

Abstract
Reported is an iridium catalyst for ortho‐selective C−H borylation of challenging secondary aromatic amide substrates, and the regioselectivity is controlled by hydrogen‐bond interactions. The BAIPy‐Ir catalyst forms three hydrogen bonds with the substrate during the crucial activation step, and allows ortho‐C−H borylation with high selectivity. The catalyst displays unprecedented ortho selectivities for a wide variety of substrates that differ in electronic and steric properties, and the catalyst tolerates various functional groups. The regioselective C−H borylation catalyst is readily accessible and converts substrates on gram scale with high selectivity and conversion.
Keywords: borylation, C−H activation, hydrogen bonds, iridium, supramolecular chemistry
Transition metal catalyzed C−H bond activation enables the functionalization of complex molecules without the need of preactivation, allowing the introduction of functional groups at a late stage of a synthesis sequence.1 The direct C−H borylation is of particular interest as the boron functional group allows further modification by a wide variety of transformations, including Suzuki coupling reactions, amination, hydroxylation, and halogenation, providing structural and functional molecular complexity.2 Crucial for the application is that the selectivity of the reaction can be controlled, and this is particularly challenging for C−H bonds that are sterically and electronically deactivated. Recently, the use of supramolecular interactions between the substrate and the ligand of a metal complex have been explored to control the selectivity,3 and this resulted in catalysts for selective meta‐ or para‐C−H borylation for electronically (un)activated substrates.4 However, ortho‐selective C−H borylation has only been reported for electronically activated arenes, such as an amine‐,5 alcohol‐,6 or thioether‐substituted7 arenes. Secondary aromatic amides are very common structural motifs in pharmaceuticals, agrochemicals, and fine chemicals,8 and the ortho‐selective C−H borylation of this class of compounds would therefore be highly interesting. However, the direct ortho‐C−H borylation of this class of compounds is highly challenging. For common iridium‐catalyzed C−H borylation the regioselectivity is largely dictated by steric factors leading to meta‐ and para‐C−H borylation of aromatic compounds (Figure 1 a).1a, 2d, 9 The use of the amide functionality as a directing group does not work as its coordination to a metal center is generally weak because of unfavorable tautomerization.10 Strategies that involve (temporary) directing groups such as silyl, iminyl, and pyridyl, operating by chelation to the metal center, have not been reported for secondary aromatic amides.11 To the best our knowledge, the group of Yu11d, 11e reported the only protocol for ortho‐C−H borylation catalyzed by palladium through a metal‐substrate chelation approach for substrates with a special electron‐withdrawing directing group to promote the enol tautomerization (Figure 1 b). Herein, we report the design of a supramolecular iridium‐based catalyst for highly ortho‐selective C−H borylation of these secondary aromatic amides and it is based on substrate orientation using hydrogen bonding.
Figure 1.
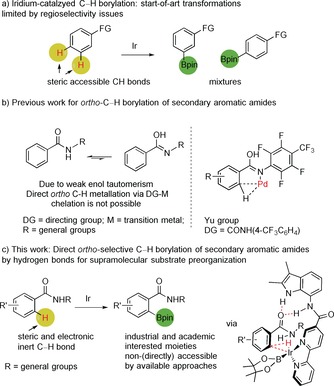
a) General regioselectivity in iridium‐catalyzed C−H borylation of aromatic compounds. b) Previous work on ortho‐selective C−H borylation of secondary aromatic amides by a chelation approach. c) Direct ortho‐selective C−H borylation of unactivated secondary aromatic amides through hydrogen bonding to the catalyst.
We designed the iridium‐based borylation catalyst reported here assuming one indole amide hydrogen‐bond motif, to preorganize the aromatic amide substrates for ortho‐C−H borylation, should be sufficient. For ortho selectivity, the motif should be close to the catalyst, and as such direct coupling of an indole amide to the ligand 2,2′‐bipyridine (BPy) was considered. DFT calculations show that the Ir complex of this ligand preorganizes N‐methylbenzamide (1 s) with the ortho‐C−H bond oriented for selective activation (Figure 2). The BAIPy ligand was prepared on multigram scale from commercially available compounds in three straightforward synthetic steps (for details see the Supporting Information).
Figure 2.
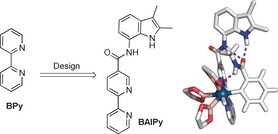
Design of the BAIPy ligand containing an indole amide functional group for substrate orientation in ortho‐selective C−H borylation of secondary aromatic amides. Support for the substrate‐catalyst complex formation is provided by DFT modeling of the substrate‐BAIPy‐trisboryl‐Ir catalyst complex (the methyl groups on the boryl ligand were omitted for clarity).
Binding studies in [D8]toluene and [D8]THF using 1H NMR spectroscopy show that 1 s is indeed bound to the free BAIPy ligand with a binding energy around 6.3 and 4.0 kJ mol−1, respectively (see Figures S1–S6, and Tables S1 and S2 in the Supporting Information).
To investigate the performance of the supramolecular catalyst we initially performed catalytic experiments in THF using 1 s as the model substrate. As expected, the catalytic reaction using the parent BPy‐Ir catalyst leads to the formation of a mixture of meta‐ and para‐borylated products, and the ortho‐borylated compound was not detected (1 so 0.0 %, Figure 3). In contrast, the BAIPy‐Ir catalyst that directs the substrate, by hydrogen bonding to the indole amide motif, shows unprecedented selective ortho‐C−H borylation (1 so, 94 %). Importantly, the supramolecular interactions direct the C−H activation to a position that is sterically and electronically unfavorable.
Figure 3.
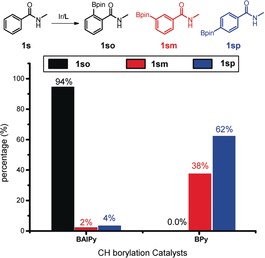
C−H borylation of N‐methylbenzamide catalyzed by BAIPy‐Ir (left) versus BPy‐Ir (right). Reaction conditions: substrate (0.6 mmol), B2Pin2 (0.9 mmol), [Ir(COD)OMe]2 (1.5 mol %), ligand (3.3 mol %), THF (0.2 m), 50 °C. 24 h. Regioselectivity was determined by GC analysis. COD=1,5‐dicyclooctadiene, Pin=pinacol, THF=tetrahydrofuran.
DFT calculations to gain more insight into the operational mode of the supramolecular BAIPy‐Ir complex show that three hydrogen bonds form (the N−O distances of 2.323–2.953 Å) when the substrate binds to the catalyst, leading to preorganization of the ortho‐C−H bond of the substrate (Figure 4; see Figures S4–S7 and Table S2). In the transition state of the oxidative activation to the iridium center, and in the iridium complex after oxidative addition, these hydrogen bonds stay in place. Importantly, the energy of the transition‐state structure TS‐AB is only around 100.8 kJ mol−1 higher than the pre‐complex A, suggesting that this sterically hindered and electronically less active ortho‐C−H bond can be activated at slightly elevated temperature, in line with the experiment. On the contrary, the meta‐ and para‐C−H borylated compounds can only form when 1 s is not bound to the BAIPy‐Ir complex through hydrogen bonds. DFT calculations show that the transition‐state structures TS‐CD and TS‐EF are higher in energy than the counterpart formed in the ortho‐borylation pathway (159.8 and 166.9 kJ mol−1 vs. 100.8 kJ mol−1; see Figures S7–S10). These calculations provide support that hydrogen‐bonding interactions orient the substrate for ortho‐selectivite C−H borylation.
Figure 4.
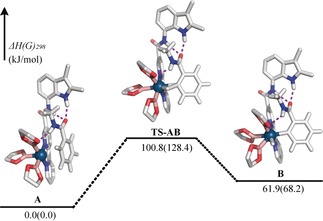
DFT calculated pathway for the C−H bond activation step in the BAIPy‐trisboryl‐Ir catalyzed ortho‐selective C−H borylation of 1 s (BP86‐D3/def2‐TZVP, enthalpy and Gibbs free energy are referenced to A, and hydrogen bonds shown in puple dotted lines; the methyl groups on the boryl ligand were deleted to reduce computation costs).
Besides the expected two hydrogen bonds formed between the indole amide motif and the carbonyl of the substrate, calculations show that an extra hydrogen bond is formed between the amide‐NH of the substrate and the oxygen atom of the boryl group. Control experiments with N,N‐dimethylbenzamide (2 s) and methyl benzoate (3 s; see Scheme S2), substrates that cannot form this extra hydrogen bond with the catalyst, show that these two substrates are converted with poor ortho‐C−H borylation selectivity (ortho 25 % and 0 % for 2 s and 3 s, respectively), suggesting that the third hydrogen bond between the substrate and the catalyst is important. The selectivity difference between 2 s and 3 s also reveals that the amide forms a stronger hydrogen‐bond interaction than the ester to preorganize the substrate, and is in line with literature.12 These control experiments also confirm that control of the regioselectivity is not a result of the classical substrate‐chelation effect.
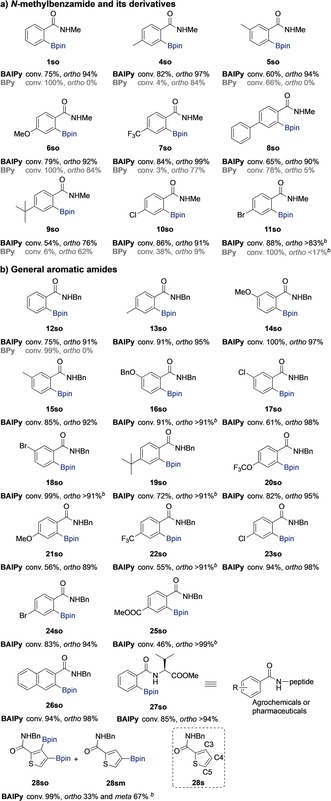
The substrate scope was explored by evaluation of various N‐methylbenzamides to study the functional‐group tolerance and the effect of variation in the sterics and electronics (Table 1 a). For the meta‐ and para‐methyl‐substituted N‐methylbenzamides 4 s and 5 s, respectively, the BAIPy‐Ir catalyst shows high ortho selectivity. The control experiment using the BPy‐Ir catalyst that does not preorganize the substrate shows no ortho‐C−H borylation for 5 s (ortho 0 %) and only moderate ortho selectivity for 4 s (ortho 84 %). Importantly, besides the enhanced selectivity, much higher conversion was obtained for 4 s when BAIPy‐Ir was used as the catalyst compared to BPy‐Ir (conversion 82 vs. 4 %), showing that substrate preorganization also affects the activity of the catalyst. We next explored a series of substrates that have different substituents at the para‐position with respect to the methyl amide (6 s–11 s) to further evaluate the substrate scope. For 6 s, having an electron‐donating methoxy group, the BAIPy‐Ir catalyst displays enhanced ortho selectivity compared to the BPy‐Ir catalyst (ortho 92 vs. 84 %). The substrate 7 s contains a widely used trifluoromethyl group and the BAIPy‐Ir catalyst also shows very high selectivity (ortho 99 % vs. 77 %) for ortho‐borylation and much higher conversion than the BPy‐Ir catalyst (conversion 84 % vs. 3 %). Also for 8 s, containing an additional aromatic ring, the BAIPy‐Ir catalyst shows unprecedented high ortho selectivity, while BPy‐Ir converts this substrate mainly into the meta‐C−H borylation product (ortho 90 % vs. 5 %). In addition to the ortho‐borylated product, some other products are formed in very small amounts, suggesting minor borylation of the appended aromatic ring (<5 % total). For 9 s, with the bulky tert‐butyl group, BAIPy‐Ir also displays higher selectivity and conversion than BPy‐Ir catalyst (ortho 76 % vs. 62 %; conversion 54 % vs. 6 %). The BAIPy‐Ir catalyst tolerates halides, as 10 s and 11 s are converted with high selectivity by BAIPy‐Ir (ortho 91 % and >83 %; conversion 86 % and 88 %), and is again much better than BPy‐Ir (ortho 9 % and <17 %; conversion 38 % and 100 %). Unfortunately, the BAIPy‐Ir catalyst is not active for ortho‐substituted substrates (for details see the Supporting Information).
Table 1.
Substrate scope of ortho‐selective borylation using the BAIPy‐Ir catalyst.[a]
|
[a] Typical reaction conditions: substrate (0.6 mmol), B2Pin2 (0.9 mmol), [Ir(COD)OMe]2 (1.5 mol %), ligand (3.3 mol %), THF (0.2 m), 50–60 °C. 24–96 h. Conversion and ortho selectivity (percentage among all the C−H borylated products) were determined by either GC analysis or 1H NMR analysis (see the Supporting Information for yields of isolated products and other details). [b] Estimated by 1H NMR analysis after chromatography.
To further demonstrate the general applicability of the BAIPy‐Ir catalyst, we extended the substrate scope to general aromatic amides (Table 1 b). Firstly, N‐benzylbenzamide (12 s) was studied in detail as a representative substrate of this subset. Interestingly, this substrate has two aromatic rings, both of which contain C−H bonds that can in principle be borylated, however the C−H bonds of the benzyl group are almost untouched (<5 %), even by BPy‐Ir, suggesting that there is sufficient difference in reactivity between the rings. As a result, 12 s can be converted by BAIPy‐Ir with high selectivity into the ortho‐C−H borylation product (ortho 91 %). In contrast, BPy‐Ir delivers only meta‐ and para‐C−H borylation products (ortho 0 %), and also quantitative conversion is obtained under these reaction conditions. We further evaluated the steric effects on the selectivity and reactivity by using either para‐ or meta‐substituted substrates (13 s–25 s), including methyl, methoxy, benzyloxy, tert‐butyl, trifluoromethoxy, ester groups, and halogens (Table 1 b). As expected, the BAIPy‐Ir catalyst shows unprecedented high selectivity for ortho‐C−H borylation and decent to good conversion (46–100 %). Importantly, extremely high selectivity was also achieved (ortho >99 %) for the substrate 25 s, which contains an ester group at the para‐position with respect to the amide. In line with the control experiments displayed in Scheme S2, the catalyst tolerates an ester group as its interaction with the indole amide unit is weaker than that with the amide. Also, a naphthalene amide (26 s) was converted in high selectivity to the ortho‐borylated product when using the BAIPy‐Ir catalyst. Next the peptide‐based aromatic amide 27 s was converted with high selectivity (conversion 85 %, ortho product >94 %). Importantly, these results demonstrate the potential of this protocol for late‐state functionalization of valuable peptide‐based aromatic compounds.
Finally, the N‐benzylthiophenecarboxamide 28 s was explored as a particular challenging substrate (Table 1 b). Generally C−H borylation is directed to C5−H because of steric and electronic effects, and the inert C3−H bond is not borylated.13 Using the BAIPy‐Ir as catalyst we surprisingly formed 67 % of the C4‐borylated product, which is usually not formed, along with some diborylated product in which both the C4‐ and C3‐positions are functionalized. Importantly, the most activated C5−H bond remains untouched. The wide substrate scope demonstrates the generality of the supramolecular approach for ortho‐selective C−H borylation of secondary aromatic amides.
We performed a gram‐scale C−H borylation reaction using 0.4–3 mol % iridium at 60 °C (see Scheme S3), forming the ortho‐C−H borylated compound 1 so and 12 so with up to 85 % yield upon isolation and a turnover number (TON) of 123. The boron functionality allows easy follow‐up chemistry to introduce various groups,2 and as one typical example, 1 so was transformed into a hydroxy group by an oxidation‐hydrolysis sequence with quantitative yield using H2O2. Thus, this readily available supramolecular iridium catalyst is feasible for large‐scale application to directly install the versatile boron moiety on the aromatic amides.
In summary, we report a readily accessible supramolecular iridium catalyst for ortho‐selective C−H borylation of valuable secondary aromatic amides, and the catalyst operates by substrate preorganization through hydrogen bonding. Catalytic experiments with N‐methylbenzamides and aromatic amides (>26 examples), including peptide‐based analogues, demonstrate that this supramolecular catalyst converts a variety of secondary aromatic amides, having a variety of functional groups at different positions on the aromatic ring, making the strategy very general. The supramolecular iridium catalyst has been applied on gram scale with high conversion and selectivity at elevated temperature. These experiments show that substrate orientation using a supramolecular catalyst is a powerful approach for controlling the regioselectivity in C−H borylation reactions.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge Dr. Vivek Sinha and Pim R. Linnebank for fruitful discussions. S.‐T. Bai thanks the China Scholarship Council for a PhD fellowship (CSC201506010269) and the RPA sustainable chemistry of the University of Amsterdam for financial support.
S.-T. Bai, C. B. Bheeter, J. N. H. Reek, Angew. Chem. Int. Ed. 2019, 58, 13039.
References
- 1.For reviews on C−H functionalization and the references cited therein, see:
- 1a. Mkhalid I. A. I., Barnard J. H., Marder T. B., Murphy J. M., Hartwig J. F., Chem. Rev. 2010, 110, 890–931; [DOI] [PubMed] [Google Scholar]
- 1b. Chen X., Engle K. M., Wang D.-H., Yu J.-Q., Angew. Chem. Int. Ed. 2009, 48, 5094–5115; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 5196–5217; [Google Scholar]
- 1c. Labinger J. A., Chem. Rev. 2017, 117, 8483–8496; [DOI] [PubMed] [Google Scholar]
- 1d. Crabtree R. H., Lei A., Chem. Rev. 2017, 117, 8481–8482; [DOI] [PubMed] [Google Scholar]
- 1e. Godula K., Sames D., Science 2006, 312, 67–72; [DOI] [PubMed] [Google Scholar]
- 1f. Wencel-Delord J., Glorius F., Nat. Chem. 2013, 5, 369–375; [DOI] [PubMed] [Google Scholar]
- 1g. Gandeepan P., Müller T., Zell D., Cera G., Warratz S., Ackermann L., Chem. Rev. 2019, 119, 2192–2452. [DOI] [PubMed] [Google Scholar]
- 2.For reviews on Ir-catalyzed C−H borylation and the reference cited therein, see:
- 2a. Ishiyama T., Miyaura N., J. Organomet. Chem. 2003, 680, 3–11; [Google Scholar]
- 2b. Ros A., Fernández R., Lassaletta J. M., Chem. Soc. Rev. 2014, 43, 3229–3243; [DOI] [PubMed] [Google Scholar]
- 2c. Hartwig J. F., Acc. Chem. Res. 2012, 45, 864–873; [DOI] [PubMed] [Google Scholar]
- 2d. Hartwig J. F., Chem. Soc. Rev. 2011, 40, 1992–2002; [DOI] [PubMed] [Google Scholar]
- 2e. Haldar C., Hoque M. E., Bisht R., Chattopadhyay B., Tetrahedron Lett. 2018, 59, 1269–1277. [Google Scholar]
- 3.For some reviews and examples using supramolecular substrate preorganization in transition-metal catalysis:
- 3a. Šmejkal T., Breit B., Angew. Chem. Int. Ed. 2008, 47, 3946–3949; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 4010–4013; [Google Scholar]
- 3b. Meeuwissen J., Reek J. N. H., Nat. Chem. 2010, 2, 615–621; [DOI] [PubMed] [Google Scholar]
- 3c. Dydio P., Reek J. N. H., Chem. Sci. 2014, 5, 2135–2145; [Google Scholar]
- 3d. Davis H. J., Phipps R. J., Chem. Sci. 2017, 8, 864–877; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3e. Nurttila S. S., Linnebank P. R., Krachko T., Reek J. N. H., ACS Catal. 2018, 8, 3469–3488; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3f. Zhang Z., Tanaka K., Yu J.-Q., Nature 2017, 543, 538–542; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3g. Das S., Incarvito C. D., Crabtree R. H., Brudvig G. W., Science 2006, 312, 1941–1943; [DOI] [PubMed] [Google Scholar]
- 3h. Bauer A., Westkämper F., Grimme S., Bach T., Nature 2005, 436, 1139–1140; [DOI] [PubMed] [Google Scholar]
- 3i. Coote S. C., Bach T., J. Am. Chem. Soc. 2013, 135, 14948–14951. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Mihai M. T., Davis H. J., Genov G. R., Phipps R. J., ACS Catal. 2018, 8, 3764–3769; [Google Scholar]
- 4b. Bisht R., Hoque M. E., Chattopadhyay B., Angew. Chem. Int. Ed. 2018, 57, 15762–15766; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 15988–15992; [Google Scholar]
- 4c. Davis H. J., Mihai M. T., Phipps R. J., J. Am. Chem. Soc. 2016, 138, 12759–12762; [DOI] [PubMed] [Google Scholar]
- 4d. Kuninobu Y., Ida H., Nishi M., Kanai M., Nat. Chem. 2015, 7, 712–717; [DOI] [PubMed] [Google Scholar]
- 4e. Davis H. J., Genov G. R., Phipps R. J., Angew. Chem. Int. Ed. 2017, 56, 13351–13355; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 13536–13540; [Google Scholar]
- 4f. Hoque M. E., Bisht R., Haldar C., Chattopadhyay B., J. Am. Chem. Soc. 2017, 139, 7745–7748. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Smith M. R., Bisht R., Haldar C., Pandey G., Dannatt J. E., Ghaffari B., Maleczka R. E., Chattopadhyay B., ACS Catal. 2018, 8, 6216–6223; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Roosen P. C., Kallepalli V. A., Chattopadhyay B., Singleton D. A., Maleczka R. E., Smith M. R., J. Am. Chem. Soc. 2012, 134, 11350–11353; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Preshlock S. M., Plattner D. L., Maligres P. E., Krska S. W., Maleczka R. E., Smith M. R., Angew. Chem. Int. Ed. 2013, 52, 12915–12919; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 13153–13157. [Google Scholar]
- 6. Chattopadhyay B., Dannatt J. E., Andujar-De Sanctis I. L., Gore K. A., Maleczka R. E., Singleton D. A., Smith M. R., J. Am. Chem. Soc. 2017, 139, 7864–7871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li H. L., Kuninobu Y., Kanai M., Angew. Chem. Int. Ed. 2017, 56, 1495–1499; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1517–1521. [Google Scholar]
- 8.
- 8a. Gerry C. J., Schreiber S. L., Nat. Rev. Drug Discovery 2018, 17, 333–352; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Blakemore D. C., Castro L., Churcher I., Rees D. C., Thomas A. W., Wilson D. M., Wood A., Nat. Chem. 2018, 10, 383–394; [DOI] [PubMed] [Google Scholar]
- 8c. Urquhart L., Nat. Rev. Drug Discovery 2018, 17, 232–232. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Fairlie D. P., Woon T. C., Wickramasinghe W. A., Willis A. C., Inorg. Chem. 1994, 33, 6425–6428; [Google Scholar]
- 9b. Shang M., Zeng S.-H., Sun S.-Z., Dai H.-X., Yu J.-Q., Org. Lett. 2013, 15, 5286–5289; [DOI] [PubMed] [Google Scholar]
- 9c. Pace V., Holzer W., Olofsson B., Adv. Synth. Catal. 2014, 356, 3697–3736. [Google Scholar]
- 10.
- 10a. Tajuddin H., Harrisson P., Bitterlich B., Collings J. C., Sim N., Batsanov A. S., Cheung M. S., Kawamorita S., Maxwell A. C., Shukla L., Morris J., Lin Z., Marder T. B., Steel P. G., Chem. Sci. 2012, 3, 3505–3515; [Google Scholar]
- 10b. Robbins D. W., Hartwig J. F., Angew. Chem. Int. Ed. 2013, 52, 933–937; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 967–971; [Google Scholar]
- 10c. Cho J.-Y., Iverson C. N., Smith M. R., J. Am. Chem. Soc. 2000, 122, 12868–12869. [Google Scholar]
- 11.For examples using the chelation approach for ortho-C−H borylation, see:
- 11a. Ishiyama T., Isou H., Kikuchi T., Miyaura N., Chem. Commun. 2010, 46, 159–161; [DOI] [PubMed] [Google Scholar]
- 11b. Boebel T. A., Hartwig J. F., J. Am. Chem. Soc. 2008, 130, 7534–7535; [DOI] [PubMed] [Google Scholar]
- 11c. Ros A., Estepa B., López-Rodríguez R., Álvarez E., Fernández R., Lassaletta J. M., Angew. Chem. Int. Ed. 2011, 50, 11724–11728; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 11928–11932; [Google Scholar]
- 11d. Dai H.-X., Yu J.-Q., J. Am. Chem. Soc. 2012, 134, 134–137; [DOI] [PubMed] [Google Scholar]
- 11e. Zhang X.-G., Dai H.-X., Wasa M., Yu J.-Q., J. Am. Chem. Soc. 2012, 134, 11948–11951; [DOI] [PubMed] [Google Scholar]
- 11f. Itoh H., Kikuchi T., Ishiyama T., Miyaura N., Chem. Lett. 2011, 40, 1007–1008; [Google Scholar]
- 11g. Ros A., López-Rodríguez R., Estepa B., Álvarez E., Fernández R., Lassaletta J. M., J. Am. Chem. Soc. 2012, 134, 4573–4576; [DOI] [PubMed] [Google Scholar]
- 11h. Crawford K. M., Ramseyer T. R., Daley C. J. A., Clark T. B., Angew. Chem. Int. Ed. 2014, 53, 7589–7593; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7719–7723; [Google Scholar]
- 11i. Roering A. J., Hale L. V. A., Squier P. A., Ringgold M. A., Wiederspan E. R., Clark T. B., Org. Lett. 2012, 14, 3558–3561; [DOI] [PubMed] [Google Scholar]
- 11j. Wang G., Liu L., Wang H., Ding Y.-S., Zhou J., Mao S., Li P., J. Am. Chem. Soc. 2017, 139, 91–94; [DOI] [PubMed] [Google Scholar]
- 11k. Kawamorita S., Ohmiya H., Hara K., Fukuoka A., Sawamura M., J. Am. Chem. Soc. 2009, 131, 5058–5059; [DOI] [PubMed] [Google Scholar]
- 11l. Bisht R., Chattopadhyay B., J. Am. Chem. Soc. 2016, 138, 84–87; [DOI] [PubMed] [Google Scholar]
- 11m. Fukuda K., Iwasawa N., Takaya J., Angew. Chem. Int. Ed. 2019, 58, 2850–2853; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 2876–2879; [Google Scholar]
- 11n. Kuninobu Y., Iwanaga T., Omura T., Takai K., Angew. Chem. Int. Ed. 2013, 52, 4431–4434; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 4527–4530; [Google Scholar]
- 11o. Niu L., Yang H., Wang R., Fu H., Org. Lett. 2012, 14, 2618–2621; [DOI] [PubMed] [Google Scholar]
- 11p. Sasaki I., Ikeda T., Amou T., Taguchi J., Ito H., Ishiyama T., Synlett 2016, 27, 1582–1586; [Google Scholar]
- 11q. Su B., Zhou T.-G., Xu P.-L., Shi Z.-J., Hartwig J. F., Angew. Chem. Int. Ed. 2017, 56, 7205–7208; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 7311–7314; [Google Scholar]
- 11r. Li H.-L., Kanai M., Kuninobu Y., Org. Lett. 2017, 19, 5944–5947. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Biswal H. S., Bhattacharyya S., Bhattacherjee A., Wategaonkar S., Int. Rev. Phys. Chem. 2015, 34, 99–160; [Google Scholar]
- 12b. Bissantz C., Kuhn B., Stahl M., J. Med. Chem. 2010, 53, 5061–5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Sasaki I., Taguchi J., Hiraki S., Ito H., Ishiyama T., Chem. Eur. J. 2015, 21, 9236–9241; [DOI] [PubMed] [Google Scholar]
- 13b. Yang L., Semba K., Nakao Y., Angew. Chem. Int. Ed. 2017, 56, 4853–4857; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4931–4935. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary