

Abstract
Protease inhibitors (PIs) are important components of treatment regimens for patients with chronic hepatitis C virus (HCV) infection. However, emergence and persistence of antiviral resistance could reduce their efficacy. Thus, defining resistance determinants is highly relevant for efforts to control HCV. Here, we investigated patterns of PI resistance–associated substitutions (RASs) for the major HCV genotypes and viral determinants for persistence of key RASs. We identified protease position 156 as a RAS hotspot for genotype 1‐4, but not 5 and 6, escape variants by resistance profiling using PIs grazoprevir and paritaprevir in infectious cell culture systems. However, except for genotype 3, engineered 156‐RASs were not maintained. For genotypes 1 and 2, persistence of 156‐RASs depended on genome‐wide substitution networks, co‐selected under continued PI treatment and identified by next‐generation sequencing with substitution linkage and haplotype reconstruction. Persistence of A156T for genotype 1 relied on compensatory substitutions increasing replication and assembly. For genotype 2, initial selection of A156V facilitated transition to 156L, persisting without compensatory substitutions. The developed genotype 1, 2, and 3 variants with persistent 156‐RASs had exceptionally high fitness and resistance to grazoprevir, paritaprevir, glecaprevir, and voxilaprevir. A156T dominated in genotype 1 glecaprevir and voxilaprevir escape variants, and pre‐existing A156T facilitated genotype 1 escape from clinically relevant combination treatments with grazoprevir/elbasvir and glecaprevir/pibrentasvir. In genotype 1 infected patients with treatment failure and 156‐RASs, we observed genome‐wide selection of substitutions under treatment. Conclusion: Comprehensive PI resistance profiling for HCV genotypes 1‐6 revealed 156‐RASs as key determinants of high‐level resistance across clinically relevant PIs. We obtained in vitro proof of concept for persistence of highly fit genotype 1‐3 156‐variants, which might pose a threat to clinically relevant combination treatments.
Abbreviations
- aa
amino acid
- DAA
direct acting antiviral
- HCV
hepatitis C virus
- NGS
next‐generation sequencing
- NS
nonstructural protein
- NS3H
NS3 helicase
- NS3P
NS3 protease
- ORF
open reading frame
- PI
protease inhibitor
- RAS
resistance‐associated substitution
- SNP
single nucleotide polymorphism
Worldwide, hepatitis C virus (HCV) is estimated to cause at least 70 million chronic infections, with 400,000 deaths annually.1 Genotypes 1‐3 cause more than 80% of infections worldwide.2 The development of efficient direct acting antivirals (DAAs) has revolutionized treatment of chronic HCV infection.3, 4, 5 The main DAA targets are the HCV nonstructural protein (NS) 3 protease (NS3P), the NS5A protein, and the NS5B polymerase. To counteract the emergence of resistance, DAAs are administered in combinations, and protease inhibitors (PIs) are important components of several DAA‐based treatment regimens.3, 4, 5, 6 The PI grazoprevir in combination with the NS5A inhibitor elbasvir, as well as the PI paritaprevir in combination with the NS5A inhibitor ombitasvir and the nonnucleosidic NS5B inhibitor dasabuvir, were approved for treatment of genotypes 1 and 4.3, 4 Recently, the PI glecaprevir in combination with the NS5A inhibitor pibrentasvir was approved for treatment of genotypes 1‐6. Further, the PI voxilaprevir in combination with the NS5A inhibitor velpatasvir and the nucleosidic NS5B inhibitor sofosbuvir received approval for all HCV genotypes, for DAA‐naïve and DAA‐experienced patients.3, 4
However, resistance to DAAs is emerging.5, 6, 7 Thus, a subset of DAA‐treated patients is experiencing treatment failure, associated with the selection of HCV resistance–associated substitutions (RASs).3, 4, 5, 6, 7, 8 Furthermore, in certain regions, specific RASs pre‐exist in up to 50% of individuals.5, 7 Currently, DAA combination therapy typically results in cure rates of more than 90%, but cure rates might be lower in difficult‐to‐treat patient groups.5, 7
Given a steep increase in the number of individuals treated with DAAs, and thus in the absolute number of individuals with treatment failure, eventually a significant number of HCV‐infected individuals can be expected to harbor DAA‐resistant HCV variants. Depending on their fitness, resistant variants might persist following end of treatment, with the potential to spread in human populations. RASs at NS3P amino acid (aa) position 156, regarded as key mediators of resistance for all clinically relevant PIs, were so far reported to be associated with high fitness costs, which has also hindered in vitro studies.5, 9 In line with these findings, 156‐RASs were reported not to persist long‐term following DAA treatment failure.5, 7
In this study, we initially aimed at carrying out PI‐resistance profiling using genotype 1‐6 infectious HCV cell culture systems. Because RASs selected at NS3P position 156 were associated with high fitness costs, we aimed at developing HCV variants allowing persistence of 156‐RASs. Applying genome‐wide next‐generation sequencing (NGS) and substitution linkage analysis, we aimed at reconstructing substitution networks identified outside NS3P, which facilitated persistence of highly resistant 156‐variants by different mechanisms. We further aimed at evaluating the effect of pre‐existing 156‐RASs on DAA combination treatment. Finally, we investigated the selection of substitutions outside NS3P in patients harboring 156‐RASs at treatment failure.
Materials and Methods
Cultured HCV
Recombinants had NS3P of the genotypes(isolates) 1a(TN),10 1b(DH1),11 2a(JFH1),12 2b(DH8),13 3a(S52),14 3a(DBN),15 4a(ED43),14 5a(SA13),14 and 6a(HK6a)14, 16 (Supporting Fig. S1). The used 4a(ED43) recombinant was further adapted as described in Supporting Fig. S1 (GenBank identifier MK600383). Substitutions were engineered by the QuikChange Site‐Directed Mutagenesis Kit (Agilent). NS3P substitutions were numbered relative to the NS3P aa sequence; other substitutions were numbered relative to the H77 polyprotein sequence (GenBank identifier AF009606) unless otherwise indicated. The entire HCV sequence of the final DNA preparation was confirmed by Sanger sequencing (Macrogen).
HUH7.5 Cell Culture
Cells were cultured as described.17 Percentage of HCV infected cells was determined by immunostaining with anti‐NS5A‐9E1012 and anti‐Core‐C7‐50 (Enzo Life Sciences and Abcam).10
The PIs grazoprevir, paritaprevir, glecaprevir (Acme Biosciences) and voxilaprevir (Gilead Sciences), NS5A inhibitors pibrentasvir, elbasvir and velpatasvir (Acme Biosciences), and NS5B inhibitor sofosbuvir (Acme Biosciences) were dissolved in DMSO. For induction of viral escape, DAA mono or combination treatment was initiated in cultures with 80%‐90% HCV‐infected cells and was administered every 2‐3 days until occurrence of viral escape (initial decrease followed by a peak in the percentage of HCV‐infected cells), control (single positive HCV‐infected cells in at least six consecutive immunostainings), or suppression (absence of HCV‐infected cells in at least six consecutive immunostainings).
Viral fitness was evaluated by monitoring viral spread kinetics and genetic stability. Spread kinetics of variants were compared to that of the original viruses following transfection of Huh7.5 cells with HCV RNA transcripts using Lipofectamine (Invitrogen)17 by immunostainings or by determination of HCV infectivity titers.16 Unless otherwise indicated, persistence of engineered RASs was evaluated by NS3P Sanger sequencing of first‐passage supernatant virus stocks.
In an evolutionary approach to stabilize 156‐RASs, 156‐variants were serially passaged, infecting naïve Huh7.5 cells with 1 mL supernatant derived from the previous passage and initiating treatment with 64‐fold EC50 grazoprevir when about 5% of cells were infected. First, six passages under PI treatment were carried out, monitoring viral spread kinetics by immunostaining. Then six PI‐free passages were done to investigate the persistence of 156‐RASs.
Single‐Cycle Infection Assays
CD81‐deficient S29 cells18 were transfected as described with 10 μg of HCV RNA transcripts using 5 μL of Lipofectamine.19 Four hours following transfection, intracellular Core protein concentration was determined; 72 hours following transfection, intracellular and extracellular infectivity titers and Core protein concentration were determined.19 HCV infectivity titers were determined as described.16
PI Sensitivity
PI concentration‐response experiments were done as described.16 Briefly, 5 × 103 cells in 96‐well plates were infected with NS3P sequence–confirmed first‐passage virus stocks. One day after infection, PI treatment was administered using a range of noncytotoxic concentrations in triplicates.20 Three days following infection, immunostaining was carried out; counts of HCV infected cells from treated wells were related to means of counts from infected, nontreated wells. Concentration‐response curves and EC50 values were obtained using GraphPad Prism.17
Patient HCV Samples
Serum or plasma from genotype 1a–infected patients with DAA treatment failure and acquisition of 156‐RASs were obtained from the European Resistance Database at the Department of Internal Medicine 1, University Hospital Frankfurt/German Center for Infection Research, External Partner Site Frankfurt, Germany (patients A, B, D, and E), and the National Reference Center for Viral Hepatitis B, C and D, Department of Virology, Henri Mondor Hospital, Créteil, France (patient C). Treatment‐naïve patients were followed at Copenhagen University Hospital, Hvidovre.21, 22 The study was conducted in accordance with the Declaration of Helsinki, and approval for the use of patient blood samples and retrospective collection of data for research purposes was obtained from the local ethics committees.
Sequence Analysis
Sanger sequencing of amplicons spanning NS3P or the complete open reading frame (ORF) is described in the Supporting Methods and Supporting Tables S1‐S4. NGS analysis of the complete ORF, referred to as genome‐wide NGS, followed the HCV RNA extraction, generation of amplicons spanning the complete ORF, and library preparation as described in the Supporting Methods and Supporting Tables S3 and S4.23, 24 Sequencing of long inserts (500‐600 bases) allowed linkage of NS3P substitutions and NS3P haplotype reconstruction using LinkGE (see Supporting Methods). Genome‐wide linkage analysis and haplotype reconstruction was based on frequency development of single nucleotide polymorphisms (SNPs) in serial viral passage and confirmed by phylogenetic analysis of subclones of ORF amplicons, including ancestral reconstruction (see Supporting Methods).
Results
PI Resistance Profiling Identified Genotype‐Specific RAS Patterns and NS3P Position 156 as a RAS Hotspot
For PI resistance profiling, Huh7.5 cells were infected with HCV recombinants with NS3P of genotypes 1a, 1b, 2a, 2b, 3a, 4a, 5a, and 6a (Supporting Fig. S1), and treated with 1‐ to 64‐fold EC50 of grazoprevir or paritaprevir. For most cultures, an initial decrease in the percentage of HCV‐infected cells was followed by viral escape (Supporting Fig. S2).
In escaped viruses, across PIs and genotypes, NGS analysis revealed RAS hotspots at NS3P aa positions 156 and 168 (Fig. 1A, Supporting Figs. S3‐S11). However, we also observed PI‐ and genotype‐specific selection patterns. For example, under grazoprevir, A156T/V/L dominated for genotypes 1a/b, 2a, 3a, and 4a; under paritaprevir, in genotype 1a R155K and in genotype 2a D168A dominated. Under both PIs, genotypes 2b, 5a and 6a did not acquire substitutions at position 156, but primarily at position 168, often in combination with other NS3P substitutions.
Figure 1.
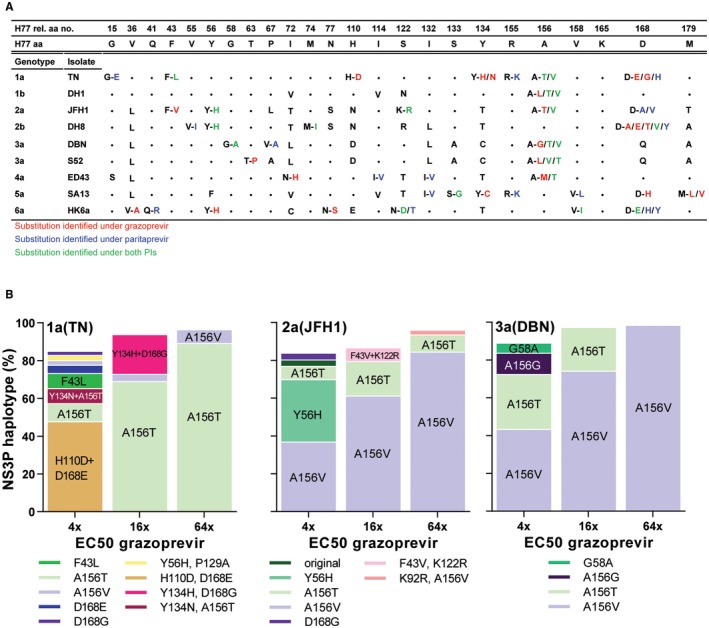
HCV genotype 1‐6 PI escape viruses harbored NS3P RASs with PI concentration‐dependent substitution patterns. (A) Putative RASs were identified by NGS. NS3P aa positions with putative RASs found in more than 5% of the viral population for at least one virus and one PI were included and numbered relative to NS3P of the H77 reference strain; H77 aa residues are specified. •, aa residues identical to that of H77; single letter, nonidentical aa residue; letters separated by dash, putative RASs indicated by the original and the mutated residues, color coded depending on the PI under which they were selected. For detailed data including NGS, see Supporting Figs. S3‐S11. (B) NS3P NGS substitution linkage analysis revealed haplotype distributions for 1a(TN), 2a(JFH1), and 3a(DBN) grazoprevir escape viruses (Supporting Figs. S3, S5, S8, S14A‐C). Haplotypes constituting greater than 2% of the viral population are included in bars; haplotypes greater than 5% are highlighted on bars.
NGS‐based NS3P linkage analysis of SNPs selected in all escape viruses (Fig. 1B, Supporting Figs. S3‐S11) and genome‐wide NGS of selected escape viruses (Supporting Fig. S3B) revealed that the complexity of substitution patterns depended on the PI concentration. Low PI concentrations selected for a variety of NS3P haplotypes and other ORF substitutions outside NS3P. In contrast, high PI concentrations created an evolutionary bottleneck and selected for fewer NS3P haplotypes dominated by few specific RASs at hotspots described previously and for fewer ORF substitutions outside NS3P. For further viral population analysis of genome‐wide NGS data, see the Supporting Results.
Engineered RASs at NS3P Position 156 Were Only Maintained for Genotype 3 and Mediated High PI Resistance
Most of the 49 engineered genotype 1‐6 HCV variants with identified NS3P substitutions showed decreased fitness. Two variants were nonviable, and 33 variants showed delayed spread kinetics following transfection. For 16 variants, the engineered substitutions were not maintained in first passage (Supporting Fig. S12). Of the nine genotype 1a/b, 2a, 3a, and 4a A156T/V‐variants, only the two 3a(DBN) 156‐variants maintained these RASs. Among the 16 genotype 1a, 2a/b, 5a, and 6a D168A/E/H/V/Y variants, 10 maintained these RASs.
Most of the 30 tested NS3P variants showed cross‐resistance to grazoprevir and paritaprevir (Supporting Fig. S12). The genotype 1a and 5a 155‐variants showed high resistance to paritaprevir (up to 70‐fold increase in EC50). The genotype 1a, 2a/b, 5a, and 6a 168‐variants as well as the genotype 3a 156‐variants showed the highest level of resistance to both PIs (up to 219‐fold and more than 423‐fold increase in EC50 for 168‐variants and 156‐variants, respectively). Given the low fitness and high resistance of 156‐variants, we concentrated on developing and characterizing viable 156‐variants for genotypes 1‐3, focusing on the 1a(TN), 2a(JFH1), and 3a(DBN) full‐length viruses, not harboring proteins of different genotypes.10, 12, 15
Persistence of A156T in Genotype 1A Escape Viruses Depended on Further Evolution Under PI Treatment Pressure
Although engineered 156‐RASs reverted (Supporting Fig. S12), 156‐RASs were found in the polyclonal escape viruses, suggesting stabilization by co‐selected fitness compensating substitutions. In an attempt to propagate 1a(TN) viruses with 156‐RASs, we passaged polyclonal escape viruses without PI treatment, resulting in a progressive reversion of A156T, whereas co‐selected substitutions did not revert, suggesting that the original 1a(TN) had been eradicated by PI treatment (Fig. 2A and Supporting Fig. S13).
Figure 2.

Persistence of 156‐RASs in HCV genotypes 1a and 2a was facilitated by further evolution under PI treatment. Haplotype frequencies were determined by NGS and substitution linkage analysis; haplotypes constituting more than 2% of the viral population are included in the bars; haplotypes with A156T/V/L constituting more than 0.5% of the viral population are included in values above the bars; haplotypes constituting more than 20% of the viral population are highlighted on the bars. (A) 1a(TN), escaping 64‐fold EC50 of grazoprevir (64 × esc.; Fig. 1B and Supporting Figs. S2 and S3) was passaged 3 times without treatment; nontreated first and third passage (NT1P, NT3P) were analyzed. (B) 1a(TN) escaping 64‐fold EC50 of grazoprevir was passaged 6 times under treatment with 64‐fold EC50 of grazoprevir and subsequently 6 times without treatment; treated third and sixth passage (64 × T3P, 64 × T6P) and nontreated ninth and twelfth passage (NT9P, NT12P) were analyzed. (C) 2a(JFH1) escaping 64‐fold EC50 of grazoprevir (different experiment than in Fig. 1B and Supporting Figs. S2 and S5) was passaged 3 times without treatment; nontreated third passage (NT3P) was analyzed (for 64 × esc., NGS was not successful; Sanger sequencing confirmed dominance of A156V). (D) 2a(JFH1) escaping 64‐fold EC50 of grazoprevir was passaged 6 times under treatment with 64‐fold EC50 of grazoprevir and subsequently 6 times without treatment; treated first, third, and sixth passage (64 × T1P, 64 × T3P, 64 × T6P) and nontreated ninth and twelfth passage (NT9P, NT12P) were analyzed. (E) 2a(JFH1) with engineered A156V, transfected in Huh7.5 cells, and escaping 64‐fold EC50 of grazoprevir was passaged as in (D); treated third and sixth passage (64 × T3P, 64 × T6P) and nontreated ninth and twelfth passage (NT9P, NT12P) were analyzed.
To stabilize 156‐RASs and putative compensatory substitutions, we passaged 1a(TN) escape variants under continued PI treatment pressure. During serial passage under grazoprevir, we observed an acceleration of viral spread kinetics; thus, on day 3 following infection, 1%, 10%, and 90% of culture cells were HCV‐infected in first, third, and sixth viral passage, respectively (data not shown). Following initial escape, the major 1a(TN) population carried A156T (Fig. 2B). In third and sixth passage under treatment, A156T was maintained in more than 90% of viral NS3P haplotypes, but additional NS3P substitutions emerged and viral populations with Y134C+A156T and A156T+D168E dominated. Following discontinuation of PI treatment, A156T showed long‐term persistence in more than 90% of viruses, mostly combined with D168E.
A Genome‐Wide Substitution Network Mediated Persistence of A156T in Genotype 1A Escape Variants by Multiple Mechanisms
Engineered 1a(TN)Y134C+A156T showed poor spread kinetics and reversion of A156T following transfection. 1a(TN)A156T+D168E spread like the original 1a(TN); A156T was maintained in first but not in third passage, where the NS3 helicase (NS3H) substitution V1656A was detected by Sanger sequencing (data not shown). Thus, the identified NS3P substitutions alone could not mediate persistence of A156T.
Genome‐wide NGS revealed that, induced by continued drug pressure, several ORF substitutions outside NS3P evolved in 1a(TN) escape virus populations from passage cultures (Supporting Fig. S14A). Evolutionary analysis using genome‐wide haplotype reconstruction based on SNP frequency development in serial viral passage suggested several lines of evolution. The viral population showing the highest prevalence at the end of the experiment (nontreated twelfth passage) evolved in three consecutive steps, termed evolutionary node 1 (N1, with acquisition of A156T, NS4BG1824D, NS5BN2651H, and NS5BE2860G), node 2 (N2, with additional acquisition of NS3HV1656A), and node 3 (N3, with additional acquisition of D168E in NS3P) (cyan in Fig. 3A). Alternative lines of evolution, branching off at N1 (purple in Fig. 3A) and N2 (blue in Fig. 3A) resulted in viral populations showing lower prevalence at the end of the experiment. Subsequently, these lines of evolution were confirmed by analysis of subclones of full ORF amplicons (Supporting Results and Supporting Fig. S15).
Figure 3.
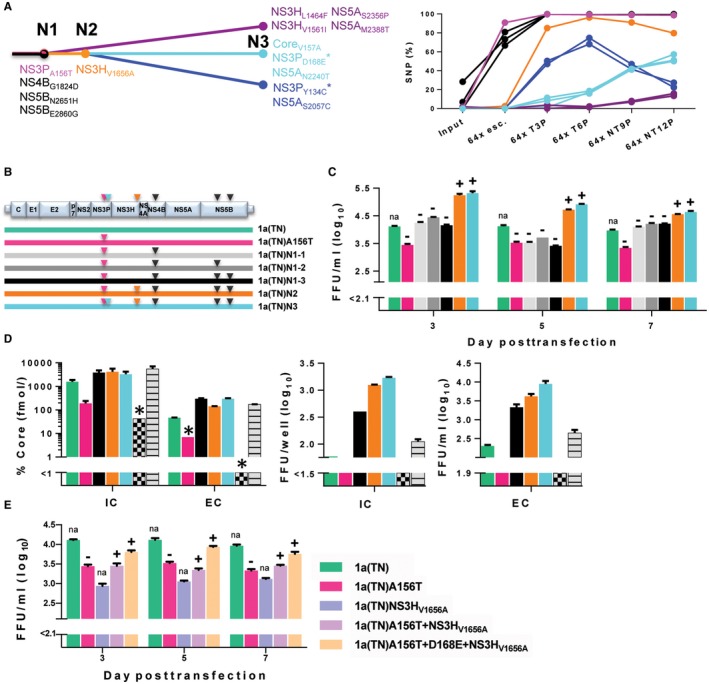
Evolution of a multimechanistic genome‐wide substitution network facilitated the persistence of A156T for HCV genotype 1a. (A) Genome‐wide linkage analysis of coding substitutions evolving in the 1a(TN) population during passage with/without treatment (Fig. 2B), based on SNP frequency development in serial viral passage (Supporting Fig. S14A). Abbreviations: N, node (major step in evolution). *NS3P linkage analysis confirmed linkage of the indicated substitution to A156T. NS4BG1824D, NS5BN2651H, and NS5BE2860G are linked. NS3HV1656A is linked to the substitution groups in blue and cyan, but not in purple. (B) Schematic overview of the 1a(TN) genome and engineered 1a(TN) recombinants. (C) Engineered recombinants were transfected in Huh7.5 cells; extracellular infectivity titers given as focus forming units per milliliter (FFU/ml) are means of triplicates with SEM. (‐) A156T reverted after first or (+) persisted after second passage. (D) Engineered recombinants were transfected in CD81‐deficient S29‐cells. Intracellular (IC) and extracellular (EC) Core levels (percentage, relative to Core concentration determined 4 hours following transfection) and intracellular infectivity titers (FFU/ml) are means of duplicates; extracellular infectivity titers (FFU/ml) are means of triplicates with SEM. *Single determinations. The bar colors in (C) and (D) match the colors of recombinants in (B). 2a(JFH1)‐GND (gray checkerboard) and 2a(JFH1) (gray stripes) are negative and positive controls, respectively. (E) 1a(TN) recombinants with NS3 substitutions engineered as indicated in the legend were transfected in Huh7.5 cells in the same experiment as the recombinants in (C); 1a(TN) and 1a(TN)A156T are identical in (C) and (E); extracellular infectivity titers (FFU/ml) are means of triplicates with SEM. (‐) A156T reverted after first or (+) persisted after second passage. Abbreviations: FFU/ml, focus forming units per milliliter; na, not applicable.
To model the evolution of N3 viruses, we constructed five 1a(TN)A156T recombinants: (1) three with different combinations of N1 substitutions in NS4B and NS5B, termed 1a(TN)N1‐1, 1a(TN)N1‐2, and 1a(TN)N1‐3; (2) one containing in addition the N2 substitution NS3HV1656A, termed 1a(TN)N2; and (3) one containing in addition the N3 substitution D168E in NS3P termed 1a(TN)N3 (Fig. 3B). Stepwise addition of these substitutions mediated an incremental increase in viral fitness, with 1a(TN)N3 showing accelerated spread kinetics and infectivity titers exceeding those of the original 1a(TN) by up to 1.2 log10 focus forming units per milliliter (Fig. 3C). In contrast to the three 1a(TN)N1 viruses, A156T persisted in 1a(TN)N2 and 1a(TN)N3 without acquisition of additional ORF substitutions.
In single‐cycle infections, allowing evaluation of the impact of genetic modifications on individual steps of the HCV life cycle,18 1a(TN)A156T showed decreased levels of intracellular Core protein compared with 1a(TN), indicating decreased replication (Fig. 3D).25 Addition of the node 1 substitutions in NS4B and NS5B restored replication, suggested by 1a(TN)N1‐3 reaching intracellular Core levels higher than those of 1a(TN). Addition of NS3HV1656A and D168E increased assembly of infectious intracellular viruses, indicated by 1a(TN)N2 and 1a(TN)N3 reaching higher intracellular infectivity titers than 1a(TN)N1‐3 (Fig. 3D).
To study whether second site substitutions in NS3 were sufficient to mediate A156T persistence, we engineered 1a(TN) with A156T+NS3HV1656A, A156T+D168E+NS3HV1656A, and for comparison, NS3HV1656A, all showing lower infectivity titers than 1a(TN), 1a(TN)N2, and 1a(TN)N3 (Fig. 3E). A156T persisted in 1a(TN)A156T+NS3HV1656A and 1a(TN)A156T+D168E+NS3HV1656A, which both acquired NS4BG1824D.
IN Genotype 2A Escape Variants, Evolution of Persistent A156L was Mediated by Initial Acquisition of A156V Associated With ORF Substitutions
Following initial escape, A156V dominated in 2a(JFH1) escape viruses, but reverted following passage without PI treatment (Fig. 2C and data not shown). Following one passage under drug pressure, the major 2a(JFH1) population carried A156V, whereas a minor population carried A156L (Fig. 2D). During subsequent passages, first with and then without treatment, A156L became the dominant substitution (Fig. 2D and Supporting Fig. S14B). Similar observations were made in two additional experiments (data not shown). In a parallel approach, we transfected and passaged engineered 2a(JFH1)A156V. During six passages under treatment and in the subsequent three drug‐free passages, more than 90% of the viral sequences maintained A156V, mostly in combination with D168E (Fig. 2E and Supporting Fig. S14D); after three further drug‐free passages, only 79% of viruses maintained A156V. In a replicate experiment, the engineered 156V changed to L (data not shown). We engineered 2a(JFH1)A156L, which showed spread kinetics and infectivity titers comparable to that of 2a(JFH1) and maintained A156L through three drug‐free passages (Supporting Fig. S14E and data not shown). For comparison we also engineered 1a(TN)A156L and 3a(DBN)A156L. Both viruses maintained A156L without drug pressure (Supporting Fig. S14E). Compared with the respective original viruses, 3a(DBN)A156L showed comparable spread, but 1a(TN)A156L showed severely impaired spread kinetics (data not shown).
As observed for 1a(TN) (Supporting Fig. S14A), genome‐wide NGS showed acquisition of various substitutions in the ORF of 2a(JFH1) escape viruses acquiring A156V with subsequent transition to A156L (Supporting Fig. S14B) and in engineered 2a(JFH1)A156V passaged under drug pressure (Supporting Fig. S14D). In contrast, 3a(DBN) escape viruses with A156V, engineered 2a(JFH1)A156L, and engineered 3a(DBN)A156L maintained 156‐RASs with no obvious selection of other ORF substitutions (Supporting Fig. S14C,E). 1a(TN)A156L acquired NS3HV1656A and NS4BG1824D, also found in passaged 1a(TN) escape variants (Supporting Fig. S14E).
A156T, A156V, and A156L RASs Conferred High Resistance for Genotypes 1‐3 Across Clinically Relevant PIs
Resistance testing showed that the developed genotype 1‐3 recombinants and polyclonal virus stocks with 156‐RASs were highly resistant to grazoprevir and paritaprevir (Fig. 4). We recently acquired the PIs glecaprevir and voxilaprevir.20 As previously observed, compared with grazoprevir and especially paritaprevir, these PIs showed increased efficacy against the original genotype 1‐3 viruses.20 However, all tested 156‐variants were highly resistant to glecaprevir and voxilaprevir (Fig. 4).
Figure 4.
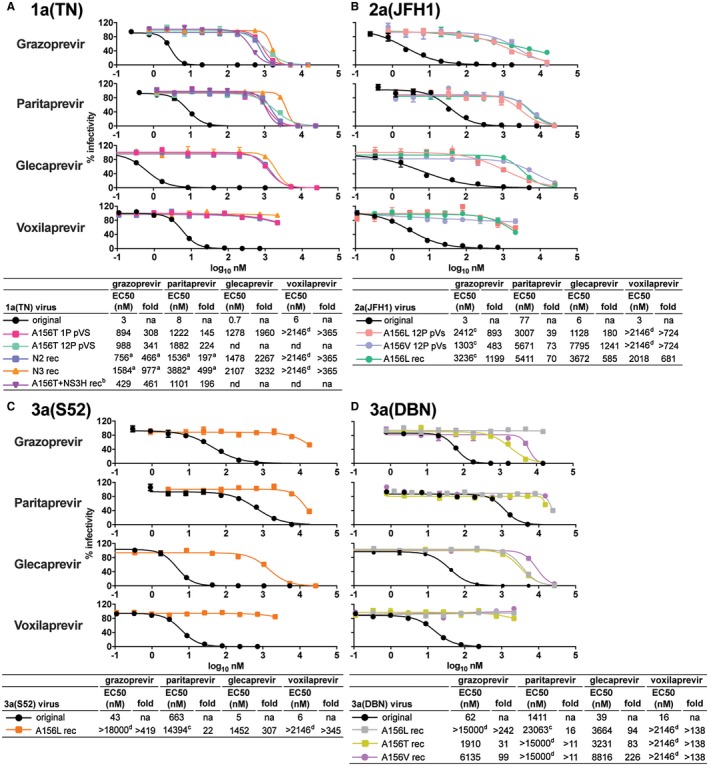
NS3P variants with 156‐RASs showed high PI resistance. For 1a(TN) (A), 2a(JFH1) (B), 3a(S52) (C), and 3a(DBN) (D) 156‐variants, PI concentration‐response experiments were carried out using grazoprevir, paritaprevir, glecaprevir, and voxilaprevir as described in the Materials and Methods section. Fold‐resistance values were calculated by relating EC50 of the indicated variants to that of the original viruses included in the same experiment; numbers are rounded off. When possible, 156‐variants were engineered as recombinants (rec) and first‐passage virus stocks were used. For 1a(TN)N2 rec, 1a(TN)N3 rec, and 1a(TN)A156T+NS3H rec (including NS3HV1656A), second‐passage virus stocks were used (Fig. 3). For 3a(S52)A156L rec, 3a(DBN)A156T rec, and 3a(DBN)A156V rec, NS3P (Supporting Fig. S12), and for 2a(JFH1)A156L rec and 3a(DBN)A156L rec (Supporting Fig. S14E), the complete ORF was sequenced. Otherwise, 156‐variants were grown as polyclonal virus stocks (pVS) from nontreated first passages (NT1P) or from nontreated twelfth passages (NT12P) of viruses from grazoprevir escape experiments (Fig. 2A,B,D,E). Abbreviations: na, not applicable; nd, not done. aResults were from a separate experiment in which the EC50s for 1a(TN) were 2 nM (grazoprevir) and 8 nM (paritaprevir). bResults were from a separate experiment in which EC50s for 1a(TN) were 1 nM (grazoprevir) and 6 nM (paritaprevir). cViruses were not fully inhibited by the highest PI concentration tested; EC50s are estimates given by GraphPad Prism. dViruses were not inhibited by at least 50% by the highest PI concentration tested; EC50s could not be estimated.
156‐RASs Mediated Escape of 1A(TN) From the PIs Glecaprevir and Voxilaprevir
In 1a(TN) viruses escaping from glecaprevir and voxilaprevir A156T/V dominated (Supporting Fig. S16A,B), without clear PI concentration‐dependent patterns as observed under grazoprevir and paritaprevir (Fig. 1B and Supporting Figs. S3‐S11). Genome‐wide NGS of escape viruses demonstrated co‐selection of node 1 (NS4BG1824D and NS5BN2651H) and node 2 (NS3HV1656A) substitutions of the previously identified substitution network (Supporting Fig. S16C).
Pre‐Existing A156T Facilitated Genotype 1 Escape From Combination Treatment
We investigated the effect of clinically relevant combination treatments with PI/NS5A inhibitor (grazoprevir/elbasvir or glecaprevir/pibrentasvir) and PI/NS5A inhibitor/NS5B inhibitor (voxilaprevir/velpatasvir/sofosbuvir) on 1a(TN)N3 harboring A156T and D168E in NS3P. In contrast to NS5A inhibitor mono‐treatments, PI mono‐treatments did not affect 1a(TN)N3 infection (Fig. 5A‐C). Furthermore, 1a(TN)N3 readily escaped both double treatments at concentrations sufficient to control the original 1a(TN) (Fig. 5A,B). Using similar concentrations, triple treatment suppressed 1a(TN) but only controlled 1a(TN)N3 (Fig. 5C). Double‐treatment escape variants acquired the NS5A‐RASs M28T or Y93H (Fig. 5D). Following termination of triple treatment, 1a(TN)N3 spread and acquired the NS5A‐RAS L31V (Fig. 5D).
Figure 5.
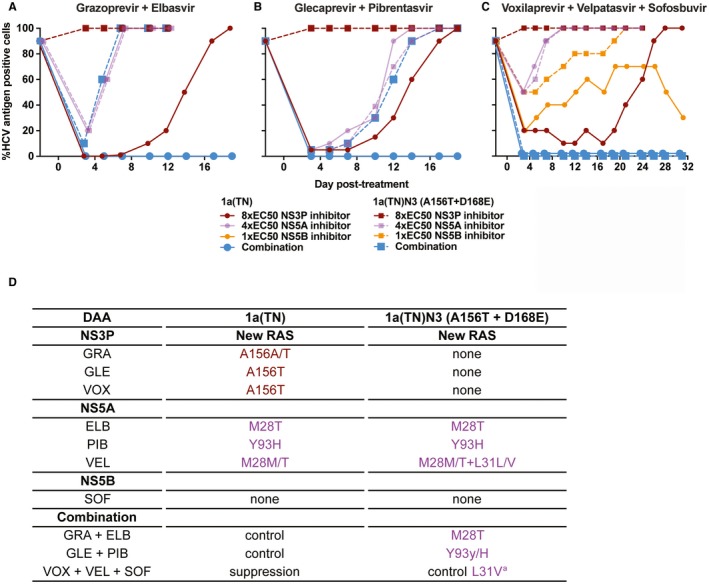
Pre‐existing A156T facilitated 1a(TN) escape from combination treatment. 1a(TN)N3 harboring A156T and D168E as well as the original 1a(TN) were subjected to mono or combination treatment with grazoprevir and elbasvir (A) or glecaprevir and pibrentasvir (B) or mono or triple treatment with voxilaprevir, velpatasvir, and sofosbuvir (C) at the indicated fold‐EC50 until viral escape, control, or suppression occurred (see Materials and Methods section). Viral control occurred for 1a(TN) under double treatment and for 1a(TN)N3 under triple treatment. Viral suppression occurred for 1a(TN) under triple treatment. (D) Sanger sequencing of DAA targets of escape variants revealed acquisition of additional RASs in NS3P (brown) and NS5A domain I (purple). aThe RAS L31V in NS5A was detected following viral spread to most of the culture cells on day 45 (data not shown), following treatment termination on day 31.
Analysis oF Genotype 1A Patient Isolates Harboring 156‐RASs at Treatment Failure Revealed Co‐Selection of Other ORF Substitutions
We studied samples from 5 genotype 1a infected patients with 156‐RASs following treatment failure (patients A to E, Fig. 6A‐E). In patients B and D with A156V, 63.7% and 8.6% of NS3P haplotypes, respectively, showed the original sequence, suggesting relatively low stability of the viral population harboring A156V. In contrast, in patient A, A156V, and in patients C and E, A156G was found in 100% of NS3P haplotypes. At least for patient C, in whom the posttreatment sample was obtained several months after treatment termination, this suggested a relatively high stability of the viral population with A156G. Genome‐wide NGS of paired samples obtained before and after treatment (patients A, B, and C) revealed that under treatment various other ORF substitutions were selected, including substitutions selected de novo. Analysis of positions mediating persistence of A156T in vitro (Fig. 3) revealed de novo substitutions at NS3P position 168 in patients B and C. Other de novo substitutions localized to NS3H (patients B and C), NS4B (patient C), and NS5B (patients A and C). NS3HT1475I and NS3HC1551S (patient B) and NS3HS1579C (patient C) localized close to NS3HL1464F and NS3HV1561I described for 1a(TN) (Fig. 3A, purple line of evolution) and were likely linked to A156V and D168E (Supporting Fig. S17). All posttreatment sequences harbored RASs in all DAA targets. For further viral population analysis of genome‐wide NGS data, see the Supporting Results.
Figure 6.
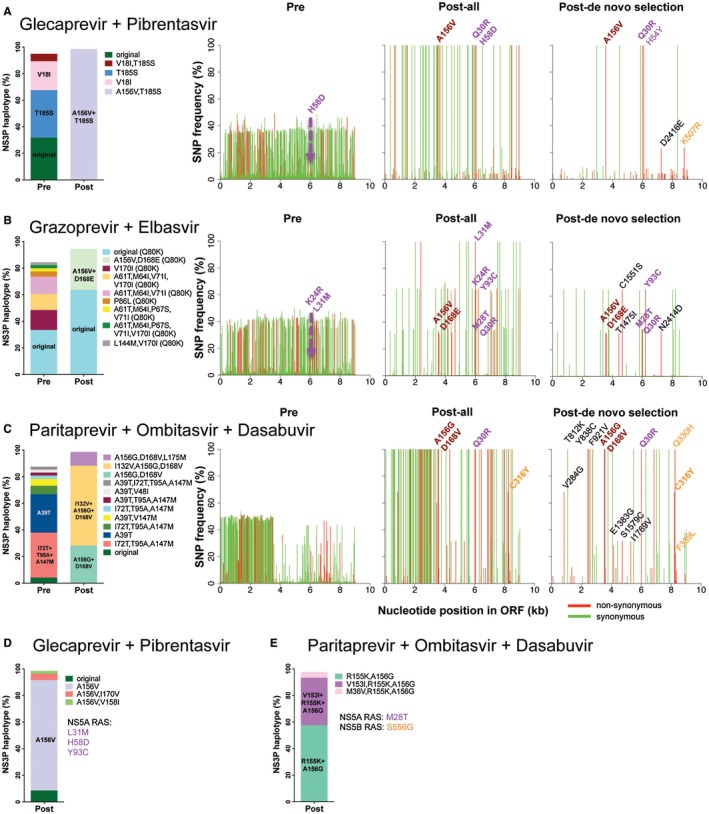
Analysis of HCV sequences of genotype 1a–infected patients failing DAA treatment and harboring A156G or V. Patients A, B, C, D, and E were treated with glecaprevir + piprentasvir (A,D), grazoprevir + elbasvir (B), or paritaprevir + ombitasvir + dasabuvir (C,E). Posttreatment samples were obtained at the given month following end of treatment: patient A, 0 months; patient B, 4.8 months; patient C, 4.6 months; patient D, 3.1 months; and patient E, information not available. Bar graphs to the left in each panel show NS3P haplotype frequencies determined by NGS and substitution linkage analysis before treatment (pre) and after treatment (post). Haplotypes constituting more than 2% of the viral population are included in the bars; haplotypes constituting more than 20% of the viral population are highlighted in the bars. Patient B had the NS3P RAS Q80K in all haplotypes detected before and after treatment. In (A‐C), original refers to the pretreatment consensus sequence. In (D), original refers to the consensus sequence without A156V. The three line graphs to the right in (A‐C) show ORF‐wide NGS, revealing SNP frequencies along the ORF. Pre, SNP frequencies in pretreatment sequences mapped against their consensus sequence (known RASs occurring in more than 0.5% of reads are specified); Post‐all, SNP frequencies in posttreatment sequences mapped to the pretreatment consensus sequence (known RASs occurring in greater than 20% of reads are specified); Post de novo selection, frequencies of de novo selected SNPs, found in less than 0.5% of reads in pretretament sequences, mapped to the pretreatment consensus sequence (all substitutions occurring in greater than 20% of reads are specified). Known RASs are indicated in bold for NS3P using relative NS3P numbers (brown), for NS5A using relative NS5A numbers (purple), and for NS5B using relative NS5B numbers (orange). Other de novo SNPs outside the drug targets are in regular font (black), with numbers relating to the polyprotein of the H77 reference strain (GenBank identifier AF009606).
Discussion
We elucidated pathways of HCV PI resistance in unprecedented detail. PI resistance profiling confirmed NS3P position 156 as a hotspot for RASs for genotypes 1‐4, but not 5 and 6. However, most identified 156‐RASs had high fitness costs, except for genotype 3. For genotypes 1 and 2, persistence of 156‐RASs depended on dynamic co‐evolution of multi‐mechanistic genome‐wide substitution networks. We developed highly fit genotype 1‐3 156‐variants showing high resistance to grazoprevir, paritaprevir, glecaprevir and voxilaprevir, readily escaping from PI/NS5A inhibitor combination treatments. In line with our in vitro findings, selection of 156‐RASs in genotype 1–infected patients experiencing DAA treatment failure was accompanied by selection of substitutions outside the drug targets.
Although antiviral resistance is a severe public health concern, the understanding of underlying mechanisms is limited.26, 27 A recently developed technique for NGS‐based substitution linkage and haplotype reconstruction allowed us to explore the nature and frequency of PI resistance–associated NS3P haplotypes in unprecedented detail.20, 28 In addition, by analyzing viruses longitudinally during evolution under drug pressure, we suggest a strategy for genome‐wide linkage analysis and haplotype reconstruction, which was validated by subclonal analysis including ancestral reconstruction.
In vitro testing is required for characterization of RASs. In constrast to enzyme and replicon assays, infectious HCV culture systems allow investigation of the complete genome and viral life cycle, and results obtained in such systems reflect in vivo data.5, 6, 7, 8, 9, 10, 11, 13, 14, 15, 16, 17, 20, 21, 22, 24, 25, 29, 30
Most previous studies focused on the characterization of RASs for genotype 1.7 While this study used genotype 1‐6 infectious culture systems for resistance profiling of grazoprevir and paritaprevir, selected RASs were in general comparable to those previously selected in replicons and in patients.5, 6, 7, 8, 31, 32, 33, 34, 35, 36, 37
In line with previous findings, we found genotype‐specific NS3P RAS selection.25, 30 Also, in contrast to genotype 3, for genotypes 1 and 2 persistence of A156T/V depended on co‐selection of genome‐wide substitution networks. For genotype 2, initial acquisition of A156V, with high resistance but low fitness, relied on co‐selection of other ORF substitutions and faciliated acquisition of A156L, with increased resistance and high fitness, without dependence on additional ORF substitutions. For all viruses, acquisition of A156L required two nucleotide changes, whereas the initial acquisition of A156V and transition from A156V to A156L each required one nucleotide change. 1a(TN) did not acquire A156L, possibly due to a bias toward initial acquisition of A156T and the transition from A156T to A156L requiring two nucleotide changes. Furthermore, in contrast to 2a(JFH1)A156L, 3a(S52)A156L and 3a(DBN)A156L, 1a(TN)A156L showed poor fitness and co‐selection of other ORF substitutions. In contrast to A156V/T, A156L was to our knowledge not described in patients,5, 6, 7, 8 possibly because PI‐containing DAA combinations available until recently were not recommended for treatment of genotypes 2 and 3.
Although patients failing DAA combination treatment often have RASs in several drug targets, NS3P positions 156 and 168 are hotspots for PI resistance.3, 5, 6, 7 In line with recent findings in replicons and patients,6 we demonstrated that 156‐RASs were essential for resistance to glecaprevir and voxilaprevir for genotype 1a (this study and Pham et al.20) and genotype 3a.20 In contrast to grazoprevir and paritaprevir, no concentration‐dependent RAS selection patterns, including RASs at other NS3P positions than at position 156, were observed for glecaprevir and voxilaprevir.20 This suggested that NS3P position 156 is the only position mediating major resistance to these PIs. In accordance with our findings, Ng et al. reported that A156T selected under glecaprevir in the 1a(H77) subgenomic replicon resulted in high resistance and severe reduction in replication efficacy.38 However, fitness‐compensating substitutions were not identified and effects on viral assembly, as described in our study, cannot be studied in replicons. We further demonstrated that pre‐existing 156‐RASs faciliated HCV escape from clinically relevant combination treatments with selection of double‐resistant escape variants.
Selection of substitutions at positions mediating persistence of A156T in 1a(TN) was neither PI nor HCV isolate‐specific. Thus, substitutions at these positions were co‐selected with 156‐RASs under grazoprevir, glecaprevir, and voxilaprevir in 1a(TN), 1a(HCV1), 2a(JFH1), 3a(S52), and 3a(DBN) (Supporting Fig. S16C) (unpublished data).20 NS4BG1824D and NS5BN2651H, increasing viral replication, might have a general fitness‐enhancing effect, as they were also detected in the 1a(TN) stock used for inoculation of initial escape experiments and selected in 1a(TN) serially pasaged without treatment (unpublished data). NS3HV1656A, mediating A156T persistence and increasing assembly of infectious viruses, might mediate interactions between NS3P and NS3H,39, 40 as it was not only acquired by 156‐escape variants, but also by engineered 1a(TN)A156T+D168E and 1a(TN)A156L, and because NS3HV1656A alone decreased the fitness of 1a(TN).
Although position 156 is highly conserved across genotypes, observed genotype‐specific differences regarding selection of 156‐RASs might be explained by differences at positions of importance for compensation of fitness costs. In 1a(TN), NS3P substitutions H110D, Y134C/H/N, and D168E/G were selected under grazoprevir, Y134C and D168E being co‐selected with A156T. H110D/Y134C/D168Q and H110N/Y134T are natural polymorphisms for genotypes 3a and 2a, respectively. In addition, all other positions of the identified substitution network in NS3H, NS4B, and NS5B showed different aa residues for genotypes 3a and 2a compared with genotype 1a (Supporting Fig. S18). Genotype 3a is difficult to treat with DAAs, due to its relatively low sensititivy to several PIs and NS5A inhibitors.7, 14, 16, 20, 29, 41 Our work suggests that an additional reason might be its greater propensity to develop resistance to PIs and sofosbuvir (this study and Ramirez et al.15).
Positions of the identified 1a(TN) substitution network were highly conserved in genotype 1a ORF sequences from databases42 (Supporting Fig. S18) and in 24 treatment‐naïve Danish genotype 1a–infected patients, analyzed by NGS (Supporting Table S6). Except at NS5B2651, the original 1a(TN) had the most prevalent aa residue; therefore, rare aa residues were selected in the substitution network mediating A156T persistence.
In databases,42 only a few sequences with 156‐RASs were available. Most genotype 1a sequences with 156‐RASs originated from clones from 2 patients treated for several days with grazoprevir and showing fast reversion of 156‐RASs following treatment.43 Given the short treatment duration, it was not surprising that there was no indication for substitutions at positions mediating 156‐RAS persistence in vitro.
Patterns of resistance in examined isolates acquiring 156‐RASs were similar to those observed in vitro. For patients with pretreatment and posttreatment samples, substitutions were acquired based on pre‐existing polymorphisms (as for 1a(TN) NS4BG1824D and NS5BN2651H) or de novo (as for the other substitutions of the identified 1a(TN) substitution network). Several de novo selected substitutions localized to NS3P position 168 as well as to NS3H, NS4B and NS5B, as described in vitro. Some NS3H substitutions localized close to 1a(TN) NS3H substitutions, introducing rare aa residues at otherwise highly conserved positions. Differences between specific substitutions selected in vitro and in vivo might be due to clinical isolates acquiring A156G/V, but not A156T, or due to 156‐RAS stabilizing substitution networks being selected under DAA combination treatment but not PI monotherapy. Finally, different alternatives for 156‐RAS stabilizing substitution networks appear to exist.
Strikingly, the fitness of highly resistant engineered genotype 1 and 2 156‐variants matched and even exceeded the fitness of the original viruses, rendering them among the HCV recombinants with the highest fitness described so far. We recently made similar observations for sofosbuvir‐resistant genotype 3a‐ and 6a‐variants.15, 24 Thus, in contrast to current perceptions, resistance might in some cases be associated with increased viral fitness.
With the development of highly fit 156‐variants for the most prevalent HCV genotypes, we have overcome a roadblock for the study of 156‐RASs, including efforts to identify PIs with efficacy against 156‐variants.
We provided evidence that evolution of highly fit and stable 156‐RAS variants was possible through selection of fitness‐compensating substitution networks and reduced efficacy of the most advanced DAA combination treatments in vitro. So far, an excellent correlation has been observed between in vitro HCV‐resistance studies and clinical data. As the number of patients treated with DAAs is increasing, it might become more likely that 156‐variants as described in this in vitro study are also observed in the clinic, and thus could pose a threat to the efficacy of DAA combination treatments.
Supporting information
Acknowledgments
We thank Anna‐Louise Sørensen and Lotte Mikkelsen (Copenhagen Univeristy Hospital, Hvidovre) for general laboratory support and technical contributions; Bjarne Ø. Lindhardt (Copenhagen University Hospital, Hvidovre) and Carsten Geisler (University of Copenhagen) for study support; John McLauchlan and Josh Singer (University of Glasgow) for review of sequences in local data collections and in the HCV‐GLUE database; Charles Rice (Rockefeller University), Robert Purcell and Suzanne Emerson (NIH), and Takaji Wakita (National institute for Infectious Diseases, Tokyo) for providing reagents; and Gilead Sciences for providing voxilaprevir.
Supported by the Danish Council for Independent Research–Medical Sciences (S.R., D.H., J.B., and J.M.G.), including a Sapere Aude advanced‐top researcher grant (J.B.); Region Hovedstadens Forskningsfond (S.R., J.B., and J.M.G.); the Lundbeck Foundation (S.R. and J.B.); the Novo Nordisk Foundation (J.B. and J.M.G.), including the Novo Nordisk Prize (J.B.), the Candys Foundation (L.V.P., A.F.P., J.B., and J.M.G), and the Innovation Fund Denmark (J.B.); Agence National de Recherche sur le SIDA et les hépatites virales (S.F. and J.‐M.P.); German Center of Infection Research, TTU Hepatitis (J.D and C.S.); Ph.D. stipends and/or bonuses from the Faculty of Health and Medical Sciences, Copenhagen University to S.B.J., L.V.P., S.B.N.S., A.F.P., J.F., and C.S.S (J.B. and J.M.G).
Potential conflict of interest: Dr. Weis advises and is on the speakers’ bureau for Bristol‐Myers Squibb, MSD, and AbbVie; she advises for GSK and is on the speakers’ bureau for Gilead. Dr. Fourati consults for AbbVie. Dr. Pawlotsky received research grants from Abbott and Abbvie; he acted as an advisor for Abbott, Abbvie, Gilead, Merck and Siemens Healthcare. Dr. Sarrazin is a speaker and advisor for AbbVie, Gilead and MSD.
References
Author names in bold designate shared co‐first authorship.
- 1. Polaris Observatory HCV Collaborators . Global prevalence and genotype distribution of hepatitis C virus infection in 2015: a modelling study. Lancet Gastroenterol Hepatol 2017;2:161‐176. [DOI] [PubMed] [Google Scholar]
- 2. Bukh J. The history of hepatitis C virus (HCV): basic research reveals unique features in phylogeny, evolution and the viral life cycle with new perspectives for epidemic control. J Hepatol 2016;65:S2‐S21. [DOI] [PubMed] [Google Scholar]
- 3. Pawlotsky JM, Negro F, Aghemo A, Berenguer M, Dalgard O, Dusheiko G, et al. EASL recommendations on treatment of hepatitis C 2018. J Hepatol 2018;69:461‐511. [DOI] [PubMed] [Google Scholar]
- 4. AASLD‐IDSA . HCV guidance: recommendations for testing, managing, and treating hepatitis C. https://www%20hcvguidelines%20org/. Accessed March 11, 2019.
- 5. Pawlotsky JM. Hepatitis C virus resistance to direct‐acting antiviral drugs in interferon‐free regimens. Gastroenterology 2016;151:70‐86. [DOI] [PubMed] [Google Scholar]
- 6. Sorbo MC, Cento V, Di Maio VC, Howe AYM, Garcia F, Perno CF, et al. Hepatitis C virus drug resistance associated substitutions and their clinical relevance: update 2018. Drug Resist Updat 2018;37:17‐39. [DOI] [PubMed] [Google Scholar]
- 7. Sarrazin C. The importance of resistance to direct antiviral drugs in HCV infection in clinical practice. J Hepatol 2016;64:486‐504. [DOI] [PubMed] [Google Scholar]
- 8. Dietz J, Susser S, Vermehren J, Peiffer KH, Grammatikos G, Berger A, et al. Patterns of resistance‐associated substitutions in patients with chronic HCV infection following treatment with direct‐acting antivirals. Gastroenterology 2018;154:976‐988. [DOI] [PubMed] [Google Scholar]
- 9. Shimakami T, Welsch C, Yamane D, McGivern DR, Yi M, Zeuzem S, et al. Protease inhibitor‐resistant hepatitis C virus mutants with reduced fitness from impaired production of infectious virus. Gastroenterology 2011;140:667‐675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li YP, Ramirez S, Jensen SB, Purcell RH, Gottwein JM, Bukh J. Highly efficient full‐length hepatitis C virus genotype 1 (strain TN) infectious culture system. Proc Natl Acad Sci U S A 2012;109:19757‐19762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pham LV, Ramirez S, Carlsen THR, Li YP, Gottwein JM, Bukh J. Efficient hepatitis C virus genotype 1b core‐NS5A recombinants permit efficacy testing of protease and NS5A inhibitors. Antimicrob Agents Chemother 2017;61:e00037‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, et al. Complete replication of hepatitis C virus in cell culture. Science 2005;309:623‐626. [DOI] [PubMed] [Google Scholar]
- 13. Ramirez S, Li YP, Jensen SB, Pedersen J, Gottwein JM, Bukh J. Highly efficient infectious cell culture of three hepatitis C virus genotype 2b strains and sensitivity to lead protease, nonstructural protein 5A, and polymerase inhibitors. Hepatology 2014;59:395‐407. [DOI] [PubMed] [Google Scholar]
- 14. Li YP, Ramirez S, Humes D, Jensen SB, Gottwein JM, Bukh J. Differential sensitivity of 5'UTR‐NS5A recombinants of hepatitis C virus genotypes 1–6 to protease and NS5A inhibitors. Gastroenterology 2014;146:812‐821. [DOI] [PubMed] [Google Scholar]
- 15. Ramirez S, Mikkelsen LS, Gottwein JM, Bukh J. Robust HCV genotype 3a infectious cell culture system permits identification of escape variants with resistance to sofosbuvir. Gastroenterology 2016;151:973‐985. [DOI] [PubMed] [Google Scholar]
- 16. Gottwein JM, Scheel TK, Jensen TB, Ghanem L, Bukh J. Differential efficacy of protease inhibitors against HCV genotypes 2a, 3a, 5a, and 6a NS3/4A protease recombinant viruses. Gastroenterology 2011;141:1067‐1079. [DOI] [PubMed] [Google Scholar]
- 17. Jensen SB, Serre SB, Humes DG, Ramirez S, Li YP, Bukh J, et al. Substitutions at NS3 residue 155, 156, or 168 of hepatitis C virus genotypes 2 to 6 induce complex patterns of protease inhibitor resistance. Antimicrob Agents Chemother 2015;59:7426‐7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Russell RS, Meunier JC, Takikawa S, Faulk K, Engle RE, Bukh J, et al. Advantages of a single‐cycle production assay to study cell culture‐adaptive mutations of hepatitis C virus. Proc Natl Acad Sci U S A 2008;105:4370‐4375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Serre SB, Krarup HB, Bukh J, Gottwein JM. Identification of alpha interferon‐induced envelope mutations of hepatitis C virus in vitro associated with increased viral fitness and interferon resistance. J Virol 2013;87:12776‐12793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pham LV, Jensen SB, Fahnoe U, Pedersen MS, Tang Q, Ghanem L, et al. HCV genotype 1–6 NS3 residue 80 substitutions impact protease inhibitor activity and promote viral escape. J Hepatol 2019;70:388‐397. [DOI] [PubMed] [Google Scholar]
- 21. Solund C, Andersen ES, Mossner B, Laursen AL, Roge BT, Kjaer MS, et al. Outcome and adverse events in patients with chronic hepatitis C treated with direct‐acting antivirals: a clinical randomized study. Eur J Gastroenterol Hepatol 2018;30:1177‐1186. [DOI] [PubMed] [Google Scholar]
- 22. Solund C, Hallager S, Pedersen MS, Fahnoe U, Ernst A, Krarup HB, et al. Direct acting antiviral treatment of chronic hepatitis C in Denmark: factors associated with and barriers to treatment initiation. Scand J Gastroenterol 2018;53:849‐856. [DOI] [PubMed] [Google Scholar]
- 23. Pedersen MS, Fahnoe U, Hansen TA, Pedersen AG, Jenssen H, Bukh J, et al. A near full‐length open reading frame next generation sequencing assay for genotyping and identification of resistance‐associated variants in hepatitis C virus. J Clin Virol 2018;105:49‐56. [DOI] [PubMed] [Google Scholar]
- 24. Pham LV, Ramirez S, Gottwein JM, Fahnoe U, Li YP, Pedersen J, et al. HCV genotype 6a escape from and resistance to velpatasvir, pibrentasvir, and sofosbuvir in robust infectious cell culture models. Gastroenterology 2018;154:2194‐2208. [DOI] [PubMed] [Google Scholar]
- 25. Serre SB, Jensen SB, Ghanem L, Humes DG, Ramirez S, Li YP, et al. Hepatitis C virus genotype 1 to 6 protease inhibitor escape variants: in vitro selection, fitness, and resistance patterns in the context of the infectious viral life cycle. Antimicrob Agents Chemother 2016;60:3563‐3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Irwin KK, Renzette N, Kowalik TF, Jensen JD. Antiviral drug resistance as an adaptive process. Virus Evol 2016;2:vew014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Margeridon‐Thermet S, Shafer RW. Comparison of the mechanisms of drug resistance among HIV, hepatitis B, and hepatitis C. Viruses 2010;2:2696‐2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wilker PR, Dinis JM, Starrett G, Imai M, Hatta M, Nelson CW, et al. Selection on haemagglutinin imposes a bottleneck during mammalian transmission of reassortant H5N1 influenza viruses. Nat Commun 2013;4:2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gottwein JM, Pham LV, Mikkelsen LS, Ghanem L, Ramirez S, Scheel TKH, et al. Efficacy of NS5A inhibitors against hepatitis C virus genotypes 1–7 and escape variants. Gastroenterology 2018;154:1435‐1448. [DOI] [PubMed] [Google Scholar]
- 30. Imhof I, Simmonds P. Genotype differences in susceptibility and resistance development of hepatitis C virus to protease inhibitors telaprevir (VX‐950) and danoprevir (ITMN‐191). Hepatology 2011;53:1090‐1099. [DOI] [PubMed] [Google Scholar]
- 31. Summa V, Ludmerer SW, McCauley JA, Fandozzi C, Burlein C, Claudio G, et al. MK‐5172, a selective inhibitor of hepatitis C virus NS3/4a protease with broad activity across genotypes and resistant variants. Antimicrob Agents Chemother 2012;56:4161‐4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pilot‐Matias T, Tripathi R, Cohen D, Gaultier I, Dekhtyar T, Lu L, et al. In vitro and in vivo antiviral activity and resistance profile of the hepatitis C virus NS3/4A protease inhibitor ABT‐450. Antimicrob Agents Chemother 2015;59:988‐997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Krishnan P, Tripathi R, Schnell G, Reisch T, Beyer J, Irvin M, et al. Resistance analysis of baseline and treatment‐emergent variants in hepatitis C virus genotype 1 in the AVIATOR study with paritaprevir‐ritonavir, ombitasvir, and dasabuvir. Antimicrob Agents Chemother 2015;59:5445‐5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gane E, Ben AZ, Mollison L, Zuckerman E, Bruck R, Baruch Y, et al. Efficacy and safety of grazoprevir + ribavirin for 12 or 24 weeks in treatment‐naive patients with hepatitis C virus genotype 1 infection. J Viral Hepat 2016;23:789‐797. [DOI] [PubMed] [Google Scholar]
- 35. Krishnan P, Schnell G, Tripathi R, Beyer J, Reisch T, Zhang X, et al. Analysis of hepatitis C virus genotype 1b resistance variants in japanese patients treated with paritaprevir‐ritonavir and ombitasvir. Antimicrob Agents Chemother 2016;60:1106‐1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jacobson IM, Lawitz E, Kwo PY, Hezode C, Peng CY, Howe AYM, et al. Safety and efficacy of elbasvir/grazoprevir in patients with hepatitis C virus infection and compensated cirrhosis: an integrated analysis. Gastroenterology 2017;152:1372‐1382. [DOI] [PubMed] [Google Scholar]
- 37. Buti M, Gordon SC, Zuckerman E, Lawitz E, Calleja JL, Hofer H, et al. Grazoprevir, elbasvir, and ribavirin for chronic hepatitis C virus genotype 1 infection after failure of pegylated interferon and ribavirin with an earlier‐generation protease inhibitor: final 24‐week results from C‐SALVAGE. Clin Infect Dis 2016;62:32‐36. [DOI] [PubMed] [Google Scholar]
- 38. Ng TI, Tripathi R, Reisch T, Lu L, Middleton T, Hopkins TA, et al. In vitro antiviral activity and resistance profile of the next‐generation hepatitis C virus NS3/4A protease inhibitor glecaprevir. Antimicrob Agents Chemother 2018;62:e01620‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ma Y, Yates J, Liang Y, Lemon SM, Yi M. NS3 helicase domains involved in infectious intracellular hepatitis C virus particle assembly. J Virol 2008;82:7624‐7639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Beran RK, Pyle AM. Hepatitis C viral NS3‐4A protease activity is enhanced by the NS3 helicase. J Biol Chem 2008;283:29929‐29937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Smith D, Magri A, Bonsall D, Ip CLC, Trebes A, Brown A, et al. Resistance analysis of genotype 3 hepatitis C virus indicates subtypes inherently resistant to nonstructural protein 5A inhibitors. Hepatology 2019;69:1861‐1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kuiken C, Yusim K, Boykin L, Richardson R. The Los Alamos hepatitis C sequence database. Bioinformatics 2005;21:379‐384. [DOI] [PubMed] [Google Scholar]
- 43. Ke R, Li H, Wang S, Ding W, Ribeiro RM, Giorgi EE, et al. Superinfection and cure of infected cells as mechanisms for hepatitis C virus adaptation and persistence. Proc Natl Acad Sci U S A 2018;115:E7139‐E7148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials