

Abstract
In neuroblastoma (NB), genetic alterations in chromatin remodeling (CRGs) and epigenetic modifier genes (EMGs) have been described. We sought to determine their frequency and clinical impact. Whole exome (WES)/whole genome sequencing (WGS) data and targeted sequencing (TSCA®) of exonic regions of 33 CRGs/EMGs were analyzed in tumor samples from 283 NB patients, with constitutional material available for 55 patients. The frequency of CRG/EMG variations in NB cases was then compared to the Genome Aggregation Database (gnomAD). The sequencing revealed SNVs/small InDels or focal CNAs of CRGs/EMGs in 20% (56/283) of all cases, occurring at a somatic level in 4 (7.2%), at a germline level in 12 (22%) cases, whereas for the remaining cases, only tumor material could be analyzed. The most frequently altered genes were ATRX (5%), SMARCA4 (2.5%), MLL3 (2.5%) and ARID1B (2.5%). Double events (SNVs/small InDels/CNAs associated with LOH) were observed in SMARCA4 (n = 3), ATRX (n = 1) and PBRM1 (n = 1). Among the 60 variations, 24 (8.4%) targeted domains of functional importance for chromatin remodeling or highly conserved domains but of unknown function. Variations in SMARCA4 and ATRX occurred more frequently in the NB as compared to the gnomAD control cohort (OR = 4.49, 95%CI: 1.63–9.97, p = 0.038; OR 3.44, 95%CI: 1.46–6.91, p = 0.043, respectively). Cases with CRG/EMG variations showed a poorer overall survival compared to cases without variations. Genetic variations of CRGs/EMGs with likely functional impact were observed in 8.4% (24/283) of NB. Our case–control approach suggests a role of SMARCA4 as a player of NB oncogenesis.
Keywords: neuroblastoma, chromatin remodeling complex, SWI/SNF, epigenetic modifier, SMARCA4
Short abstract
What's new?
Mutations that affect chromatin remodeling can lead to cancer. In this paper, the authors investigated the impact of variations in chromatin remodeling genes and epigenetic modifier genes on neuroblastoma patients. They compared the frequency of these variations in NB cases with data from the Genome Aggregation Database (gnomAD). Neuroblastoma cases had a higher frequency of SMARCA4 and ATRX gene variations than the general population. Furthermore, NB patients with CRG/EMG mutations had poorer overall survival than NB cases without such mutations. These findings highlight the importance of chromatin remodeling in neuroblastoma as an avenue for new therapeutics.
Abbreviations
- aCGH
array‐comparative genomic hybridization
- CNAs
copy number alterations
- CRGs
chromatin remodeling genes
- EMGs
epigenetic modifiers genes
- OS
overall survival
- PFS
progression‐free survival
- SNVs
single nucleotide variants
- TSCA
TruSeq® Custom Amplicon
- WES
whole exome sequencing
- WGS
whole genome sequencing
Introduction
Alterations in chromatin remodeling genes (CRGs) have emerged as an important cause of cancer; in particular, the identification of inactivating genetic alterations in the SMARCB1 gene in rhabdoid tumors (RTs) has highlighted the involvement of the SWI/SNF complex in tumor formation.1, 2 Indeed, SWI/SNF gene mutations and alterations in other epigenetic modifier genes (EMGs) are thought to occur in 20% of human cancers.3
In neuroblastoma (NB), the most common extracranial solid tumor in children, genetic alterations concern predominantly copy number alterations, with MYCN amplification and segmental chromosome alterations associated with a poor outcome.4 Only few genes have been shown to be altered recurrently by genetic events, including ALK.4 Genes involved in chromatin remodeling such as ATRX, DAXX, ARID1A and ARID1B genes have also been shown to be targeted by mutations or structural rearrangements,5, 6, 7, 8, 9 but the frequency of CRG/EMG alterations has not been described in detail in NB.
The aim of our study was to determine the frequency of CRGs/EMGs variations in NB patients and to correlate findings with clinical parameters and outcome.
Materials and Methods
Study series
The study consisted of 283 NB patients, constituting a clinically representative cohort (Table 1, Supporting Information Table S1; Supporting Information Fig. S1).
Table 1.
Cases showing SNVs, InDels and focal CNAs in CRGs/EMGs in 283 neuroblastoma patients and 30 neuroblastoma cell lines
| Cohort | MYCN status | Stage | Age at diagnosis | Analytical technique | Cases with pathogenic/likely pathogenic variations in CRGs/EMGs | Number of pathogenic/likely pathogenic variations in CRGs/EMGs | Pathogenic/likely pathogenic variations in CRG/EMGs | Origin of variations |
|---|---|---|---|---|---|---|---|---|
| Patients n = 283 | MNA n = 64; MN‐NA n = 219 | Stage 4 n = 128;Localized n = 130; Stage 4s n = 25 | <18 months n = 159; ≥18 months n = 124 | WES/WGS n = 55 | 16 | 18 | ATRX n = 5 | 2/5 germline; 3/5 Somatic |
| ARID1B n = 5 | Germline | |||||||
| MLL3 n = 3 | 2/3 Germline; 1/3 Somatic | |||||||
| MLL2 n = 2 | Gemline | |||||||
| PBRM1 n = 1 | Somatic | |||||||
| ARID1A n = 1 | Germline | |||||||
| SMARCA4 n = 1 | Germline | |||||||
| TSCA n = 228 | 40 | 42 | ATRX n = 9 | Unknown | ||||
| SMARCA4 n = 7 | ||||||||
| MLL2 n = 1 | ||||||||
| MLL3 n = 4 | ||||||||
| ARID1A n = 2 | ||||||||
| ARID2 n = 4 | ||||||||
| CHD7 n = 5 | ||||||||
| SMARCC1 n = 1 | ||||||||
| SMARCA2 n = 2 | ||||||||
| SMARCD3 n = 1 | ||||||||
| PBRM1 n = 2 | ||||||||
| DAXX n = 1 | ||||||||
| ARID1B n = 2 | ||||||||
| BCL11A n = 1 | ||||||||
| Cell lines n = 30 | MNA n = 22 MN‐NA n = 8 | TSCA n = 30 | 7 | 7 | ATRX n = 1 | |||
| MLL2 n = 2 | ||||||||
| MLL3 n = 1 | ||||||||
| SMARCA4 n = 2 | ||||||||
| BCL11A n = 1 |
In the 55 samples analyzed by WES/WGS, 7 CRG/EMG genes show at least one variation. In the 248 samples analyzed by the TSCA approach, 14 CRG/EMG genes show a variation. Five genes harbor a variation in NB cell lines.
Patients were treated in French pediatric oncology centers according to the relevant national/international protocols. Written informed consent was obtained from parents/guardians according to national law. Our study was authorized by the ethics committees “Comité de Protection des Personnes Sud‐Est IV”, references L07–95/L12–171, and “Comité de Protection des Personnes Ile de France”, reference 0811728.
In 55 patients for whom paired normal and tumor tissue was available, whole‐exome sequencing (WES)/whole‐genome sequencing (WGS) techniques were performed, whereas for 248 diagnostic tumor samples, a TruSeq® Custom Amplicon (TSCA) approach was used. Twenty patients are common to the two subsets (Supporting Information Table S1).
Furthermore, 30 NB cell lines (Supporting Information Table S1) and six germline controls from healthy donors were studied.
To analyze the frequency of CRG/EMG variations, 28 major genes involved in chromatin remodeling processes were selected: ACTB, ACTL6A, ACTL6B, ARID1A, ARID1B, ARID2, BCL11A, BCL11B, BCL7A, BCL7B, BCL7C, BRD7, BRD9, DPF1, DPF2, DPF3, PBRM1, PHF10, SMARCA2, SMARCA4, SMARCB1, SMARCC1, SMARCC2, SMARCD1, SMARCD2, SMARCD3, SMARCE1, SS18. Five epigenetic modifier genes, ATRX, DAXX, CHD7, MLL2 (KMT2D), MLL3 (KMT2C), were also selected based to their involvement in a wide variety of human malignancies (Supporting Information Table S2).
DNA extraction and aCGH analysis
DNA was extracted from diagnostic tumor samples harboring >20% of tumor cells using standard procedures. Genomic copy number profiles were determined by aCGH, and MYCN copy number status was confirmed by FISH.
Whole exome/whole genome sequencing
Paired diagnostic tumor and normal DNA from 55 patients were whole‐genome (n = 16) and/or whole‐exome (n = 39) sequenced (Illumina® Hiseq2500; average coverage: 80×—100× per sample, respectively10). WGS for eight cases has been reported previously.10
TruSeq® amplicon panel sequencing
Tumor DNA from 248 NB cases and six healthy donor germline DNA samples (negative controls) were analyzed using a TSCA® panel approach covering 33 CRGs/EMGs, (average coverage >1,500×; TSCA®v1.5, Illumina Inc., San Diego, CA; Supporting Information Table S2).
Bioinformatics analysis
Bioinformatics approaches depended on the genetic alteration (single nucleotide variations [SNVs]), small or large InDels; focal or extended copy number alterations (CNAs) and the sequencing technique.
Whole‐exome and whole‐genome sequencing analyses
The WGS/WES sequencing raw reads were mapped to the reference human genome using BWA (assembly GRCh37/hg19)11, 12 followed by analysis of the exons of the 33 CRGs/EMGs (Supporting Information Table S2).
SNVs/InDels <30 bp were called using GATK's HaplotypeCaller v3.5 algorithm.12, 13 The Manta tool (1.0.3) was used to detect structural variations and large InDels.14 All SNVs/small InDels with a VAF >20% were retained.
Tumor copy number profiles were calculated with FREEC (0.9) using constitutional DNA as reference,15, 16, 17 with annotations of CRGs/EMGs to highlight focal variations.18
Genetic variations were termed “somatic” if no evidence of these variations was observed in the constitutional reads.
TSCA® analysis
The TSCA® sequencing reads were mapped to the reference human genome with Bowtie.19 To prevent strand or coverage bias due to noncomplete overlap of forward (FW) and reverse (RV) reads of amplicons, the bam files were split to FW and RV read bam(s).
An adapted approach was developed to enable variant calling in TSCA® data.20
For SNVs, coverage analysis was performed at each base position (GATK DepthOfCoverage). The background of noise (variability) of each amplicon was then analyzed in the six negative controls.
All possible variants (e.g., ref = A; A > T, A > C, A > G, A > ‐) and their alternative allele fraction (AF) were calculated for each position, and Fisher's exact two‐sided tests with a Bonferroni correction were performed to compare percentages of variant allele fraction (VAF) for a given base between a case and the negative controls. Significant variations were filtered‐in in case of an increase in the percentage of variant base (5% significance level). Only positions with total depth of coverage higher than 100× were considered for variant analysis. All SNVs/small InDels with a VAF >5% were retained.
Recurrent variants and variants with VAF >20% detected in more than two samples were filtered‐out as they most likely indicate polymorphisms. Finally, FW and RV results were confronted and ambiguous cases were filtered‐out.
Amplicon InDels Hunter tool was used to detect large InDels.21 Recurrent InDels detected in more than two samples were filtered‐out.
For focal CNAs, the depth of coverage of targeted regions was calculated for FW and RV bams (GATK DepthOfCoverage). For each sample, the coverage of each amplicon was normalized by the median of sample depth followed by comparison to negative controls. The FW and RV normalized data were then merged to reconstruct copy number profiles. The copy number profile obtained from TSCA data using our in‐house developed pipeline was also confirmed by another technique ONCOCNV, a method that includes a multifactor normalization with respect to library size, CG‐content and target length to detection of large copy number changes from amplicon deep sequencing data.22
Finally, the list of SNVs and InDels was manually curated and visually inspected in IGV (Integrated Genome Viewer).23
Variant annotation, prioritization and classification
All SNVs/InDels were annotated using SnpEff/SnpSift.24 Annotation was performed using canonical transcripts and publicly available databases dbSNPv150,25 gnomADv2.0.2 (http://gnomad.broadinstitute.org/),26 COSMICv74,27 ClinVar(25‐02‐2018),28 dbNSFPv2.9.318 and known cancer Hotspots.29 Loss of function (LoF) assessment was performed using the snpEff tool. SNVs/InDels were discarded as benign/likely benign/polymorphic based on population minor allele frequency (popmax) ≥0.1% from gnomAD database. However, SNVs/inDels reported as pathogenic/likely pathogenic in ClinVar were retained. In addition to COSMIC annotations, six prediction algorithms VEST3,30 MetaSVM,31 REVEL,32 MutationTaster,33 M_CAP34 and CADD35 were used to classify all likely protein‐altering SNVs/InDels (frameshift, nonsense, splice donor/acceptor, nonsynonymous and in‐frame insertions/deletions) as pathogenic, likely pathogenic. The flowchart of variant classification is indicated in Supporting Information Figure S4A and the prediction score thresholds in Supporting Information Figure S4B.
SNVs/small InDels validation
All retained SNVs/small InDels with VAF >20% were confirmed by Sanger sequencing. SNVs with VAF <20% were validated by NGS/targeted sequencing in an independent second experiment.36
Comparison of CRG/EMG variation frequencies between NB and gnomAD reference cohort
We compared the CRG/EMG variation frequencies from the NB cohort (Supporting Information Table S3) with those reported in a large unrelated population (gnomAD database; n = ~123,000). The gnomAD VCF file was intersected with the CRG/EMG TSCA® panel bed file and the corresponding subset of variants from gnomAD were extracted. Using the same approach as in the NB cohort, SNVs/InDels from gnomAD were annotated/classified into pathogenic/likely pathogenic and their frequency was calculated by dividing the sum of the allele count by the median of total alleles. To compare the frequencies of CRG/EMG variations in the NB vs. the control cohort, for each gene an Odds Ratios was calculated: OR = (NB cases with pathogenic/likely pathogenic variant/cases without pathogenic or likely pathogenic variant)/(gnomAD control with pathogenic or likely pathogenic variant/gnomAD control without pathogenic or likely pathogenic variant) along with 95% confidence intervals. p‐Value and OR were obtained using two‐sided Fisher's test performed in R.37
Statistical analysis
Correlation analyses using chi‐squared test was done with MedCalc (Medical Calculator 13.3.0.0). Progression‐free survival (PFS) and overall survival (OS) were estimated using the Kaplan–Meier method, and comparisons were made using log‐rank tests.
Western blot
Immunoblots were done as reported previously using monoclonal rabbit SMARCA4/BRG1 (EPR3912, #GTX62750, Genetex, Irvine, CA) and HRP‐conjugated GAPDH (#HRP‐60004, ProteinTech, Rosemont, IL) antibodies.
Results
Genetic variations of CRGs/EMGs detected in 283 NB patients and in 30 NB cell lines
In a cohort of 283 patients studied by WGS/WES (n = 35) or by TSCA® (n = 228), or both (n = 20), pathogenic/likely pathogenic genetic variations (focal CNAs, InDels, SNVs) in at least one CRG/EMG were identified in 56/283 cases (20%). Altogether 60 pathogenic/likely pathogenic genetic variations were identified (Table 1, Fig. 1 and Supporting Information Table S3). The most frequently altered genes were ATRX, SMARCA4, MLL3 and ARID1B.
Figure 1.
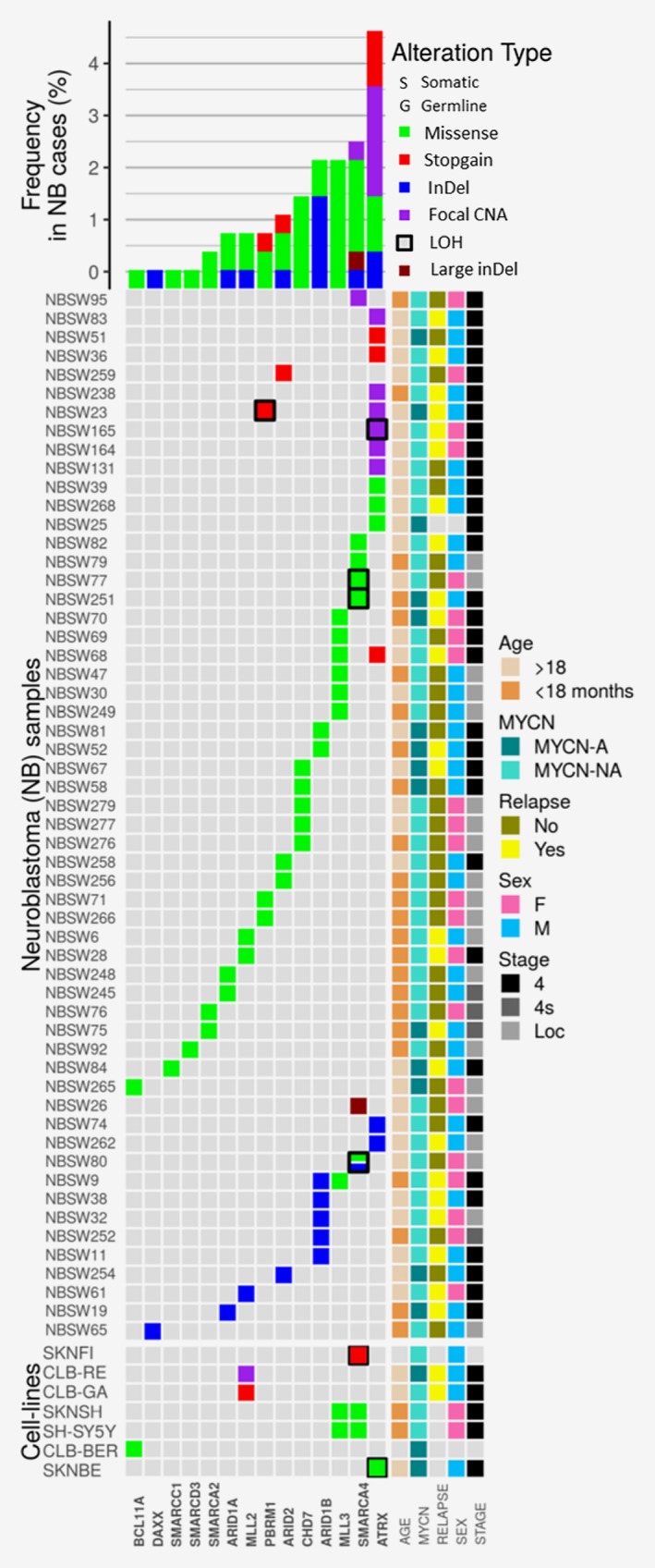
Genetic variations in chromatin remodeling and epigenetic modifier genes across a series of 283 NB patients and 30 NB cell lines analyzed by TSCA and/or WGS/WES approaches. In the lower part of the figure, genes found to be altered (n = 14) are arranged in rows; cases for whom a genetic variation is detected in the studied genes (n = 56) are arranged in columns, respectively. The 60 SNVs, InDels, and focal CNAs detected in the 56 cases are represented by colored cases. Double events (SNV/inDels and LOH) are represented by colored cases surrounded by a black square. In the lower part of the graph, the 9 SNVs, InDels and CNAs detected in 7 NB cell lines are represented. The overall frequencies are indicated in the upper half of the figure. Nonsynonymous SNVs are represented in green, stop‐gain SNVs are represented in red, InDels are represented in blue, focal CNA in purple and large deletions in brown. The right data grid summarizes clinical information of each neuroblastoma sample.
ATRX was targeted by focal CNAs, small InDels and SNVs in 6, 2 and 6 cases, respectively (Supporting Information Table S3 and Fig. 1). SMARCA4 was altered by 6 SNVs/InDel, 1 focal CNA and 1 large deletion. MLL3 was targeted by SNVs in seven cases (Supporting Information Table S3, Fig. 1). ARID1A and ARID1B genes were altered in altogether 10/283 (3.5%) cases, with two SNVs and one InDel in ARID1A and five inDels and two SNVs in ARID1B.9
Variations in two different CRGs/EMGs were observed in three cases (Supporting Information Table S3, in bold; Fig. 1), targeting MLL3 and ATRX, MLL3 and ARID1B, and PBRM1 and ATRX, respectively.
Double events encompassing both SNV/small InDels, focal CNAs and additional LOH/copy number loss of the same gene were observed in six cases (Fig. 1). The SMARCA4 gene was targeted by double events in three cases (NBSW80, NBSW251 and NBSW77) with either a SNV and/or InDel (3 bp) in the presence of only 1 copy of the SMARCA4 gene, corresponding to focal deletions (Figs. 2 a and 2 b).
Figure 2.
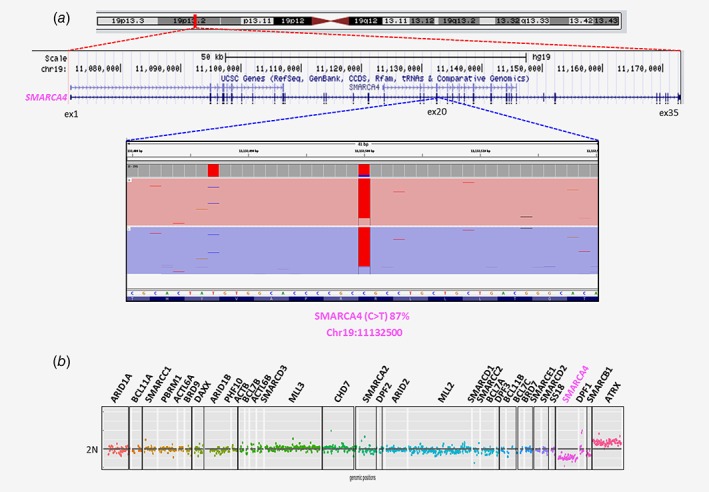
Example of double event detection (SNV and copy number loss in SMARCA4 gene) by TSCA and aCGH data analysis in NBSW80. (a) The IGV profile from TSCA analysis revealed the presence of one SNV in SMARCA4 (C>T; chr19: 11132500) gene with variant allele fraction of 87%. (b) The comparison between the copy number profiles obtained by TSCA revealed a second event with copy number loss in the SMARCA4 gene.
Case NBSW165 showed a loss of one copy and a focal CNA covering a region from EX2 to EX9 of the second copy of ATRX (Fig. 1; Supporting Information Table S3). A copy number loss involving the PBRM1 gene is observed in case NBSW23; a somatic SNV is also observed in the same gene (Fig. 1).
In another patient (NBSW26) presenting with speech delay, attention deficit and hyperactivity disorder as well as an adrenal stage INSS stage 1, INRG L1 NB diagnosed at 4 years of age, a germline deletion of 1.7 Mb at chromosome band 19p13.2 (chr19:10462524–12157782) was detected (Supporting Information Fig. S2). The germline deleted region encompassing among others the SMARCA4 gene was confirmed by aCGH on peripheral blood lymphocytes (Supporting Information Table S2).
Targeted TSCA® sequencing of the 33 CRGs/EMGs was performed in 30 NB cell lines (Supporting Information Table S1). Nine variations were detected in seven NB cell lines (Table 1, Supporting Information Table S3, Fig. 1), most frequently targeting SMARCA4 (3 cases), MLL2 (2 cases) and MLL3 (2 cases) (Fig. 1, Supporting Information Table S3).
Interestingly, a genetic event consisting of partial loss of the MLL2 gene was detected in the NB cell line CLB‐Re (Fig. 3 a). SNP6 analysis in the same NB cell line revealed a large region of copy number loss in chr12 encompassing the MLL2 gene at the position chr12:49449107–58196639, confirming the observation obtained by TSCA sequencing (Fig. 3 b).
Figure 3.
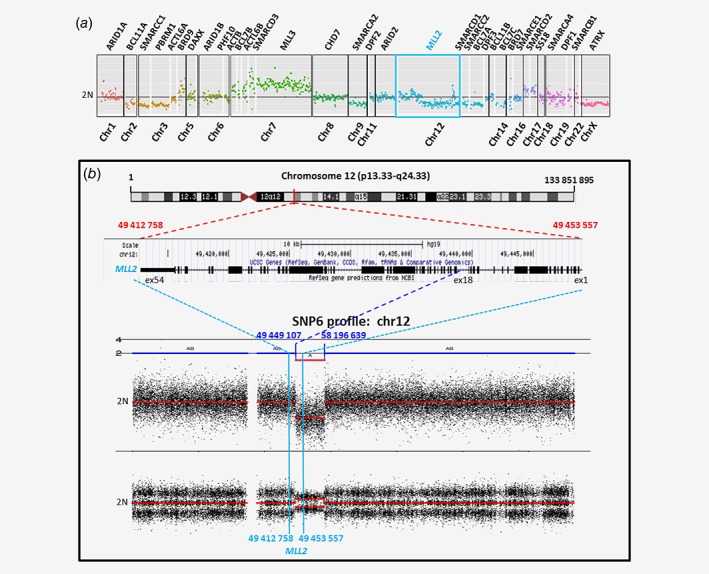
Partial loss event detected in MLL2 gene in CLB‐Re NB cell line. (a) The copy number profile obtained by TSCA shows the presence of a partial loss starting in MLL2 gene. (b) SNP6.0 array‐based copy number encompassing the Chromosome 12 confirms the presence of a large loss with a breakpoint within the MLL2 gene.
In the SKNFI cell line, a double event (SNV and LOH) was observed in SMARCA4 (Figs. 4 a and 4 b). The absence of expression of SMARCA4 was confirmed by expression analysis and western blot. On the other hand, expression of SMARCA4 was maintained in the SKNSH cell line which harbors a single SNV event in this gene (Figs. 4 c and 4 d).
Figure 4.
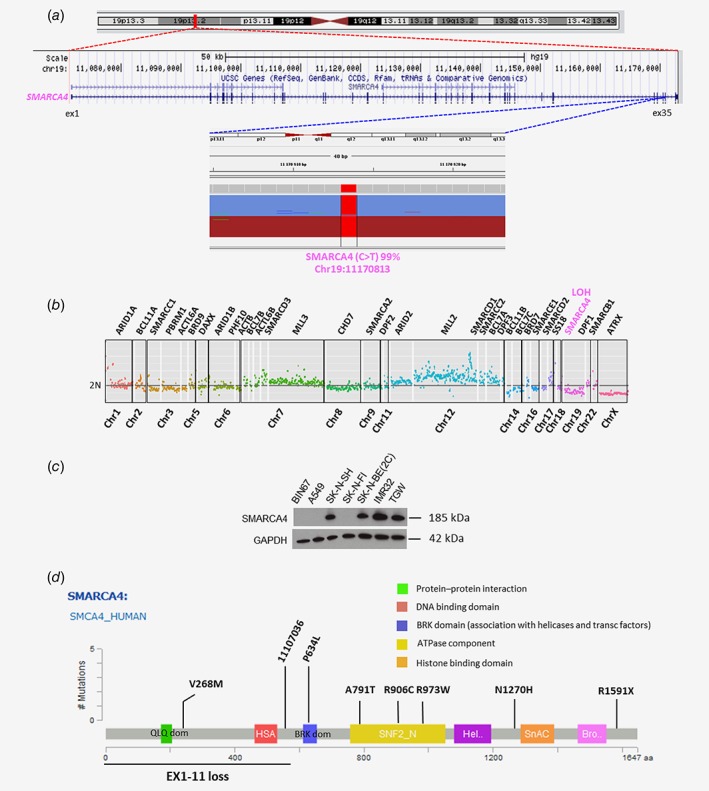
Example of double event (LOH and SNV in SMARCA4 gene) in SKNFI NB cell line. (a) The IGV profile from TSCA ampliseq analysis revealed the presence of one SNV in SMARCA4 gene (C>T; Chr19: 11170813) with variant allele fraction of 99%. (b) The copy number profile obtained by TSCA shows the presence of a second event with copy number loss in the same gene. (c) Immunoblot of SMARCA4 expression in different neuroblastoma and nonneuroblastoma cell lines. BIN67: Small cell carcinoma of the ovary of hypercalcemic type; A549: lung carcinoma; SKNSH, SKNFI, SKNBE(2C), IMR32 and TGW: neuroblastoma. (d) SNVs, inDels and focal CNAs identified in SMARCA4 gene mapping functional domains in patients enrolled in the study.
Germline vs. somatic occurrence of variations
Altogether, among the 60 pathogenic/likely pathogenic CRG/EMG variations, the variation occurred at a somatic level in five cases, targeting ATRX, PBRM1 or MLL3, whereas a germline variation could be confirmed in 13 other cases, targeting ARID1B, MLL3, MLL2, ATRX, ARID1A and SMARCA4 (Supporting Information Table S3; Fig. 1). For the remaining 42 variations detected in tumor samples without paired germline material available, a distinction between germline and somatic variation could not be made.
We then compared the frequencies of pathogenic/likely pathogenic SNVs/inDels of the 33 studied CRGs/EMGs in the NB cohort to those observed in gnomAD as a control cohort. The analysis revealed a significantly higher frequency of variations (p < 0.05 after Bonferroni correction) in two CRGs, SMARCA4 (OR 4.49, 95% CI: 1.63–9.97, p = 0.038) and ATRX (OR 3.44, 95% CI: 1.46–6.91, p = 0.043) in NB cases when compared to gnomAD, independent of whether the observed SNVs were of known germline, somatic or unknown origin, indicating an enrichment for variations in the genes SMARCA4 and ATRX in NB cases. All other CRGs/EMGs included in our study were not altered with a higher frequency than in the general population (Supporting Information Table S4).
Potential functional impact
The potential functional impact of the observed variations was analyzed in silico. For a given variation, within the studied gene, the localization of the amino acid change was studied with regards to domains of functional importance for chromatin remodeling or highly conserved domains but with unknown function.
Among the 30 variations detected in CRGs, 12 variations were localized in these domains (9 SNVs, 1 focal CNA, 1 InDel and 1 large inDel; Supporting Information Table S3). Their position was then analyzed with regards to protein‐specific domains (DNA binding, bromodomain, histone binding, protein–protein interaction domains). In the SMARCA4 gene, four identified variations mapped to these functional domains (Fig. 4 d), including three missense point mutations in the ATPase domain of SMARCA4 gene (Fig. 4 d).
Among the 30 variations observed in EMGs, 12 variations were localized in domains of functional importance (10 SNVs, 1 focal CNA, 1 InDel; Supporting Information Table S3).
Overall, among all 60 variations observed in CRGs/EMGs, 24 mapped to domains of functional importance at a protein level (Supporting Information Table S3).
Correlation of CRGs/EMGs variations with clinical parameters
There were no statistically significant correlations between the presence of a CRG/EMG variation and the main clinical prognostic parameters of NB (data not shown). Furthermore, among patients whose tumors were analyzed by WGS (n = 8),10 the presence of CRG/EMG variations, was not associated with an overall increase in the tumor mutational burden (data not shown).
A statistically significant poorer overall survival was observed for patients whose tumors harbored CRG/EMB variations in the overall cohort (Supporting Information Fig. S3A). However, there was no statistically significant difference of overall survival between the two patient groups in patient subgroups defined by age, stage or MYCN status, nor among all high‐risk patients only (n = 124; Supporting Information Fig. S3C). Furthermore, no statistically significant difference in PFS was observed (Supporting Information Fig. S3B).
Discussion
Recent reports have highlighted chromatin remodeling as an important player in oncogenesis, with the main mechanisms of action consisting in tumor suppression, but variations in CRG/EMGs might also play a role in oncogenesis via gain of function.38, 39 CRG alterations have previously been identified in NB patients, targeting ARIDs, ATRX, DAXX or SMARCA4.7, 8, 9, 40, 41
Our study now focused on the genetic status of 33 CRGs/EMGs in tumor samples of 283 NB patients. Altogether, CRG/EMG variations (focal CNA, InDels, SNVs) were identified in 56 cases (20%), mapping to CRG/EMG functional domains in 8.4% (24/283) of cases and most frequently targeting ATRX, SMARCA4, MLL3 and ARID1B. Other CRGs and many more EMGs, not covered by the panel used in this targeted analysis, exist throughout the genome,42 and thus it is possible that variations targeting other CRG/EMGs not taken into account in our study might also be present in the analyzed samples.
In NB, ATRX alterations have been associated with activated alternative lengthening of telomere (ALT).8 In our study, 5% (14/283) of cases showed an ATRX variation (SNV/InDel or focal CNA). Further studies will be required to establish the role of these variations on patient survival.
The MLL3 gene is rarely altered in NB, with two MLL3 variations (A293V and P309L) reported outside of annotated protein domains.43 In our cohort, seven missense SNVs were detected, four of them within MLL3 functional domains.
Alterations in ARID1A/ARID1B have been described in 11% of NB patients,9 with the presence of variations in a single allele of ARID1A/ARID1B possibly corresponding to a dominant tumor suppressor.41 Ten cases showed ARID1A/ARID1B variations, with two SNV/inDels occurring in functional domains.
Altogether, among the 56 cases with CRG/EMG variations, four patients harbored somatic and 12 showed germline variations. For the remaining 40 cases, due to the absence of paired germline DNA, it could not be established whether the observed variations were somatic or might concern private constitutional polymorphisms. Yet constitutional variations of potential functional impact might also be of oncogenic importance.
Indeed, germline events in CRGs such as SMARCB1, SMARCA4 or SMARCE1 have been described as predisposition syndromes for other cancer types, including RT.1 In our series, germline events in CRGs/EMGs were observed in 23.6% (13/55) of patients with available germline material.
In RT, the recurrent germline loss of one SMARCB1 allele followed by somatic loss of the second allele indicates a classic tumor suppressor, with loss of critical subunits of SWI/SNF.2 Given the possible suppressor behavior of CRG/EMGs, we searched for double events possibly causing loss of function, with double events identified in 2.1% (6/283) of patients, most frequently in SMARCA4 (3/283).
SMARCA4 has been shown to play a role in the oncogenesis of different tumors and thus are not specific to NB. Indeed, germline alterations in SMARCA4 conferring predisposition to SCCOHT and RT.39, 44, 45 At a somatic level, missense point mutations in SMARCA4 mapping to the ATPase domain might cause loss of direct binding between BAF and PRC1 which could contribute to oncogenesis or epigenetic plasticity during tumor development.46 Such SMARCA4 missense point mutations have been described in medulloblastoma.47, 48
In our study, 5/8 SMARCA4 variations concerned missense point mutations. Although it has been suggested that SMARCA4 might function as an oncogene in NB, and the overexpression of SMARCA4 in Stage 4 NB patients is associated with poorer outcomes,49 the functional impact of genetic variations can be difficult to determine without in‐depth functional studies which are beyond the scope of this article.
Even though the somatic or germline origin of SMARCA4 variations in our study remained undetermined, our data suggest that variations in SMARCA4 might play a role in the oncogenesis of NB, and several arguments now underline its role. First, we describe a case of NB within the context of a germline deletion encompassing SMARCA4. Second, our findings highlight a significantly higher frequency of variations in SMARCA4 in NB cases when compared to the gnomAD reference population. Third, we identified four NB cases showing double events targeting SMARCA4. In addition, a double event (stop‐gain variation and LOH) of SMARCA4 was observed in the SKNFI NB cell line causing the absence of SMARCA4 expression.
Further functional studies are needed for a deeper understanding of SMARCA4 variations in NB in order to guide the development of more effective therapies.
Altogether, our data demonstrate CRG/EMG variations with likely functional impact in 8.4% (24/283) of all NB patients.
Although no difference in PFS between cases with or without CRG/EMG variations was observed, a poorer overall survival in cases harboring CRG/EMG variations was observed. This suggests that cases harboring CRG/EMG variations might present more aggressive disease or that salvage treatment used at relapse might potentially be more efficient in cases showing no CRG/EMG variation. Correlations between the presence of variations in CRG/EMG and other genetic factors associated with poor outcome in NB should be determined in future studies and larger series, such as the overall mutational burden50 or factors contributing to a mechanistical classification of NB including mutations of the RAS:MAPK pathway and telomere maintenance mechanisms.51
New treatment strategies are necessary for patients with high‐risk NB, and our data further highlight the potential interest of drugs modulating chromatin remodeling processes. Histone deacetylase inhibitors (HDACi) or DNA methyltransferase inhibitors were the first epigenetic compounds to reach clinical trials with potential benefits in some patients with CRG/EMG variations.52 Further preclinical development of therapeutic approaches involving CRGs/EMGs and in particular SMARCA4 will be warranted.
Supporting information
Figure S1: Kaplan–Meier curves and log‐rank analysis for (OS) overall survival of 283 NB cases according to the main clinical parameters for NB (A, according to MYCN status; B, according to INSS disease stages; C, according to age at the diagnosis). (A) OS for MYCN amplified NB patients vs. MYCN non amplified patients. MYCN amplification: n = 64 (23%). The 5‐year overall survival for patients with MYCN amplified NB was 24% vs. 84% for patients with non‐MYCN amplified. (B) OS for stage 4 patients vs. stage 4 s vs. Loc NB patients. INSS stage 4: n = 152 (53.7%); INSS stage 4 s: n = 25 (8.8%); INSS Localized disease: n = 130 (45%). (C) OS for patients with an age at diagnosis <18 months vs. patients with an age at diagnosis ≥18 months. >18 months: n = 159 (56%); ≥18 months: n = 124 (44%).
Figure S2: Large germline deletion (1.7 Mb) encompassing CDKN2D, ILF3, SMARCA4 and CNN1 genes is detected in chr19 (chr19:10462524–12,157,782) in the patient NBSW26. No genes of interest are included in the 2 small deletions detected at the end of chr19 (chr19:52271802–52,588,019 and chr19:55294257–55,370,554).
Figure S3: (A) Kaplan–Meier curves and log‐rank analysis for overall (OS) and progression‐free survival (PFS) (B) of 283 NB cases comparing tumors with a CRG/EMG variation and tumors without CRG/EMG variations. A statistically significant lower OS was observed for patients whose tumors harbor CRG/EMG variations (5 year OS, 58% vs. 74%, p = 0.0302, log‐rank test) (C) Kaplan–Meier curves and log‐rank analysis for overall survival (OS) of 124 high‐risk NB patients included in the cohort comparing tumors with CRG/EMG variation and tumors without CRG/EMG variations. A statistically significant difference between the two patient groups was observed.
Figure S4: (A) Variation (SNVs/InDels) annotation, prioritization and classification workflow. (B) A table with six in silico prediction tools/algorithms used to prioritize and classify variations (SNVs) into pathogenic/likely pathogenic and variation of unknown significance (VUS).
Table S1: summary of all studied NB cases. (A) NB patients. NB patients were chosen to substitute a clinically representative cohort: (INSS stage 4: n = 128 (45%); INSS stage 4 s: n = 25 (8.8%); localized disease, n = 130 (45%); MYCN amplification: n = 64 (23%). (B) NB cell lines. Legend A: DOD, dead of disease; MD, Missing Data; NA, not applicable. Genomic profile: MNA, MYCN amplification; MN‐NA, MYCN Not‐amplified. Genomic profile: 17q G, 17q gain; 1pD, 1p deletion. Technique of sequencing: WGS, Whole Genome Sequencing; WES, Whole Exome Sequencing; TSCA, TruSeq® Custom Amplicon. NB high‐risk patient subgroup has been defined according to the current SIOPEN treatment strategy. Legend B: F: female; M: male; Loc: localized; MD, missing data. MYCN status: MNA, MYCN amplification; MN‐NA, MYCN Not‐amplified; ALK mutation, wt: wild type; ALK status: ALK‐NA, ALK not amplified; ALK A, ALK amplified; Genomic profile: 17q G, 17q gain; 1pD, 1p deletion
Table S2: TSCA® design covering 33 CRGs/EMGs, consisting of a total of 1,070 amplicons (262,756 bp) (average coverage: <1,500X; TSCA®v1.5, Illumina Inc., San Diego, CA, USA). All exonic regions of targeted genes are completely covered except for 240 bp of exonic regions for SMARCC2, DAXX and SMARCC1 genes, indicated in bold in the table.
Table S3: Summary of all SNV, InDels and focal CNAs detected in NB cases. (A) SNVs/InDels/focal CNAs detected in NB patients. (B) SNVs/InDels/focal CNAs detected in NB cell lines. Legend: For a given coordinate, the base corresponding to the reference genome (Human Genome Browser, http://genome.ucsc.edu/; hg19) is indicated. In bold: NB cases showing multiple variations in CRGs/EMGs. Abbreviations: D, Deleterious; T, Tolerated; NA, Not available; AA, amino acid; CNA copy number alteration. Technique of sequencing: WGS, Whole Genome Sequencing; WES, Whole Exome Sequencing; TSCA, TruSeq® Custom Amplicon.
Table S4: Odds ratios and Fisher's Exact test results testing for significance of the excess of pathogenic or likely pathogenic SNVs and InDels in NB cases vs. gnomAD controls (Fisher's Exact test level of significance = 0.05)
Acknowledgements
Our study was supported by the Annenberg Foundation, the Nelia and Amadeo Barletta Foundation (FNAB) and the Association Hubert Gouin Enfance et Cancer. Our study was also funded by the Associations Enfants et Santé, Les Bagouz à Manon, Les amis de Claire. Funding was obtained from SiRIC/INCa (Grant INCa‐DGOS‐4654), as well as from the CEST of Institute Curie, and Programme Hospitalier de recherche en Cancérologie (PHRC) IC2007‐09 grant. High‐throughput sequencing was performed by the ICGex NGS platform of the Institut Curie supported by the grants ANR‐10‐EQPX‐03 (Equipex) and ANR‐10‐INBS‐09‐08 (France Génomique Consortium) from the Agence Nationale de la Recherche (“Investissements d'Avenir” program), by the Canceropole Ile‐de‐France and by the SiRIC‐Curie program‐SiRIC Grant “INCa‐DGOS‐4654”, as well as the grant of collaboration with CEA/IG/CNG financed by France Génomique infrastructure, as part of the 517 program “Investissements d'Avenir” from the Agence Nationale pour la Recherche (contract ANR‐10‐INBS‐518 09).
Conflict of interest: The authors declare no conflict of interest.
References
- 1. Masliah‐Planchon J, Bièche I, Guinebretière J‐M, et al. SWI/SNF chromatin remodeling and human malignancies. Annu Rev Pathol Mech Dis 2015;10:145–71. [DOI] [PubMed] [Google Scholar]
- 2. Versteege I, Sévenet N, Lange J, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998;394:203–6. [DOI] [PubMed] [Google Scholar]
- 3. Kadoch C, Hargreaves DC, Hodges C, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 2013;45:592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Matthay KK, Maris JM, Schleiermacher G, et al. Neuroblastoma. Nat Rev Dis Primer 2016;2:16078. [DOI] [PubMed] [Google Scholar]
- 5. Molenaar JJ, Koster J, Zwijnenburg DA, et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 2012;483:589–93. [DOI] [PubMed] [Google Scholar]
- 6. Cheung N‐KV, Dyer MA. Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer 2013;13:397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pugh TJ, Morozova O, Attiyeh EF, et al. The genetic landscape of high‐risk neuroblastoma. Nat Genet 2013;45:279–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kurihara S, Hiyama E, Onitake Y, et al. Clinical features of ATRX or DAXX mutated neuroblastoma. J Pediatr Surg 2014;49:1835–8. [DOI] [PubMed] [Google Scholar]
- 9. Sausen M, Leary RJ, Jones S, et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet 2013;45:12–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eleveld TF, Oldridge DA, Bernard V, et al. Relapsed neuroblastomas show frequent RAS‐MAPK pathway mutations. Nat Genet 2015;47:864–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res 2010;20:1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat Genet 2011;43:491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen X, Schulz‐Trieglaff O, Shaw R, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016;32:1220–2. [DOI] [PubMed] [Google Scholar]
- 15. Boeva V, Popova T, Bleakley K, et al. Control‐FREEC: a tool for assessing copy number and allelic content using next‐generation sequencing data. Bioinformatics 2012;28:423–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012;22:568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Seshan V, Olshen A. DNAcopy: DNA copy number data analysis. R package version 1.46.0. Vienna, Austria: R Foundation for Statistical Computing, 2016. [Google Scholar]
- 18. Liu X, Wu C, Li C, et al. dbNSFP v3.0: a one‐stop database of functional predictions and annotations for human nonsynonymous and splice‐site SNVs. Hum Mutat 2016;37:235–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Langmead B, Salzberg SL. Fast gapped‐read alignment with bowtie 2. Nat Methods 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011;27:2987–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kadri S, Zhen CJ, Wurst MN, et al. Amplicon Indel hunter is a novel bioinformatics tool to detect large somatic insertion/deletion mutations in amplicon‐based next‐generation sequencing data. J Mol Diagn 2015;17:635–43. [DOI] [PubMed] [Google Scholar]
- 22. Boeva V, Popova T, Lienard M, et al. Multi‐factor data normalization enables the detection of copy number aberrations in amplicon sequencing data. Bioinformatics 2014;30:3443–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Robinson JT, Thorvaldsdóttir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol 2011;29:24–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cingolani P, Platts A, Wang LL, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly (Austin) 2012;6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sherry ST, Ward MH, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 2001;29:308–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature 2016;536:285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Forbes SA, Beare D, Boutselakis H, et al. COSMIC: somatic cancer genetics at high‐resolution. Nucleic Acids Res 2017;45:D777–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016;44:D862–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chang MT, Bhattarai TS, Schram AM, et al. Accelerating discovery of functional mutant alleles in cancer. Cancer Discov 2018;8:174–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Carter H, Douville C, Stenson PD, et al. Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genomics 2013;14(Suppl 3):S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dong C, Wei P, Jian X, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet 2015;24:2125–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet 2016;99:877–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schwarz JM, Cooper DN, Schuelke M, et al. MutationTaster2: mutation prediction for the deep‐sequencing age. Nat Methods 2014;11:361–2. [DOI] [PubMed] [Google Scholar]
- 34. Jagadeesh KA, Wenger AM, Berger MJ, et al. M‐CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet 2016;48:1581–6. [DOI] [PubMed] [Google Scholar]
- 35. Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bellini A, Bernard V, Leroy Q, et al. Deep sequencing reveals occurrence of subclonal ALK mutations in neuroblastoma at diagnosis. Clin Cancer Res 2015;21:4913–21. [DOI] [PubMed] [Google Scholar]
- 37. R Core Team (2018). R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; Available from: https://www.gbif.org/tool/81287/r-a-language-and-environment-for-statistical-computing [date last accessed June 18, 2018]. [Google Scholar]
- 38. Biegel JA, Busse TM, Weissman BE. SWI/SNF chromatin remodeling complexes and cancer. Am J Med Genet C Semin Med Genet 2014;166C:350–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Holsten T, Bens S, Oyen F, et al. Germline variants in SMARCB1 and other members of the BAF chromatin‐remodeling complex across human disease entities: a meta‐analysis. Eur J Hum Genet 2018;26:1083–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Peifer M, Hertwig F, Roels F, et al. Telomerase activation by genomic rearrangements in high‐risk neuroblastoma. Nature 2015;526:700–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cancer Genome Atlas Research Network , Kandoth C, Schultz N, et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013;497:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: mechanistic insights gained from human genomics. Sci Adv 2015;1:e1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chmielecki J, Bailey M, He J, et al. Genomic profiling of a large set of diverse pediatric cancers identifies known and novel mutations across tumor spectra. Cancer Res 2017;77:509–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Witkowski L, Goudie C, Foulkes WD, et al. Small‐cell carcinoma of the ovary of hypercalcemic type (malignant Rhabdoid tumor of the ovary): a review with recent developments on pathogenesis. Surg Pathol Clin 2016;9:215–26. [DOI] [PubMed] [Google Scholar]
- 45. Witkowski L, Carrot‐Zhang J, Albrecht S, et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat Genet 2014;46:438–43. [DOI] [PubMed] [Google Scholar]
- 46. Stanton BZ, Hodges C, Calarco JP, et al. Smarca4 ATPase mutations disrupt direct eviction of PRC1 from chromatin. Nat Genet 2017;49:282–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Robinson G, Parker M, Kranenburg TA, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature 2012;488:43–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pugh TJ, Weeraratne SD, Archer TC, et al. Medulloblastoma exome sequencing uncovers subtype‐specific somatic mutations. Nature 2012;488:106–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jubierre L, Soriano A, Planells‐Ferrer L, et al. BRG1/SMARCA4 is essential for neuroblastoma cell viability through modulation of cell death and survival pathways. Oncogene 2016;35:5179–90. [DOI] [PubMed] [Google Scholar]
- 50. Gröbner SN, Worst BC, Weischenfeldt J, et al. The landscape of genomic alterations across childhood cancers. Nature 2018;555:321–7. [DOI] [PubMed] [Google Scholar]
- 51. Ackermann S, Cartolano M, Hero B, et al. A mechanistic classification of clinical phenotypes in neuroblastoma. Science 2018;362:1165–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gryder BE, Sodji QH, Oyelere AK. Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Med Chem 2012;4:505–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Kaplan–Meier curves and log‐rank analysis for (OS) overall survival of 283 NB cases according to the main clinical parameters for NB (A, according to MYCN status; B, according to INSS disease stages; C, according to age at the diagnosis). (A) OS for MYCN amplified NB patients vs. MYCN non amplified patients. MYCN amplification: n = 64 (23%). The 5‐year overall survival for patients with MYCN amplified NB was 24% vs. 84% for patients with non‐MYCN amplified. (B) OS for stage 4 patients vs. stage 4 s vs. Loc NB patients. INSS stage 4: n = 152 (53.7%); INSS stage 4 s: n = 25 (8.8%); INSS Localized disease: n = 130 (45%). (C) OS for patients with an age at diagnosis <18 months vs. patients with an age at diagnosis ≥18 months. >18 months: n = 159 (56%); ≥18 months: n = 124 (44%).
Figure S2: Large germline deletion (1.7 Mb) encompassing CDKN2D, ILF3, SMARCA4 and CNN1 genes is detected in chr19 (chr19:10462524–12,157,782) in the patient NBSW26. No genes of interest are included in the 2 small deletions detected at the end of chr19 (chr19:52271802–52,588,019 and chr19:55294257–55,370,554).
Figure S3: (A) Kaplan–Meier curves and log‐rank analysis for overall (OS) and progression‐free survival (PFS) (B) of 283 NB cases comparing tumors with a CRG/EMG variation and tumors without CRG/EMG variations. A statistically significant lower OS was observed for patients whose tumors harbor CRG/EMG variations (5 year OS, 58% vs. 74%, p = 0.0302, log‐rank test) (C) Kaplan–Meier curves and log‐rank analysis for overall survival (OS) of 124 high‐risk NB patients included in the cohort comparing tumors with CRG/EMG variation and tumors without CRG/EMG variations. A statistically significant difference between the two patient groups was observed.
Figure S4: (A) Variation (SNVs/InDels) annotation, prioritization and classification workflow. (B) A table with six in silico prediction tools/algorithms used to prioritize and classify variations (SNVs) into pathogenic/likely pathogenic and variation of unknown significance (VUS).
Table S1: summary of all studied NB cases. (A) NB patients. NB patients were chosen to substitute a clinically representative cohort: (INSS stage 4: n = 128 (45%); INSS stage 4 s: n = 25 (8.8%); localized disease, n = 130 (45%); MYCN amplification: n = 64 (23%). (B) NB cell lines. Legend A: DOD, dead of disease; MD, Missing Data; NA, not applicable. Genomic profile: MNA, MYCN amplification; MN‐NA, MYCN Not‐amplified. Genomic profile: 17q G, 17q gain; 1pD, 1p deletion. Technique of sequencing: WGS, Whole Genome Sequencing; WES, Whole Exome Sequencing; TSCA, TruSeq® Custom Amplicon. NB high‐risk patient subgroup has been defined according to the current SIOPEN treatment strategy. Legend B: F: female; M: male; Loc: localized; MD, missing data. MYCN status: MNA, MYCN amplification; MN‐NA, MYCN Not‐amplified; ALK mutation, wt: wild type; ALK status: ALK‐NA, ALK not amplified; ALK A, ALK amplified; Genomic profile: 17q G, 17q gain; 1pD, 1p deletion
Table S2: TSCA® design covering 33 CRGs/EMGs, consisting of a total of 1,070 amplicons (262,756 bp) (average coverage: <1,500X; TSCA®v1.5, Illumina Inc., San Diego, CA, USA). All exonic regions of targeted genes are completely covered except for 240 bp of exonic regions for SMARCC2, DAXX and SMARCC1 genes, indicated in bold in the table.
Table S3: Summary of all SNV, InDels and focal CNAs detected in NB cases. (A) SNVs/InDels/focal CNAs detected in NB patients. (B) SNVs/InDels/focal CNAs detected in NB cell lines. Legend: For a given coordinate, the base corresponding to the reference genome (Human Genome Browser, http://genome.ucsc.edu/; hg19) is indicated. In bold: NB cases showing multiple variations in CRGs/EMGs. Abbreviations: D, Deleterious; T, Tolerated; NA, Not available; AA, amino acid; CNA copy number alteration. Technique of sequencing: WGS, Whole Genome Sequencing; WES, Whole Exome Sequencing; TSCA, TruSeq® Custom Amplicon.
Table S4: Odds ratios and Fisher's Exact test results testing for significance of the excess of pathogenic or likely pathogenic SNVs and InDels in NB cases vs. gnomAD controls (Fisher's Exact test level of significance = 0.05)