

Abstract
We investigated the cellular mechanisms underlying the Ca2+-dependent release of glutamate from cultured astrocytes isolated from rat hippocampus. Using Ca2+imaging and electrophysiological techniques, we analyzed the effects of disrupting astrocytic vesicle proteins on the ability of astrocytes to release glutamate and to cause neuronal electrophysiological responses, i.e., a slow inward current (SIC) and/or an increase in the frequency of miniature synaptic currents. We found that the Ca2+-dependent glutamate release from astrocytes is not caused by the reverse operation of glutamate transporters, because the astrocyte-induced glutamate-mediated responses in neurons were affected neither by inhibitors of glutamate transporters (β-threo-hydroxyaspartate, dihydrokainate, andl-trans-pyrrolidine-2,4-dicarboxylate) nor by replacement of extracellular sodium with lithium. We show that Ca2+-dependent glutamate release from astrocytes requires an electrochemical gradient necessary for glutamate uptake in vesicles, because bafilomycin A1, a vacuolar-type H+-ATPase inhibitor, reduced glutamate release from astrocytes. Injection of astrocytes with the light chain of the neurotoxin Botulinum B that selectively cleaves the vesicle-associated SNARE protein synaptobrevin inhibited the astrocyte-induced glutamate response in neurons. Therefore, the Ca2+-dependent glutamate release from astrocytes is a SNARE protein-dependent process that requires the presence of functional vesicle-associated proteins, suggesting that astrocytes store glutamate in vesicles and that it is released through an exocytotic pathway.
Keywords: astrocyte-neuron signaling, astrocyte calcium waves, transmitter release, Botulinum neurotoxin, bafilomycin, exocytosis, SNARE protein, V-ATPase
Recent evidence suggests that astrocytes are involved in the active control of neuronal activity and synaptic transmission. Astrocytes can respond to neuronal activity with increased intracellular Ca2+ (Dani et al., 1992; Porter and McCarthy, 1996; Pasti et al., 1997) that can propagate to neighboring astrocytes as a Ca2+ wave (Cornell-Bell et al., 1990; Charles et al., 1991; Newman and Zahs, 1997). Furthermore, an elevation of astrocyte Ca2+ can signal to neurons, inducing neuronal Ca2+ elevations (Nedergaard, 1994; Parpura et al., 1994; Hassinger et al., 1995; Pasti et al., 1997;Bezzi et al., 1998), regulating neuronal excitability (Araque et al., 1998a, 1999b; Newman and Zahs, 1998), and modulating synaptic transmission (Araque et al., 1998a,b; Kang et al., 1998). Therefore, astrocytes and neurons may function as a network where bi-directional communication takes place (Pasti et al., 1997; for review, see Araque et al., 1999a).
Glutamate released from astrocytes is the signal responsible for the modulation of neuronal activity and synaptic transmission by astrocytes in cultured hippocampal cells (Araque et al., 1998a,b, 1999b; Sanzgiri et al., 1999), hippocampal slices (Pasti et al., 1997; Bezzi et al., 1998; Kang et al., 1998), and intact retina (Newman and Zahs, 1998). Although astrocytes can release glutamate through the reverse operation of glutamate transporters (Szatkowski et al., 1990) or by a swelling-induced mechanism (Kimelberg et al., 1990), astrocyte-induced modulation of neuronal activity is attributable to glutamate released through a Ca2+-dependent process (Araque et al., 1998a,b; Bezzi et al., 1998; Newman and Zahs, 1998). Although prolonged incubation with tetanus toxin can reduce the release of glutamate from astrocytes (Bezzi et al., 1998), the cellular mechanism responsible for this Ca2+-dependent release of glutamate from astrocytes is poorly understood.
It has been shown that core proteins of the SNARE complex, synaptobrevin II and syntaxin, are expressed in astrocytes (Parpura et al., 1995), raising the possibility that Ca2+-dependent glutamate release is mediated by a vesicular mechanism. To begin to test this possibility, we now use pharmacological tools that interact with the function of two vesicle-associated proteins, vacuolar-type H+-ATPase (V-ATPase) and synaptobrevin, that are essential in neurotransmitter release (Schiavo et al., 1992;Liu and Edwards, 1997). We monitored the glutamate release from astrocytes using well characterized glutamate-dependent electrophysiological responses to stimulation of astrocytes, i.e., a neuronal slow inward current (SIC) and/or an increase in the frequency of miniature postsynaptic currents (mPSCs) (Araque et al., 1998a,b;Kang et al., 1998). We demonstrate that the Ca2+-dependent glutamate release from astrocytes is not caused by the reverse operation of glutamate transporters. However, bafilomycin A1, a V-ATPase inhibitor that dissipates the electrochemical proton gradient necessary for glutamate uptake into vesicles (Maycox et al., 1988), reduced the astrocyte-induced neuronal responses without affecting astrocyte Ca2+ waves. Furthermore, injection of astrocytes with the light chain of the neurotoxin Botulinum B (BoNT/B), which selectively cleaves the vesicle-associated SNARE protein synaptobrevin (Schiavo et al., 1992), did not prevent Ca2+ signaling in astrocytes but strongly reduced the astrocyte-induced responses in neurons. We conclude that Ca2+-dependent glutamate release from astrocytes requires the presence of functional vesicle-associated proteins.
MATERIALS AND METHODS
Preparation. Primary cultures of mixed hippocampal neurons and astrocytes from 1- to 3-d-old postnatal rats were prepared as described previously (Araque et al., 1998a) and used after 6–25 d in culture. At the time of use, astrocytes were confluent in these cultures. In some experiments, cells were plated on coverslips coated with 0.15% agarose and sprayed with poly-d-lysine (0.3 mg/ml) and collagen (0.4 mg/ml) using an atomizer, which produced microdrops where cells grew forming microislands of astrocytes and neurons.
Electrophysiology. Whole-cell patch-clamp recordings were obtained from neurons with an Axopatch-1C amplifier and pClamp software (Axon Instruments, Foster City, CA). Currents were filtered at 1–2 kHz and sampled above 1 kHz. External control solution contained (in mm): 140 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, 10 glucose, and 6 sucrose, pH 7.35. Because Araque et al. (1998a) demonstrated that the SIC is partly mediated through NMDA glutamate receptors, in some experiments NMDA receptor activation was optimized by omitting Mg2+ and adding 10 μmglycine to the solution. The patch pipette solution contained (in mm): 140 K-gluconate, 10 EGTA, 4 Mg-ATP, 0.2 Tris-GTP, and 10 HEPES, pH 7.35. The membrane potential was held at −60 mV.
The morphological identification of neurons was confirmed electrophysiologically by their ability to generate TTX-sensitive Na+-mediated action potentials and by the presence of fast synaptic currents. Astrocytes were stimulated mechanically using glass micropipettes filled with external saline (Charles, 1994; Nedergaard, 1994; Araque et al., 1998a). We have previously shown that contact between a patch pipette and an astrocyte causes the Ca2+-dependent release of glutamate and a glutamate-dependent SIC in adjacent neurons (Araque et al., 1998a). This SIC is caused by the regulated release of glutamate from astrocytes, because attenuation of the astrocyte Ca2+ signal by either injection of the Ca2+ chelator BAPTA or treatment with thapsigargin prevents the appearance of the SIC in neurons (Araque et al., 1998a). In some experiments, astrocytes were stimulated by focal application of norepinephrine (10 mm) delivered by pressure ejection (0.5 sec, 10–20 psi) (Narishige IM-200, Narishige, Greenvale, NY) from a micropipette, which was positioned using an Eppendorf micromanipulator. Unless stated otherwise, the incidence of astrocyte-induced responses was defined as the proportion of responses relative to the total number of astrocytes stimulated in each experiment; at least eight astrocytes were stimulated in each parallel control and test condition, and data were obtained from at least three different experiments (i.e., at least 24 astrocytes were stimulated in each condition). Likewise, to determine the number of neurons eliciting postsynaptic currents, at least 11 putative presynaptic neurons were stimulated in each parallel control and test condition, and data were obtained from at least three different experiments. Therefore, for these variables, n values correspond to number of preparations, whereas for the other variables, such as the amplitude of the SIC, n represents the number of cells examined. Statistical differences were established using the Student'st test, unless stated otherwise. All experiments were performed at room temperature (20–23°C). Data are expressed as mean ± SEM.
Calcium measurements. The ability of stimuli to evoke a wave of elevated Ca2+ in astrocytes was monitored by fluorescence microscopy using the Ca2+ indicators fluo-3 or fluo-4. Cultures were incubated at 37°C for 15–30 min with the acetoxymethyl ester of those indicators (10 μg/ml; Molecular Probes, Eugene, OR), and then washed for 20–30 min. Coverslips containing either fluo-3- or fluo-4-loaded cells were visualized using a silicon intensified target camera (Hamamatsu Photonic System, Bridgewater, NJ) or IC-300 intensified CCD camera (Photon Technology International, Monmouth Junction, NJ) attached to a Nikon 300 inverted microscope and a NeDLC optical workstation (Prairie Technologies, LLC, Waunakee, WI). Quantitative fluorescence measurements were made using the NeDLC video software.
Microinjection into astrocytes. Individual astrocytes were microinjected with fluoro-ruby, the light chain of the neurotoxin BoNT/B (List Biological Laboratories, Campbell, CA) and the Ca2+ chelator BAPTA, separately or in combination, as described elsewhere (Araque et al., 1998a). Microinjection pipettes (tip diameter of ∼400 nm) were pulled from Kwik-Fil borosilicate glass capillaries (World Precision Instruments, Sarasota, FL) using a Sutter P-2000 micropipette puller (Sutter Instrument Co., Novato, CA), and they were filled with 1 mmfluoro-ruby, 83.3 μg/ml BoNT/B, and 0.1 m BAPTA. Because fluoro-ruby contains a dextran moiety that prevents its passage through gap junctions, the dye was retained within the injected cell, allowing the identification of the injected cells. Pipette solutions were pressure-injected into single astrocytes by a 1–15 sec pulse of 0.1–5 psi using an Eppendorf micromanipulator and a Narishige IM-200 microinjector. Based on quantification of the fluoro-ruby fluorescence (Araque et al., 1998a), the final intracellular concentrations were estimated to be 333 ng/ml for BoNT/B and 0.4 mm for BAPTA. After injections, cells were allowed to recover for 45–60 min before electrophysiology, Ca2+ imaging experiments, or immunocytochemistry was performed.
Immunocytochemistry. Cells were fixed with 4% paraformaldehyde in PBS at room temperature for 30 min. Synaptobrevin II was immunocytochemically detected using a monoclonal antibody (clone 69.1, 1:500; provided by Dr. R. Jahn, Max Planck Institute) visualized by using an FITC-conjugated secondary antibody. After washing, cells were mounted in n-propyl gallate glycerol and examined using conventional epifluorescence microscopy and laser-scanning confocal microscopy.
RESULTS
The astrocyte-induced SIC is not mediated by reverse operation of glutamate transporters
To study the astrocyte-to-neuron signaling, we have used mechanical stimulation to evoke Ca2+elevations in astrocytes (Charles et al., 1991; Nedergaard, 1994;Araque et al., 1998a,b). This Ca2+elevation is both necessary and sufficient to cause the release of glutamate from astrocytes (Araque et al., 1998a,b), which in turn causes CNQX- and AP5-sensitive SICs in adjacent neurons (Araque et al., 1998a,b). This time course is a characteristic of the response, because a similar time course has been observed when different stimuli have been used, e.g., photorelease of Ca2+after NP-EGTA injection (Araque et al., 1998b) or astrocyte stimulation with PGE2 (Sanzgiri et al., 1999). Although mechanical stimulation is not physiological, it does mimic the action of endogenous ligands (Sanzgiri et al., 1999) and photorelease of Ca2+ (Araque et al., 1998b) in its ability to stimulate Ca2+-dependent glutamate release from astrocytes and affords the advantage that the stimulus can be directly applied to astrocytes. Consequently, for Ca2+ waves and astrocyte-neuron signaling studies, mechanical stimuli have become the experimental stimulus of choice (Araque et al., 1999a).
Szatkowski et al. (1990) reported that glutamate can be released from glia through the reverse operation of the glutamate transporters. We investigated whether a similar mechanism is responsible for the Ca2+-dependent glutamate-mediated electrophysiological responses detected in neurons after Ca2+ elevations in astrocytes. We analyzed the effects of the inhibitors of the glutamate transportersl-trans-pyrrolidine-2,4-dicarboxylate (t-PDC; 1 mm), β-threo-hydroxyaspartate (β-HA; 0.3–1 mm), and dihydrokainate (DHK; 1 mm) on the astrocyte-induced responses in adjacent neurons. The proportion of mechanically stimulated astrocytes that evoked neuronal responses and the amplitude of the astrocyte-induced SIC were unaffected by t-PDC, DHK, or β-HA (Table1). Because the glutamate transporters also co-transport sodium (but not lithium) across the membrane, we asked whether the replacement of the extracellular sodium by lithium would increase the SIC amplitude by increasing the reverse operation of the sodium-mediated glutamate carriers. We found that neither the incidence nor the amplitude of the astrocyte-mediated SIC in neurons was significantly affected by substitution of Na+ with Li+(Table 1). These results indicate that the glutamate transporters are not required for Ca2+-dependent glutamate release from astrocytes.
Table 1.
Astrocyte-induced neuronal responses were unaffected by glutamate transporter inhibitors
| Control | t-PDC | Control | β-HA | Control | DHK | Sodium | Lithium | |
|---|---|---|---|---|---|---|---|---|
| Astrocytes that evoked neuronal responses (%) | 61.0 ± 3.9 | 66.8 ± 8.6 | 54.8 ± 2.4 | 63.5 ± 5.4 | 75.4 ± 4.0 | 76.4 ± 3.6 | 74.5 ± 5.5 | 76.1 ± 3.9 |
| (n = 3) | (n= 3) | (n = 3) | (n = 3) | (n = 3) | (n = 3) | (n = 3) | (n = 3) | |
| SIC amplitude (pA) | 39.3 ± 10.0 | 42.9 ± 7.0 | 30.6 ± 7.7 | 28.5 ± 3.5 | 37.1 ± 5.3 | 43.9 ± 9.1 | 159.7 ± 25.2 | 193.5 ± 29.0 |
| (n = 15) | (n = 17) | (n = 21) | (n = 19) | (n = 23) | (n = 24) | (n = 23) | (n = 23) |
Control solutions include 2 mm Mg2+, except for extracellular sodium and lithium in which extracellular Mg2+ was omitted. No significant differences were found between control and test conditions (Student's ttest).
Bafilomycin A1 reduces the magnitude of synaptic transmission and astrocyte–neuron signaling
Vesicular uptake of neurotransmitters requires a transmembrane electrochemical proton gradient maintained by a V-ATPase that is potently and selectively inhibited by bafilomycin A1 (Bowman et al., 1988; Maycox et al., 1988;Carlson et al., 1989; Hanada et al., 1990; Moriyama et al., 1990;Roseth et al., 1995). Therefore, to investigate the cellular mechanisms involved in the glutamate release from astrocytes we have used bafilomycin A1 as a pharmacological tool that prevents the uptake and storage of glutamate in vesicles.
We first investigated the effects of bafilomycin on synaptic transmission, a well known vesicle-mediated exocytotic process. We extracellularly stimulated up to three putative presynaptic neurons (physically in contact with a process of a potential postsynaptic neuron) while recording from a postsynaptic neuron. Ten stimuli were delivered at 1 Hz, and the presence of postsynaptic currents was recorded. In control conditions, stimulation of most of the putative presynaptic neurons (97.4 ± 2.6%; n = 25 cell pairs in three preparations) evoked synaptic currents in the potential postsynaptic neuron (Fig.1A), indicating that most of the neurons in physical contact were synaptically connected and their synaptic machinery was functional. In parallel cultures incubated with 1–5 μm bafilomycin for 60–90 min, the number of presynaptic neurons that elicited synaptic currents was significantly reduced (from 97.4 ± 2.6% in control to 47.0 ± 13.6% in bafilomycin; n = 35 cell pairs in three preparations; p < 0.05), without modifying the ability of neurons to exhibit all-or-none action potentials (data not shown). The mean amplitude of the evoked EPSCs was strongly reduced after incubation with bafilomycin (Fig.1B–D), and the relative number of synaptic failures (i.e., absence of evoked EPSCs) was dramatically increased by bafilomycin treatment (Fig.1A,B,E). In support of the possibility that bafilomycin acts on the secretory machinery, the frequency of miniature EPSCs (mEPSCs) was significantly reduced by bafilomycin. Those mEPSCs still detected in bafilomycin had an amplitude similar to control mEPSCs (Fig.1F,H), indicating a presynaptic mechanism of action of bafilomycin. Because bafilomycin decreases vesicle filling by reducing the vesicular electrochemical gradient, one might expect to obtain a reduction of the mPSC amplitude, a result that we did not observe. However, it is likely that smaller mPSCs were beneath the detection level in our experiments, a result that is in agreement with the observed reduction of the mPSC frequency. Taken together, these results indicate that bafilomycin reduced the probability of transmitter release, and they are in agreement with the predicted effects of bafilomycin by preventing the uptake and storage of neurotransmitter into synaptic vesicles.
Fig. 1.

Bafilomycin A1 reduced the probability of synaptic transmitter release. A,B, Representative traces of evoked EPSCs in control cultures and after incubation with bafilomycin. In many cases, EPSCs were absent in bafilomycin-treated cultures. When present, the amplitude of EPSCs in bafilomycin was smaller than in control, and the number of synaptic failures was increased by bafilomycin. C, Averaged (n = 10) EPSCs in control and bafilomycin. Shown is the same pair of pre-postsynaptic neurons as in B. D,Mean evoked EPSC amplitude in control and bafilomycin-treated cultures (n = 25 and 35 presynaptic neurons, respectively).E, Mean percentage of synaptic failures observed during trials of 10 stimuli delivered at 1 Hz in control and after incubation with bafilomycin (n = 25 and 35 presynaptic neurons, respectively). F, mEPSCs in control and after incubation with bafilomycin. G, Mean frequency of mEPSCs measured during 1–4 min in control and in bafilomycin-treated cells (n = 9). H, Average cumulative probability plots of the mEPSC amplitude in control (n = 9) and after incubation with bafilomycin (n = 8). Cumulative probability plots were calculated in 1 pA bins and were not significantly different between control and bafilomycin-treated cells (Kolmogorov–Smirnov test). Holding potential was −60 mV. Significant differences with respect to control were established by the Student's t test atp < 0.02 (**) and p < 0.001 (***).
Because incubation with bafilomycin was effective in reducing the amount of neurotransmitter released from synaptic terminals, we asked whether it also reduced glutamate release from astrocytes by determining the effects of bafilomycin on the glutamate-dependent neuronal responses elicited by mechanical stimulation of astrocytes. In control conditions, neuronal responses were elicited by 63.4 ± 2.8% of the stimulated astrocytes (n = 46 stimulated astrocytes in three preparations), whereas their incidence was significantly reduced (p < 0.001) after bafilomycin treatment (32.5 ± 2.8%; n = 48 stimulated astrocytes in three preparations) (Fig.2B). Just as in our synaptic recordings, the amplitude of the astrocyte-induced SIC was also significantly reduced by bafilomycin (Fig.2A,C). One concern with the interpretation of these results is whether the SIC reduction is secondary to the effects of bafilomycin on synaptic transmission. This is unlikely to be the case because our previous studies (Araque et al., 1998a) have shown that incubation of cultures in tetanus toxin holoprotein, which blocked synaptic transmission, did not impact the SIC that is caused by glutamate release from astrocytes. Thus, it is likely that the reduction in incidence and amplitude of the SIC after bafilomycin treatment is attributable to direct effects of the V-ATPase inhibitor on astrocytes.
Fig. 2.
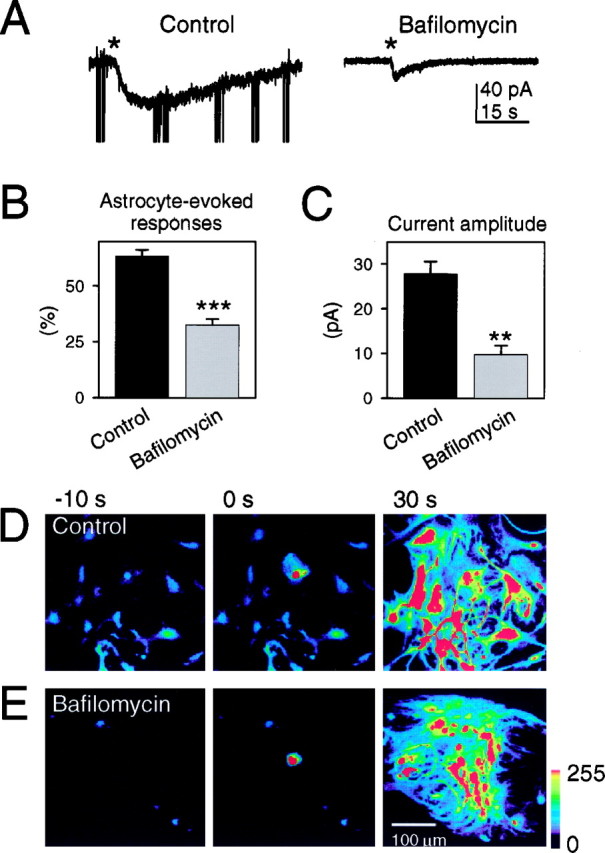
Bafilomycin reduced astrocyte-evoked neuronal responses but did not prevent Ca2+ waves in astrocytes. A, Representative whole-cell current neuronal responses to astrocyte stimulation in control (left) and after incubation with bafilomycin (right). Mechanical stimulation of astrocytes is indicated by asterisk. Fast, high-amplitude synaptic currents have been truncated. B, Proportion of mechanically stimulated astrocytes that evoked glutamate-dependent responses in adjacent neurons in control and bafilomycin-treated cultures (n = 3 different cultures).C, Current amplitude recorded in neurons after mechanical stimulation of adjacent astrocytes in control and bafilomycin-treated cells (n = 37 and 45 stimulated astrocytes, respectively). Significant differences with respect to control were established by the Student's t test atp < 0.01 (**) and p < 0.001 (***). D,E, Mechanically induced Ca2+ waves recorded in cultures loaded with the Ca2+ indicator fluo-3 in control conditions and after incubation with bafilomycin. Left to right panels show pseudocolor images representing intensity of fluo-3 emission, taken before, during, and after mechanical stimulation, at the times indicated. Zero time corresponds to the time of astrocyte stimulation. Mechanical stimulation increased the intracellular Ca2+ in the injected cell as well as in neighboring nonstimulated astrocytes.
Although these data are consistent with the possibility that the glutamate released from astrocytes is stored in an acidic compartment that is sensitive to bafilomycin, it is important to confirm the specificity of bafilomycin action. Although bafilomycin is reported to have a higher affinity for the V-ATPase than the Ca2+-ATPase, it is necessary to confirm that bafilomycin does not reduce glutamate release as a result of inhibiting the Ca2+-ATPase and thus the Ca2+ signal that is necessary to stimulate glutamate release. Mechanically induced Ca2+ waves in astrocytes were elicited by 12 of 13 stimulated astrocytes in control conditions (Fig.2D), and by eight of eight astrocytes after incubation with bafilomycin (Fig. 2E), indicating that the effects of bafilomycin on the SIC were not caused by the impairment of the stimulus-induced Ca2+elevation in astrocytes. Taken together, these results indicate that the release of glutamate from astrocytes requires the presence of the functional V-ATPase, a vesicle-associated protein necessary for the uptake and storage of neurotransmitters in vesicles.
The vesicle-associated protein synaptobrevin is required for glutamate release from astrocytes
Synaptobrevin is a vesicle-associated SNARE protein involved in exocytosis from synaptic terminals that has been demonstrated to be expressed by both neurons (Schiavo et al., 1992;Fernández-Chacón and Südhof, 1999) and astrocytes (Parpura et al., 1995; Araque et al., 1998a; Maienschein et al., 1999). Synaptobrevin can be selectively cleaved by Botulinum toxin B (Schiavo et al., 1992), a clostridial toxin constituted by a heavy and a light chain with receptor binding and proteolytic properties, respectively (Ahnert-Hilger and Bigalke, 1995). Because high concentrations of clostridial toxin receptors are present on nerve cells but are practically absent on glial cells (Ahnert-Hilger and Bigalke, 1995), the holotoxin acts selectively on neurons (Araque et al., 1998a). Therefore, to investigate the involvement of synaptobrevin on glutamate release from astrocytes, we bypassed the steps required for the Botulinum B holotoxin to become intracellularly active (i.e., binding, internalization, and reduction) by directly microinjecting the light chain of the neurotoxin BoNT/B into single astrocytes. In addition, we concurrently injected the fluorescent indicator fluoro-ruby to label the injected cell.
To confirm that synaptobrevin was cleaved after injection with BoNT/B, we performed immunocytochemistry using an antibody against synaptobrevin II that was visualized using an FITC-conjugated secondary antibody (Fig.3A,B). Although the anti-synaptobrevin fluorescent signal was similar in control uninjected cells and in cells injected with fluoro-ruby alone (n = 4) (Fig. 3C), it was markedly reduced in cells injected with BoNT/B (n = 6) (Fig.3D).
Fig. 3.

Microinjection of the light chain of the Botulinum toxin B (BoNT/B) into astrocytes reduces synaptobrevin immunofluorescence. An individual astrocyte in a microisland was microinjected with fluoro-ruby (A) together with the light chain of BoNT/B. Subsequent immunostaining of the preparation with anti-synaptobrevin II showed a reduced immunoreactivity in the microinjected cell (B). Images inA and B were constructed from a stack of 16 successive images (2 μm deep) obtained with laser scanning confocal microscopy. Boxes show regions used to compute average linescan fluorescent intensities (20 lines wide) that are presented in D. In C andD, the normalized intensities of fluoro-ruby and anti-synaptobrevin immunoreactivity are shown as average line scans. In BoNT/B-injected astrocytes (D), the intensity of the anti-synaptobrevin immunoreactivity (green line) is reduced in the region that corresponds to the injected cell (red line), whereas in astrocytes injected with fluoro-ruby alone (C, original micrographs are not shown), it remains constant in comparison to neighboring uninjected cells.
We determined whether the injection of BoNT/B affected Ca2+ signaling in astrocytes by loading cells with the Ca2+ indicator fluo-4 (Fig.4). Mechanical stimulation of uninjected astrocytes (18 of 21) or astrocytes injected with fluoro-ruby alone (five of six) increased the intracellular Ca2+ in the directly stimulated cell and evoked Ca2+ waves in neighboring astrocytes. These results confirm that neither the injection procedure per se nor the injection of fluoro-ruby affects the ability of astrocytes to respond to direct stimulation or to evoke Ca2+ waves (Araque et al., 1998a,b). Mechanically induced Ca2+ wave generation was also unaffected by injection of BoNT/B into the stimulated astrocytes (10 of 11). However, the Ca2+elevation in astrocytes and the generation of Ca2+ waves were practically prevented when BAPTA was coinjected (only one of eight injected astrocytes evoked Ca2+ waves), confirming the suitability of the injection procedure.
Fig. 4.

Astrocyte Ca2+ wave propagation was unaffected by injection of BoNT/B. A,Mechanically induced Ca2+ wave recorded in astrocytes loaded with the Ca2+ indicator fluo-4 in control conditions. Panels show images in pseudocolor mode representing intensity of fluo-4 emission, taken before, during, and after mechanical stimulation, at the times indicated. Zero time corresponds to the time of astrocyte stimulation. Mechanical stimulation increases intracellular Ca2+ in the injected cell as well as in neighboring non-stimulated astrocytes.B, Stimulation of a single astrocyte microinjected with fluoro-ruby and BoNT/B (left panel) increases the intracellular Ca2+ in the injected cell as well as in neighboring unstimulated astrocytes. Right panels show pseudocolor images representing intensity of fluo-4 emission at the times indicated in A. Zero time corresponds to the time of astrocyte stimulation. C, As in B but with a single astrocyte microinjected with fluoro-ruby, BoNT/B, and BAPTA (left panel). Mechanical stimulation of the injected cell did not change the fluorescent emission of fluo-4 in either the stimulated or neighboring astrocytes (right panels).
To further confirm that the Ca2+ signal was preserved in BoNT/B-injected astrocytes, we analyzed the involvement of the injected astrocytes in Ca2+ waves generated by adjacent astrocytes and the ability of those astrocytes to generate ligand-evoked Ca2+ waves. We found that eight of nine BoNT/B-injected astrocytes participated in Ca2+ waves initiated by adjacent astrocytes that were stimulated either mechanically or by pressure ejection of norepinephrine (NE). Furthermore, the susceptibility of astrocytes to directly respond to NE was unmodified by injection of BoNT/B. Indeed, Ca2+ waves were similarly generated by focal application of NE to uninjected or BoNT/B-injected astrocytes (six of six, and five of five astrocytes, respectively). Taken together, these results indicate that both the signal transduction and the related intracellular second messenger system underlying the generation and propagation of Ca2+ waves were still functional after microinjection of BoNT/B, again confirming the healthy condition of the injected astrocytes.
After determining that microinjected BoNT/B cleaves synaptobrevin without affecting the overall physiology of astrocytes, we asked whether it prevents glutamate release from astrocytes by monitoring the SIC in neurons cultured on top of astrocytes. BoNT/B is unlikely to pass through the gap junctions that interconnect astrocytes. Therefore, to ensure that all astrocytes which make contact with neurons contained BoNT/B and fluoro-ruby, we used microisland cultures containing a maximum of four astrocytes and microinjected BoNT/B into each cell (Fig. 5A). Mechanical stimulation of uninjected astrocytes reliably induced a SIC in adjacent neurons (8 of 11 stimulated astrocytes; mean current amplitude: 96.8 ± 26.6 pA). Similarly, 8 of 13 astrocytes that had been injected with fluoro-ruby alone evoked neuronal SIC after astrocyte stimulation (mean current amplitude: 69.2 ± 22.4 pA; which is not significantly different from uninjected astrocytes). However, only 1 of 16 astrocytes injected with fluoro-ruby and BoNT/B induced a SIC in adjacent neurons (mean current amplitude: 5.6 ± 5.6 pA; which is significantly different from fluoro-ruby-injected astrocytes;p < 0.01) (Fig. 5B–D). These results indicate that microinjection into astrocytes of BoNT/B, which selectively cleaves the SNARE protein synaptobrevin, prevented the stimulus-induced Ca2+-dependent glutamate release from astrocytes that is detected in associated neurons as a SIC.
Fig. 5.

BoNT/B prevents glutamate-mediated SIC in adjacent neurons. A, Left panel, Phase-contrast image of a microisland containing two astrocytes and two neurons (somas indicated by arrows). Right panel, Epifluorescence image of the same microisland after the microinjection of both astrocytes with fluoro-ruby. B, Representative whole-cell currents recorded from neurons in microislands in which the astrocytes were microinjected with either fluoro-ruby (left) or fluoro-ruby and BoNT/B (right). Although mechanically stimulated astrocytes injected with fluoro-ruby alone reliably evoked SIC in adjacent neurons, those injected with BoNT/B failed to evoke SIC. Mechanical stimulation of the astrocyte is indicated by the asterisk. Note the noise increase during the SIC, which is probably caused by the activation of NMDA receptors (Araque et al., 1998a). C, Proportion of mechanically stimulated astrocytes that evoked glutamate-dependent SIC in adjacent neurons. D, Current amplitude recorded in neurons after mechanical stimulation of adjacent astrocytes. Significant differences were established by the Student'st test at p < 0.01 (**).
DISCUSSION
Our present results suggest that vesicle-associated mechanisms underlie the Ca2+-dependent glutamate release from astrocytes that is involved in the astrocyte-to-neuron signaling. Indeed, we have found that the Ca2+-dependent, glutamate-mediated neuronal electrophysiological responses elicited by mechanical stimulation of astrocytes were not mediated by the reverse operation of glutamate transporters, which is in agreement with previous findings using Ca2+ imaging techniques and enzymatic assays as well as different stimuli (e.g., bradykinin, glutamate receptor agonists, photorelease of Ca2+) (Parpura et al., 1994; Araque et al., 1998b; Bezzi et al., 1998).
By disrupting the function of two vesicle-associated proteins, V-ATPase and synaptobrevin, that are known to be essential in neurotransmitter release from synaptic terminals (Schiavo et al., 1992; Simpson et al., 1994; Cousin et al., 1995; Pocock et al., 1995; Liu and Edwards, 1997;Stevens and Forgac, 1997; Deitcher et al., 1998; Nonet et al., 1998;Fernández-Chacón and Südhof, 1999), we examined the participation of these vesicle-associated proteins in glutamate release from astrocytes.
We have found that bafilomycin A1, a V-ATPase inhibitor that dissipates the electrochemical proton gradient necessary for glutamate uptake in vesicles (Bowman et al., 1988; Maycox et al., 1988; Carlson et al., 1989; Moriyama et al., 1990), reduced the ability of astrocyte to release glutamate. Interestingly, although astrocyte Ca2+ waves require functional internal Ca2+ stores that can be depleted after inhibition of the Ca2+-ATPase by thapsigargin (Charles et al., 1993; Newman and Zahs, 1997; Araque et al., 1998a,b), Ca2+ waves in astrocytes were unaffected by the V-ATPase inhibitor, confirming that bafilomycin did not affect other ATPases but selectively inhibited the V-ATPase (Bowman et al., 1988). These results indicate that Ca2+-dependent glutamate release from astrocytes requires the functional presence of the vesicle-associated protein V-ATPase, suggesting that the glutamate released by stimulation of astrocytes is stored in a vesicular compartment.
We have also demonstrated that glutamate release from astrocytes was strongly reduced after microinjection of astrocytes with the light chain of the neurotoxin Botulinum B, which cleaved the vesicle-associated SNARE protein synaptobrevin. Our data are in agreement with previous studies showing that chronic treatment with tetanus toxin (>12 hr) reduced the amount of glutamate released from astrocytes (Jeftinija et al., 1997; Bezzi et al., 1998). However, because of the prolonged incubation times used in these previous studies, the physiological integrity of the astrocytes was uncertain. In this study we microinjected the active chain of Botulinum toxin to perform experiments on the time frame of ∼1 hr after microinjection. What is more, Ca2+ imaging studies support the specificity of toxin action and demonstrate the physiological integrity of astrocytes. Because our previous studies have shown that astrocytes express synaptobrevin II and syntaxin (Parpura et al., 1995), and because BoNT/B blocks the Ca2+-dependent SIC, our studies indicate that the Ca2+-dependent release of glutamate from astrocytes is a SNARE protein-dependent process. Although these results strongly implicate a vesicular mode of glutamate release, further studies are required, however, that must include an ultrastructural analysis before it is possible to conclude that this release pathway is mediated by exocytosis.
ATP released from astrocytes acts as a diffusible extracellular messenger that is critical for the propagation of Ca2+ waves between cultured astrocytes (Cotrina et al., 1998; Guthrie et al., 1999). Although we did not study ATP release per se, the fact that Ca2+waves appeared unaffected by either BoNT/B injection or by bafilomycin treatment suggests that ATP is still released from astrocytes. Consequently, it is likely that ATP is released through a distinct mechanism from the glutamate release pathway.
The combined conclusions based on bafilomycin and Botulinum toxin studies strongly implicate a vesicular compartment as mediating the Ca2+-dependent release of glutamate from astrocytes. Furthermore, because this Ca2+-dependent glutamate signaling pathway is present in acutely isolated preparations of brain slice (Bezzi et al., 1998), it is not likely to be a peculiarity simply associated with astrocytes in cell culture. Assuming that this property is present in brain tissue, what structural form does it take? Characteristically, we think of vesicle-mediated transmitter release as being associated with a cloud of vesicles as is often found in nerve terminals. However, the processes of astrocytes that enwrap nerve terminals do not contain an abundance of these structures. Thus, further evidence is needed before it could be concluded that vesicles mediate glutamate release from astrocytes. However, it is likely that few vesicles need be present to mediate such release. At the developing neuromuscular junction, for example, significant quantal release of acetylcholine, presumably released from vesicles, can be detected at times when clearly defined ultrastructural correlates of the synapse are lacking (Buchanan et al., 1989).
In conclusion, our data support a requirement for SNARE proteins and a vesicular electrochemical proton gradient for the release of glutamate from astrocytes, raising the hypothesis that vesicle exocytosis mediates this transmitter release.
Footnotes
This work was supported by grants from National Institutes of Health (NS24233 and NS37585) and from the Iowa State University Biotechnology Council to P.G.H., and by a long-term postdoctoral fellowship from the Human Frontier Science Program to A.A. We thank Drs. W. Buño and J. Lerma for their comments on this manuscript.
Correspondence should be addressed to Dr. Araque at his present address: Instituto Cajal, Consejo Superior de Investigaciones Cientificas, Doctor Arce 37, Madrid 28002, Spain. E-mail:araque@cajal.csic.es.
REFERENCES
- 1.Ahnert-Hilger G, Bigalke H. Molecular aspects of tetanus and botulinum neurotoxin poisoning. Prog Neurobiol. 1995;46:83–96. doi: 10.1016/0301-0082(95)00003-e. [DOI] [PubMed] [Google Scholar]
- 2.Araque A, Parpura V, Sanzgiri RP, Haydon PG. Glutamate-dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. Eur J Neurosci. 1998a;10:2129–2142. doi: 10.1046/j.1460-9568.1998.00221.x. [DOI] [PubMed] [Google Scholar]
- 3.Araque A, Sanzgiri RP, Parpura V, Haydon PG. Calcium elevation in astrocytes causes an NMDA receptor-dependent increase in the frequency of miniature synaptic currents in cultured hippocampal neurons. J Neurosci. 1998b;18:6822–6829. doi: 10.1523/JNEUROSCI.18-17-06822.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999a;22:208–215. doi: 10.1016/s0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- 5.Araque A, Sanzgiri RP, Parpura V, Haydon PG. Astrocyte-induced modulation of synaptic transmission. Can J Physiol Pharmacol. 1999b;77:699–706. [PubMed] [Google Scholar]
- 6.Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Lodi Rizzini B, Pozzan T, Volterra A. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature. 1998;391:281–285. doi: 10.1038/34651. [DOI] [PubMed] [Google Scholar]
- 7.Bowman EJ, Siebers A, Altendorf K. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells and plant cells. Proc Natl Acad Sci USA. 1988;85:7972–7976. doi: 10.1073/pnas.85.21.7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buchanan J, Sun YA, Poo MM. Studies of nerve-muscle interactions in Xenopus cell culture: fine structure of early functional contacts. J Neurosci. 1989;9:1540–1554. doi: 10.1523/JNEUROSCI.09-05-01540.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carlson MD, Kish PE, Ueda T. Characterization of the solubilized and reconstituted ATP-dependent vesicular glutamate uptake system. J Biol Chem. 1989;264:7369–7376. [PubMed] [Google Scholar]
- 10.Charles AC. Glia-neuron intercellular calcium signaling. Dev Neurosci. 1994;16:196–206. doi: 10.1159/000112107. [DOI] [PubMed] [Google Scholar]
- 11.Charles AC, Merrill JE, Dirksen ER, Sanderson MJ. Intercellular signaling in glial cells: calcium waves and oscillations in response to mechanical stimulation and glutamate. Neuron. 1991;6:983–992. doi: 10.1016/0896-6273(91)90238-u. [DOI] [PubMed] [Google Scholar]
- 12.Charles AC, Dirksen ER, Merrill JE, Sanderson MJ. Mechanisms of intercellular calcium signaling in glial cells studied with dantrolene and thapsigargin. Glia. 1993;7:134–145. doi: 10.1002/glia.440070203. [DOI] [PubMed] [Google Scholar]
- 13.Cornell-Bell AH, Finkbeiner SM, Cooper MS, Smith SJ. Glutamate induces calcium waves in cultured astrocytes: long-range glial signalling. Science. 1990;247:470–473. doi: 10.1126/science.1967852. [DOI] [PubMed] [Google Scholar]
- 14.Cotrina ML, Lin JH, Alves-Rodrigues A, Liu S, Li J, Azmi-Ghadimi H, Kang J, Naus CC, Nedergaard M. Connexins regulate calcium signaling by controlling ATP release. Proc Natl Acad Sci USA. 1998;95:15735–15740. doi: 10.1073/pnas.95.26.15735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cousin MA, Nicholls DG, Pocock JM. Modulation of ion gradients and glutamate release in cultured cerebellar granule cells by ouabain. J Neurochem. 1995;64:2097–2104. doi: 10.1046/j.1471-4159.1995.64052097.x. [DOI] [PubMed] [Google Scholar]
- 16.Dani JW, Chernjavsky A, Smith SJ. Neuronal activity triggers calcium waves in hippocampal astrocyte networks. Neuron. 1992;8:429–440. doi: 10.1016/0896-6273(92)90271-e. [DOI] [PubMed] [Google Scholar]
- 17.Deitcher DL, Ueda A, Stewart BA, Burgess RW, Kidokoro Y, Schwarz TL. Distinct requirements for evoked and spontaneous release of neurotransmitter are revealed by mutations in the Drosophila gene neuronal-synaptobrevin. J Neurosci. 1998;18:2028–2039. doi: 10.1523/JNEUROSCI.18-06-02028.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fernández-Chacón R, Südhof TC. Genetics of synaptic vesicle function: toward the complete functional anatomy of an organelle. Annu Rev Physiol. 1999;61:753–776. doi: 10.1146/annurev.physiol.61.1.753. [DOI] [PubMed] [Google Scholar]
- 19.Guthrie PB, Knappenberger J, Segal M, Bennet MVL, Charles AC, Kater SB. ATP released from astrocytes mediates glial calcium waves. J Neurosci. 1999;19:520–528. doi: 10.1523/JNEUROSCI.19-02-00520.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanada H, Moriyama Y, Maeda M, Futai M. Kinetic studies of chromaffin granule H+-ATPase and effects of bafilomycin A1. Biochem Biophys Res Commun. 1990;170:873–878. doi: 10.1016/0006-291x(90)92172-v. [DOI] [PubMed] [Google Scholar]
- 21.Hassinger TD, Atkinson PB, Strecker GJ, Whalen LR, Dudek FE, Koseel AH, Kater SB. Evidence for glutamate-mediated activation of hippocampal neurons by glial calcium waves. J Neurobiol. 1995;28:159–170. doi: 10.1002/neu.480280204. [DOI] [PubMed] [Google Scholar]
- 22.Jeftinija SD, Jeftinija KV, Stefanovic G. Cultured astrocytes express proteins involved in vesicular glutamate release. Brain Res. 1997;750:41–47. doi: 10.1016/s0006-8993(96)00610-5. [DOI] [PubMed] [Google Scholar]
- 23.Kang J, Jiang L, Goldman SA, Nedergaard M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci. 1998;1:683–692. doi: 10.1038/3684. [DOI] [PubMed] [Google Scholar]
- 24.Kimelberg HK, Goderie SK, Higman S, Pang S, Waniewski RA. Swelling-induced release of glutamate, aspartate, and taurine from astrocytes cultures. J Neurosci. 1990;10:1583–1591. doi: 10.1523/JNEUROSCI.10-05-01583.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Y, Edwards RH. The role of vesicular transport proteins in synaptic transmission and neural degeneration. Annu Rev Neurosci. 1997;20:125–156. doi: 10.1146/annurev.neuro.20.1.125. [DOI] [PubMed] [Google Scholar]
- 26.Maienschein V, Marxen M, Volknandt W, Zimmermann H. A plethora of presynaptic proteins associated with ATP-storing organelles in cultured astrocytes. Glia. 1999;26:233–244. doi: 10.1002/(sici)1098-1136(199905)26:3<233::aid-glia5>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 27.Maycox PR, Deckwerth T, Hell JW, Jahn R. Glutamate uptake by brain synaptic vesicles. Energy dependence of transport and functional reconstitution in proteoliposomes. J Biol Chem. 1988;263:15423–15428. [PubMed] [Google Scholar]
- 28.Moriyama Y, Maeda M, Futai M. Energy coupling of l-glutamate transport and vacuolar H+-ATPase in brain synaptic vesicles. J Biochem. 1990;108:689–693. doi: 10.1093/oxfordjournals.jbchem.a123264. [DOI] [PubMed] [Google Scholar]
- 29.Nedergaard M. Direct signaling from astrocytes to neurons in cultures of mammalian brain cells. Science. 1994;263:1768–1771. doi: 10.1126/science.8134839. [DOI] [PubMed] [Google Scholar]
- 30.Newman EA, Zahs KR. Calcium waves in retinal glial cells. Science. 1997;275:844–847. doi: 10.1126/science.275.5301.844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Newman EA, Zahs KR. Modulation of neuronal activity by glial cells in the retina. J Neurosci. 1998;18:4022–4028. doi: 10.1523/JNEUROSCI.18-11-04022.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nonet ML, Saifee O, Zhao H, Rand JB, Wei L. Synaptic transmission deficits in Caenorhabditis elegans synaptobrevin mutants. J Neurosci. 1998;18:70–80. doi: 10.1523/JNEUROSCI.18-01-00070.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369:744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- 34.Parpura V, Fang Y, Basarsky TA, Jahn R, Haydon PG. Expression of synaptobrevin II, cellubrevin and syntaxin but not SNAP-25 in cultured astrocytes. FEBS Lett. 1995;377:489–492. doi: 10.1016/0014-5793(95)01401-2. [DOI] [PubMed] [Google Scholar]
- 35.Pasti L, Volterra A, Pozzan T, Carmignoto G. Intracellular calcium oscillations in astrocytes: a highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J Neurosci. 1997;17:7817–7830. doi: 10.1523/JNEUROSCI.17-20-07817.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pocock JM, Cousin MA, Parkin J, Nicholls DG. Glutamate exocytosis from cerebellar granule cells: the mechanism of a transition to an L-type Ca2+ channel coupling. Neuroscience. 1995;67:595–607. doi: 10.1016/0306-4522(95)00061-m. [DOI] [PubMed] [Google Scholar]
- 37.Porter JT, McCarthy KD. Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J Neurosci. 1996;16:5073–5081. doi: 10.1523/JNEUROSCI.16-16-05073.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roseth S, Fykse EM, Fonnum F. Uptake of L-glutamate into rat brain synaptic vesicles: effect of inhibitors that bind specifically to the glutamate transporter. J Neurochem. 1995;65:96–103. doi: 10.1046/j.1471-4159.1995.65010096.x. [DOI] [PubMed] [Google Scholar]
- 39.Sanzgiri RP, Araque A, Haydon PG. Prostaglandin E2 stimulates glutamate receptor-dependent astrocyte neuromodulation in cultured hippocampal cells. J Neurobiol. 1999;41:221–229. [PubMed] [Google Scholar]
- 40.Schiavo G, Benfenati F, Poulain B, Rossetto O, Polverino de Laureto P, DasGupta BR, Montecucco C. Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature. 1992;359:832–835. doi: 10.1038/359832a0. [DOI] [PubMed] [Google Scholar]
- 41.Simpson LL, Coffield JA, Bakry N. Inhibition of vacuolar adenosine triphosphatase antagonizes the effects of clostridial neurotoxins but not phospholipase A2 neurotoxins. J Pharmacol Exp Ther. 1994;269:256–262. [PubMed] [Google Scholar]
- 42.Stevens TH, Forgac M. Structure, function and regulation of the vacuolar (H+)-ATPase. Annu Rev Cell Dev Biol. 1997;13:779–808. doi: 10.1146/annurev.cellbio.13.1.779. [DOI] [PubMed] [Google Scholar]
- 43.Szatkowski M, Barbour B, Attwell D. Non-vesicular release of glutamate from glial cells by reversed electrogenic glutamate uptake. Nature. 1990;348:443–447. doi: 10.1038/348443a0. [DOI] [PubMed] [Google Scholar]