

Abstract
Canonical Wnt signaling, which is transduced by β‐catenin and lymphoid enhancer factor 1/T cell‐specific transcription factors (LEF1/TCFs), regulates many aspects of metazoan development and tissue renewal. Although much evidence has associated canonical Wnt/β‐catenin signaling with mood disorders, the mechanistic links are still unknown. Many components of the canonical Wnt pathway are involved in cellular processes that are unrelated to classical canonical Wnt signaling, thus further blurring the picture. The present review critically evaluates the involvement of classical Wnt/β‐catenin signaling in developmental processes that putatively underlie the pathology of mental illnesses. Particular attention is given to the roles of LEF1/TCFs, which have been discussed surprisingly rarely in this context. Highlighting recent discoveries, we propose that alterations in the activity of LEF1/TCFs, and particularly of transcription factor 7‐like 2 (TCF7L2), result in defects previously associated with neuropsychiatric disorders, including imbalances in neurogenesis and oligodendrogenesis, the functional disruption of thalamocortical circuitry and dysfunction of the habenula.
Keywords: beta‐catenin, brain development, habenula, mental disorders, neurogenesis, oligodendrogenesis, postmitotic differentiation, TCF7L2, thalamus, Wnt pathway
Abbreviations
ADHD, attention‐deficit/hyperactivity disorder
ASD, autism spectrum disorder
BD, bipolar disorder
CA, Cornu Amonis
DG, dentate gyrus
DRD 2, dopamine D2 receptor
DISC1, disrupted in schizophrenia 1
DVL, Dishevelled
GSK3α/β, Glycogen Synthase Kinase 3α or β
HMG, high mobility group
iOLs, immature premyelinating oligodendrocytes
IPs, intermediate progenitors
iPSCs, induced pluripotent stem cells
LRP5/6, lipoprotein receptor‐related protein 5 or 6
LEF1/TCFs, lymphoid enhancer factor 1 and T cell‐specific transcription factors
MD, major depression
mOLs, myelin‐producing oligodendrocytes
NDD, neurodevelopmental disorder
OPCs, oligodenrocyte precursor cells
RG, radial glia
SCZ, schizophrenia
SNP, single nucleotide polymorphisms
SVZ, subventricular zone
TCF7L2, transcription factor 7‐like 2
VZ, ventricular zone
Mental disorders: current view on causes
The present view posits that the causes of mental disorders are multifactorial and involve an interplay between various innate vulnerabilities and biological and social environmental risk factors that eventually manifest as mood swings, social deficits, and alterations of the perception of reality. Accumulating evidence suggests that mental disorders that are diagnosed by psychiatrists as different entities are closely related from a biological point of view. Genetic risk factors overlap across these disorders 1, 2, and common structural changes in the brain and deficits in cognitive circuits are seen despite potentially different etiologies 3, 4. On the other hand, advanced brain imaging and high‐throughput genomic studies of psychiatric patients have provided evidence that each of these major mental disorders can be stratified into different biological subclasses based on specific genetic components and so called endophenotypes, i.e., quantitative biological traits, such as gene expression, anatomical alterations, specific behavior, etc. Endophenotype‐based classifications of disorders and links between endophenotypes and specific sets of genes may provide new opportunities to diagnose psychiatric conditions and provide personalized treatment 5, thus underscoring the need to understand the molecular mechanisms that underlie anatomical and functional brain disruptions in mental disorders. The present review seeks to identify some of these mechanisms.
Canonical Wnt/β‐catenin signaling and LEF1/TCF transcription factors
The first demonstration of an association between the canonical Wnt pathway and mental disorders was the discovery that the classical mood stabilizer lithium activated β‐catenin by inhibiting GSK3α/β 6, which is a key enzyme in the canonical Wnt pathway 7. Many Wnt pathway genes have been associated with mental illnesses during the last 15 years, further implicating the contribution of Wnt signaling in pathogenesis of these disorders. The present review discusses several possible links between functional alterations of canonical Wnt signaling and endophenotypes that have been associated with mental disorders.
The term ‘canonical Wnt signaling’ has been used as an umbrella term to describe diverse cellular pathways that are activated by Wnt ligands and diverge at the level of Glycogen Synthase Kinase 3α or β (GSK3α/β) inhibition (Fig. 1, Box 1), thus causing ambiguity about the specific pathways to which the authors of various studies refer. Defining the actual pathway is not always straightforward. For example, the terms ‘canonical Wnt’ and ‘Wnt/β‐catenin’ are often used interchangeably to describe a Wnt pathway that is mediated by low‐density lipoprotein receptor‐related protein 5 (LRP5) or LRP6, and GSK3α/β. This can be misleading because one needs to assume that GSK3α/β inhibition in the destruction complex is followed by the stabilization of β‐catenin. In fact, GSK3α/β has many other targets and is involved in many molecular processes. Therefore, the term Wnt/GSK3α/β or upstream canonical Wnt would be more accurate when the downstream effectors are not identified. Even the term ‘Wnt/β‐catenin’ is misleading when considering that the accumulation of β‐catenin is not always followed by its nuclear translocation. The activation of this pathway can also increase other activities of β‐catenin, such as its functions in cell adhesion and protein scaffolding. To help clarify these ambiguities, the present review focuses specifically on the classical canonical Wnt pathway, which is characterized by the translocation of β‐catenin to the nucleus, where it acts as a co‐activator of lymphoid enhancer factor 1 (LEF1)/TCF transcription factors (LEF1, TCF7, TCF7L1, TCF7L2) that bind DNA through the high mobility group domain (HMG). This pathway is further referred to here as the canonical Wnt/β‐catenin pathway.
Figure 1.
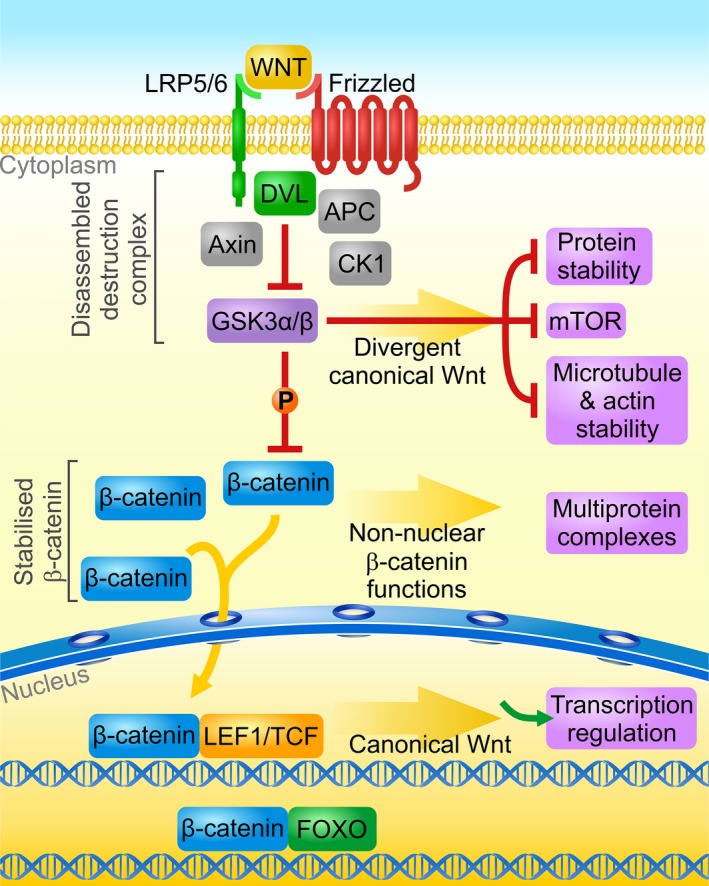
Canonical Wnt/β‐catenin and divergent signaling pathways. The binding of a Wnt ligand to a Frizzled receptor and LRP5/6 co‐receptor, followed by the recruitment of DVL to the receptor complex, leads to translocation and inhibition of the destruction complex, which consists of the kinase GSK3α/β, Adenomatous Polyposis Coli (APC) and Axin. The phosphorylation of β‐catenin by GSK3α/β in the active destruction complex primes this protein for proteasomal degradation (not shown). Upon inhibition of the complex, β‐catenin accumulates in the cytoplasm and translocates into the nucleus where it activates gene transcription as a co‐activator of LEF1/TCFs. This canonical pathway can branch downstream (a) GSK3α/β inhibition (e.g., to slow protein degradation during mitosis, activate the mTOR pathway, or stabilize the cytoskeleton), (b) β‐catenin stabilization (e.g., to stabilize cell adhesion with cadherins or facilitate the assembly of PDZ domain‐containing proteins), and (c) β‐catenin nuclear translocation (to activate gene transcription by interacting with nuclear receptors, FOXO and other transcription factors).
Box 1. WNT signaling pathways.
Wnt signaling is traditionally divided into three main branches: the canonical, planar cell polarity (Wnt/PCP) and Wnt/Ca2+ 143 pathways. All Wnt signaling pathways are activated by a common mechanism that requires the binding of Wnt‐protein ligands to the cell‐surface receptors Frizzled, and subsequent recruitment of Dishevelled (DVL) proteins. In the canonical Wnt pathway, signaling is transduced by Frizzled receptors together with LRP5 and LRP6 co‐receptors (Fig. 1). This is followed by the disassembly of the so‐called destruction complex, which comprises GSK3α/β and two scaffolding proteins: Axin and Adenomatous Polyposis Coli (APC). At the core of the canonical pathway is inhibition of the GSK3α/β‐mediated phosphorylation of proteins. In the classical view, this leads to the stabilization of β‐catenin (canonical Wnt/β‐catenin pathway), which otherwise is directed to proteasomal degradation upon the phosphorylation by GSK3α/β 7. However, the canonical Wnt pathway can diverge downstream of GSK3α/β to slow down protein degradation in the proteasome (Wnt/STOP pathway), activate mammalian/mechanistic target of rapamycin (mTOR), or stabilize microtubules and the actin cytoskeleton in neurons 144. In the canonical Wnt/β‐catenin pathway, β‐catenin accumulates in the cytoplasm and translocates to the nucleus, where it acts as a transcription co‐activator of LEF1/TCFs 13. Mammals express four members of the LEF1/TCF family (LEF1, TCF7, TCF7L1 which mainly acts as a transcription repressor, and TCF7L2; Fig. 2), which belong to HMG superfamily of transcription factors 145. LEF/TCFs and nuclear β‐catenin can also act independently of each other by interacting with other transcription factors 146, but the majority of β‐catenin binds to chromatin via LEF/TCFs 147. β‐catenin, in addition to its function in the nucleus, is involved in multiprotein assembly and cadherin‐mediated cell‐cell adhesion at the membrane. Although the submembranous pool of β‐catenin is largely independent of Wnt signaling, Wnts can sometimes increase the levels of this pool. For example, in neurons, the Wnt/β‐catenin pathway can diverge to modulate dendritogenesis, synaptogenesis, and synaptic vesicle localization independently of nuclear functions of β‐catenin by increasing its interactions with cadherins and PDZ‐containing proteins at the plasma membrane.
Recent reviews have broadly discussed the role of Wnt signaling in the development of pathologies that are associated with mental disorders. However, they focused more either on the role of the divergent GSK3α/β and divergent Wnt/β‐catenin pathways in dendritogenesis, synaptogenesis, and synaptic plasticity, or on upstream Wnt signaling 8, 9, 10, 11, 12, with relatively little coverage of the role of the downstream effectors LEF/TCFs in brain pathologies. We focus on the contribution of these downstream components of the canonical Wnt/β‐catenin pathway and nuclear β‐catenin to the pathogenesis of mental disorders 13. Particular attention is given to the role of TCF7L2, as it has been recently identified as having a central role in neural stem cell differentiation and postmitotic differentiation of some brain regions, impairments of which might contribute to mental disorders.
Evidence of alterations of canonical Wnt pathway activity and TCF7L2 in mental disorders
Human genetic studies
The vulnerability to mental illnesses is exacerbated by genetic factors. Although a single causal gene is unlikely to be identified, rates of common polymorphisms and the occurrence of de novo mutations in large populations of patients can help to identify molecular pathways that contribute to the pathogenesis of psychiatric disorders 14. Both upstream and downstream elements of the canonical Wnt pathway have been associated with different psychiatric conditions: WNT1 and WNT2 with autism spectrum disorder (ASD), WNT2B with bipolar disorder (BD), WNT7A with ASD and BD, WNT7B with schizophrenia (SCZ), LRP5 with SCZ, attention‐deficit/hyperactivity disorder (ADHD) and major depression (MD), and LRP6 with ADHD 15, 16, 17, 18, 19, 20, 21. The CTNNB1 gene, which encodes β‐catenin, has also been associated with ASD and SCZ 16, 22, 23. These examples suggest possible impairments in the canonical Wnt pathway across several major psychiatric disorders.
Among genes that encode downstream nuclear effectors of the canonical Wnt/β‐catenin pathway, only TCF7L2 has been repeatedly associated with mental disorders in both gene candidate and genome‐wide association studies (Fig. 2). Notably, TCF7L2, which is located on chromosome 10 and has been historically denoted T cell‐specific transcription factor 4 (abbreviated as TCF4), should not be confused with transcription factor 4 (TCF4; located on chromosome 18), which is a SCZ risk factor itself but does not belong to the LEF1/TCF family. Several common intronic polymorphisms and polymorphisms in the proximity of the TCF7L2 locus have been associated with SCZ 24, 25, 26, 27, 28, 29. Another common variant in an intronic region of TCF7L2 was identified as a BD susceptibility factor in patients with elevated body mass 30. Some of these polymorphisms (e.g., rs7903146) confer high genetic risk for developing type 2 diabetes, providing a possible molecular explanation for the prevalence of metabolic disorders in SCZ and BP 31. Finally, de novo splice site mutations of TCF7L2 were found in ASD patients 32, and stop‐gain and substitution mutations were observed in patients with neurodevelopmental disorders (NDD) 33, 34, 35. These genetic data are a strong indication that TCF7L2 is involved in pathological processes that result in mental disorders. Still unknown, however, are the specific processes because associations of TCF7L2 with specific endophenotypes have not yet been tested. New data from animal studies, discussed below, may shed some light on this issue.
Figure 2.
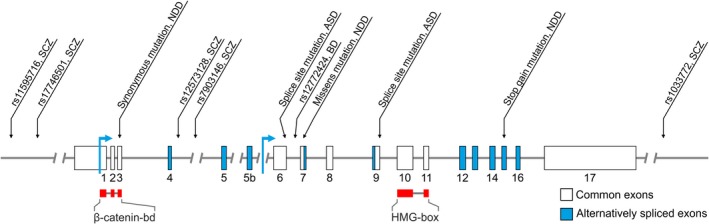
Location of single nucleotide polymorphisms (SNPs) in the human TCF7L2 gene. Representative intron–exon structure of the TCF7L2 gene. Long introns are represented by a double slash. Blue boxes indicate the alternatively spliced exons, blue arrows represent alternative transcription start sites. Important protein domains are marked by red boxes: β‐catenin binding domain, and DNA binding domain ‐ HMG‐box. Black arrows indicate the location of SNPs or mutations.
Animal model studies
Manipulations of individual components of the canonical Wnt pathway in animal models (Table 1) resulted in behavioral changes and provided further evidence of the involvement of this pathway in the pathogenesis of mental disorders. GSK3α/β is the most studied canonical Wnt‐related component with regard to behavioral alterations in animals, because it is a putative target of the classic mood stabilizer lithium. The genetic 36, 37 and chemical 37, 38, 39 downregulation of GSK3α/β (total, or in regions that receive dopaminergic input) decreased the vulnerability to depressive‐like behavior in the forced swim test, tail suspension test or sucrose preference test in mice, whereas Gsk3b overexpression in a dopaminergic region 37 had the opposite effect. Other studies implicated low GSK3 activity in mania and psychosis. Gsk3b gene knockout in dopamine D2 receptor (DRD2)‐expressing neurons, which are affected in SCZ, decreased amphetamine‐induced hyperlocomotion and prepulse inhibition, mimicking antipsychotic actions 40. Conversely, the overactivity of GSK3 (by Gsk3b overexpression or genetic blocking of inhibitory phosphorylation of GSK3) increased amphetamine‐induced hyperactivity 41, 42, decreased sensorimotor gating 42, 43, and altered the sleep‐wake cycle 44, all of which are considered notable features of mania in BP and psychosis in SCZ. Finally, mice with deficiency in Dishevelled (DVL) activity (by Dvl1 knockout), which is inhibitory for the GSK3α/β containing destruction complex in the canonical Wnt pathway, exhibited social deficit and stereotyped behavior, reproducing aspects of ASD 43, 45. Because GSK3α/β is a negative regulator of β‐catenin, these studies imply that reducing activity of the Wnt pathway increases the vulnerability to depression, mania, psychosis, and autistic behavior.
Table 1.
Wnt pathway genes and genetic modifications mentioned in this review. LOF, loss‐of‐function mutation; GOF, gain‐of‐function mutation.
| Mutation | Explanation | Targeted Wnt pathways | Expected effect on the canonical Wnt/β‐catenin pathway |
|---|---|---|---|
| Upstream components of the canonical Wnt pathway | |||
| Wnt3 overexpression 98 | GOF | Canonical and divergent canonical | Upregulation |
| Wnt3a overexpression 75 | GOF | Canonical and divergent canonical | Upregulation |
| Wnt3a knockout 65 | LOF | Canonical and divergent canonical | Downregulation |
| Lrp6 knockout 77, 95 | LOF | Canonical and divergent canonical | Downregulation |
| Dkk1 overexpression 94, 133 | GOF; Dkk1 encodes an antagonist of LRP5/6 | Canonical and divergent canonical | Downregulation |
| GSK3α/β, a component of β‐catenin destruction complex | |||
| Gsk3b knockout 36, 40 | LOF | Canonical and divergent canonical | Upregulation |
| GSK3β K85A/K86A 37 | LOF, dominan negative mutant; lysine‐to‐alanine mutations to produce catalytically inactive GSK3β | Canonical and divergent canonical | Upregulation |
| Gsk3b overexpression 37, 42, 44 | GOF | Canonical and divergent canonical | Downregulation |
| GSK3α S21A and GSK3/β S9A 41 | GOF; serine‐to‐alanine mutations to block inhibitory serine phosphorylation of GSK3α and β | Canonical and divergent canonical | Downregulation |
| GSK3β S9A 44 | GOF; serine‐to‐alanine mutation to block inhibitory serine phosphorylation of GSK3β | Canonical and divergent canonical | Downregulation |
| Dishevelled (DVL), an inhibitor of β‐catenin destruction complex | |||
| Dvl1 knockout 43, 45 | LOF | Canonical and divergent canonical | Downregulation |
| β‐catenin (Ctnnb1) | |||
| Ctnnb1 knockout 62, 63, 70, 74 | LOF | Canonical Wnt/β‐catenin, divergent Wnt/β‐catenin, Wnt‐independent | Downregulation |
| β‐catenindm (Ctnnb1 dm) 74 | LOF, transcriptionally nonfunctional β‐catenin with intact adhesive function; deletions of N‐terminal and C‐terminal transactivation domains | Canonical Wnt/β‐catenin | Downregulation |
| β‐cateninΔexon3 40, 70, 72, 137, 141 | GOF, a stabilized form of β‐catenin; deletion of exon3 that encodes the GSK3α/β phosphorylation sites | Canonical Wnt/β‐catenin and divergent Wnt/β‐catenin | Upregulation |
| β‐cateninΔ90 71, 73 | GOF, a stabilized form of β‐catenin; deletion of N‐terminal fragment that contains GSK3α/β phosphorylation sites | Canonical Wnt/β‐catenin and divergent Wnt/β‐catenin | Upregulation |
| β‐cateninΔ29–48 75 | GOF, a stabilized form of β‐catenin with; deletion of 29–48 aminoacid residues that contain GSK3α/β phosphorylation sites | Canonical Wnt/β‐catenin and divergent Wnt/β‐catenin | Upregulation |
| β‐catenin S37F or S37Y 46, 47 | GOF, a stabilized form of β‐catenin; serine‐to‐phenylalanine or serine‐to‐tyrosine mutation in GSK3α/β phosphorylation sites | Canonical Wnt/β‐catenin and divergent Wnt/β‐catenin | Upregulation |
| Constitutively active β‐catenin 70 | GOF; β‐catenin activation domain coupled with LEF1 | Canonical Wnt/β‐catenin | Upregulation |
| LEF/TCF transcription factors | |||
| Tcf7l2 knockout 49, 50, 61, 114, 115, 136, 140 | LOF | Canonical Wnt/β‐catenin and Wnt‐independent | Downregulation |
| Lef1 knockout 51, 77, 95 | LOF | Canonical Wnt/β‐catenin and Wnt‐independent | Downregulation |
| Tcf7l1 knockout 61 | LOF | Canonical Wnt/β‐catenin and Wnt‐independent | Upregulation |
| dnLEF1 96, 133 | LOF, a dominant negative form of LEF1; LEF1 with a truncated β‐catenin‐binding domain | Canonical Wnt/β‐catenin | Downregulation |
| dnTCF7L2 63, 69, 78, 96 | LOF, a dominant negative form of TCF7L2; TCF7L2 with a truncated β‐catenin‐binding domain | Canonical Wnt/β‐catenin | Downregulation |
| TCF7L2ΔHMG 139 | LOF; TCF7L2 without the DNA binding domain (HMG), likely act as a dominant negative mutant | Canonical Wnt/β‐catenin and Wnt‐independent | Downregulation |
| LEF1ΔHMG 68 | LOF; LEF1 without the DNA binding domain (HMG), likely act as a dominant negative mutant | Canonical Wnt/β‐catenin and Wnt‐independent | Downregulation |
| Dominant‐active LEF1 68, 75, 77, 95 | GOF; LEF1 fused to the herpes simplex virus VP16 transactivation domain | Canonical Wnt/β‐catenin and Wnt‐independent | Upregulation |
The involvement of alterations of GSK3α/β or upstream Wnt signaling activity does not prove the involvement of β‐catenin, not to mention nuclear β‐catenin and LEF1/TCFs. For example, the expression of a stabilized form of β‐catenin that is resistant to GSK3α/β‐mediated degradation (Table 1) did not produce similar antipsychotic effects in aforementioned mice with Gsk3b knockout in DRD2 neurons, thus excluding such a contribution in this case 40. However, mice with stabilized β‐catenin (in the forebrain and cerebellum or in regions that receive dopaminergic input) exhibited a decrease in the vulnerability to depressive‐like behavior in the forced swim test 46, 47, which was consistent with the aforementioned effects of GSK3α/β inhibition. This suggested the role of β‐catenin in antidepressant‐like response to GSK3α/β inhibition.
The role of LEF1/TCF in behavioral regulation has only recently begun to be investigated. Several experiments tentatively implicated TCF7L2 deficiency in anxiety and depression. The silencing of Tcf7l2 in zebrafish by morpholinos antagonized the effect of lithium on dark‐induced locomotion 48, suggesting the involvement of TCF7L2 in the behavioral response to this medication for BD. Tcf7l2 haploinsufficiency increased anxiety‐like phenotypes in the light‐dark box test and contextual fear conditioning 49 and reduced exploratory activity in some mouse strains 50. LEF1 was not shown to be a risk factor in mental disorders. Nevertheless, a recent study found that hypothalamus‐specific Lef1 knockout increased anxiety‐like behavior in both zebrafish and mice 51, suggesting the possible role of LEF1 in hypothalamic‐pituitary‐adrenal axis activity and the stress response.
Altogether, genetic alterations in individuals with mental disorders and behavioral deficits in genetically modified animals have provided convincing evidence that the canonical Wnt/β‐catenin pathway and LEF1/TCFs (particularly TCF7L2) are involved in the pathogenesis of mental disorders.
Developmental neurogenesis in the neocortex
Neocortical abnormalities in mental disorders
The cerebral cortex is the outer layer of the brain. The main part of the cortex in mammals is the neocortex, which processes and integrates emotional and sensory information, controls problem solving and decision‐making. Development of the gyrated human neocortex requires the particularly intensive expansion of neural progenitor populations to produce sufficient numbers of neurons.
Neuroimaging showed changes in cortical thickness and the cortical surface in patients who suffer from psychiatric disorders. These phenotypes may result from excessive or insufficient synaptic pruning or myelination that occurs in adolescents, but also from impaired neurogenesis, e.g., alterations of the ratio of symmetrical cell divisions and asymmetrical cell divisions of neural progenitors. In adult individuals who were diagnosed with MD, a decrease in the thickness of cortical gray matter was observed specifically in the frontal and temporal lobes 52. In patients with BD 53 or SCZ 54, such cortical thinning was more widespread, with the greatest differences in the frontal and temporal lobes. In the ADHD group, a lower volume was also observed predominantly in the frontal cortex, with a reduction of the cortical surface but no changes in cortical thickness 55. By contrast, in the ASD group, the cortical surface was unaffected, but an increase in cortical thickness was observed, especially in the frontal cortex and limbic lobe regions 56. Thus, changes in cortical volume and size are a common endophenotypes in individuals with mental disorders. Unresolved issues include whether such alterations are causes or consequences of the disorder, whether they underlie impairments in neurogenesis, and what are the molecular mechanisms that are involved in these pathologies.
Canonical Wnt/β‐catenin signaling in the neurogenic niche
Neurons in the neocortex are generated in the ventricular zone (VZ) of the dorsal telencephalon from uncommited radial glia (RG) and in the subventricular zone (SVZ) from neural intermediate progenitors (IPs) 57, 58. Symmetrical cell divisions of RG expand this population, leading to the generation of more ‘radial units’ and consequently surface expansion, whereas asymmetrical cell divisions maintain neural progenitor populations and simultaneously produce neurons that migrate within ‘columnar units’ to generate cortical thickness 59. At later embryogenesis, RG progenitors switch to astrogenic divisions.
The canonical Wnt/β‐catenin signaling pathway is active in the cortical VZ during embryogenesis, as demonstrated by studies in Wnt reporter mice, measurement of Wnt7a expression, and the presence of LEF1, TCF7L1, and TCF7L2 60, 61, 62. The activity of Wnt/β‐catenin signaling is high in neurogenic niches but decreases in the intermediate zone that is populated by migrating and differentiating cells 63, 64. Moreover, caudomedially located hem and laterally located antihem, which are two signaling centers of the developing cortex (neocortex and hippocampus), secrete Wnt signals (WNT3A and WNT2B) 65, and Wnt signaling inhibitor SFRP2 (secreted frizzled‐related protein 2 that binds to WNT proteins) 66. This creates a caudomedial to rostrolateral gradient of canonical Wnt/β‐catenin signaling, which is opposite to the gradient of neurogenesis 67. LEF1 expression in the VZ also gradually fades from the medial edge toward the lateral edge, whereas the expression gradient of the transcriptional repressor TCF7L1 is opposite to LEF1 61, 68, presumably contributing to establishment of the gradient. In addition to this spatial pattern, a gradual decrease in Wnt/β‐catenin signaling occurs as development of the cortex progresses 69, 70. Below we discuss how these dynamic spatiotemporal patterns of Wnt/β‐catenin signaling control the timing and progression of neocortical neurogenesis.
Regulation of proliferation versus differentiation of neural progenitors
Many studies have investigated the role of β‐catenin in different aspects of neurogenesis and neural cell migration. Much effort has been expended to differentiate its function between the regulation of cell adhesion and gene transcription (a focus of the present review), by using genetic modifications in mice (Table 1).
Enhancing canonical Wnt signaling by β‐catenin stabilization has consistently led to the expansion of a progenitor pool that starts at midgestation followed by neocortical hyperplasia 70, 71, 72, 73. Blocking Wnt signaling by Ctnnb1 knockout had the opposite effect, in which it prematurely decreased the number of proliferating cells in the VZ and led to smaller cortical hemispheres at birth 62. This expansion or reduction of the neocortical neuroepithelium progenitor pool was not attributable to changes in the proliferation rate but rather to alterations of the ratio between cell‐cycle exit and reentry 71, 72. This phenotype was specific for RG, and not for IPs 74, 75, and therefore might be attributable to the cytoskeletal function of β‐catenin because intact apical junctions are critical for maintaining RG polarity and vertical cleavage during mitosis, which result in self‐expanding divisions 58, 76. However, expression of transcriptionally nonfunctional β‐catenindm with intact adhesive function (Ctnnb1 dm mutation, β‐catenin allele that lacks N‐ and C‐terminal transcriptional outputs) also resulted in the depletion of progenitor cells and the precocious production of neurons, despite normal VZ architecture 74. Likewise, the focal expression of dominant negative TCF7L2 (dnTCF7L2, lacking β‐catenin binding domain) in the developing neocortex stimulated cell‐cycle exit in progenitors 63, confirming the role of nuclear β‐catenin and LEF1/TCFs in the regulation of neural progenitor pool size in the neocortex.
Studies in which the uncommitted RG and committed IPs were analyzed as separate populations revealed opposite effects of canonical Wnt/β‐catenin signaling in each population (Fig. 3). In mice with Lrp6 knockout, Ctnnb1 knockout or a Ctnnb1 dm mutation SVZ and postmitotic layer transiently thickened during early neurogenesis, before the depletion of RG finally led to a dramatic decrease in the number of neurons at late neurogenesis or postnatally 62, 74, 77. This was caused by an increase in proliferation of IPs, which are transit‐amplifying cells. Consistent with these findings, the ectopic expression of Wnt3a, dominant active LEF1 (LEF1 fused to the herpes simplex virus VP16 transactivation domain), or stabilized β‐catenin (β‐cateninΔ29‐48) in the developing neocortex promoted the expansion of RG on the one hand, and differentiation of IPs on the other hand 75. This evidence implies a dual role of Wnt/β‐catenin signaling in regulating cell‐cycle mode in uncommitted and committed progenitor populations, in which it maintains RG proliferative (symmetric) divisions and promotes IP differentiative divisions.
Figure 3.
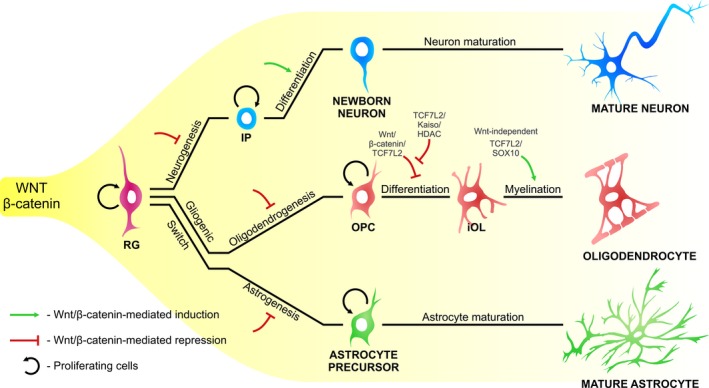
Role of Wnt/β‐catenin signaling and TCF7L2 in neurogenesis and gliogenesis in the neocortex. The process of developmental neurogenesis and gliogenesis in the neocortex is opposite to the temporal gradient of Wnt signaling (shown in yellow). Genetic manipulation to downregulate Wnt/β‐catenin signaling causes premature neurogenic divisions of uncommitted progenitors (RG) and a shorter time of neurogenesis as a consequence of precocious astrogenesis. Wnt/β‐catenin signaling inhibits oligodendrocyte precursor cell (OPC) differentiation. This is antagonized by interactions between TCF7L2 and the Kaiso co‐repressor. TCF7L2 interacts with the SOX10 to promote the further differentiation of iOL into mOL in a β‐catenin‐independent manner. Steps in this process that were shown to be activated or inhibited by Wnt/β‐catenin signaling or TCF7L2 are indicated in green with arrows and red bar‐headed lines, respectively. GP, glial progenitor
A recent study shed some light on which transcription factors of the LEF/TCF family mediate the above effects. Neither LEF1 77, nor TCF7L1 61, appeared to be necessary for development of the neocortex in single knockout studies. Mice with a conditional knockout of Tcf7l2 exhibited a decrease in progenitor proliferation at late neurogenesis and had a smaller cortex 61, which resembled impairments that were observed in mice with transcriptionally nonfunctional β‐catenindm 74. This identified TCF7L2 as a regulator of cortical neurogenesis. However, these impairments were already manifested at early and mid‐neurogenesis in Ctnnb1 dm mutant mice, whereas development of the cortex was unaffected at this stage in Tcf7l2 knockout mice, and the ratio between RG and IPs was unaltered. This suggests that another transcription factor from the LEF1/TCF family, in addition to TCF7L2, is a mediator of the canonical Wnt/β‐catenin pathway in the developing cortex. Tcf7l1/Tcf7l2 double‐knockout did not deteriorate or rescue the phenotype of single Tcf7l2 knockout, thus excluding TCF7L1. LEF1 might compensate for the loss of TCF7L2 during neurogenesis in the neocortex, but this hypothesis needs to be tested in Lef1/Tcf7l2 double‐knockout mice.
Regulation of the neurogenesis timing
Further complicating the role of canonical Wnt signaling in neurogenesis is the involvement of this pathway in the regulation of the timing and duration of neurogenesis. Lrp6 knockout, Ctnnb1 knockout, and a Ctnnb1 dm mutation caused a precocious switch from neurogenesis to astrogenesis in the neocortex 62, 74, 77. Thus, in addition to RG depletion, a shortening of the neurogenic phase could contribute to neocortical hypoplasia in mice with inhibited Wnt/β‐catenin signaling. Moreover, Wnt/β‐catenin signaling possibly regulates the timing of generating successive neuronal populations, which migrate in the neocortical mantle zone in an inside‐out manner, in which later born neurons travel to more superficial parts of the neocortex past early neuron layers 57. The ectopic expression of stabilized β‐catenin starting at mid‐neurogenesis significantly decreased the fraction of late‐born (upper‐layer) neurons 69, 72. Conversely, the fraction of late‐born neurons increased upon the expression of dnTCF7L2 69. Ctnnb1 knockout and a Ctnnb1 dm mutation accelerated the production of upper‐layer neurons 74. Overall, these findings suggest that Wnt/β‐catenin signaling prevents a premature switch of neural progenitors to the production of subsequent neuronal populations and regulates the duration of neurogenesis by suppressing the switch from neurogenesis to astrogenesis.
Regulation of cell migration during neocortical organogenesis
Finally, some studies have investigated the role of nuclear β‐catenin in the establishment of ordered cortical structure. Both the expression of stabilized β‐catenin and Ctnnb1 knockout in the cortical neuroepithelium led to the early loss of RG apical adherens junctions 62, 70, 74, followed by RG scaffold disassembly 62, 72, a severe migration defect in newborn neurons 62, and the loss of neocortical laminar structure 62, 70, 72, 73, 74, 75. This was likely caused by the loss of β‐catenin function in cell adhesion because polarized architecture of the RG, which is necessary for the radial migration of new neurons, is based on β‐catenin‐mediated interactions between N‐cadherin and the cytoskeleton in apical junctions 76. Indeed, apical junctions and laminar structures were intact during neurogenesis, when canonical Wnt signaling was attenuated downstream of β‐catenin stabilization, by a Ctnnb1 dm mutation or the knockout of Lef1, Tcf7l1, or Tcf7l2 61, 74, 77. On the other hand, the ectopic expression of in utero‐delivered dnTCF7L2 reduced migratory speed in cortical layers, but it did not affect RG scaffolds 78, whereas expression of the β‐catenin activation domain coupled with LEF1 (constitutively active β‐catenin) to activate nuclear β‐catenin signaling, caused cell intermingling between the VZ and SVZ 70. This confirmed the role of canonical Wnt/β‐catenin signaling in some aspects of neuronal migration, but not in the radial organization of migration. Therefore, although integrity of the neuroepithelium during neuronal generation and the initial radial migration of neurons require an intact adhesive function of β‐catenin, maintaining RG assembly and borders between different layers at later stages, as well as new neuron migration along RG basal processes, is at least partly controlled by the nuclear activity of β‐catenin. This is further supported by overrepresentation of cell adhesion genes in putative target genes of LEF/TCFs 79.
Implications for mental disorders
The association between Wnt signaling, cortical size, and adult behavioral anomalies has been demonstrated in mouse studies. Mice with deficiency in DVL activity (Dvl1 −/− mice), in which Wnt signaling was decreased but not entirely abolished, exhibited transient increases in brain weight and cortical thickness at mid‐gestation, accompanied by a reduction of RG and transient expansion of IP population and deep layer neurons. In adulthood, the cortex of these mice was smaller than controls (consistent with RG depletion), but the frontal cortex was thicker 45, resembling the cortical endophenotype of ASD patients. These mice exhibited social deficits and stereotyped behaviors, which were rescued by transient administration of the canonical Wnt agonist CHIR99021 (a selective inhibitor of GSK3β) at mid‐gestation.
Another link is chromodomain helicase DNA‐binding protein 8 (CHD8), a chromatin remodeling factor which regulates Wnt signaling. De novo mutations of CHD8 are one of the most replicated genetic alterations in ASD. Chd8 knockdown in mice by the in utero delivery of shRNA caused the depletion of RG and premature production of neurons during development, and behavioral alterations in adulthood, including social deficits 80. Both developmental and behavioral impairments were rescued by the simultaneous expression of stabilized β‐catenin.
Human induced pluripotent stem cells (iPSCs), in vitro cell differentiation, and brain organoid technologies have provided novel opportunities to model SCZ and BD. The causal role of canonical Wnt pathway downregulation was investigated in neural lineage cells that were derived from two related BD patients 81. These cells were characterized by impairments in proliferation and neurogenesis and WNT7B downregulation compared to cells derived from healthy relatives. The treatment of these cells with the canonical Wnt agonist CHIR99021 significantly increased LEF1 levels and rescued proliferation deficits. Finally, XAV939, an inhibitor of the canonical Wnt/β‐catenin pathway (a stabilizer of Axin in the β‐catenin destruction complex), rescued morphological impairments in iPSC‐derived cerebral organoids with mutations of the disrupted in schizophrenia 1 (DISC1) gene 82, the first gene that was identified as a risk factor for SCZ, BP, and MD. Whether these effects indeed depended on LEF/TCFs was not examined. These findings preliminarily supported the canonical Wnt and neurogenesis hypothesis of mental disorders, but such studies are still too scarce to draw more definitive conclusions.
Neurogenesis in the hippocampus
Hippocampal abnormalities and hippocampal neurogenesis in mental disorders
The hippocampus is the main part of the evolutionary old allocortex that is composed of two layers of neurons: granule neurons in the dentate gyrus (DG) and pyramidal neurons in Cornu Amonis (CA) fields. This structure is involved in episodic memory formation and the consolidation of emotional memories. An interesting feature of the hippocampal DG is its ability to produce new neurons throughout life. Adult neurogenesis in the hippocampus is presumed to occur in both humans and rodents and play a role in cognitive plasticity.
Neuroimaging studies have repeatedly reported the loss of hippocampal volume in individuals who are diagnosed with ADHD, BD, SCZ, ASD, and MD 56, 83, 84, 85, 86. However, a large study of MD patients suggested that hippocampal atrophy is a consequence rather than a cause of mental disorders because such atrophy occurred in patients after multiple depressive episodes, whereas people who experienced only a single depressive episode did not present this abnormality 84. Several postmortem studies have shown a lower number of proliferating cells in the hippocampus in deceased SCZ patients 87, but technical issues (low rate of adult neurogenesis, as well as low number and quality of samples) have limited the conclusiveness of these findings. To date, no strong evidence has been reported that hippocampal shrinkage is caused by impairments in developmental or adult neurogenesis.
Despite growing interest in adult hippocampal neurogenesis in the context of mental disorders, its causative role is still controversial. Studies on animals are also inconclusive. For example, neogenin‐deficient mice exhibited depressive‐like behavior 88, whereas cyclin D2‐deficient mice did not exhibit any major behavioral deficits 89, even though adult neurogenesis was compromised in both strains. A number of antipsychotic and antidepressant drugs boosted adult neurogenesis in rodents, but many negative results were also reported 90, 91. Last but not least, some controversy exists about the extent to which adult neurogenesis occurs in humans 92. Some studies concluded that in humans neurogenesis in the hippocampus is negligible after infancy, whereas other studies reported evidence that it does not substantially decline with age.
Regulation of hippocampus development by canonical Wnt/β‐catenin signaling and LEF1/TCF
In general, canonical Wnt signaling is implicated in similar processes in the hippocampus as in the neocortex; therefore we focus on differences between the two regions. The developing hippocampus is located within direct proximity to the hem, which secretes Wnt ligands 66, 93. The expression of Lef1 is higher in hippocampal progenitors in both the hippocampal VZ and migratory stream compared with the neocortical VZ 61, 68. Thus, developmental neurogenesis in the hippocampus, particularly in the DG, occurs under conditions of much higher canonical Wnt/β‐catenin sinagling activity than in the neocortex and is mostly regulated by Lef1.
Consistent with high activity of the canonical Wnt/β‐catenin pathway, the inhibition of canonical Wnt signaling at the selective level of signal transduction or LEF1/TCFs had more pronounced effects in the hippocampus than in the neocortex. Wnt3a −/− mice with conditional Ctnnb1 knockout, or mice that express LEF1 without the DNA binding domain (LEF1ΔHMG; likely act as a dominant negative mutant for LEF1/TCFs) the hippocampal VZ was truncated at midgestation, and the hippocampus was virtually absent at the perinatal stage 62, 65, 68, 74. Ectopic expression of Dkk1, which encodes a conanical Wnt signaling inhibitor Dickkopf 1 that binds to LRP5/6, Lrp6 knockout or Lef1 knockout, which only partially inhibited the canonical Wnt pathway, had less severe effects. Nevertheless, the DG, whose progenitor domain is located in direct vicinity to the hem, was smaller 94, 95. Conversely, canonical Wnt gain‐of‐function mutant mice with stabilized β‐catenin had enlarged and poorly organized hippocampi 62, 73, 96. The specific roles of LEF1, TCF7L2, and TCF7L1 in hippocampal developmental neurogenesis have been investigated in gene knockout studies. In Lef1 −/− mice, granule cells were absent in the DG, despite a normal number of proliferating cells in the primary neurogenic niche in the VZ 68, 95. By contrast, CA neurons, which originate from a part of the hippocampal neuroepithelium with lower Lef1 expression, were normally produced in these mice, suggesting a redundant role for LEF1 and other LEF1/TCF family members in this area, similar to the neocortex. Tcf7l1 knockout did not result in any hippocampal alterations, but the size of both the granule and pyramidal cell layers of the hippocampus was markedly reduced in Tcf7l2 knockout mice 61, implying a role for TCF7L2 in the regulation of the expansion of hippocampal progenitor, as well as neocortical progenitors. Altogether, these studies demonstrated that canonical Wnt/β‐catenin signaling, activated by WNT3A and mediated predominantly by LEF1 together with TCF7L2 are indispensable for maintaining proliferating hippocampal progenitors.
Regulation of adult neurogenesis in the hippocampus by canonical Wnt/β‐catenin signaling
Postnatally, the population of hippocampal progenitors becomes restricted to the subgranular zone of the DG and transforms into adult neural stem cells that continue to divide, although at a much lower rate than in embryos. Adult neurogenesis is not simply an extension of a developmental process into the adulthood because the environment in the postnatal brain is not more neurogenic 97, implying that other mechanisms may be involved in the postnatal regulation of granule cell generation. However, in the adult hippocampus, Wnt/β‐catenin signaling is still active, both in the SGZ and DG, which has been demonstrated in Wnt reporter mice 64, 98. The source of Wnt signaling could be hippocampal astrocytes that reside in the SGZ, which expresses Wnt3 98, or cells in the hilus of the DG, which expresses Wnt7a 99. The expression of LEF1/TCF7L2 is undetectable or very low in the adult hippocampus 99, 100. Some transient expression of these factors may be found in the SGZ and DG, but high reporter gene activity in these regions in Wnt reporter mice is difficult to explain. Genetic modifications of both upstream and downstream (activating and inhibitory) elements of the canonical Wnt/β‐catenin pathway, including the ectopic expression of Wnt3 or dominant negative LEF1 (dnLEF1) in the SGZ, confirmed its involvement in adult neurogenesis in the hippocampus 96, 98.
Implications for mental disorders
Growing evidence implicates canonical Wnt signaling‐associated defects in adult hippocampal neurogenesis in pathogenesis of psychiatric conditions. Nonetheless, much of this evidence is indirect and correlative, and, so far, no solid evidence has linked canonical Wnt‐regulated adult neurogenesis and mental disorders. Many studies reported changes in the expression of the upstream and downstream components of canonical Wnt signaling in the hippocampus of animals that were treated with antidepressant and antipsychotic drugs. For example, chronic administration of selective serotonin reuptake inhibitors increased Wnt2, Fz9, Frzb, Lef1, Ctnnb1, and Dvl1 expression in the hippocampus 101. However, the fact that psychiatric drugs affect the canonical Wnt pathway in the hippocampus does not necessarily mean that this connection is crucial for neurogenesis and the treatment of depression.
The connection between adult hippocampal neurogenesis, the canonical Wnt/β‐catenin pathway, and mental disorders has been demonstrated by investigations of DISC1, which can stabilize β‐catenin through direct GSK3β inhibition. In both the developing and adult DG, DISC1 maintains neural progenitor proliferation 102. The expression of stabilized β‐catenin rescued the impairment of RG proliferation caused by Disc1 silencing, and the canonical Wnt agonist SB216763 (an inhibitor of GSK3β) normalized depression‐like behavior in these mice. This indicates that DISC1 acts through the canonical Wnt pathway to regulate neurogenesis and modulate mental homeostasis. However, whether the protective role of β‐catenin depends on the rescue of neurogenesis, and whether downstream Wnt signaling (nuclear of β‐catenin and LEF/TCFs) is involved has not been directly tested.
Postmitotic development of the diencephalon
Thalamus and habenula in mental disorders
The thalamo‐habenular region is a subcortical part of the brain derived from a common developmental progenitor domain. The thalamus relays sensorimotor information to the cortex, process this information in cortico‐thalamic network, and produce goal‐directed behaviors through cortico–basal ganglia–thalamic circuits 103. The habenula controls reward‐ and aversion‐driven behaviors by connecting the prefrontal cortex, limbic system, and basal ganglia with the brainstem monoaminergic system 104, which is a part of the reward system.
Psychiatric disorders have been repeatedly linked with disturbances in thalamic anatomy and connectivity. Decreases in the volume of the thalamus, as well as reductions of functional connectivity between the thalamus and prefrontal region, with an increase in functional connectivity with the motor cortex are commonly detected in SCZ and BD 85, 86, 105, 106. In ASD, this pattern is partially reversed, with thalamic hypoconnectivity to the prefrontal cortex, temporal lobe, and sensory and motor cortices 107. Unfortunately, neuroimaging data from the habenula are scarce and often do not reach statistical significance, because isolating signal from this small region is difficult. Nevertheless, habenular impairments, and mainly impairments in the lateral habenular part, have been associated with behavioral alterations (e.g., depressive‐like behavior and decreased sensory gating) in animal studies, using lesions or deep stimulation of the habenula 108.
TCF7L2, LEF1 and nuclear β‐catenin in the postnatal diencephalon
Abundant expression of Tcf7l2 and Lef1 was specifically observed during postmitotic development and in adulthood in the thalamus, habenula, and some midbrain structures in vertebrates, including primates 48, 100, 109, 110. These TCF7L2‐positive brain regions exhibited massive nuclear β‐catenin accumulation 48, 100, 111, and high reporter gene activity in Wnt reporter mice 111, 112. This accumulation appears to be independent of the upstream components of the Wnt signaling, but dependent of TCF7L2. In thalamic cells, the silencing of Tcf7l2 in thalamic neurons prevented β‐catenin from entering the nucleus 48, whereas inhibition of upstream canonical Wnt signaling had no effect on the localization of β‐catenin 113. The role of canonical Wnt/β‐catenin signaling in postmitotic neurons in the thalamo‐habenular domain is little understood compared with its role in cortical neural progenitors.
TCF7L2 in the postmitotic development and maintenance of thalamo‐habenular regions
TCF7L2 is not required for initial specification and neurogenesis of the thalamo‐habenular region but its role is important at later developmental stages 114, 115. Recent knockout studies in mice showed a dual role of TCF7L2 in regulating postmitotic development of this region during embryogenesis and postnatally (Fig. 4). During embryogenesis, the lack of TCF7L2 resulted in the loss of anatomical boundaries between the thalamus, habenula, and adjacent structures. The nuclear organization and molecular postmitotic identity of thalamic and habenular neurons was also lost. Furthermore, thalamocortical axons and fasciculus retroflexus, which is the main bundle of habenular efferent axons, were missing. These defects were most likely underlined by decreases in the expression of a number cell adhesion genes and axon‐navigating genes, which were observed in Tcf7l2−/− brain, many of them specific for the thalamus and habenula 115. In mice with the postnatal knockout of Tcf7l2 (thalamus‐specific), the expression of these genes was not altered, but the expression of many genes that are induced in this region postnatally was downregulated, among them ion channel and neurotransmitter genes. These results imply that both postmitotic developmental genetic program and postnatal switch in gene expression in the thalamo‐habenular region are controlled by TCF7L2. Further research should address the mechanism of differential gene expression regulation by TCF7L2 in the two developmental stages.
Figure 4.
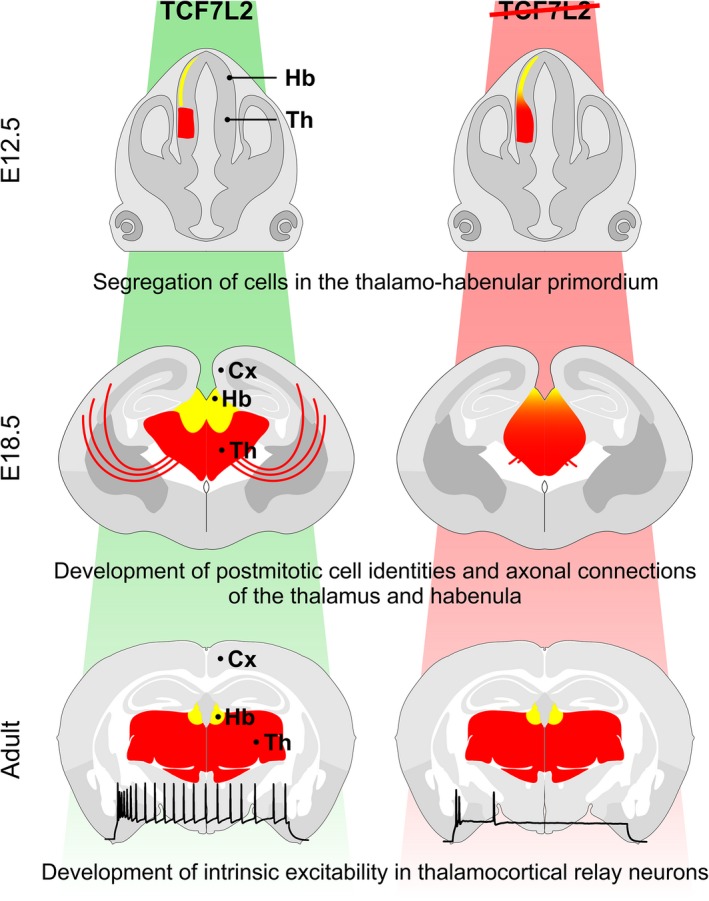
Role of TCF7L2 in the developing thalamus and habenula. Early knockout of Tcf7l2 impairs segregation of cells in the thalamo‐habenular primordium (upper and middle panel), and disrupts axon growth and regional cell identities (middle panel). Conditional postnatal knockout of Tcf7l2 impairs intrinsic excitability of thalamo‐cortical relay neurons (bottom panel).
Implications for mental disorders
Some thalamic dysfunctions might arise from improper neuronal migration and aberrant thalamocortical circuit development, considering that many risk factor genes for psychiatric diseases are involved in this process. For example, numerous genes that are associated with SCZ and ASD encode axon guidance molecules that belong to the families of SLIT and ROBO, Ephrins and EPH receptors, DCC and UNC5, Semaphorins and Plexins, and Neuregulin 116, 117, 118, all of which are involved in axon guidance. Genes from these families were downregulated in the thalamus in TCF7L2‐deficient mice during embryogenesis, including Dcc, Unc5, Robo3, Epha3, Epha8, Slit3, Nrp1, and Ntng1 115. Cell adhesion genes (e.g., Cntn4, Cntn6, and Reln), which potentially regulate cell migration and axon outgrowth, are another group that is affected by Tcf7l2 knockout. All of these genes have been linked with SCZ 119, 120, what shows that TCF7L2 might play a role in the etiology of mental disorders.
Ion channels and neurotransmitter genes, some of which were dysregulated in mice with postnatal Tcf7l2 knockout, are also often associated with psychiatric disorders. Among ion channels whose expression is altered in the adult TCF7L2‐deficent thalamus is CACNA1C, which encodes a Ca2+ channel. Mutations of these genes are associated with SCZ disorder with the third highest score in the largest genome wide association study to date 121. Mutations of KCNIP4 are associated with personality disorders and ASD 122. CACNA1G was one of the most downregulated genes in neurons derived from BD patients 81. Expression of the serotonin transporter gene SLC6A4 has been linked with ASD 123, 124, and was also altered in the adult TCF7L2‐deficent thalamus. Functional connectivity impairments in the TCF7L2‐deficent adult thalamus may also be caused by improper synapse formation within target structures of thalamocortical axons. For example, these mice abnormally express the brain‐derived neurotrophic factor (BDNF) gene, which is also associated with SCZ through its involvement in synapse regulation and plasticity 125.
These multiple genetic overlaps between embryonic and adult TCF7L2‐target genes in the thalamus and psychiatric conditions imply TCF7L2‐mediated thalamic alterations in the pathogenesis of mental disorders. This hypothesis has not been yet directly addressed. Future research should explain the relationship between risk variants of TCF7L2 and endophenotypes of the thalamus, and more thoroughly investigate the role of this transcription factor in behavior regulation in animal models.
Developmental oligodendrogenesis in the cortex
Oligodendrogenesis and myelination in the cortex and mental disorders
Oligodendrocytes are myelinating cells in the vertebrate central nervous system. Their role is to provide trophic support and electric insulation to axons to increase the velocity of nerve signal conduction and maintain axonal integrity. The loss of oligodendroglia and cortical demyelination, followed by the demage of axons are the primary pathology in multiple sclerosis and other leukodystrophies. Slowly accumulating evidence implicates disturbances in oligodendrogenesis and impairments in myelination also in the development of mental disorders. However, when compared to neurogenesis, the possible involvement of oligodendrogenesis in the etiology of psychiatric illnesses has been little investigated. Most of this research has focused on SCZ, but some evidence suggests similar endophenotypes in BP. The levels of oligodendrocyte‐ and myelin‐related transcripts were altered in cortical samples from SCZ 126 and BP 127 patients in postmortem studies. This could simply reflect hypomyelination, which is often observed in SCZ patients 128. Nonetheless, a decrease in the density of oligodendrocytes despite a normal number of oligodenrocyte precursor cells (OPCs) in the prefrontal cortex in SCZ patients 129 suggests that defects in the differentiation of oligodendrocyte lineage cells might be one cause of hypomyelination. This was further corroborated by the lower production of OPCs from iPSCs that were derived from SCZ patient fibroblasts 130. These postmortem and iPSCs studies included a small number of samples; therefore, the statistical power of these findings is low. However, several large‐cohort genomic studies that associated markers of oligodendrocytes (OLIG2 and SOX10) and genes that encode myelin proteins (MBP, PLP1, MOG, MOBP, CNP, and MAG) with SCZ 128, 131 suggested that the differentiation of oligodendroglia might be one of primary impairments in the etiology of this disorder.
Regulation of cortical oligodendrogenesis by Wnt/β‐catenin signaling and TCF7L2
Oligodendroglia is produced from RG in the VZ/SVZ of the spinal cord and several niches in the brain during embryonic development and postnatally, after neurogenesis is completed. In rodents, most of oligodendrocytes in the adult cortex are derived from the cortical VZ/SVZ of the dorsal telencephalon. OPCs are produced in this niche postnatally and migrate radially into the corpus callosum (a nerve tract beneath the cortex) and cortex. The generation of functional oligodendrocytes occurs through several intermediate stages. Proliferating OPCs generate immature premyelinating oligodendrocytes (iOLs), which finally maturate into myelin‐producing oligodendrocytes (mOLs) (Fig. 3) 132.
Oligodendrocyte precursor cells begin to be produced in the cortical VZ/SVZ when the level of canonical Wnt/β‐catenin signaling activity, which is high during neurogenesis, decreases 133. This downregulation is critical for the switch between neurogenesis and oligodendrogenesis, demonstrated by the loss of OPCs and mOLs after the ectopic expression of stabilized β‐catenin in early oligodendroglia progenitors 134, 135, and the precocious generation of OPCs after the early ablation of Ctnnb1 in oligodendroglia lineage cells 135. This was further supported by an in utero injection of Dkk1 or a construct that encoded dnLEF1 to downregulate Wnt/β‐catenin signaling, both of which significantly increased the number of OPCs perinatally in the cortex 133. These data demonstrated that the activity of canonical Wnt/β‐catenin signaling prevents premature oligodendrogenesis from RG, similar to the inhibition of premature astrogenesis.
At subsequent stages of oligodendrogenesis that follow the generation of OPCs, the activity of canonical Wnt/β‐catenin signaling was detected in subsets of differentiating and maturating cells 136, 137. Among LEF1/TCFs, only TCF7L2 is present in oligodendroglia lineage cells at higher levels 134, 138. In a single‐cell RNA‐seq study, Tcf7l2 transcripts were first observed in OPCs, increased to the highest levels in iOLs, and then decreased to lower levels in mOLs 138. Its expression subsequently fades, but reappears during remyelination in animal models of myelin damage both in the spinal cord and corpus callosum 136, 137, 139. However, the pattern of TCF7L2 protein expression and Wnt reporter gene activity little overlapped 136, 137. Studies on genetically modified mice with impaired β‐catenin and TCF7L2 functions in oligodendroglia suggested opposite roles of Wnt/β‐catenin and TCF7L2. In both oligodendroglia‐specific TCF7L2‐deficient mice (with Tcf7l2 knockout or TCF7L2ΔHMG mutation) and in mice with the ectopic expression of stabilized (β‐cateninΔexon3) in maturing oligodendroglia, myelin did not form properly in the corpus callosum or spinal cord during development and in demyelination lesions 136, 137, 139, 140, 141.
These puzzling findings were resolved by the genome‐wide mapping of TCF7L2 chromatin occupancy at different stages of oligodendrocyte differentiation in vitro 139. This analysis identified two factors whose binding regions overlapped with sites that were occupied by TCF7L2 in two developmental stages: Kaiso (a co‐repressor of transcription, whose expression is high in OPCs) and SOX10. Further investigations revealed that Kaiso, together with histone deacetylases HDAC1/2 (which form a repressive complex with Kaiso), blocks the potential interaction between TCF7L2 and β‐catenin during the transition of OPCs into iOLs, resulting in inhibition of the expression of canonical Wnt/β‐catenin target genes 134, 139, 142. Subsequently, during oligodendrocyte maturation, TCF7L2 interacts with SOX10 at regulatory elements of myelination‐associated genes and activates their expression in a Wnt‐independent manner 139. Thus, TCF7L2 plays a dual role in oligodendrogenesis by inhibiting the canonical Wnt/β‐catenin pathway during OPC differentiation and Wnt‐independently regulating myelin genes in oligodendrocytes.
In summary, after the generation of OPCs, further steps of oligodendrogenesis are sequentially regulated by TCF7L2 (Fig. 3). Wnt/β‐catenin signaling must be inhibited by TCF7L2, together with Kaiso and HDAC1/2, to enable the further differentiation of OPCs, followed by the Wnt‐independent interaction between TCF7L2 and SOX10 to induce myelination.
Possible direct links between mental disorders, Wnt, TCF7L2, and oligodendrogenesis have not been investigated. The only indication that such links exist comes from the aforementioned genetic studies that associated mental disorders with SOX10 and myelin‐associated genes 128, which are directly regulated by TCF7L2 and SOX10.
Conclusions
Changes in the volume of cortical areas, thalamo‐cortical dysconnectivity, and white matter microstructural alterations are commonly identified endophenotypes in psychiatric research. Evidence from animal studies strongly implicates canonical Wnt/β‐catenin signaling and TCF7L2‐dependent transcription in the development of these impairments, and human genetic studies have found associations between this pathway and major mental disorders. Variants of TCF7L2 are among the strongest risk factors for diabetes, and TCF7L2 has been extensively studied in the context of peripheral metabolism regulation, but studies on its role in brain development and pathologies have been relatively scarce. Future research on animals with conditional mutations of Tcf7l2 may help confirm putative links between TCF7L2, brain phenotypes, and behavioral deficits. Identifying relationships between molecular and cellular events, structural and functional alterations in the brain, and behavioral deficits might contribute to the development of personalized treatments for mental disorders.
Acknowledgements
The authors were supported by the Polish National Science Centre: 2015/19/B/NZ3/02949 and 2017/25/B/NZ3/01665 to MBW, 2015/19/B/NZ4/03571 to AN, and 2017/24/C/NZ3/00447 to ŁMS.
Edited by Maria Papatriantafyllou
All authors contributed equally to this article
The copyright line for this article was changed on 4 September 2019 after original online publication.
References
- 1. Gandal MJ, Haney JR, Parikshak NN, Leppa V, Ramaswami G, Hartl C, Schork AJ, Appadurai V, Buil A, Werge TM et al (2018) Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 359, 693–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anttila V, Bulik‐Sullivan B, Finucane HK, Walters RK, Bras J, Duncan L, Escott‐Price V, Falcone GJ, Gormley P, Malik R et al (2018) Analysis of shared heritability in common disorders of the brain. Science 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Goodkind M, Eickhoff SB, Oathes DJ, Ying J, Andrew C, Jones‐Hagata LB, Ortega BN, Zaiko YV, Roach EL, Korgaonkar MS et al (2015) Identification of a common neurobiological substrate for mental illness. JAMA Psychiatry 72, 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sprooten E, Rasgon A, Goodman M, Carlin A, Leibu E, Lee WH and Frangou S (2017) Addressing reverse inference in psychiatric neuroimaging: meta‐analyses of task‐related brain activation in common mental disorders. Hum Brain Mapp 38, 1846–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yager J and Feinstein RE (2017) Potential applications of the National Institute of Mental Health's Research Domain Criteria (RDoC) to clinical psychiatric practice: how RDoC might be used in assessment, diagnostic processes, case formulation, treatment planning, and clinical notes. J Clin Psychiatry 78, 423–432. [DOI] [PubMed] [Google Scholar]
- 6. Stambolic V, Ruel L and Woodgett JR (1996) Lithium inhibits glycogen synthase kinase‐3 activity and mimics wingless signalling in intact cells. Curr Biol 6, 1664–1668. [DOI] [PubMed] [Google Scholar]
- 7. Wiese KE, Nusse R and van Amerongen R (2018) Wnt signalling: conquering complexity. Development 145, dev165902. [DOI] [PubMed] [Google Scholar]
- 8. Mulligan KA and Cheyette BN (2017) Neurodevelopmental perspectives on Wnt signaling in psychiatry. Mol Neuropsychiatry 2, 219–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oliva CA, Montecinos‐Oliva C and Inestrosa NC (2018) Wnt signaling in the central nervous system: new insights in health and disease. Prog Mol Biol Transl Sci 153, 81–130. [DOI] [PubMed] [Google Scholar]
- 10. Stanganello S, Zahavi EE, Burute M, Smits J, Jordens I, Maurice MM, Kapitein LC and Hoogenraad CC (2019) Wnt signaling directs neuronal polarity and axonal growth. iScience 13, 318–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. He CW, Liao CP and Pan CL (2018) Wnt signalling in the development of axon, dendrites and synapses. Open Biol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dickins EM and Salinas PC (2013) Wnts in action: from synapse formation to synaptic maintenance. Front Cell Neurosci 7, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Archbold HC, Yang YX, Chen L and Cadigan KM (2012) How do they do Wnt they do?: regulation of transcription by the Wnt/β‐catenin pathway. Acta Physiol 204, 74–109. [DOI] [PubMed] [Google Scholar]
- 14. Martin AR, Daly MJ, Robinson EB, Hyman SE, Neale BM (2018) Predicting polygenic risk of psychiatric disorders. Biol Psychiatry, 10.1016/j.biopsych.2018.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zandi PP, Belmonte PL, Willour VL, Goes FS, Badner JA, Simpson SG, Gershon ES, McMahon FJ, DePaulo JR Jr, Potash JB et al (2008) Association study of Wnt signaling pathway genes in bipolar disorder. Arch Gen Psychiatry 65, 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Levchenko A, Davtian S, Freylichman O, Zagrivnaya M, Kostareva A and Malashichev Y (2015) Beta‐catenin in schizophrenia: possibly deleterious novel mutation. Psychiatry Res 228, 843–848. [DOI] [PubMed] [Google Scholar]
- 17. Wassink TH, Joseph P, Vieland VJ, Jian H, Swiderski RE, Jennifer P, Terry B, Gretel B, Folstein SE and Haines JL (2001) Evidence supporting WNT2 as an autism susceptibility gene. Am J Med Genet 105, 406–413. [DOI] [PubMed] [Google Scholar]
- 18. Martin PM, Yang X, Robin N, Lam E, Rabinowitz JS, Erdman CA, Quinn J, Weiss LA, Hamilton SP, Kwok P‐y et al (2013) A rare WNT1 missense variant overrepresented in ASD leads to increased Wnt signal pathway activation. Transl Psychiatry 3, e301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Turner TN, Hormozdiari F, Duyzend Michael H, McClymont Sarah A, Hook Paul W, Iossifov I, Raja A, Baker C, Hoekzema K, Stessman Holly A et al (2016) Genome sequencing of autism‐affected families reveals disruption of putative noncoding regulatory DNA. Am J Hum Genet 98, 58–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grünblatt E, Nemoda Z, Werling AM, Roth A, Angyal N, Tarnok Z, Thomsen H, Peters T, Hinney A, Hebebrand J et al (2018) The involvement of the canonical Wnt‐signaling receptor LRP5 and LRP6 gene variants with ADHD and sexual dimorphism: association study and meta‐analysis. Am J Med Genet B Neuropsychiatr Genet, 10.1002/ajmg.b.32695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao H and Nyholt DR (2017) Gene‐based analyses reveal novel genetic overlap and allelic heterogeneity across five major psychiatric disorders. Hum Genet 136, 263–274. [DOI] [PubMed] [Google Scholar]
- 22. Guo X, Yang J, Huang J, Chen Z, Wu X, Zhu L, Huang G, Long J and Su L (2018) Influence of CTNNB1 rs2953 polymorphism on schizophrenia susceptibility in Chinese Han population through modifying miR‐485 binding to CTNNB1. Genes Brain Behav 18, e12524. [DOI] [PubMed] [Google Scholar]
- 23. O'Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD et al (2012) Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alkelai A, Greenbaum L, Lupoli S, Kohn Y, Sarner‐Kanyas K, Ben‐Asher E, Lancet D, Macciardi F and Lerer B (2012) Association of the type 2 diabetes mellitus susceptibility gene, TCF7L2, with schizophrenia in an Arab‐Israeli family sample. PLoS ONE 7, e29228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hansen T, Ingason A, Djurovic S, Melle I, Fenger M, Gustafsson O, Jakobsen KD, Rasmussen HB, Tosato S, Rietschel M et al (2011) At‐risk variant in TCF7L2 for type II diabetes increases risk of schizophrenia. Biol Psychiatry 70, 59–63. [DOI] [PubMed] [Google Scholar]
- 26. Liu L, Li J, Yan M, Li J, Chen J, Zhang Y, Zhu X, Wang L, Kang L and Yuan D(2017) TCF7L2 polymorphisms and the risk of schizophrenia in the Chinese Han population. Oncotarget 8, 28614–28620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Need AC, Ge D, Weale ME, Maia J, Feng S, Heinzen EL, Shianna KV, Yoon W, Kasperaviciūte D, Gennarelli M et al (2009) A genome‐wide investigation of SNPs and NVs in schizophrenia. PLoS Genet 5, e1000373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ben‐David E, Shifman S and International Schizophrenia Consortium (2010) Further investigation of the association between rs7341475 and rs17746501 and schizophrenia. Am J Med Genet B Neuropsychiatr Genet 153B, 1244–1247. [DOI] [PubMed] [Google Scholar]
- 29. Shifman S, Johannesson M, Bronstein M, Chen SX, Collier DA, Craddock NJ, Kendler KS, Li T, O'Donovan M, O'Neill FA et al (2008) Genome‐wide association identifies a common variant in the reelin gene that increases the risk of schizophrenia only in women. PLoS Genet 4, e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cuellar‐Barboza AB, Winham SJ, McElroy SL, Geske JR, Jenkins GD, Colby CL, Prieto ML, Ryu E, Cunningham JM, Frye MA et al (2016) Accumulating evidence for a role of TCF7L2 variants in bipolar disorder with elevated body mass index. Bipolar Disord 18, 124–135. [DOI] [PubMed] [Google Scholar]
- 31. Nagalski A, Kozinski K and Wisniewska MB (2016) Metabolic pathways in the periphery and brain: contribution to mental disorders? Int J Biochem Cell Biol 80, 19–30. [DOI] [PubMed] [Google Scholar]
- 32. Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE et al (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu Z, Zhang N, Zhang Y, Du Y, Zhang T, Li Z, Wu J and Wang X (2018) Prioritized high‐confidence risk genes for intellectual disability reveal molecular convergence during brain development. Front Genet 9, 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Deciphering Developmental Disorders Study (2017) Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Deciphering Developmental Disorders Study (2015) Large‐scale discovery of novel genetic causes of developmental disorders. Nature 519, 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. O'Brien WT, Harper AD, Jové F, Woodgett JR, Maretto S, Piccolo S and Klein PS (2004) Glycogen synthase kinase‐3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J Neurosci 24, 6791–6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wilkinson MB, Dias C, Magida J, Mazei‐Robison M, Lobo M, Kennedy P, Dietz D, Covington H 3rd, Russo S, Neve R et al (2011) A novel role of the WNT‐dishevelled‐GSK3β signaling cascade in the mouse nucleus accumbens in a social defeat model of depression. J Neurosci 31, 9084–9092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gould TD, Einat H, Bhat R and Manji H (2004) AR‐A014418, a selective GSK‐3 inhibitor, produces antidepressant‐like effects in the forced swim test. Int J Neuropsychopharmacol 7, 387–390. [DOI] [PubMed] [Google Scholar]
- 39. Kaidanovich‐Beilin O, Milman A, Weizman A, Pick CG and Eldar‐Finkelman H (2004) Rapid antidepressive‐like activity of specific glycogen synthase kinase‐3 inhibitor and its effect on beta‐catenin in mouse hippocampus. Biol Psychiatry 55, 781–784. [DOI] [PubMed] [Google Scholar]
- 40. Urs NM, Snyder JC, Jacobsen JP, Peterson SM and Caron MG (2012) Deletion of GSK3β in D2R‐expressing neurons reveals distinct roles for β‐arrestin signaling in antipsychotic and lithium action. Proc Natl Acad Sci USA 109, 20732–20737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Polter A, Beurel E, Yang S, Garner R, Song L, Miller CA, Sweatt DJ, McMahon L, Bartolucci AA, Li X et al (2010) Deficiency in the inhibitory serine‐phosphorylation of glycogen synthase kinase‐3 increases sensitivity to mood disturbances. Neuropsychopharmacology 35, 1761–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Prickaerts J, Moechars D, Cryns K, Lenaerts I, van Craenendonck H, Goris I, Daneels G, Bouwknecht JA and Steckler T (2006) Transgenic mice overexpressing glycogen synthase kinase 3beta: a putative model of hyperactivity and mania. J Neurosci 26, 9022–9029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lijam N, Paylor R, McDonald MP, Crawley JN, Deng CX, Herrup K, Stevens KE, Maccaferri G, McBain CJ, Sussman DJ et al (1997) Social interaction and sensorimotor gating abnormalities in mice lacking Dvl1. Cell 90, 895–905. [DOI] [PubMed] [Google Scholar]
- 44. Ahnaou A and Drinkenburg WH (2011) Disruption of glycogen synthase kinase‐3‐beta activity leads to abnormalities in physiological measures in mice. Behav Brain Res 221, 246–252. [DOI] [PubMed] [Google Scholar]
- 45. Belinson H, Nakatani J, Babineau BA, Birnbaum RY, Ellegood J, Bershteyn M, McEvilly RJ, Long JM, Willert K, Klein OD et al (2016) Prenatal β‐catenin/Brn2/Tbr2 transcriptional cascade regulates adult social and stereotypic behaviors. Mol Psychiatry 21, 1417–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gould TD, Einat H, O'Donnell KC, Picchini AM, Schloesser RJ and Manji HK (2007) Beta‐catenin overexpression in the mouse brain phenocopies lithium‐sensitive behaviors. Neuropsychopharmacology 32, 2173–2183. [DOI] [PubMed] [Google Scholar]
- 47. Dias C, Feng J, Sun H, Shao N, Mazei-Robison MS, Damez-Werno D, Scobie K, Bagot R, LaBonté B, Ribeiro E et al (2014) β-catenin mediates stress resilience through Dicer1/microRNA regulation. Nature 516, 51–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Misztal K, Brozko N, Nagalski A, Szewczyk LM, Krolak M, Brzozowska K, Kuznicki Jand Wisniewska MB (2017) TCF7L2 mediates the cellular and behavioral response to chronic lithium treatment in animal models. Neuropharmacology 113 , 490–501. [DOI] [PubMed] [Google Scholar]
- 49. Savic D, Distler MG, Sokoloff G, Shanahan NA, Dulawa SC, Palmer AA and Nobrega MA(2011) Modulation ofTcf7l2 expression alters behavior in mice. PLoS ONE 6, e26897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sittig Laura J, Carbonetto P, Engel KA, Krauss KS, Barrios‐Camacho CM and Palmer AA (2016) Genetic background limits generalizability of genotype‐phenotype relationships. Neuron 91, 1253–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xie Y, Kaufmann D, Moulton MJ, Panahi S, Gaynes JA, Watters HN, Zhou D, Xue HH, Fung CM, Levine EM et al (2017) Lef1‐dependent hypothalamic neurogenesis inhibits anxiety. PLoS Biol 15, e2002257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schmaal L, Hibar DP, Sämann PG, Hall GB, Baune BT, Jahanshad N, Cheung JW, van Erp TGM, Bos D, Ikram MA et al (2017) Cortical abnormalities in adults and adolescents with major depression based on brain scans from 20 cohorts worldwide in the ENIGMA Major Depressive Disorder Working Group. Mol Psychiatry 22, 900–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hibar DP, Westlye LT, Doan NT, Jahanshad N, Cheung JW, Ching CRK, Versace A, Bilderbeck AC, Uhlmann A, Mwangi B et al (2018) Cortical abnormalities in bipolar disorder: an MRI analysis of 6503 individuals from the ENIGMA Bipolar Disorder Working Group. Mol Psychiatry 23, 932–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. van Erp Theo GM, Walton E, Hibar Derrek P, Schmaal L, Jiang W, Glahn DC, Pearlson GD, Yao N, Fukunaga M, Hashimoto R et al (2018) Cortical Brain Abnormalities in 4474 individuals with schizophrenia and 5098 control subjects via the Enhancing Neuro Imaging Genetics Through Meta Analysis (ENIGMA) Consortium. Biol Psychiatry 84, 644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ambrosino S, de Zeeuw P, Wierenga LM, van Dijk S and Durston S (2017) What can cortical development in attention‐deficit/hyperactivity disorder teach us about the early developmental mechanisms involved? Cereb Cortex 27, 4624–4634. [DOI] [PubMed] [Google Scholar]
- 56. van Rooij D, Anagnostou E, Arango C, Auzias G, Behrmann M, Busatto GF, Calderoni S, Daly E, Deruelle C, Di Martino A et al (2018) Cortical and subcortical brain morphometry differences between patients with autism spectrum disorder and healthy individuals across the lifespan: results from the ENIGMA ASD Working Group. Am J Psychiatry 175, 359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Beattie R and Hippenmeyer S (2017) Mechanisms of radial glia progenitor cell lineage progression. FEBS Lett 591, 3993–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Arai Y and Taverna E (2017) Neural progenitor cell polarity and cortical development. Front Cell Neurosci 11, 384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lui JH, Hansen DV and Kriegstein AR (2011) Development and evolution of the human neocortex. Cell 146, 18–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Grove EA, Tole S, Limon J, Yip L and Ragsdale CW (1998) The hem of the embryonic cerebral cortex is defined by the expression of multiple Wnt genes and is compromised in Gli3‐deficient mice. Development 125, 2315–2325. [DOI] [PubMed] [Google Scholar]
- 61. Chodelkova O, Masek J, Korinek V, Kozmik Z and Machon O (2018) Tcf7L2 is essential for neurogenesis in the developing mouse neocortex. Neural Dev 13, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Machon O, van den Bout CJ, Backman M, Kemler R and Krauss S (2003) Role of beta‐catenin in the developing cortical and hippocampal neuroepithelium. Neuroscience 122, 129–143. [DOI] [PubMed] [Google Scholar]
- 63. Woodhead GJ, Mutch CA, Olson EC and Chenn A (2006) Cell‐autonomous beta‐catenin signaling regulates cortical precursor proliferation. J Neurosci 26, 12620–12630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Garbe DS and Ring RH (2012) Investigating tonic Wnt signaling throughout the adult CNS and in the hippocampal neurogenic niche of BatGal and ins‐TopGal mice. Cell Mol Neurobiol 32, 1159–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lee SM, Tole S, Grove E and McMahon A (2000) A local Wnt‐3a signal is required for development of the mammalian hippocampus. Development 127, 457–467. [DOI] [PubMed] [Google Scholar]
- 66. Subramanian L, Remedios R, Shetty A and Tole S (2009) Signals from the edges: the cortical hem and antihem in telencephalic development. Semin Cell Dev Biol 20, 712–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Caviness VS, Nowakowski RS and Bhide PG (2009) Neocortical neurogenesis: morphogenetic gradients and beyond. Trends Neurosci 32, 443–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Galceran J, Miyashita‐Lin EM, Devaney E, Rubenstein JL and Grosschedl R (2000) Hippocampus development and generation of dentate gyrus granule cells is regulated by LEF1. Development 127, 469–482. [DOI] [PubMed] [Google Scholar]
- 69. Mutch CA, Funatsu N, Monuki ES and Chenn A (2009) Beta‐catenin signaling levels in progenitors influence the laminar cell fates of projection neurons. J Neurosci 29, 13710–13719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Machon O, Backman M, Machonova O, Kozmik Z, Vacik T, Andersen L and Krauss S (2007) A dynamic gradient of Wnt signaling controls initiation of neurogenesis in the mammalian cortex and cellular specification in the hippocampus. Dev Biol 311, 223–237. [DOI] [PubMed] [Google Scholar]
- 71. Chenn A and Walsh CA (2002) Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 297, 365–369. [DOI] [PubMed] [Google Scholar]
- 72. Wrobel CN, Mutch CA, Swaminathan S, Taketo MM and Chenn A (2007) Persistent expression of stabilized beta‐catenin delays maturation of radial glial cells into intermediate progenitors. Dev Biol 309, 285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chenn A and Walsh CA (2003) Increased neuronal production, enlarged forebrains and cytoarchitectural distortions in beta‐catenin overexpressing transgenic mice. Cereb Cortex 13, 599–606. [DOI] [PubMed] [Google Scholar]
- 74. Draganova K, Zemke M, Zurkirchen L, Valenta T, Cantù C, Okoniewski M, Schmid MT, Hoffmans R, Götz M, Basler K et al (2015) Wnt/β‐catenin signaling regulates sequential fate decisions of murine cortical precursor cells. Stem Cells 33, 170–182. [DOI] [PubMed] [Google Scholar]
- 75. Munji RN, Choe Y, Li G, Siegenthaler JA and Pleasure SJ (2011) Wnt signaling regulates neuronal differentiation of cortical intermediate progenitors. J Neurosci 31, 1676–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Miyamoto Y, Sakane F and Hashimoto K (2015) N‐cadherin‐based adherens junction regulates the maintenance, proliferation, and differentiation of neural progenitor cells during development. Cell Adh Migr 9, 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhou C‐J, Borello U, Rubenstein JLR and Pleasure SJ (2006) Neuronal production and precursor proliferation defects in the neocortex of mice with loss of function in the canonical Wnt signaling pathway. Neuroscience 142, 1119–1131. [DOI] [PubMed] [Google Scholar]
- 78. Bocchi R, Egervari K, Carol‐Perdiguer L, Viale B, Quairiaux C, De Roo M, Boitard M, Oskouie S, Salmon P and Kiss JZ (2017) Perturbed Wnt signaling leads to neuronal migration delay, altered interhemispheric connections and impaired social behavior. Nat Commun 8, 1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wisniewska MB, Nagalski A, Dabrowski M, Misztal K and Kuznicki J (2012) Novel β‐catenin target genes identified in thalamic neurons encode modulators of neuronal excitability. BMC Genom 13, 635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Durak O, Gao F, Kaeser‐Woo TJ, Rueda R, Martorell AJ, Nott A, Liu CY, Watson WA and Tsai LH (2016) Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat Neurosci 19, 1477–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Madison JM, Zhou F, Nigam A, Hussain A, Barker DD, Nehme R, van der Ven K, Hsu J, Wolf P, Fleishman M et al (2015) Characterization of bipolar disorder patient‐specific induced pluripotent stem cells from a family reveals neurodevelopmental and mRNA expression abnormalities. Mol Psychiatry 20, 703–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Srikanth P, Lagomarsino VN, Muratore CR, Ryu SC, He A, Taylor WM, Zhou C, Arellano M and Young‐Pearse TL (2018) Shared effects of DISC1 disruption and elevated WNT signaling in human cerebral organoids. Transl Psychiatry 8, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hoogman M, Bralten J, Hibar DP, Mennes M, Zwiers MP, Schweren LSJ, van Hulzen KJE, Medland SE, Shumskaya E et al (2017) Subcortical brain volume differences in participants with attention deficit hyperactivity disorder in children and adults: a cross‐sectional mega‐analysis. Lancet Psychiatry 4, 310–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Schmaal L, Veltman DJ, van Erp TGM, Sämann PG, Frodl T, Jahanshad N, Loehrer E, Tiemeier H, Hofman A, Niessen WJ et al (2016) Subcortical brain alterations in major depressive disorder: findings from the ENIGMA Major Depressive Disorder working group. Mol Psychiatry 21, 806–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. van Erp TGM, Hibar DP, Rasmussen JM, Glahn DC, Pearlson GD, Andreassen OA, Agartz I, Westlye LT, Haukvik UK, Dale AM et al (2016) Subcortical brain volume abnormalities in 2028 individuals with schizophrenia and 2540 healthy controls via the ENIGMA consortium. Mol Psychiatry 21, 547–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hibar DP, Westlye LT, van Erp TGM, Rasmussen J, Leonardo CD, Faskowitz J, Haukvik UK, Hartberg CB et al (2016) Subcortical volumetric abnormalities in bipolar disorder. Mol Psychiatry 21, 1710–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Allen KM, Fung SJ and Weickert CS (2016) Cell proliferation is reduced in the hippocampus in schizophrenia. Aust N Z J Psychiatry 50, 473–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sun D, Sun XD, Zhao L, Lee DH, Hu JX, Tang FL, Pan JX, Mei L, Zhu XJ and Xiong WC (2018) Neogenin, a regulator of adult hippocampal neurogenesis, prevents depressive‐like behavior. Cell Death Dis 9, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Filipkowski RK and Kaczmarek L (2018) Severely impaired adult brain neurogenesis in cyclin D2 knock‐out mice produces very limited phenotypic changes. Prog Neuropsychopharmacol Biol Psychiatry 80 , 63–67. [DOI] [PubMed] [Google Scholar]
- 90. Chikama K, Yamada H, Tsukamoto T, Kajitani K, Nakabeppu Y and Uchimura N (2017) Chronic atypical antipsychotics, but not haloperidol, increase neurogenesis in the hippocampus of adult mouse. Brain Res 1676, 77–82. [DOI] [PubMed] [Google Scholar]
- 91. Miller BR and Hen R (2015) The current state of the neurogenic theory of depression and anxiety. Curr Opin Neurobiol 30, 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Baptista P and Andrade JP (2018) Adult hippocampal neurogenesis: regulation and possible functional and clinical correlates. Front Neuroanat 12, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Hsu LC‐L, Nam S, Cui Y, Chang C‐P, Wang C‐F, Kuo H‐C, Touboul JD, Chou S‐J et al (2015) Lhx2 regulates the timing of β‐catenin‐dependent cortical neurogenesis. Proc Natl Acad Sci USA 112, 12199–12204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Solberg N, Machon O and Krauss S (2008) Effect of canonical Wnt inhibition in the neurogenic cortex, hippocampus, and premigratory dentate gyrus progenitor pool. Dev Dyn 237, 1799–1811. [DOI] [PubMed] [Google Scholar]
- 95. Zhou CJ, Zhao C and Pleasure SJ (2004) Wnt signaling mutants have decreased dentate granule cell production and radial glial scaffolding abnormalities. J Neurosci 24, 121–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Karalay O, Doberauer K, Vadodaria KC, Knobloch M, Berti L, Miquelajauregui A, Schwark M, Jagasia R, Taketo MM, Tarabykin V et al (2011) Prospero‐related homeobox 1 gene (Prox1) is regulated by canonical Wnt signaling and has a stage‐specific role in adult hippocampal neurogenesis. Proc Natl Acad Sci USA 108, 5807–5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Götz M, Nakafuku M and Petrik D (2016) Neurogenesis in the Developing and Adult Brain‐Similarities and Key Differences. Cold Spring Harb Perspect Biol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Lie DC, Colamarino SA, Song HJ, Désiré L, Mira H, Consiglio A, Lein ES, Jessberger S, Lansford H, Dearie AR et al (2005) Wnt signalling regulates adult hippocampal neurogenesis. Nature 437, 1370–1375. [DOI] [PubMed] [Google Scholar]
- 99. Shimogori T, VanSant J, Paik E and Grove EA (2004) Members of the Wnt, Fz, and Frp gene families expressed in postnatal mouse cerebral cortex. J Comp Neurol 473, 496–510. [DOI] [PubMed] [Google Scholar]
- 100. Nagalski A, Irimia M, Szewczyk L, Ferran JL, Misztal K, Kuznicki J and Wisniewska MB (2013) Postnatal isoform switch and protein localization of LEF1 and TCF7L2 transcription factors in cortical, thalamic, and mesencephalic regions of the adult mouse brain. Brain Struct Funct 218, 1531–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Okamoto H, Voleti B, Banasr M, Sarhan M, Duric V, Girgenti MJ, DiLeone RJ, Newton SS and Duman RS (2010) Wnt2 expression and signaling is increased by different classes of antidepressant treatments. Biol Psychiatry 68, 521–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK, Tassa C, Berry EM, Soda T, Singh KK et al (2009) Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta‐catenin signaling. Cell 136, 1017–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Wolff M and Vann SD (2019) The cognitive thalamus as a gateway to mental representations. J Neurosci 39, 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Fakhoury M (2017) The habenula in psychiatric disorders: more than three decades of translational investigation. Neurosci Biobehav Rev 83, 721–735. [DOI] [PubMed] [Google Scholar]
- 105. Boedhoe PSW, Heymans MW, Schmaal L, Abe Y, Alonso P, Ameis SH, Anticevic A, Arnold PD, Batistuzzo MC and Benedetti F (2018) An empirical comparison of meta‐ and mega‐analysis with data From the ENIGMA obsessive‐compulsive Disorder Working Group. Front Neuroinform 12, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Woodward ND and Heckers S (2016) Mapping thalamocortical functional connectivity in chronic and early stages of psychotic disorders. Biol Psychiatry 79, 1016–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Woodward ND, Giraldo‐Chica M, Rogers B and Carissa JC (2017) Thalamocortical dysconnectivity in autism spectrum disorder: an analysis of the autism brain imaging data exchange. Biol Psychiatry Cogn Neurosci Neuroimaging 2, 76–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Shabel SJ, Wang C, Monk B, Aronson S and Malinow R (2019) Stress transforms lateral habenula reward responses into punishment signals. Proc Natl Acad Sci USA 116, 12488–12493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Shimizu N, Kawakami K and Ishitani T (2012) Visualization and exploration of Tcf/Lef function using a highly responsive Wnt/β‐catenin signaling‐reporter transgenic zebrafish. Dev Biol 370, 71–85. [DOI] [PubMed] [Google Scholar]
- 110. Murray KD, Choudary PV and Jones EG (2007) Nucleus‐ and cell‐specific gene expression in monkey thalamus. Proc Natl Acad Sci USA 104, 1989–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Pratt T, Davey JW, Nowakowski TJ, Raasumaa C, Rawlik K, McBride D, Clinton M, Mason JO and Priceet DJ (2012) The expression and activity of β‐catenin in the thalamus and its projections to the cerebral cortex in the mouse embryo. BMC Neurosci 13, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Maretto S, Cordenonsi M, Dupont S, Braghetta P, Broccoli V, Hassan AB, Volpin D, Bressan GM and Piccolo S (2003) Mapping Wnt/beta‐catenin signaling during mouse development and in colorectal tumors. Proc Natl Acad Sci USA 100, 3299–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Misztal K, Wisniewska MB, Ambrozkiewicz M, Nagalski A and Kuznicki J (2011) WNT protein‐independent constitutive nuclear localization of beta‐catenin protein and its low degradation rate in thalamic neurons. J Biol Chem 286, 31781–31788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Lee M, Yoon J, Song H, Lee B, Lam DT, Yoon J, Baek K, Clevers H and Jeong Y (2017) Tcf7l2 plays crucial roles in forebrain development through regulation of thalamic and habenular neuron identity and connectivity. Dev Biol 424, 62–76. [DOI] [PubMed] [Google Scholar]
- 115. Lipiec MA, Kozinski K, Zajkowski T, Dabrowski M, Chakraborty C, Toval A, Ferran J, Nagalski A and Wisniewska MB (2019) The transcription factor TCF7L2 functions as a terminal selector in thalamic and habenular regions of the brain. bioRxiv. [PREPRINT] [Google Scholar]
- 116. Wang Z, Li P, Wu T, Zhu S, Deng L and Cui G (2018) Axon guidance pathway genes are associated with schizophrenia risk. Exp Ther Med 16, 4519–4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. McFadden K and Minshew NJ (2013) Evidence for dysregulation of axonal growth and guidance in the etiology of ASD. Front Hum Neurosci 7, 671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Mostaid MS, Lloyd D, Liberg B, Sundram S, Pereira A, Pantelis C, Karl T, Weickert CS, Everall IP and Bousman CA (2016) Neuregulin‐1 and schizophrenia in the genome‐wide association study era. Neurosci Biobehav Rev 68, 387–409. [DOI] [PubMed] [Google Scholar]
- 119. Guidotti A, Grayson DR and Caruncho HJ (2016) Epigenetic RELN dysfunction in schizophrenia and related neuropsychiatric disorders. Front Cell Neurosci 10, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Zhao Z, Webb BT, Jia P, Bigdeli TB, Maher BS, van den Oord E, Bergen SE, Amdur RL, O'Neill FA, Walsh D et al (2013) Association study of 167 candidate genes for schizophrenia selected by a multi‐domain evidence‐based prioritization algorithm and neurodevelopmental hypothesis. PLoS ONE 8, e67776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014) Biological insights from 108 schizophrenia‐associated genetic loci. Nature 511, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Weißflog L, Scholz CJ, Jacob CP, Nguyen TT, Zamzow K, Groß‐Lesch S, Renner TJ, Romanos M, Rujescu D, Walitza S et al (2013) KCNIP4 as a candidate gene for personality disorders and adult ADHD. Eur Neuropsychopharmacol 23, 436–447. [DOI] [PubMed] [Google Scholar]
- 123. Veenstra‐VanderWeele J, Muller CL, Iwamoto H, Sauer JE, Owens WA, Shah CR, Cohen J, Mannangatti P, Jessen T, Thompson BJ et al (2012) Autism gene variant causes hyperserotonemia, serotonin receptor hypersensitivity, social impairment and repetitive behavior. Proc Natl Acad Sci USA 109, 5469–5474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Muller CL, Anacker AMJ and Veenstra‐VanderWeele J (2016) The serotonin system in autism spectrum disorder: from biomarker to animal models. Neuroscience 321, 24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Nieto R, Kukuljan M and Silva H (2013) BDNF and schizophrenia: from neurodevelopment to neuronal plasticity, learning, and memory. Front Psychiatry 4, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Hakak Y, Walker JR, Li C, Wong WH, Davis KL, Buxbaum JD, Haroutunian V and Fienberg AA (2001) Genome‐wide expression analysis reveals dysregulation of myelination‐related genes in chronic schizophrenia. Proc Natl Acad Sci USA 98, 4746–4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Tkachev D, Mimmack ML, Ryan MM, Wayland M, Freeman T, Jones Peter B, Starkey M, Webster MJ, Yolken RH andBahn S (2003) Oligodendrocyte dysfunction in schizophrenia and bipolar disorder. Lancet 362, 798–805. [DOI] [PubMed] [Google Scholar]
- 128. Chen X, Duan H, Xiao L and Gan J (2018) Genetic and epigenetic alterations underlie oligodendroglia susceptibility and white matter etiology in psychiatric disorders. Front Genet 9, 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Mauney SA, Pietersen CY, Sonntag K‐C and Woo Tsung‐Ung W (2015) Differentiation of oligodendrocyte precursors is impaired in the prefrontal cortex in schizophrenia. Schizophr Res 169 , 374–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. McPhie DL, Nehme R, Ravichandran C, Babb SM, Ghosh SD, Staskus A, Kalinowski A, Kaur R, Douvaras P, Du F et al (2018) Oligodendrocyte differentiation of induced pluripotent stem cells derived from subjects with schizophrenias implicate abnormalities in development. Transl Psychiatry 8, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Roussos P and Haroutunian V (2014) Schizophrenia: susceptibility genes and oligodendroglial and myelin related abnormalities. Front Cell Neurosci 8, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Goldman SA and Kuypers NJ (2015) How to make an oligodendrocyte. Development 142, 3983–3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Langseth AJ, Munji RN, Choe Y, Huynh T, Pozniak CD and Pleasure SJ (2010) Wnts influence the timing and efficiency of oligodendrocyte precursor cell generation in the telencephalon. J Neurosci 30, 13367–13372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Ye F, Chen Y, Hoang T, Montgomery RL, Zhao XH, Bu H, Hu T, Taketo MM, van Es JH, Clevers H et al (2009) HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta‐catenin‐TCF interaction. Nat Neurosci 12, 829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Dai ZM, Sun S, Wang C, Huang H, Hu X, Zhang Z, Lu QR and Qiu M (2014) Stage‐specific regulation of oligodendrocyte development by Wnt/β‐catenin signaling. J Neurosci 34, 8467–8473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Hammond E, Lang J, Maeda Y, Pleasure D, Angus‐Hill M, Xu J, Horiuchi M, Deng W and Guo F (2015) The Wnt effector transcription factor 7‐like 2 positively regulates oligodendrocyte differentiation in a manner independent of Wnt/β‐catenin signaling. J Neurosci 35, 5007–5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Fancy SPJ, Baranzini SE, Zhao C, Yuk D‐I, Irvine K‐A, Kaing S, Sanai N, Franklin RJM and Rowitch DH (2009) Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian . CNS Genes Dev 23, 1571–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Rosenberg AB, Roco CM, Muscat RA, Kuchina A, Sample P, Yao Z, Graybuck LT, Peeler DJ, Mukherjee S, Chen W et al (2018) Single‐cell profiling of the developing mouse brain and spinal cord with split‐pool barcoding. Science 360, 176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Zhao C, Deng Y, Liu L, Yu K, Zhang L, Wang H, He X, Wang J, Lu C, Wu LN et al (2016) Dual regulatory switch through interactions of Tcf7l2/Tcf4 with stage‐specific partners propels oligodendroglial maturation. Nat Commun 7, 10883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Weng C, Ding M, Fan S, Cao Q and Lu Z (2017) Transcription factor 7 like 2 promotes oligodendrocyte differentiation and remyelination. Mol Med Rep 16, 1864–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Feigenson K, Reid M, See J, Crenshaw EB and Grinspan JB (2009) Wnt signaling is sufficient to perturb oligodendrocyte maturation. Mol Cell Neurosci 42, 255–265. [DOI] [PubMed] [Google Scholar]
- 142. Iioka H, Doerner SK and Tamai K (2009) Kaiso is a bimodal modulator for Wnt/beta‐catenin signaling. FEBS Lett 583, 627–632. [DOI] [PubMed] [Google Scholar]
- 143. van Amerongen R (2012) Alternative Wnt pathways and receptors. Cold Spring Harb Perspect Biol 4, a007914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Acebron SP and Niehrs C (2016) β‐Catenin‐Independent Roles of Wnt/LRP6 Signaling. Trends Cell Biol 26, 956–967. [DOI] [PubMed] [Google Scholar]
- 145. Cadigan KM and Waterman ML (2012) TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb Perspect Biol 4, a007906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Valenta T, Hausmann G and Basler K (2012) The many faces and functions of β‐catenin. EMBO J 31, 2714–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Schuijers J, Mokry M, Hatzis P, Cuppen E and Clevers H (2014) Wnt‐induced transcriptional activation is exclusively mediated by TCF/LEF. EMBO J 33, 146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]