

Abstract
Aim
To assess the efficacy and safety of once‐daily ipragliflozin 50 mg versus placebo in Japanese people with type 1 diabetes mellitus (T1DM) inadequately controlled with insulin.
Materials and methods
We conducted a multicentre, double‐blind, parallel‐group, placebo‐controlled phase 3 study. Participants (N = 175) were randomized (2:1) to receive once‐daily ipragliflozin 50 mg (n = 115) or placebo (n = 60), combined with insulin, for 24 weeks. The primary endpoint was change in glycated haemoglobin (HbA1c); key secondary endpoints included change in insulin dose and body weight. Treatment‐emergent adverse events (TEAEs) were evaluated.
Results
The ipragliflozin group demonstrated a significant decrease in HbA1c from baseline to end of treatment versus the placebo group: adjusted mean difference (AMD) −3.8 mmol/mol (95% confidence interval [CI] −6.2, −1.5) or − 0.36% (95% CI −0.57, −0.14; P = 0.001). Significant reductions in total daily insulin dose (AMD −7.35 IU [95% CI −9.09, −5.61]; P < 0.001) and body weight (AMD −2.87 kg [95% CI −3.58, −2.16]; P < 0.001) were observed for the ipragliflozin group versus placebo. Two serious TEAEs occurred (major hypoglycaemia and abdominal abscess); both were in the placebo group. All other TEAEs were mild or moderate in severity. Four cases of study discontinuation occurred; three in the placebo group and one in the ipragliflozin group. No diabetic ketoacidosis was reported for any participant in this study.
Conclusions
Daily ipragliflozin 50 mg in combination with insulin significantly reduced HbA1c, daily insulin dose and body weight versus placebo in people with T1DM. No safety concerns were identified after 24 weeks of treatment. Overall, once‐daily ipragliflozin 50 mg was both efficacious and well tolerated.
Keywords: antidiabetic drug, clinical trial, insulin therapy, phase III study, SGLT2 inhibitor, type 1 diabetes
1. INTRODUCTION
Type 1 diabetes mellitus (T1DM) develops mainly because of autoimmune destruction of insulin‐producing pancreatic β cells, resulting in hyperglycaemia.1, 2 Mean glycated haemoglobin (HbA1c) concentration tends to be higher in people with T1DM (~61 mmol/mol [7.82%]) than those with type 2 diabetes mellitus (T2DM; ~52 mmol/mol [7.03%]).3, 4 Higher HbA1c levels confer a greater risk of developing diabetes‐related complications.5 A recent study of Japanese people with T1DM reported that those with a high body mass index (BMI; ≥23 kg/m2) had significantly higher HbA1c levels and weight‐adjusted daily insulin doses than those with BMI <23 kg/m2. Additionally, being overweight was associated with higher blood pressure and dyslipidaemia.6
Insulin therapy, the current standard of care for people with T1DM, can lead to hypoglycaemia and weight gain. This not only hinders optimal blood glucose control but is a major hurdle for successfully treating T1DM.1, 2, 7 Better treatment options are therefore needed for these patients. Novel therapies that control blood glucose without inducing hypoglycaemia or weight gain and that result in better metabolic control may improve patient outcomes.7
Sodium‐glucose co‐transporter‐2 (SGLT2) is a sodium‐dependent glucose transport protein and the main protein responsible for renal reabsorption of glucose. SGLT2, expressed primarily in the proximal renal tubules of the renal cortex,8 is a therapeutic target for blood glucose control. Ipragliflozin (ASP1941) is an SGLT2‐selective inhibitor discovered by Astellas Pharma Inc. and Kotobuki Pharmaceutical Co., Ltd.9 Ipragliflozin lowers blood glucose in an insulin‐independent manner, by inhibiting glucose uptake in the kidney and promoting excretion in the urine.10, 11 It was approved for the treatment of T2DM in Japan in 2014 and is manufactured as an orally active drug (trade name Suglat).9
Owing to its insulin‐independent mechanism of action, ipragliflozin is expected to be efficacious in T1DM. A 2‐week pharmacokinetic/pharmacodynamic study of ipragliflozin, including 42 Japanese people with T1DM who received a daily oral dose of placebo or ipragliflozin (25, 50 or 100 mg) concomitant with insulin, showed dose‐dependent increases in area under the curve and maximum plasma concentration for patients taking ipragliflozin; the pharmacodynamic data showed a dose‐dependent increase in urinary glucose excretion.12 Ipragliflozin exposure and dose‐dependent increases were similar among people with T1DM and those with T2DM in a recent trial studying the pharmacodynamic effects of daily ipragliflozin. Renal glucose clearance increased from baseline in a dose‐dependent manner for both patient groups and there was no substantial difference in the exposure–response relationship (data on file). These results suggest that the ipragliflozin dose regimen used in people with T2DM (50 mg/d starting dose) is suitable for people with T1DM.
The present phase 3 study aimed to assess the efficacy and safety of once‐daily oral ipragliflozin 50 mg compared with placebo over 24 weeks in people with T1DM inadequately controlled (HbA1c 58–96 mmol/mol [7.5%–11%]) with insulin therapy.
2. METHODS
2.1. Participants
Men or women aged ≥20 years, diagnosed with T1DM, and who had been receiving insulin therapy for ≥12 weeks prior to visit 1 (−6 weeks), were eligible for the present study. Patients with HbA1c levels between 58 and 96 mmol/mol (7.5% and 11%) at screening, fasting blood C‐peptide levels <0.1987 nmol/L and a body mass index (BMI) of 20.0 to 35.0 kg/m2 were included. Patients were excluded if they had experienced major hypoglycaemia (requiring the assistance of a caregiver) or diabetic ketoacidosis (DKA) within 12 weeks prior to visit 1 (−6 weeks); or had received hypoglycaemic agents other than insulin or α‐glucosidase inhibitors (α‐GIs) within 8 weeks prior to visit 1 (−6 weeks). Complete inclusion and exclusion criteria are described in the Supporting Information. All participants provided written informed consent.
2.2. Study design, treatments and blinding
This study was conducted in accordance with the ethical principles of the Declaration of Helsinki, Good Clinical Practice, guidelines of the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use and all applicable laws and regulations. Study approval was obtained from the institutional review board at each study site. The study was registered at clinicaltrials.gov: NCT02897219.
This was a multicentre, double‐blind, parallel‐group, placebo‐controlled study conducted at 36 sites in Japan from August 29, 2016, to July 22, 2017. The study design is shown in Figure S1. Participants taking an α‐GI concomitantly with insulin were required to undergo a 4‐week washout (washout visit [−10 weeks] to visit 1 [−6 weeks]) after providing written consent and completing the temporary registration. All participants completed an observation period consisting of a 4‐week screening period (visit 1 [−6 weeks] to visit 2 [−2 weeks]), followed by a 2‐week single‐blind placebo run‐in period (visit 2 [−2 weeks] to visit 3 [0 weeks]). Participants receiving insulin alone started the observation period after providing written consent and completing temporary registration.
Participants were randomized (2:1) to receive ipragliflozin or placebo. During the treatment period (double‐blind, 24 weeks, visit 3 [0 weeks] to visit 10 [24 weeks]), participants received once‐daily ipragliflozin 50 mg or placebo in combination with insulin. Visits were scheduled for weeks 0 (visit 3), 2, 4, 8, 12, 16, 20 and 24. Interruption of the study treatment was allowed if a substantial adjustment of insulin dose resulting from an inability to consume food was needed; participants with treatment interruptions lasting >2 consecutive weeks were withdrawn from the study. One tablet of ipragliflozin or placebo (identical tablets and packaging) was administered orally before or after breakfast. Insulin therapy rules were applied from the start of visit 1 (−6 weeks) to the end of treatment (EOT; ie, week 24 or the time of study drug discontinuation). Introduction of new insulin therapy or switching between subcutaneous insulin infusion and multiple daily injections was not allowed (continuous subcutaneous insulin infusion could be switched to multiple daily injections if the duration was <2 weeks).
A 15% reduction of the total daily dose of insulin reported at visit 1 (−6 weeks) was suggested at visit 3 (0 weeks); insulin reduction was considered if self‐monitored blood glucose (SMBG) levels were < 4.44 mmol/L (80 mg/dL). Insulin dose adjustments were performed in the usual clinical setting under the guidance of a physician. Certain concomitant treatments were prohibited during the study period, including hypoglycaemic agents other than insulin preparations and continuous systemic corticosteroid treatment or immunosuppressants (unless used temporarily or applied topically). Treatment for hypoglycaemia, such as glucose (unless taken orally) and glucagon, was prohibited during the trial period; if treatment for hypoglycaemia was necessary, the participant was removed from the study. Temporary use of such treatments for reasons other than hypoglycaemia was allowed. Participants requiring hospitalization were to be withdrawn from the study. Treatment compliance was verified by quantity of unused study drugs at each visit.
2.3. Efficacy outcomes
Data collected at visit 3 (0 weeks) were used for baseline efficacy assessments. The primary efficacy outcome was the change in HbA1c from baseline to EOT; that is, at week 24 or at study drug discontinuation for participants discontinuing prior to week 24.
Secondary outcomes included the percentage of participants with HbA1c <64 mmol/mol (<8.0%) at EOT; changes in fasting plasma glucose (FPG), insulin doses (basal/bolus/total), each time point of the seven‐point SMBG, and body weight vs placebo. Subgroup analyses included change in HbA1c from baseline to EOT according to sex, age, BMI, duration of T1DM, estimated glomerular filtration rate (eGFR [mL/min/1.73 m2] = 194 × serum creatinine [mg/dL]−1.094 × age [years]−0.287 [× 0.739 if female]),13 use of α‐GIs, dosing method of insulin, dose reduction of insulin, baseline HbA1c, and total daily insulin dose.
2.4. Safety
Safety outcomes included vital signs, treatment‐emergent adverse events (TEAEs), general laboratory tests (haematology, blood chemistry and urine analysis) and ECG findings. Regarding hypoglycaemia, blood glucose levels ≤3.89 mmol/L (70 mg/dL), as measured by SMBG or central/local laboratory, were handled as adverse events (AEs). Probable symptomatic hypoglycaemia (hypoglycaemic symptoms estimated to be caused by blood glucose ≤3.89 mmol/L but not confirmed by measurement), and relative hypoglycaemia (presence of hypoglycaemic symptoms but with blood glucose >3.89 mmol/L) were also handled as AEs. Participants underwent screening for ketone bodies at each visit; those with total ketone body measurements ≥5000 μmol/L or those who developed DKA were to be withdrawn from the study.
2.5. Statistical methods
Sample size was determined using published data from an 8‐week clinical trial with empagliflozin, an SGLT2 inhibitor, in which the drug decreased HbA1c by 0.4% in people with T1DM.14 After consideration of study differences and assuming a 0% decrease in the placebo group and a shared SD of 0.7% with an assignment ratio of 2:1, the numbers of participants required to detect a 0.4% difference between groups with a power of 90% were calculated to be 49 and 97 in the placebo and treatment groups, respectively. We therefore planned to enrol 50 and 100 participants in the respective groups.
The full analysis set (FAS; primary analysis) included all participants who received at least one dose of the study drug during the treatment period and for whom at least one efficacy variable was measured after administration of the study drug. The per‐protocol set (PPS) (secondary analysis) consisted of participants included in the FAS who met the inclusion criteria and did not meet any of the exclusion criteria, received the study drug for at least 56 days, had ≥80% compliance and no major protocol deviations. The safety analysis set included all participants who received at least one dose of the study drug.
Primary and secondary endpoints were examined descriptively and using analysis of covariance (ANCOVA) with variable value at baseline as a covariate to calculate adjusted mean differences (AMDs) compared with placebo and 95% confidence intervals (CIs). Subgroup analyses for the primary endpoint were performed using stratification according to the subgroups described previously. The primary endpoint of HbA1c used an unadjusted α‐value of 0.05 to control for type 1 error. Adjustments for multiplicity were not applied for other endpoints. The number and percentage of participants with AEs and drug‐related AEs and descriptive statistics were calculated for laboratory test values using the safety analysis set. A two‐sided significance level of 0.05 was used. Data were analysed using SAS Drug Development software (ver. 4.5 or higher; SAS Inc., Cary, North Carolina) and SAS® software (version 9.4 or higher; SAS Inc.).
3. RESULTS
3.1. Participants
The participant disposition is summarized in Figure S2. A total of 210 people consented to participate in the study; 196 started the run‐in period and 175 were randomized and received either placebo (n = 60) or ipragliflozin 50 mg (n = 115). Six participants in the placebo group discontinued before completion of the treatment period because of TEAEs (n = 3), protocol deviations (n = 2) or withdrawal of consent (n = 1). Three participants from the ipragliflozin group discontinued because of TEAEs (n = 1) or withdrawal of consent (n = 2). The safety analysis set included 175 participants who received the study drug. The FAS consisted of all participants in the safety analysis set, except for one participant in the placebo group who was excluded because efficacy variables were not measured after study drug administration. The PPS consisted of 57 and 114 participants in the placebo and ipragliflozin groups, respectively.
Baseline participant characteristics, including age, sex and body weight, were generally comparable between the two groups (Table 1). Baseline HbA1c and basal/bolus/total daily doses of insulin were similar in both groups; similar percentages of participants in each group had reduced their insulin dose (as directed by a study investigator) at visit 3 (0 weeks). The mean duration of study treatment for the placebo and ipragliflozin groups was 161.3 and 167.1 days, respectively. The mean treatment compliance between groups was similar: 96.3% and 98.3% for the placebo and ipragliflozin groups, respectively.
Table 1.
Baseline patient characteristics
| Placebo N = 59 | Ipragliflozin 50 mg N = 115 | |
|---|---|---|
| Men, n (%) | 27 (45.8) | 54 (47.0) |
| Women, n (%) | 32 (54.2) | 61 (53.0) |
| Age, years | 48.3 ± 12.8 | 49.7 ± 13.1 |
| Age group, n (%) | ||
| <65 years | 52 (88.1) | 96 (83.5) |
| ≥65 years | 7 (11.9) | 19 (16.5) |
| Body weight, kg | 64.68 ± 9.07 | 66.06 ± 11.39 |
| BMI, kg/m2 | 24.21 ± 2.82 | 24.67 ± 2.95 |
| BMI, n (%) | ||
| <25 kg/m2 | 35 (59.3) | 69 (60.0) |
| ≥25 kg/m2 | 24 (40.7) | 46 (40.0) |
| Underwent α‐GI washout, n (%) | 3 (5.1) | 7 (6.1) |
| Route of insulin injection, n (%) | ||
| CSII | 2 (3.4) | 8 (7.0) |
| MDI | 57 (96.6) | 107 (93.0) |
| eGFR, mL/min/1.73 m2 | 93.85 ± 21.55 | 93.76 ± 20.92 |
| eGFR, n (%) | ||
| 30 to <60 mL/min/1.73 m2 | 1 (1.7) | 3 (2.6) |
| 60 to <90 mL/min/1.73 m2 | 27 (45.8) | 48 (41.7) |
| ≥90 mL/min/1.73 m2 | 31 (52.5) | 64 (55.7) |
| HbA1c, mmol/mol | 71.3 ± 8.6 | 71.4 ± 9.0 |
| HbA1c, % | 8.67 ± 0.79 | 8.68 ± 0.81 |
| HbA1c, n (%) | ||
| <63 mmol/mol | 12 (20.3) | 21 (18.3) |
| ≥63 mmol/mol | 47 (79.7) | 94 (81.7) |
| FPG, mmol/L | 10.90 ± 3.93 | 10.65 ± 3.83 |
| FPG, mg/dL | 196.4 ± 70.9 | 191.8 ± 69.0 |
| Basal insulin dose, IU/d | 18.94 ± 9.94 | 19.15 ± 9.80 |
| Bolus insulin dose, IU/d | 31.54 ± 17.46 | 30.09 ± 15.62 |
| Total insulin dose, IU/d | 50.48 ± 24.95 | 49.24 ± 22.58 |
| Total insulin dose | ||
| <50 IU/d | 38 (64.4) | 72 (62.6) |
| ≥50 IU/d | 21 (35.6) | 43 (37.4) |
| Total insulin dose, IU/kg·d | 0.76 ± 0.29 | 0.74 ± 0.28 |
| Total insulin dose, n (%) | ||
| <0.3 IU/kg·d | 0 | 1 (0.9) |
| ≥0.3 IU/kg·d | 59 (100) | 114 (99.1) |
| Reduction of daily dose of insulin preparation (Visit 3 [0 weeks]) | 29 (49.2) | 53 (46.1) |
Abbreviations: BMI, body mass index; eGFR, estimated glomerular filtration rate; FPG, fasting plasma glucose; HbA1c, glycated haemoglobin; α‐GI, α‐glucosidase inhibitor, CSII, continuous subcutaneous insulin infusion; MDI, multiple daily injections.
Values are mean ± SD, unless otherwise indicated.
3.2. Primary efficacy outcome
A greater decrease in HbA1c from baseline to EOT was observed in the ipragliflozin group (−5.1 mmol/mol [−0.47%]) compared with the placebo group (−1.3 mmol/mol [−0.11%]). The AMD versus placebo was −3.8 mmol/mol (95% CI −6.2, −1.5]; −0.36% [95% CI −0.57, −0.14]), showing a statistically significant decrease in the ipragliflozin group compared with placebo (P = 0.001; ANCOVA [Figure 1 and Table S1 ]). The mean HbA1c in the placebo group remained relatively unchanged, while the mean HbA1c decreased from baseline starting at week 4 in the ipragliflozin group and remained stable until week 24 (Figure 1). Similarly to the FAS analysis, the PPS analysis showed a statistically significant decrease in HbA1c levels for ipragliflozin compared with placebo (P = 0.001, data not shown).
Figure 1.
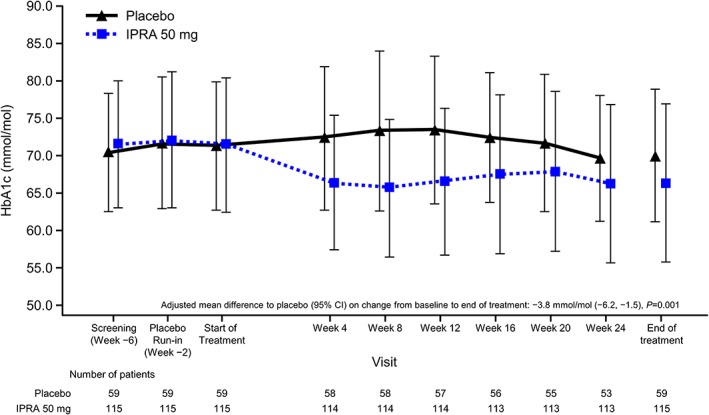
Time course of glycated haemoglobin (HbA1c). Data are shown as mean ± standard deviation in National Glycohemoglobin standardization Program units. CI, confidence interval; IPRA, ipragliflozin
3.3. Secondary efficacy outcomes
The percentage of participants reaching an HbA1c target of <63 mmol/mol (8%) at EOT was lower for placebo (23.7%) compared with ipragliflozin treatment (47.8%); percentages at baseline were similar for the two groups (placebo, 20.3%; ipragliflozin, 18.3% [Table S1]). Subgroup analyses for change from baseline to EOT in HbA1c levels were conducted using the described subgroups. No substantial differences were observed among subgroups. Assessment was considered problematic for the following subgroups because of the small number of participants: age ≥ 65 years; eGFR≥30 to <60 mL/min/1.73 m2; participation in washout; and continuous subcutaneous insulin infusion (Figure S3).
The mean change in body weight from baseline to EOT was greater with ipragliflozin treatment (−2.92 kg) compared with placebo (−0.04 kg); AMD versus placebo was −2.87 kg (95% CI −3.58, −2.16; P < 0.001 [Figure 2 and Table S1]). The mean change from baseline to EOT for FPG was −0.08 mmol/L (−1.4 mg/dL) for placebo and − 2.51 mmol/L (−45.2 mg/dL) for ipragliflozin treatment. The AMD versus placebo was −2.63 mmol/L (95% CI −3.74, −1.53); −47.4 mg/dL [95% CI −67.3, −27.5]), showing a statistically significant decrease for ipragliflozin treatment compared with placebo (P < 0.001; ANCOVA [Table S1]).
Figure 2.
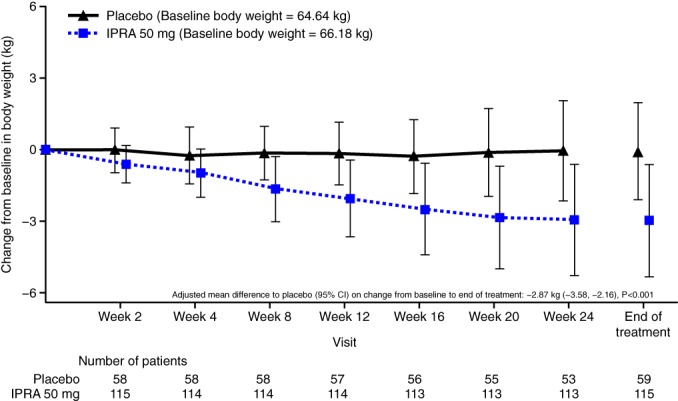
Time course of changes in body weight. Data are shown as mean ± SD
CI, confidence interval; IPRA, ipragliflozin
During the treatment period, the basal, bolus and total insulin doses decreased by week 2, followed by a gradual increase until week 24 for the placebo group. For the ipragliflozin group, the basal daily dose of insulin decreased until week 16, and the bolus and total insulin daily doses decreased until week 8; these decreases were maintained to week 24 (Figure 3A−C). The mean changes from baseline to EOT in basal, bolus and total insulin daily doses were 0.38, 0.25 and 0.63 IU, respectively, for the placebo group and − 3.38, −3.25 and − 6.64 IU, respectively, for the ipragliflozin group. The AMD versus placebo was −3.75 IU (95% CI −4.73, −2.76) for basal −3.66 IU (95% CI −5.13, −2.19) for bolus and − 7.35 IU (95% CI −9.09, −5.61) for total insulin. There was a significant reduction from baseline to EOT for basal, bolus and total daily insulin doses (all P < 0.001; ANCOVA) in the ipragliflozin group compared with the placebo group. The mean percent change from baseline to EOT in basal, bolus and total insulin daily doses were 3.47%, 1.14% and 1.49%, respectively, for the placebo group and − 18.68%, −9.43% and − 13.77%, respectively, for the ipragliflozin group (Table S1).
Figure 3.
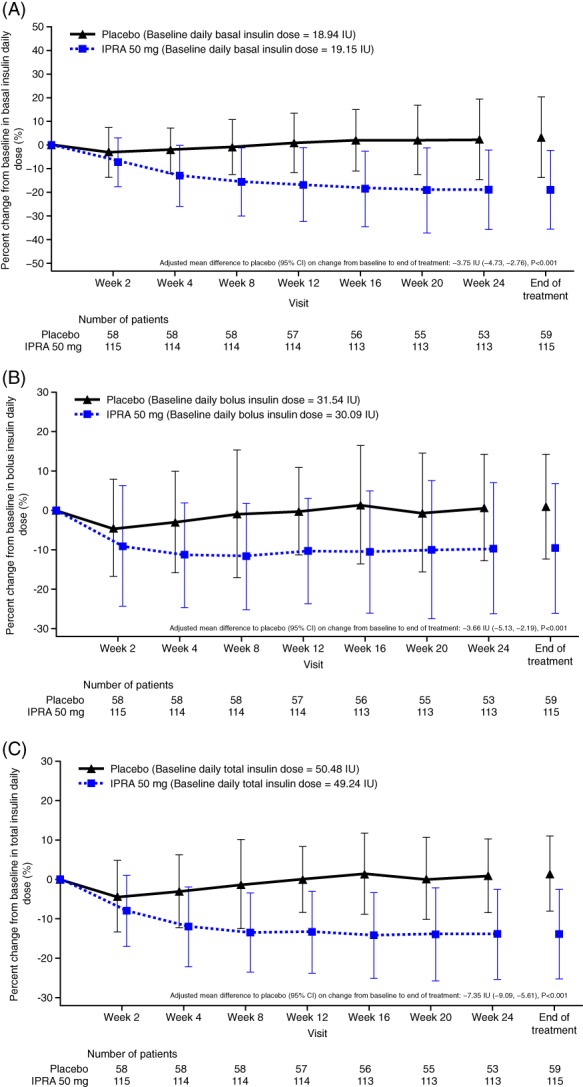
Time course of percent changes in daily insulin dose (A) basal insulin, (B) bolus insulin, (C) total insulin. Data are shown as mean ± SD. CI, confidence interval; IPRA, ipragliflozin
The time course of seven‐point SMBG included monitoring before breakfast, 1 hour after the start of breakfast, before lunch, 1 hour after the start of lunch, before dinner, 1 hour after the start of dinner and before bedtime. The AMD versus placebo of SMBG from baseline to EOT showed a statistically significant decrease for four time points (before breakfast [P < 0.001], 1 hour after the start of breakfast [P = 0.027], before lunch [P = 0.001] and before dinner [P < 0.001]; Figure S4).
3.4. Safety
3.4.1. Adverse events
The TEAEs with the highest incidence in the ipragliflozin group were hypoglycaemia, nasopharyngitis, blood ketone body increase, and headache. The incidence of hypoglycaemia was high in both groups (placebo, 93.3%; ipragliflozin, 98.3%). One case of major hypoglycaemia (requiring the assistance from another person) was reported in one participant in the placebo group; all other cases were minor. The incidence of nasopharyngitis was comparable between the two groups; all other common TEAEs had higher incidences in the treatment group. Two serious TEAEs occurred (hypoglycaemia and abdominal abscess), both in the placebo group; all other TEAEs were mild or moderate. No deaths were reported (Table 2).
Table 2.
Treatment‐emergent adverse events
| Adverse event | Placebo N = 60, PY = 26.8, n (%) events [/PY] | Ipragliflozin 50 mg N = 115, PY = 53.1, n (%) events [/PY] |
|---|---|---|
| TEAEs | 59 (98.3) 1523 | 114 (99.1) 3644 |
| Drug‐related TEAEs | 52 (86.7) 1193 | 113 (98.3) 3424 |
| Serious TEAEs | 2 (3.3) 2 | 0 |
| Drug‐related serious TEAEs | 1 (1.7) 1 | 0 |
| TEAEs leading to permanent discontinuation of study drug | 3 (5.0) 3 | 1 (0.9) 1 |
| Drug‐related TEAEs leading to permanent discontinuation of study drug | 3 (5.0) 3 | 1 (0.9) 1 |
| Deaths | 0 | 0 |
| TEAEs not related to hypoglycaemia | 39 (65.0) 124 | 73 (63.5) 232 |
| Drug‐related | 15 (25.0) 23 | 41 (35.7) 89 |
| TEAEs related to hypoglycaemiaa | 56 (93.3) 1399 [52.2] | 113 (98.3) 3412 [64.3] |
| Drug‐related | 51 (85.0) 1170 [43.7] | 112 (97.4) 3335 [62.8] |
| Mild | 56 (93.3) 1398 [52.2] | 113 (98.3) 3405 [64.1] |
| Moderate | 0 | 2 (1.7) 7 [0.1] |
| Severe | 1 (1.7) 1 [0.0] | 0 |
| Major hypoglycaemiab | 1 (1.7) 1 [0.0] | 0 |
| Documented symptomatic hypoglycaemiac | 48 (80.0) 742 [27.7] | 103 (89.6) 1648 [31.0] |
| With blood glucose ≤50 mg/dL (≤2.77 mmol/L) | 37 (61.7) 205 [7.7] | 75 (65.2) 470 [8.9] |
| Asymptomatic hypoglycaemiad | 51 (85.0) 599 [22.4] | 97 (84.3) 1676 [31.6] |
| With blood glucose ≤50 mg/dL (≤2.77 mmol/L) | 18 (30.0) 59 [2.2] | 50 (43.5) 218 [4.1] |
| Probable symptomatic hypoglycaemiae | 2 (3.3) 4 [0.1] | 11 (9.6) 49 [0.9] |
| Relative hypoglycaemiaf | 8 (13.3) 54 [2.0] | 17 (14.8) 39 [0.7] |
| TEAEs related to UTI | 6 (10.0) 7 | 4 (3.5) 4 |
| TEAEs related to genital infection | 0 | 6 (5.2) 6 |
| TEAEs related to polyuria/pollakiuria | 2 (3.3) 2 | 7 (6.1) 7 |
| TEAEs related to volume depletion | 3 (5.0) 3 | 7 (6.1) 7 |
| TEAEs related to increased ketone bodies | 2 (3.3) 2 | 15 (13.0) 16 |
Abbreviations: PY, participant‐year; TEAE, treatment‐emergent adverse event; UTI, urinary tract infection.
Number of events per PY is available only for TEAEs related to hypoglycaemia.
Hypoglycaemia requiring the assistance of another person.
Typical hypoglycaemic symptoms present and blood glucose level ≤ 3.89 mmol/L (≤70 mg/dL).
Typical hypoglycaemic symptoms absent and blood glucose level ≤ 3.89 mmol/L (≤70 mg/dL).
Blood glucose level not measured and presence of hypoglycaemic symptoms that can be estimated as caused by a drop in blood glucose level to ≤3.89 mmol/L (≤70 mg/dL).
Typical hypoglycaemic symptoms estimated to be present and blood glucose level > 3.89 mmol/L (>70 mg/dL).
There were four cases of study discontinuation because of TEAEs; three in the placebo group (food craving, urethritis and hypoglycaemia) and one in the treatment group (drug eruption; Table 2). TEAEs of interest included hypoglycaemia (described previously) and urinary tract infection (UTI)‐related, body fluid decreased‐related, genital infection‐related and ketone body increase‐related TEAEs. UTI‐related TEAEs were higher in the placebo group (10.0%) than the ipragliflozin group (3.5%), and the incidence of body fluid decrease‐related TEAEs was similar. Genital infection and ketone body increase‐related TEAEs were higher in the ipragliflozin group (5.2% and 13.0%, respectively) than in the placebo group (0% and 3.3%, respectively; Table 2). No incidence of DKA was reported in either group, and no participant had total ketone body measurements ≥5000 μmol/L.
3.4.2. Laboratory variables
No significant changes in eGFR were observed in either group from baseline to EOT (Table S2). The incidence of increased blood ketone bodies and ketosis was lower in the placebo group (1.7% for both) compared with the ipragliflozin group (9.6% and 3.5%, respectively). The change from baseline (± SD) at EOT in the ipragliflozin group was 182.47 ± 336.70 μmol/L for total ketone bodies, 44.81 ± 80.40 μmol/L for acetoacetic acid, and 137.60 ± 259.57 μmol/L for 3‐hydroxybutyric acid (Table S2).
3.4.3. Blood pressure
Greater decreases were observed in sitting diastolic/systolic blood pressure and sitting pulse rate in the ipragliflozin group compared with placebo at EOT, but changes from baseline were not clinically significant.
4. DISCUSSION
Ipragliflozin treatment significantly reduced HbA1c, insulin dose and body weight from baseline. Such reductions were not observed in the placebo group. While most participants experienced TEAEs in both the placebo and treatment groups, most events were mild or moderate and only two serious TEAEs occurred, both in the placebo group. One case of major hypoglycaemia (requiring the assistance of another person) was reported in the placebo group; none was reported in the treatment group.
Total ketone bodies were generally higher in the treatment group, and levels >3000 μmol/L were reached in some participants, but DKA was not reported in the present study. Ketone body measurements at baseline are reported to be higher in participants with T1DM versus those with T2DM15; this would be expected to influence the magnitude of change at EOT for those with T1DM versus those with T2DM. However, it has been shown that people with T1DM and those with T2DM have similar ketone body results with regard to percent change from baseline with ipragliflozin treatment.7 In the present study, we measured ketone bodies in a fasting state, whereas in the published literature, ketone bodies were tested in a non‐fasting state. It should be noted that measurement in a fasting state results in higher readings. SGLT2 inhibitors are associated with an increase in blood ketone bodies15 and this was observed in the present study, although it is important to note that the potential benefits of improved glycaemic control and reductions in insulin dose and body weight may outweigh the risks of ketone body‐related events that may not be of clinical significance.
Both UTI‐related and genital infection‐related TEAEs were more common in women, similar to reports from studies in people with T2DM. There were no noticeable differences in TEAEs in the present study compared with those in studies in people with T2DM taking ipragliflozin.16, 17, 18 In general, the incidence of overall TEAEs was relatively high, which may be related to the high incidence of hypoglycaemia in both groups, while the incidences of nasopharyngitis and genital infection‐related and UTI‐related TEAEs were low and similar in the ipragliflozin and placebo groups.
An 18‐week, phase II clinical trial involving people with T1DM who received the SGLT2 inhibitor canagliflozin reported a reduction in HbA1c and significantly more participants with no increase in body weight in the canagliflozin‐treated groups compared with placebo. Reductions in body weight and insulin dose were also reported in canagliflozin‐treated participants. Despite similar efficacy results in that trial to those in the present study, canagliflozin treatment resulted in several incidences of DKA (100 mg, 4.3%; 300 mg, 6.0%; placebo, 0%).19 A phase 3, 24‐week study of dapagliflozin in people with T1DM reported similar results to the present study of ipragliflozin, including a significant reduction in HbA1c and daily insulin dose from baseline to EOT and similar incidences of TEAEs. DKA was reported; however, the incidence was low and similar among the groups (5 mg, 1%; 10 mg, 2%; placebo, 1%). Regarding major hypoglycaemia, the numbers were slightly higher than in the present study but similar among the groups.20 A second phase 3, 24‐week study of dapagliflozin confirmed the results of the first study and reported DKA in 2.6%, 2.2% and 0% of patients taking 5‐mg, 10‐mg or placebo, respectively.21 Recently, the results of two phase 3 (26‐ and 52‐week) studies of empagliflozin in people with T1DM reported significant reductions in HbA1c and daily insulin dose for all doses of empagliflozin (2.5 mg, 10 mg and 25 mg), and an incidence of DKA of 3.3%, 4.3%, 0.8% and 1.2% for 25 mg, 10 mg, 2.5 mg empagliflozin and placebo, respectively; the incidence of major hypoglycaemia was low and comparable among the groups.22 While the present study did not report any incidences of DKA, there are reports of DKA in people with T1DM taking SGLT2 inhibitors, although the incidence of DKA is often associated with missed insulin doses or pump failure.19, 20, 21, 22 Larger studies are needed to understand the potential for DKA in people with T1DM taking ipragliflozin.
The present study was limited in that patients with major hypoglycaemia or DKA 3 months prior to study enrolment were excluded for safety reasons; exclusion of these patients could have affected the overall safety results.
In conclusion, the present phase 3 study in Japanese people with T1DM and inadequate glycaemic control on insulin therapy demonstrated a statistically significant decrease in HbA1c versus placebo for once‐daily ipragliflozin 50 mg in combination with insulin for 24 weeks. Importantly, statistically significant reductions in insulin dose and body weight were observed in participants treated with ipragliflozin versus placebo. There were no safety concerns with the administration of ipragliflozin for 24 weeks. Overall, a once‐daily 50‐mg dose of ipragliflozin was shown to be both efficacious and well tolerated.
CONFLICT OF INTEREST
H.I., T.S. and J.T. are employees of the study sponsor (Astellas Pharma Inc.). K.K. has received advisory fees from Sanwa Kagaku Kenkyusho, Kissei Pharmaceutical, Novo Nordisk Pharma and Takeda Pharmaceutical; honoraria from Astellas Pharma, AstraZeneca, Daiichi Sankyo, MSD, Ono Pharmaceutical, Novo Nordisk Pharma, Boehringer Ingelheim Japan, Taisho Toyama Pharmaceutical, Takeda Pharmaceutical, and Mitsubishi Tanabe Pharma; and scholarship/donation fees from Boehringer Ingelheim Japan, Taisho Toyama Pharmaceutical, and Mitsubishi Tanabe Pharma.
AUTHOR CONTRIBUTIONS
K.K. contributed to conception and design of the study, interpretation of data, and drafting and critically revising the manuscript. H.I. contributed to conception and design of the study, acquisition and interpretation of data, and drafting and critically revising the manuscript. T.S. contributed to conception and design of the study, analysis and interpretation of data, and drafting and critically revising the manuscript. J.T. contributed to design of the study, analysis of data, and critically revising the manuscript. All authors gave final approval of the version to be published and agree to be accountable for the accuracy and integrity of the manuscript.
Data sharing and data accessibility
Studies conducted with product indications or formulations that remain in development are assessed after study completion to determine if individual participant data can be shared. The plan to share individual participant data is based on the status of product approval or termination of the compound, in addition to other study‐specific criteria described on www.clinicalstudydatarequest.com under “Sponsor Specific Details for Astellas”.
Supporting information
Figure 1. Study flow: (A) description of study periods; (B) description of treatment schedules.
Figure 2. Disposition of patients.
Figure 3. HbA1c reduction stratified by patient characteristics. Numbers in parentheses for each parameter indicate the number of patients in the placebo and ipragliflozin groups, respectively. Data are from the FAS.
Figure 4. Time‐course of seven‐point self‐monitored blood glucose: (A) placebo; (B) ipragliflozin 50 mg. Data are shown as mean ± standard deviation.
Table S1. Efficacy and safety parameters
Table S2. Ketone‐related parameters and eGFR
Appendix S1: Complete eligibility criteria.
ACKNOWLEDGMENTS
This study was funded by Astellas Pharma Inc. We thank Sarah Bubeck, PhD, of Edanz Medical Writing for providing medical writing support, which was funded by Astellas Pharma Inc. through EMC K.K. in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Kaku K, Isaka H, Sakatani T, Toyoshima J. Efficacy and safety of ipragliflozin add‐on therapy to insulin in Japanese patients with type 1 diabetes mellitus: A randomized, double‐blind, phase 3 trial. Diabetes Obes Metab. 2019;21:2284–2293. 10.1111/dom.13807
Funding information This study was funded by Astellas Pharma Inc. Medical writing support was provided by Sarah Bubeck, PhD, of Edanz Medical Writing; this was funded by Astellas Pharma Inc. through EMC K.K. in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
The peer review history for this article is available at https://publons.com/publon/10.1111/dom.13807.
REFERENCES
- 1. Paschou SA, Papadopoulou‐Marketou N, Chrousos GP, Kanaka‐Gantenbein C. On type 1 diabetes mellitus pathogenesis. Endocr Connect. 2018;7:R38‐R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maneda M, Noda M, Origasa H, et al. Japanese Clinical Practice Guideline for Diabetes 2016. J Diabetes Investig. 2018;9:657‐697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Japan Diabetes Clinical Data Management Study Group, Basic tabulation materials . 2018; http://jddm.jp/data/index-2017.html. Accessed October 22.
- 4. Yokoyama H, Araki SI, Kawai K, et al. Declining trends of diabetic neuropathy, retinopathy and neuropathy with improving diabetes care indicators in Japanese patients with type 2 and type 1 diabetes (JDDM 46). BMJ Open Diabetes Res Care. 2018;6:e000521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. The Diabetes Control and Complications Trial Research Group . The relationship of glycemic exposure (HbA1c) to the risk of development and progression of retinopathy in the diabetes control and complications trial. Diabetes. 1995;44:968‐983. [PubMed] [Google Scholar]
- 6. Arai K, Yokoyama H, Okoguchi F, et al. Association between body mass index and core components of metabolic syndrome in 1486 patients with type 1 diabetes mellitus in Japan (JDDM 13). Endocr J. 2008;55:1025‐1032. [DOI] [PubMed] [Google Scholar]
- 7. Chillaron JJ, Flores Le‐Roux JA, Benaiges D, Pedro‐Botet J. Type 1 diabetes, metabolic syndrome and cardiovascular risk. Metabolism. 2014;63:181‐187. [DOI] [PubMed] [Google Scholar]
- 8. Chen J, Williams S, Ho S, et al. Quantitative PCR tissue expression profiling of the human SGLT2 gene and related family members. Diabetes Ther. 2010;1:57‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Poole RM, Dungo RT. Ipragliflozin: first global approval. Drugs. 2014;74:611‐617. [DOI] [PubMed] [Google Scholar]
- 10. Kashiwagi A, Kazuta K, Yoshida S, Nagase I. Randomized, placebo‐controlled, double‐blind glycemic control trial of novel sodium‐dependent glucose cotransporter 2 inhibitor ipragliflozin in Japanese patients with type 2 diabetes mellitus. J Diabetes Investig. 2014;5:328‐391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tahara A, Kurosaki E, Yokono M, et al. Pharmacological profile of ipragliflozin (ASP1941), a novel selective SGLT2 inhibitor, in vitro and in vivo. Naunyn Schmiedebergs Arch Pharmacol. 2012;385:423‐436. [DOI] [PubMed] [Google Scholar]
- 12. Kaku K, Hiroyuki I, Toyoshima J, Sakatani T. Clinical pharmacology study of ipragliflozin in Japanese patients with type 1 diabetes mellitus: A phase 2, randomized, placebo‐controlled trial. Diabetes Obes Metab. 2019;21(6):1445‐1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Japanese Society of Nephrology . Essential points from Evidence‐based Clinical Practice Guidelines for Chronic Kidney Disease 2018. Clin Exp Nephrol. 2019;23(1):1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Perkins BA, Cherney DZ, Partridge H, et al. Sodium‐glucose cotransporter 2 inhibition and glycemic control in type 1 diabetes: results of an 8‐week open‐label proof‐of‐concept trial. Diabetes Care. 2014;37:1480‐1483. [DOI] [PubMed] [Google Scholar]
- 15. Polidori D, Iijima H, Goda M, Maruyama N, Inagaki N, Crawford P. Intra‐ and inter‐subject variability for increases in serum ketone bodies in patients with type 2 diabetes treated with the sodium glucose co‐transporter 2 inhibitor canagliflozin. Diabetes Obes Metab. 2018;20:1321‐1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kashiwagi A, Kazuta K, Takinami Y, Yoshida S, Utsuno A, Nagase I. Ipragliflozin improves glycemic control in Japanese patients with type 2 diabetes mellitus: the BRIGHTEN study. Diabetol Int. 2015;6:8‐18. [Google Scholar]
- 17. Kashiwagi A, Takahashi H, Ishikawa H, et al. A randomized, double‐blind, placebo‐controlled study on long‐term efficacy and safety of ipragliflozin treatment in patients with type 2 diabetes mellitus and renal impairment: results of the Long‐Term ASP1941 Safety Evaluation in Patients with Type 2 Diabetes with Renal Impairment (LANTERN) study. Diabetes Obes Metab. 2015;17:152‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kashiwagi A, Yoshida S, Nakamura I, et al. Efficacy and safety of ipragliflozin in Japanese patients with type 2 diabetes stratified by body mass index: A subgroup analysis of five randomized clinical trials. J Diabetes Investig. 2016;7:544‐554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Henry RR, Thakkar P, Tong C, Polidori D, Alba M. Efficacy and safety of canagliflozin, a sodium glucose cotransporter 2 inhibitor as add‐on to insulin in patients with type 1 diabetes. Diabetes Care. 2015;38:2258‐2265. [DOI] [PubMed] [Google Scholar]
- 20. Dandona P, Mathieu C, Phillip M, et al. Efficacy and safety of dapagliflozin in patients with inadequately controlled type 1 diabetes (DEPICT‐1): 24 week results from a multicenter, double‐blind, phase 3, randomized controlled trial. Lancet Diabetes Endocrinol. 2017;5:864‐876. [DOI] [PubMed] [Google Scholar]
- 21. Mathieu C, Dandona P, Gillard P, et al. Efficacy and safety of dapagliflozin in patients with inadequately controlled type 1 diabetes (the DEPICT‐2 study): 24‐week results from a randomized controlled trial. Diabetes Care. 2018;41:1938‐−1946. [DOI] [PubMed] [Google Scholar]
- 22. Rosenstock J, Marquard J, Laffel LM, et al. Empagliflozin as adjunctive to insulin therapy in type 1 diabetes: the EASE trials. Diabetes Care. 2018;41:2560‐2569. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1. Study flow: (A) description of study periods; (B) description of treatment schedules.
Figure 2. Disposition of patients.
Figure 3. HbA1c reduction stratified by patient characteristics. Numbers in parentheses for each parameter indicate the number of patients in the placebo and ipragliflozin groups, respectively. Data are from the FAS.
Figure 4. Time‐course of seven‐point self‐monitored blood glucose: (A) placebo; (B) ipragliflozin 50 mg. Data are shown as mean ± standard deviation.
Table S1. Efficacy and safety parameters
Table S2. Ketone‐related parameters and eGFR
Appendix S1: Complete eligibility criteria.
Data Availability Statement
Studies conducted with product indications or formulations that remain in development are assessed after study completion to determine if individual participant data can be shared. The plan to share individual participant data is based on the status of product approval or termination of the compound, in addition to other study‐specific criteria described on www.clinicalstudydatarequest.com under “Sponsor Specific Details for Astellas”.