmoser A., Arigoni D., Helv. Chim. Acta 2005, 88, 3011–3048. [Google Scholar]
- 2. Cornforth J. W., Pure Appl. Chem. 1961, 2, 607–630. [Google Scholar]
- 3. Nicolaou K. C., Sorensen E. J., Classics in Total Synthesis, Wiley-VCH, Weinheim, 1996; [Google Scholar]; Nicolaou K. C., Snyder S. A., Classics in Total Synthesis II, Wiley-VCH, Weinheim, 2003. [Google Scholar]
- 4. Stork G., Burgstahler A. W., J. Am. Chem. Soc. 1955, 77, 5068–5077. [Google Scholar]
- 5.For excellent reviews of acyclic conformational analysis, see
- 5a. Hoffmann R. W., Angew. Chem. Int. Ed. Engl. 1992, 31, 1124–1134; [Google Scholar]; Angew. Chem. 1992, 104, 1147–1157; [Google Scholar]
- 5b. Hoffmann R. W., Angew. Chem. Int. Ed. 2000, 39, 2054–2070; [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 2134–2150. [Google Scholar]
- 6.
- 6a. Müller K., Faeh C., Diederich F., Science 2007, 317, 1881–1886; [DOI] [PubMed] [Google Scholar]
- 6b. Salwiczek M., Nyakatura E. K., Gerling U. I. M., Ye S., Koksch B., Chem. Soc. Rev. 2012, 41, 2135–2171; [DOI] [PubMed] [Google Scholar]
- 6c. Gillis E. P., Eastman K. J., Hill M. D., Donnelly D. J., Meanwell N. A., J. Med. Chem. 2015, 58, 8315–8359; [DOI] [PubMed] [Google Scholar]
- 6d. Harsanyi A., Sandford G., Green Chem. 2015, 17, 2081–2086. [Google Scholar]
- 7.
- 7a. Huchet Q. A., Kuhn B., Wagner B., Kratochwil N. A., Fischer H., Kansy M., Zimmerli D., Carreira E. M., Müller K., J. Med. Chem. 2015, 58, 9041–9060; [DOI] [PubMed] [Google Scholar]
- 7b. Molnár I. G., Thiehoff C., Holland M. C., Gilmour R., ACS Catal. 2016, 6, 7167–7173; [Google Scholar]
- 7c. Meanwell N. A., J. Med. Chem. 2018, 61, 5822–5880; [DOI] [PubMed] [Google Scholar]
- 7d. Erdeljac N., Kehr G., Ahlqvist M., Knerr L., Gilmour R., Chem. Commun. 2018, 54, 12002–12005. [DOI] [PubMed] [Google Scholar]
- 8. Berkowitz D. B., Karukurichi K. R., de la Salud-Bea R., Nelson D. L., McCune C. D., J. Fluorine Chem. 2008, 129, 731–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Zimmer L. E., Sparr C., Gilmour R., Angew. Chem. Int. Ed. 2011, 50, 11860–11871; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 12062–12074; [Google Scholar]
- 9b. Scheidt F., Selter P., Santschi N., Holland M. C., Dudenko D. V., Daniliuc C., Mück-Lichtenfeld C., Hansen M. R., Gilmour R., Chem. Eur. J. 2017, 23, 6142–6149. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. O'Hagan D., Chem. Soc. Rev. 2008, 37, 308–319; [DOI] [PubMed] [Google Scholar]
- 10b. Thiehoff C., Rey Y. P., Gilmour R., Isr. J. Chem. 2017, 57, 92–100; [Google Scholar]
- 10c. Aufiero M., Gilmour R., Acc. Chem. Res. 2018, 51, 1701–1710. [DOI] [PubMed] [Google Scholar]
- 11.For direct, catalytic approaches to generate this motif see:
- 11a. Banik S. M., Medley J. W., Jacobsen E. N., J. Am. Chem. Soc. 2016, 138, 5000–5003; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Molnár I. G., Gilmour R., J. Am. Chem. Soc. 2016, 138, 5004–5007; [DOI] [PubMed] [Google Scholar]
- 11c. Scheidt F., Schäfer M., Sarie J. C., Daniliuc C. G., Molloy J. J., Gilmour R., Angew. Chem. Int. Ed. 2018, 57, 16431–16435; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 16669–16673; [Google Scholar]
- 11d. Haj M. K., Banik S. M., Jacobsen E. N., Org. Lett. 2019, 10.1021/acs.orglett.9b00938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For an example of a surprisingly high level of discrimination for F-acetyl-CoA versus acetyl-CoA (i.e. single F versus H), see:
- 12a. Weeks A. W., Chang M. C. Y., Proc. Natl. Acad. Sci. USA 2012, 109, 19667–19672; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Weeks A. M., Keddie N. S., Wadoux R. D. P., O'Hagan D., Chang M. C. Y., Biochemistry 2014, 53, 2053–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bonini C., Racioppi R., Viggiani L., Righi G., Rossi L., Tetrahedron: Asymmetry 1993, 4, 793–805. [Google Scholar]
- 14.For an application in the synthesis of the C1–C13 fragment of amphotericin B, see Bonini C., Chiummiento L., Martuscelli A., Viggiani L., Tetrahedron: Asymmetry 2004, 45, 2177–2179. [Google Scholar]
- 15.For selected examples of stereochemically rich fluoroalkanes, see
- 15a. Farran D., Slawin A. M. Z., Kirsch P., O'Hagan D., J. Org. Chem. 2009, 74, 7168–7171; [DOI] [PubMed] [Google Scholar]
- 15b. Hunter L., Slawin A. M. Z., Kirsch P., O'Hagan D., Angew. Chem. Int. Ed. 2007, 46, 7887–7890; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 8033–8036; [Google Scholar]
- 15c. O'Hagan D., J. Org. Chem. 2012, 77, 3689–3699. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Roth B. D., Prog. Med. Chem. 2008, 40, 1–22; [DOI] [PubMed] [Google Scholar]
- 16b. Roth B. D., Blankley C. J., Chucholowski A. W., Ferguson E., Hoefle M. L., Ortwine D. F., Newton R. S., Sekerke C. S., Sliskovic D. R., Wilson M., J. Med. Chem. 1991, 34, 357–366; [DOI] [PubMed] [Google Scholar]
- 16c. Tabernero L., Bochar D. A., Rodwell V. W., Stauffacher C. V., Proc. Natl. Acad. Sci. USA 1999, 96, 7167–7171; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16d. Istvan E. S., Deisenhofer J., Science 2001, 292, 1160–1164; [DOI] [PubMed] [Google Scholar]
- 16e. Müller M., Angew. Chem. Int. Ed. 2005, 44, 362–365; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 366–369; [Google Scholar]
- 16f. Yasukouchi H., Machida K., Nishiyama A., Mitsuda M., Org. Process Res. Dev. 2019, 23, 654–659. [Google Scholar]
- 17. Fischer S., Huwyler N., Wolfrum S., Carreira E. M., Angew. Chem. Int. Ed. 2016, 55, 2555–2558; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2601–2604. [Google Scholar]
- 18.PyMol-Schrödinger: The PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC.
- 19.
- 19a.BioSolveIT GmbH, Sankt Augustin. http://www.biosolveit.de, LeadIT, version 2.3.2;
- 19b.BioSolveIT GmbH, Sankt Augustin. http://www.biosolveit.de, SeeSAR, version 8.1.
- 20.For an example of a mono-fluorinated analogue of this drug chemotype, see Saadi J., Wennemers H., Nat. Chem. 2016, 8, 276–280. [DOI] [PubMed] [Google Scholar]
- 21. Lovering F., Bikker J., Humblet C., J. Med. Chem. 2009, 52, 6752–6756; [DOI] [PubMed] [Google Scholar]; Lovering F., MedChemComm 2013, 4, 515–519. [Google Scholar]
- 22.
- 22a. O'Hagan D., Schaffrath C., Cobb S. L., Hamilton J. T., Murphy C. D., Nature 2002, 416, 279; [DOI] [PubMed] [Google Scholar]
- 22b. Dong C., Huang F., Deng H., Schaffrath C., Spencer J. B., O'Hagan D., Naismith J. D., Nature 2004, 427, 561–565. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary