

Abstract
NMDA receptors interact via the extended intracellular C-terminal domain of the NR2 subunits with constituents of the postsynaptic density for purposes of retention, clustering, and functional regulation at central excitatory synapses. To examine the role of the C-terminal domain of NR2A in the synaptic localization and function of NR2A-containing NMDA receptors in hippocampal Schaffer collateral–CA1 pyramidal cell synapses, we analyzed mice which express NR2A only in its C-terminally truncated form. In CA1 cell somata, the levels, activation, and deactivation kinetics of extrasynaptic NMDA receptor channels were comparable in wild-type and mutant NR2AΔC/ΔCmice. At CA1 cell synapses, however, the truncated receptors were less concentrated than their full-length counterparts, as indicated by immunodetection in cultured neurons, synaptosomes, and postsynaptic densities. In the mutant, the NMDA component of evoked EPSCs was reduced in a developmentally progressing manner and was even more reduced in miniature EPSCs (mEPSCs) elicited by spontaneous glutamate release. Moreover, pharmacologically isolated NMDA currents evoked by synaptic stimulation had longer latencies and displayed slower rise and decay times, even in the presence of an NR2B-specific antagonist. These data strongly suggest that the C-terminal domain of NR2A subunits is important for the precise synaptic arrangement of NMDA receptors.
Keywords: immunocytochemistry; Western blotting; patch clamp; hippocampal culture and slice; mice expressing C-terminally truncated NR2A subunits; nucleated patches; evoked EPSCs; miniature currents; NR2B-specific antagonists (CP-101,606)
NMDA receptors are critical players in excitatory synaptic transmission as well as in synaptic plasticity, which may underlie memory acquisition and recall (Bliss and Collingridge, 1993). Several NMDA receptor subtypes exist, defined by the particular NR2 subunits that assemble with the principal NR1 subunit. The NR2 subunits impart on receptor channels distinct kinetic, gating, divalent ion block, ion permeation, and pharmacological properties (Hollmann and Heinemann, 1994; McBain and Mayer, 1994;Dingledine et al., 1999). Furthermore, they possess extended intracellular C-terminal domains by which they interact with diverse proteins of the postsynaptic density (PSD) for purposes of synaptic localization, clustering, and signal transduction (Sheng, 1996;Kennedy, 1997, 1998; Kornau et al., 1997; Kim and Huganir, 1999).
The importance of the C termini of NMDA receptor subunits is further emphasized by NR2ΔC/ΔCmice expressing C-terminally truncated NR2 subunits (Sprengel et al., 1998), which phenotypically resemble the respective NR2 knockout mice. Mice expressing C-terminally truncated NR2BΔC subunits died perinatally (Mori et al., 1998; Sprengel et al., 1998), whereas mice expressing C-terminally truncated NR2AΔC subunits were ataxic and failed to develop long-term potentiation (LTP) (Sprengel et al., 1998). The protein level of NR2ΔC subunits in total brain appeared normal (Mori et al., 1998; Sprengel et al., 1998). Thus, the most likely explanation for the correspondence in phenotype of mice expressing a C-terminally truncated NR2 subunit, or not expressing the NR2 subunit at all, was that absence of the C-terminal tail prevented the proper linkage of synaptic NMDA receptors to signal transduction pathways, thus amounting to a lack of NMDA receptor function (Sprengel et al., 1998). In addition, Mori and colleagues (1998) found a greatly reduced contribution of NR2BΔC subunits at perinatal hippocampal CA1 synapses.
CA1 neurons from adult animals express NMDA receptors containing NR2A and/or NR2B subunits. Their relative contribution to synaptic transmission and plasticity is unclear and may result from distinct regulation by intracellular signaling molecules and/or differential subcellular localization. The C-terminal tails of NR2A and NR2B are potential targets for tyrosine kinases, CaM kinase II, and protein kinases A and C (Moon et al., 1994; Wang and Salter, 1994; Köhr and Seeburg, 1996; Omkumar et al., 1996; Leonard and Hell, 1997;Tingley et al., 1997). The regulation of NMDA receptor activity can be caused by a direct phosphorylation of receptor protein or may involve the phosphorylation of associated postsynaptic proteins (Zheng et al., 1999). Although in cultured neurons NMDA receptors containing NR2A localize preferentially at synaptic sites and NMDA receptors containing NR2B subunits can also localize extrasynaptically (Li et al., 1998;Tovar and Westbrook, 1999), both NR2A- and NR2B-containing receptors are activated by Schaffer collateral stimulation in hippocampal slices of adult mice (Kirson and Yaari, 1996), consistent with the presence of both NR2A and NR2B subunits in postsynaptic densities (Kennedy, 1997,1998).
We have studied NR2AΔC/ΔCmice by immunocytochemistry, biochemistry, and electrophysiology. We found a reduced number of synaptic NR2AΔC-containing NMDA receptors compared with wild type, although the number of somatic, i.e., extrasynaptic mutant receptors appeared unchanged. In addition, the slower kinetic properties of evoked synaptic NMDA receptor currents in the mutants suggest that the truncated mutant receptors are located, on average, farther from release sites than wild-type receptors.
MATERIALS AND METHODS
Immunocytochemistry. Cultures of mouse hippocampal neurons (embryonic day 16) were grown as described (Brewer et al., 1993) from 10 wild-type and 11 NR2AΔC/ΔCembryos. After 21 d in vitro, cultures were fixed and labeled (Kornau et al., 1995) with an affinity-purified rabbit antiserum raised against an N-terminal epitope of NR2A (8.5 μg/ml). Cultures from 14 embryos were labeled in parallel with anti-NR2B C-terminal rabbit antiserum diluted 1:300 (Kornau et al., 1995). Cultures from one wild-type and two mutant embryos were double-labeled with anti-NR2A or anti-NR2B, respectively, and mouse anti-synapsin I (1:250; Chemicon, Temecula, CA) antibodies. After incubation with secondary antibodies (Alexa 488-conjugated goat anti-rabbit IgG and Alexa 568-conjugated goat anti-mouse IgG; Molecular Probes, Eugene, OR), 8–22 individual neurons per embryo were selected under phase optics, then imaged confocally (Zeiss LSM 310) at identical contrast and brightness settings.
Images for computer analysis were chosen based on normal morphological appearance of pyramidal neurons, independent of their staining patterns. Dendrites were selected in Adobe Photoshop 5.0.2, and their length was measured using Canvas 5.0 (Daneba Systems). In NIH Image 1.62 (developed at National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/), pictures were inverted and an identical threshold intensity (130) was set to automatically analyze intensity, area, and number of the clusters. For background analysis (pixels below threshold), a “density slice” range was set from 254 to 130. Neither background intensity nor cluster staining intensity differed between genotypes (background, NR2A: wild type, 36 ± 1, n = 12; mutant, 33 ± 1,n = 12; NR2B: wild type, 39 ± 1,n = 6; mutant, 36 ± 2, n = 6; cluster intensity, NR2A: wild type, 163 ± 2, n = 12; mutant, 157 ± 1, n = 12: NR2B, wild type, 170 ± 9, n = 6; mutant, 183 ± 5,n = 6).
Western blot analysis. Homogenates, synaptosomes, and PSD fractions were prepared as described (Carlin et al., 1980) from postnatal day 30 (P30) mouse forebrains (PSD fraction of mutants in NR2A blot, P42) except that the Triton X-100-treated synaptosomes were not purified on a second sucrose density gradient but were centrifuged (1 hr, 70,000 × g) to obtain the PSD fraction. For synaptosomes and PSD preparations, eight brains (for homogenates, two brains) were pooled and homogenized. Protein concentration was determined by a Lowry assay after deoxycholic acid-trichloroacetic acid precipitation.
Proteins were separated by 10% SDS-PAGE (for NR2A) or 8% SDS-PAGE (for NR1 and NR2B) and blotted onto nitrocellulose membranes (BA 85, Schleicher & Schuell) using a wet transfer device. Primary antibodies and dilutions were the following: mouse monoclonal, N-terminal anti-NR2A, 1:500 (2H9.24A6, Boehringer Mannheim, Mannheim, Germany); rabbit polyclonal, C-terminal anti-NR2B, 1:20000 (see above); rabbit polyclonal, C-terminal anti-NR1, 1:300 (Chemicon). Signals were generated by alkaline phosphatase with appropriate species-matched secondary antibodies (anti-mouse, Boehringer Mannheim; anti-rabbit, Jackson ImmunoResearch, West Grove PA). Immunoblots were scanned and signals were quantified in Image Gauge 3.0. To document specific enrichment of PSD protein complexes, an antibody against a cytosolic protein was applied (rabbit polyclonal anti-Dendrin, crude bleed, 1:2000). Dendrin was present in homogenates, reduced in synaptosomes, but undetectable in PSDs (data not shown).
Electrophysiology. Transverse hippocampal slices (250 μm) were cut from brains of 13- to 15-d-old (P15) and 28- to 30-d-old (P30) C57BL6 wild-type and NR2AΔC/ΔCmice. CA1 pyramidal cells were identified by infrared differential interference contrast microscopy (Stuart et al., 1993). All measurements were performed at room temperature (22°C) using an EPC-9 amplifier (HEKA Elektronik). Somatic recordings were performed with a piezo-controlled fast application system (solution exchange time, measured with an open patch pipette, was 100–200 μsec) to activate nucleated whole-soma patches in solution containing (in mm): 135 NaCl, 5.4 KCl, 1.8 CaCl2, 5 HEPES, pH 7.25, NaOH. The intracellular solution consisted of (in mm): 140 CsCl, 2 MgCl2, 2 Na2-ATP, 10 EGTA, 10 HEPES, pH 7.25, CsOH. AMPA or NMDA receptor currents were activated by glutamate (1 mm; 10 or 50 msec application) in the presence of AP5 (30 μm, Tocris, Ballwin, MO) or NBQX (5 μm, Tocris), glycine (10 μm, Sigma, St. Louis, MO) and 0 mm Mg2+ from −100 mV to +100 mV in 20 mV increments. The ratio of NMDA/AMPA receptor currents was derived from the peaks recorded at −40 mV and −80 mV. Desensitization was investigated at −80 mV and +80 mV using 400 msec glutamate pulses, and the recovery from desensitization was investigated by 50 msec pulses at −80 mV using a paired-pulse protocol.
For synaptic currents, the recording chamber was perfused with an extracellular low Mg2+ solution (Ringer's) consisting of (in mm): 125 NaCl, 2.5 KCl, 25 glucose, 25 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 0.1 MgCl2, 0.01 bicuculline (Sigma), and 0.01 glycine, continuously bubbled with 5% CO2–95% O2. Intracellular solution for EPSC recordings contained (in mm): 125 Cs-gluconate, 20 CsCl, 10 NaCl, 10 HEPES, 0.2 EGTA, 4 ATP-Mg, 0.3 GTP-Na, and 2.5 QX-314 (Calbiochem, La Jolla CA), pH 7.25, CsOH. Pipette resistance was 3–5 MΩ. A bipolar tungsten electrode was placed in the stratum radiatum to stimulate at 0.05 Hz. Series resistance compensation was set to 60–90% in whole-cell configuration. To monitor series resistance, a 5 mV hyperpolarizing voltage step was applied before each stimulation, and cells with changes >15% were discarded. The amplitudes of the AMPA and NMDA components of EPSCs were determined at −60 mV in the absence and presence of NBQX (5–10 μm). The presence of residual Mg2+ did not affect the kinetics of NMDA EPSCs recorded at −60 mV, because the kinetics were in good agreement with those recorded at + 40 mV in 1.0 mmMg2+-containing Ringer's solution. For analyses and example traces in Figures, the averages of four to eight single stimulations were used. Ifenprodil (0.3, 3, or 10 μm; Sigma) reversibly reduced somatic NMDA receptor currents. We used CP-101,606 (CP, 10 μm) in a concentration known to selectively reduce NR2B-mediated NMDA receptor-mediated currents in slices (Stocca and Vicini, 1998). The reduction of NMDA EPSCs was only partially reversible even after 40 min of washout. CP did not affect AMPA-mediated currents, which was tested in control experiments. In both genotypes, AP5 (30 μm) completely blocked NMDA EPSCs (data not shown).
Miniature EPSCs containing AMPA and NMDA components were recorded at −70 mV in low Mg2+ Ringer's solution (see above) containing additionally Ca2+(4 mm final concentration) and TTX (1 μm; Molecular Probes). Subsequently, AP5 (30–50 μm) was added to record AMPA-only mEPSCs from the same cell. Recordings were analyzed off-line (filtering, 2.9 kHz; sampling, 5 kHz) from 3 min sections using an event detection program (kindly provided by Prof. Misgeld, Institute of Physiology, University of Heidelberg). For event detection, trigger level was set at approximately two to three times baseline noise, and false positive events (e.g., artifacts or events overlapping in time) were excluded by subsequent raw data inspection. Rise time, peak, and decay of averaged mEPSCs per 3 min section were analyzed, and frequencies were calculated. The NMDA-only component was estimated by subtracting the integral of the pharmacologically isolated AMPA mEPSC from the integral of the total mEPSC. During 30 min recordings, the frequencies of the mEPSCs reduced to the same extent in wild type and mutant (data not shown). Miniature EPSCs were monoexponentially and biexponentially fitted. The slow components of biexponential fits comprised <25% in wild type and mutant, and they were not selectively affected by AP5. Therefore, the effects on monoexponentially fitted mEPSCs are presented.
Values are expressed as mean ± SEM, and p values represent the result of independent two-tailed t tests.
RESULTS
Reduced NR2A staining in hippocampal dendrites of NR2AΔC/ΔCmice
To investigate the localization of NMDA receptors containing the truncated NR2AΔC subunit, we first compared by immunocytochemistry the staining intensity (see Materials and Methods) and distribution of NR2A in hippocampal cultures of wild-type and NR2AΔC/ΔCmice. Cultured neurons from 18 mice were fluorescently labeled with an antibody against the N terminus of the NR2A subunit and were imaged by confocal microscopy. Examples of neuronal dendrites from wild-type and NR2AΔC/ΔCmice are depicted in Figure1A. Most wild-type neurons had a large number of intensely NR2A-stained clusters along the entire length of their dendrites. These clusters were located in spines, as demonstrated by double-staining with an antibody against the synaptic vesicle protein synapsin I (Fig. 1A,panels 1–3). Most NR2AΔC/ΔCneurons had fewer intensely NR2A-stained clusters (panel 4), with the consequence that the synapsin I staining dominated in the overlay (panel 5). Because of these characteristic differences, evaluators blinded to genotype correctly classified seven out of nine wild-type and nine out of nine NR2AΔC/ΔCcultures.
Fig. 1.
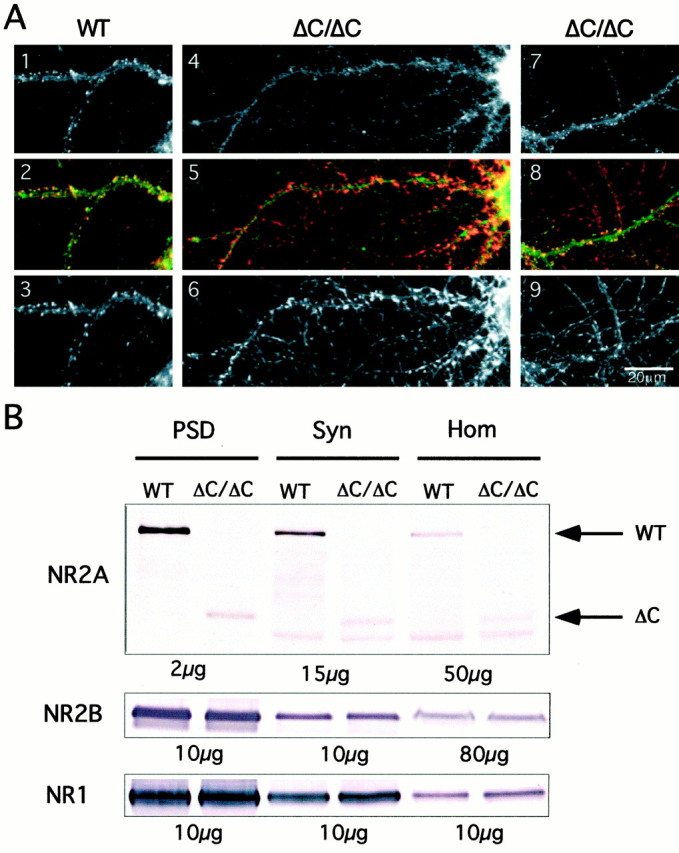
Reduced synaptic enrichment of NR2AΔC protein.A, Immunocytochemistry of primary hippocampal cultures (21 d in vitro). Panels 1–3, Representative neuron from wild-type mouse (WT), double-stained for anti-NR2A (1,green in2) and anti-synapsin I (3,red in 2); panels 4–6,representative neuron from an NR2AΔC/ΔCmouse (ΔC/ΔC), double-stained for anti-NR2A (4,green in 5) and anti-synapsin I (6, red in5); panels 7–9, representative neuron from ΔC/ΔC mouse, double-stained with anti-NR2B (7,green in 8) and anti-synapsin I (9, red in 8). There are fewer brightly stained spines opposite synapsin I-containing terminals in ΔC/ΔC neurons. B, Immunoblots on postsynaptic densities (PSD), synaptosomes (Syn), and homogenate (Hom) from WT and ΔC/ΔC forebrains incubated withNR2A, NR2B, or NR1antibody. Enrichment of full-length NR2A from Hom to PSD was approximately four times more efficient than that of truncated NR2A in ΔC/ΔC mice. Also, the NR2AΔC protein was slightly reduced in the homogenate compared with full-length NR2A. NR2B and NR1 were similarly enriched in both genotypes; NR2B is not upregulated in ΔC/ΔC. Thearrows indicate the WT 175 kDa and the truncated 100 kDa NR2A protein.
Cluster frequencies were quantified for 24 neurons from NR2A-stained cultures (three mice per genotype), and 12 neurons were stained with an NR2B antibody (five mice per genotype). The analysis was restricted to dendrites, and a total of 20.6 mm of wild-type and 20.8 mm of mutant dendrites were analyzed for NR2A-stained clusters, and 16 mm of wild-type and 14 mm of mutant dendrites were analyzed for NR2B-stained clusters. The NR2A cluster frequency was reduced by 30% from 33 ± 4 clusters per 100 μm dendrite (mean ± SEM,n = 12) in wild-type neurons to 23 ± 3 (n = 12) in NR2AΔC/ΔCneurons (p < 0.05) (Fig. 1A), whereas the NR2B cluster frequency was similar in wild-type (40 ± 3, n = 6) and NR2AΔC/ΔCneurons (39 ± 3, n = 6) (Fig.1A). Thus, the cluster frequencies of NR2AΔC-containing NMDA receptors are reduced in dendrites of cultured hippocampal neurons.
Reduced enrichment of NR2AΔC in postsynaptic densities
To elucidate whether the reduced amount of NR2AΔC in hippocampal cultures is caused by lower expression or altered subcellular distribution, we compared the amount of NR2A in homogenates, synaptosomes, and PSDs, prepared from forebrains of wild-type and NR2AΔC/ΔCmice (4–6 week old). Immunoblots were analyzed with an antibody against the N terminus of NR2A and with antibodies against NR2B and NR1 as controls.
In wild-type mice, NR2A, NR2B, and NR1 subunits were enriched in synaptosomes and PSDs, when compared with their levels in homogenates (Fig. 1B). The enrichment from homogenate to PSD of NR2AΔC in NR2AΔC/ΔCmice was approximately fourfold less than that of full-length NR2A in wild type, but enrichment of NR2B and NR1 was not altered. We also noticed slightly lower levels of NR2AΔC in NR2AΔC/ΔChomogenates when compared with full-length NR2A in homogenates from wild-type mice. The immunoblots did not detect a compensatory increase in the expression of NR2B (Fig. 1B).
Normal NR2AΔC-containing NMDA receptor levels in CA1 cell somata
To investigate whether somatic, i.e., extrasynaptic NMDA receptors are reduced in NR2AΔC/ΔCmice, we activated pharmacologically isolated NMDA receptor currents by fast application of glutamate in nucleated whole-soma patches of CA1 pyramidal neurons (Sather et al., 1992). Peak currents through NMDA receptor channels were recorded at −40 mV in nucleated patches of 12 wild-type and 7 NR2AΔC/ΔCmice (2 week old = P15) and were −196 ± 25 pA (n = 16) and −254 ± 56 pA (n = 10), respectively (Fig.2A). To evaluate this difference, we related the NMDA peak currents to pharmacologically isolated AMPA peak currents, which were −1098 ± 59 pA in wild-type (n = 16) and −1206 ± 98pA in NR2AΔC/ΔCmice (n = 10). Hence, the ratio of currents through NMDA and AMPA receptors was the same in both genotypes (wild type, 0.18 ± 0.02, n = 16; NR2AΔC/ΔC, 0.2 ± 0.03, n = 10). Similar results were obtained at −80 mV (data not shown), demonstrating comparable levels of functional NMDA receptor channels in the somata of CA1 neurons of both genotypes.
Fig. 2.

Somatic NMDA receptor currents show normal levels of functional NR2AΔC-containing receptors in ΔC/ΔC mice.A, Glutamate was fast-applied for 10 msec to nucleated whole-soma patches of P15 WT (filled symbols) and P15 ΔC/ΔC mice (open symbols) in the presence of AP5 to activate AMPA receptor currents (▪/■) or for 50 msec in the presence of NBQX and glycine but in the absence of Mg2+ to activate NMDA receptor currents (●/○) at −40 mV. The amplitude ratio of NMDA/AMPA receptor currents was comparable (right panel). B, The NR2B-specific antagonist ifenprodil (3 μm) reduced NMDA receptor currents at −80 mV by ∼50% in both genotypes.C, NMDA receptor currents desensitized faster (*p < 0.002) in ΔC/ΔC (○) than in WT (●) during prolonged glutamate application (400 msec; at −80 mV). Representative mutant trace was enlarged by a factor of 1.25.D, Slower recovery from desensitization in ΔC/ΔC mice (○, *p < 0.006) compared with WT (●), investigated by stimulation with two 50 msec glutamate pulses (interpulse intervals: 0.1, 0.2, 0.3, 0.5, 1, 2, 3, 5, or 10 sec).Inset shows superimposed NMDA receptor currents, activated at −80 mV with interpulse intervals of 0.2 (1), 0.5 (2), 1 (3), and 2 sec (4). Data were pooled (WT, n = 7–10; ΔC/ΔC, n = 4–6;n, number of investigated patches per interpulse intervals) and fitted biexponentially (WT, 0.08 and 1.8 msec; ΔC/ΔC, 0.44 and 3.0 msec).
We used the NR2B-specific antagonist ifenprodil (Williams, 1993) to investigate the contribution of NR2B-containing receptor channels to the somatic currents. Application of 3 μm ifenprodil reduced NMDA receptor-mediated peak currents by 48 ± 3% in wild-type mice (n = 5) and by 50 ± 7% in NR2AΔC/ΔCmice (n = 3) (Fig. 2B). The similar effect of ifenprodil on peak NMDA currents in wild-type and NR2AΔC/ΔCmice indicates a similar somatic ratio of functional NR2A- and NR2B-type NMDA receptors in the two genotypes.
The extent of desensitization during glutamate application in NR2AΔC/ΔCmice provided additional proof for the presence of NR2AΔC-containing NMDA receptor channels. As determined for recombinant receptors (Köhr and Seeburg, 1996), NMDA receptor currents desensitized faster in NR2AΔC/ΔCthan in wild-type mice, with time constants of 96 ± 10 msec (mutant, n = 13) and 211 ± 25 msec (wild type,n = 17), respectively, when recorded at −80 mV (p < 0.001) (Fig. 2C). Accordingly, the second glutamate response in a paired-pulse protocol was smaller in the mutant because of slower recovery from desensitization (Fig.2D).
Thus, comparable size and ifenprodil sensitivity but faster desensitization of NMDA receptor currents in the mutant demonstrate that NR2AΔC-containing receptors contribute to the somatic NMDA receptor population to the same extent as full-length NR2A-containing receptors in wild type. Therefore, the relatively small reduction in NR2AΔC subunit levels revealed by immunoblots of homogenates does not affect the number of somatic NMDA receptors.
Smaller amplitudes of stimulus-evoked NMDA EPSCs in NR2AΔC/ΔCmice
To determine whether the reduced synaptic enrichment of NR2AΔC protein as revealed by immunomethods affects NMDA receptor-mediated currents at synaptic sites, we analyzed EPSCs mediated by NMDA receptor channels (NMDA EPSCs) in CA1 pyramidal cells of wild-type and mutant mice (P15). Synaptic EPSCs were evoked by stimulation of Schaffer collaterals in 0.1 mm Mg2+ at −60 mV in the presence of the GABAA receptor antagonist bicuculline (10 μm). The NMDA and AMPA components of the EPSCs were determined in the presence and absence, respectively, of the AMPA receptor antagonist NBQX (Fig.3A). In both genotypes, NBQX blocked the fast EPSC component mediated by AMPA receptors and also reduced the late component of the EPSC caused by residual Mg2+ block (0.1 mm), which was relieved as long as AMPA receptors were not blocked. NMDA EPSCs were −102 ± 19 pA (n = 8) in wild-type slices but −49 ± 6 pA (n = 12) in mutant slices (p < 0.05). The AMPA components of EPSCs were −192 ± 34 pA (n = 8) in wild-type slices and −135 ± 18 pA (n = 12) in mutant slices (p = 0.16). Thus, the ratio of NMDA to AMPA EPSCs was 0.55 ± 0.05 in wild-type mice but 0.4 ± 0.04 in NR2AΔC/ΔCmice (Fig. 3A). Hence, in P15 mutant mice, NMDA EPSC amplitudes were reduced by 27%. In P30 mutants, the reduction reached 43% (Fig. 3A), consistent with less NR2B contribution in older animals (Sans et al., 2000). In previous experiments (Sprengel et al., 1998), this reduction was not observed, because the NMDA component was determined in pharmacologically nonisolated EPSCs 50 msec after the AMPA peak, and the slower deactivation kinetics of NMDA EPSCs (see below) implied larger amplitudes in the mutant.
Fig. 3.

Synaptically evoked NMDA EPSCs have smaller amplitudes and slower kinetics in ΔC/ΔC mice. A,EPSCs were evoked in bicuculline (10 μm), glycine (10 μm), and low Mg2+ (0.1 mm) at −60 mV in the absence or presence of NBQX to determine the peaks of the AMPA (▪/■) or NMDA (●/○) components. The amplitude ratio of NMDA/AMPA components (right panel) was decreased in P15 and P30 (*p < 0.02) ΔC/ΔC mice, indicating reduced levels of synaptic NMDA receptors in a developmentally progressing manner. B, Superimposed NMDA EPSCs evoked in bicuculline, glycine, and normal Mg2+ (1 mm) at +40 mV showing slower rise (see inset) and decay time constants in P15 ΔC/ΔC (○) compared with P15 WT (●). Mutant trace was enlarged by a factor of 1.15. Rise and decay times of all measurements are depicted in the right panels (*p < 0.001). Consistent results were obtained at −60 mV in low Mg2+ (Table 1).
Thus, during evoked synaptic transmission, fewer NMDA receptors are activated in NR2AΔC/ΔCthan wild-type mice.
Slower kinetics and delayed onset of stimulus-evoked NMDA EPSCs in NR2AΔC/ΔCmice
After AMPA receptor blockade, we noticed slower rise and decay times for the NMDA EPSCs in P15 and P30 NR2AΔC/ΔCmice at −60 mV in 0.1 mm Mg2+and at +40 mV in 1 mm Mg2+(p < 0.001 for both) (Fig. 3, Table1).
Table 1.
Peaks and kinetics of evoked NMDA EPSCs
| Rise time (msec) | Peak (pA) | Decay (msec) | Vh(mV) | n | ||
|---|---|---|---|---|---|---|
| WT | P15 | 6.9 ± 0.4 | −125 ± 14 | 126 ± 10 | −60 | 27 (10) |
| P30 | 4.5 ± 0.31-213 | −130 ± 24 | 73 ± 51-159 | −60 | 8 (4) | |
| ΔC/ΔC | P15 | 9.1 ± 0.61-160 | −81 ± 7* | 243 ± 161-165 | −60 | 35 (17) |
| P30 | 6.7 ± 0.51-1651-213 | −66 ± 8 | 158 ± 101-165 | −60 | 15 (5) | |
| WT | P15 | 5.7 ± 0.2 | 145 ± 16 | 171 ± 9 | +40 | 15 (9) |
| ΔC/ΔC | P15 | 7.7 ± 0.31-165 | 116 ± 12 | 292 ± 111-165 | +40 | 19 (12) |
Between genotypes:
F1-165: p < 0.001,
F1-160: p < 0.005,
p < 0.05; within genotype (P15 vs P30):
F1-213: p < 0.005,
F1-159: p < 0.05.
NMDA EPSCs were evoked in CA1 pyramidal cells by Schaffer collateral stimulation in bicuculline (10 μm), NBQX (10 μm), and glycine (10 μm) at −60 mV in 0.1 mm Mg2+ and at +40 mV in 1 mm Mg2+, in 2- and 4-week-old wild-type (WT) and NR2AΔC/ΔC mice (P15 and P30). Values are mean ± SEM. Numbers of recorded cells and analyzed animals (in parentheses) are indicated. Statistical significance was weighted by a two-sample t test between wild-type and NR2AΔC/ΔC mice (*) or between P15 and P30 within one genotype (#).
To determine whether the slower decay kinetics of NMDA EPSCs in the mutant can be explained by an increased contribution of slower deactivating NR2B-containing receptors, given that NR2AΔC-containing receptors are reduced at synaptic sites, we blocked NR2B-containing receptors with the ifenprodil derivative CP-101,606. In P15 NR2AΔC/ΔCmice, application of 10 μm CP reduced NMDA EPSCs by 59 ± 3% (n = 9) and in wild-type mice by 44 ± 5% (n = 9) (p < 0.05) (Fig.4A,B). An increased synaptic contribution of NR2B-containing NMDA receptors in NR2AΔC/ΔCmice was also demonstrated by more accelerated decay time constants of the NMDA EPSCs after NR2B blockade in P15 NR2AΔC/ΔC(from 295 ± 14 to 172 ± 9 msec, n = 9,p < 0.001) relative to wild-type mice (from 173 ± 12 to 129 ± 13 msec, n = 9, p< 0.01) (Fig. 4B). The more pronounced effect of CP on EPSC decay kinetics than on amplitudes in P15 mice appears to derive from the lower peak open probability of NR2B- than NR2A-containing receptor channels (Chen et al., 1999). Consistent with increased synaptic NR2B contribution in the mutant and the gradual postnatal decline in hippocampal NR2B expression (Sans et al., 2000), CP accelerated NMDA EPSCs in P15 and P30 mutants but only in P15 wild-type animals (Fig. 4B). In P30 wild types, CP reduced the amplitudes of NMDA EPSCs without accelerating the kinetics, possibly because of an increased formation of NR1–NR2A and NR1–NR2A–NR2B receptor channels. In the latter triheteromeric channels, the presence of NR2A may result in fast-decaying NMDA EPSCs, and the presence of NR2B may explain the CP effect on amplitudes.
Fig. 4.

Increased NR2B contribution to NMDA EPSCs in ΔC/ΔC mice. A, The NR2B-specific antagonist CP 101,606 (CP, 10 μm) reduced evoked NMDA EPSCs more (*p < 0.05) in P15 ΔC/ΔC than in P15 WT and accelerated their deactivation time course (WT, control 222 msec, CP 161 msec; ΔC/ΔC, control 355 msec,CP, 190 msec). CP did not affect the significantly different rise times (WT, 5.9 msec; ΔC/ΔC, 7.5 msec). B,CP reduced amplitudes of evoked NMDA EPSCs more in ΔC/ΔC than WT mice (left panel), at P15 (*p< 0.05) and P30 (p > 0.05). As expected from the declining expression of NR2B during postnatal development, CP accelerated the deactivation of NMDA EPSCs in WT mice at P15 (*p < 0.05; see Results) but not at P30 [control 92 ± 8 msec (●), CP 86 ± 5 msec (▾),n = 11]. In contrast, CP accelerated the deactivation in ΔC/ΔC mice at P15 (**p < 0.001; see Results) and at P30 [*p < 0.01, ΔC/ΔC, control 177 ± 13 msec (○), CP 125 ± 9 msec; (▿), n = 11], consistent with increased NR2B contribution in the mutant. Notably, the decay times remained slower in mutants than in wild types (P15, p < 0.02; P30,p < 0.005). Results obtained at +40 mV (Mg2+, 1 mm) and at −60 mV (Mg2+, 0.1 mm) showed no significant difference and were pooled.
Surprisingly, after NR2B blockade by CP, the decay times of NMDA EPSCs remained slower in mutant than wild type for both age groups (Fig.4B) (P15, p < 0.02; P30,p < 0.005), and CP did not affect the significantly different rise times (Table 1). Thus, NR2AΔC contributes to the slower rise and decay times of NMDA EPSCs in NR2AΔC/ΔCmice. Indeed, C-terminal truncation could have affected affinity, gating, and/or the localization of synaptic NR2AΔC-containing receptors.
With regard to changes in affinity for glutamate, or in channel gating, these are unlikely considering that C-terminal truncation of NR2A subunits did not alter the rise (data not shown) and decay kinetics of NMDA receptor currents either in HEK293 cells (Köhr and Seeburg, 1996) or in the nucleated whole-soma patches from area CA1 of the mutant mice. The rise time of somatic NMDA currents was 4.8 ± 0.2 msec (n = 17) in wild-type and 4.0 ± 0.3 msec (n = 13) in NR2AΔC/ΔCmice, and the decay time constant for wild type was 66.9 ± 2.8 msec (n = 17) and 63.5 ± 3.6 msec (n = 13) for NR2AΔC/ΔC.
The slower kinetics of NR2AΔC receptor-mediated NMDA EPSCs may be better explained by a localization of NR2AΔC-containing NMDA receptors more distant from release sites in the mutant compared with wild type, which should delay the onset of NMDA EPSCs relative to AMPA EPSCs. Indeed, the interval between onset of the AMPA and NMDA components was 1.1 ± 0.2 msec (n = 10) in P15 and 1.0 ± 0.2 msec (n = 8) in P30 wild-type mice, and 1.7 ± 0.2 msec (n = 10) in P15 and 1.5 ± 0.1 msec (n = 14) in P30 NR2AΔC/ΔCmice (p < 0.05 for P15 and P30 mice).
In summary, the slower rise and decay times of NMDA EPSCs, which persisted after NR2B blockade, together with the delayed onset of the NMDA component relative to the AMPA component, indicate that NR2AΔC-containing NMDA receptors appear to localize more distant from release sites than wild-type receptors (see Discussion).
Strongly reduced NMDA component of mEPSCs in NR2AΔC/ΔCmice
Further evidence for altered synaptic localization of NR2AΔC-containing NMDA receptors was obtained by recording mEPSCs, given that glutamate released spontaneously in the absence of action potentials should activate only AMPA and NMDA receptors located close to active release sites (Yuste and Denk, 1995; Mainen et al., 1999;Murthy et al., 2000).
mEPSCs in P15 NR2AΔC/ΔCand wild-type mice were recorded at −70 mV in TTX (1 μm), bicuculline (10 μm), glycine (10 μm), and low Mg2+ (0.1 mm). The AMPA component was obtained in the presence of the NMDA receptor antagonist AP5. As expected (McBain and Dingledine, 1992), AP5 reduced the peak and blocked the late component of mEPSCs. The NMDA component could not be isolated in the presence of NBQX, because it was too small for detection, even at +40 mV in normal Mg2+ (1.0 mm). Therefore, the NMDA component was estimated by subtracting the AMPA component from the total mEPSC integral.
In both genotypes, mEPSCs with both AMPA and NMDA components displayed comparable rise times and amplitudes (n = 7 recordings each) (Fig. 5A, Table2). The decay time course of the mEPSC was slower in wild type because of a prominent NMDA component that was absent in NR2AΔC/ΔCmice (Fig. 5A). Consistently, AP5 (30 μm)-mediated blockade of NMDA receptors accelerated the decay of mEPSCs in wild type but not in the mutant (Table 2). Indeed, in wild-type mice, the pharmacologically isolated AMPA mEPSCs decayed as fast as the mutant mEPSCs recorded in control solution. The presence of a small NMDA component in mutant mEPSCs was solely indicated by an effect of AP5 on amplitude (Fig. 5A). The averaged integral of the calculated NMDA component was more than twice as large in wild type (0.14 ± 0.024 pC; n = 7) as in the mutant (0.063 ± 0.019 pC; n = 7) (p < 0.05) (Table 2). Moreover, the ratio of NMDA to AMPA integrals was 2.3 ± 0.6 in wild-type and 0.7 ± 0.2 in NR2AΔC/ΔCmice (p < 0.005). Hence, the NMDA component of mEPSCs is reduced in the mutant by 70%. The remaining NMDA component of mEPSCs was CP insensitive (data not shown), suggesting that it was mediated mainly by NR2A-containing receptors.
Fig. 5.

NMDA components of miniature EPSCs are strongly reduced in ΔC/ΔC mice. mEPSCs were recorded in TTX (1 μm), bicuculline (10 μm), glycine (10 μm), and low Mg2+ (0.1 mm) at −70 mV. mEPSCs had comparable rise times and amplitudes, but their deactivation time course was faster in ΔC/ΔC than in WT mice (see also Table 2). NMDA receptor blockade (AP5) accelerated the deactivation only in WT and reduced the amplitude in both genotypes. The calculated ratio of NMDA/AMPA integrals (right panel) was decreased in the mutant by 70% (*p < 0.005). The current traces in control and AP5-containing solution are averages (WT,n = 158 and n = 97; ΔC/ΔC, n = 120 and n = 32).
Table 2.
Peaks, kinetics, and integrals of miniature EPSCs
| Rise time (msec) | Peak (pA) | Decay (msec) | Integral (pC) | n | |
|---|---|---|---|---|---|
| WT | 1.3 ± 0.1 | −13.0 ± 1.5 | 9.3 ± 0.9 | 0.21 ± 0.02 | 7 (3) |
| + AP5 | 1.2 ± 0.1 | −9.3 ± 0.4 | 6.1 ± 0.82-159 | 0.07 ± 0.012-214 | |
| ΔC/ΔC | 1.2 ± 0.1 | −14.4 ± 1.6 | 6.5 ± 0.8* | 0.16 ± 0.02 | 7 (3) |
| + AP5 | 1.4 ± 0.2 | −11.3 ± 0.7*2-159 | 6.3 ± 0.6 | 0.10 ± 0.012-159 |
Between genotypes:
p < 0.05; within genotype (control vs AP5):
F2-214: p< 0.001,
F2-159: p < 0.05.
mEPSCs were recorded in CA1 pyramidal cells in TTX (1 μm), bicuculline (10 μm), and glycine (10 μm) at −70mV in 0.1 mmMg2+. The effect of AP5 (30 μm) was studied in 2-week-old wild-type (WT) and NR2AΔC/ΔC mice. Values are mean ± SEM. Numbers of recorded cells and analyzed animals (in parentheses) are indicated. Statistical significance was weighted by a two-samplet test between wild-type and NR2AΔC/ΔC mice (*) or between control and AP5-containing solution within one genotype (#).
The NMDA component in P15 NR2AΔC/ΔCmice is thus reduced more in mEPSCs (70%) than in evoked EPSCs (27%, see above) but remains unchanged in nucleated patches, supporting the notion that NR2AΔC-containing receptors are located, on average, farther away from release sites than wild-type receptors.
DISCUSSION
We asked in this study to what extent absence of the intracellular C-terminal domain of the NR2A subunit impairs the contribution of NR2A-containing NMDA receptors to synaptic transmission in NR2AΔC/ΔCmice. We show by immunocytochemistry that dendritic NR2AΔC clusters are less frequent in cultured hippocampal neurons from the mutant when compared with wild type. Western blots prepared from mouse forebrain indicate that steady-state levels of the NR2AΔC subunit are lower in the mutant relative to NR2A in wild type, and that the characteristic high enrichment of NR2A in PSDs is not achieved in the mutant. Electrophysiological studies reveal that NR2AΔC/ΔCmice have normal NMDA currents in somata, somewhat reduced NMDA components in evoked EPSCs, but drastically reduced NMDA components in mEPSCs. Evoked NMDA EPSCs show a delayed onset relative to AMPA EPSCs and have slower rise and decay times. Thus, our data suggest that synaptic NR2AΔC-containing NMDA receptors are specifically reduced in number directly under release sites and, in addition, are more distant from release sites, on average, than full-length NR2A receptors in the vicinity of release sites.
C-terminal truncation of NR2A did not affect somatic NMDA receptor currents in mutant compared with wild type, except for increased desensitization, consistent with the relatively small reduction of NR2AΔC in forebrain homogenates. However, the amplitudes of evoked synaptic NMDA currents were reduced by one-fourth (P15) to one-half (P30) in the mutant. This finding and the substantial decline of the NMDA component of mEPSCs by 70% is consistent with the drastically reduced enrichment of NR2AΔC in the PSD fraction. The apparent reduction of NR2AΔC at synaptic sites could be caused by impaired targeting, transport, or anchoring of NR2AΔC subunits to synaptic sites as a consequence of C-terminal truncation (see also Sattler et al., 2000). Our experiments cannot distinguish among these possibilities. Nevertheless, NR2AΔC still enriches in the PSD fraction, although less than full-length NR2A, which may be attributed to its association with the NR1 or NR2B subunits in heteromeric receptors.
In addition to the reduction of NR2AΔC at synaptic sites, evoked NMDA EPSCs had a delayed onset relative to AMPA EPSCs and rose and deactivated more slowly in mutant than wild type. Perfusion of a NR2B-specific antagonist revealed an increased NR2B contribution to NMDA EPSCs that was not compensatory, because no evidence for an upregulation of NR2B expression was found in immunoblots. However, after NR2B blockade, NR2AΔC-containing NMDA receptors themselves must be responsible for the remaining slower NMDA EPSCs in NR2AΔC/ΔC mice. How can this finding be explained?
Although a slower recovery from desensitization may contribute to the slow deactivation of EPSCs (Lester and Jahr, 1992), it cannot explain the slower rise times of NMDA EPSCs in NR2AΔC/ΔCmice. Changes in affinity for glutamate or channel gating could result in slower deactivation of NMDA EPSCs. However, C-terminal truncation does not affect affinity for glutamate or channel gating in general, because the deactivation of NMDA currents in nucleated whole-soma patches was comparable in wild type and mutant. Alternatively, full-length NR2A-containing NMDA receptors may undergo a synapse-specific arrangement to achieve a higher receptor packing density, which might result in apparent higher agonist affinities of NMDA receptor channels, as previously seen for recombinantly expressed glycine receptors (Taleb and Betz, 1994). This explanation seems unlikely, however, because if C-terminal truncation prevents an apparent increase in receptor affinity at synaptic sites, the NMDA EPSCs in the mutant should decay more quickly, not more slowly, than in wild type.
Our results are better explained by the following scenario (Fig.6). Full-length NR2A-containing receptors, either as NR1-NR2A or NR1-NR2A-NR2B hetero-oligomers, localize preferentially close to release sites in PSDs. Truncation of the C terminus of NR2A shifts the average localization of NR2AΔC-containing NMDA receptors so that they are less tightly linked to release sites and to other PSD proteins, consistent with the reduced enrichment of NR2AΔC in the PSD fraction and the delayed onset of NMDA EPSCs. This explanation relies on the assumption that less glutamate accumulates in the synaptic cleft when glutamate is released independently of action potentials than when it is released by presynaptic stimulation. This assumption seems justified because synaptic stimulation releases glutamate simultaneously from many presynaptic terminals. In addition, glutamate is less efficiently cleared from the synaptic cleft in our recordings at room temperature than in vivo, because glutamate reuptake is temperature dependent (Bergles and Jahr, 1998). The different glutamate concentrations in the cleft associated with mEPSCs versus evoked EPSCs are reflected in the slower decay kinetics of evoked EPSCs compared with mEPSCs in our experiments (see also Burgard and Hablitz, 1993). We hypothesize that action potential-independent glutamate release activates only receptors within the PSD, thus resulting in the strongly reduced NMDA component of mEPSCs in the mutant. The higher glutamate concentrations produced by electrical stimulation also activate the receptors localized outside the PSD.
Fig. 6.

A scheme explaining our results by an improper synaptic localization of NR2AΔC-containing NMDA receptors in hippocampal CA1 synapses of ΔC/ΔC mice. Wild-type NR2A-containing receptors are concentrated in the PSD (gray). The truncated NR2A-containing receptors in ΔC/ΔC mice are specifically reduced within the PSD but localize more distantly from the release site. mEPSC, Glutamate action potential-independent release activates only receptors in the PSD, reflected by the drastically reduced NMDA component of mEPSCs in ΔC/ΔC mice.eEPSC, During synaptic stimulation (evoked), more glutamate is released (either from additional active zones or from neighboring terminals) and activates NMDA receptors in the PSD and nearby. Therefore, in ΔC/ΔC mice more NMDA receptors distant from release site are activated with an increased latency, resulting in NMDA EPSCs with slower rise and decay times, even in the presence of NR2B antagonist.
Our schematic depiction may also apply to the situation early in development when only NR2B-containing receptors are expressed in the CA1 synapse. Truncating the C terminus of NR2B may also shift the localization of synaptic NR2BΔC-containing receptors toward positions more distant from release sites, because NMDA EPSCs are reduced to one-third in NR2BΔC/ΔCmice, although the amount of NR2BΔC protein is unchanged compared with full-length NR2B in the wild type (Mori et al., 1998). These data and our data both support the hypothesis that the C termini of NR2 subunits are necessary for NMDA receptor channels to achieve appropriate densities near release sites.
In adult NR2AΔC/ΔC mice, hippocampal CA1 LTP is drastically reduced (Sprengel et al., 1998), which we initially explained by hypothesizing that the missing C-terminal domain of NR2A in the NMDA receptor precludes the assembly of constituents of signal transduction pathways to monitor and appropriately process the Ca2+ transients elicited by NMDA receptor stimulation. In the present study, we found that NMDA EPSC amplitudes were reduced by ∼50% in the NR2AΔC/ΔCmice (P30 mutants) and that the activated NR2AΔC-containing NMDA receptors appear to be inappropriately localized. Thus, we propose that the previously observed reduced level of LTP in NR2AΔC/ΔCmice is caused in part by impaired synaptic localization of NR2AΔC-containing receptors.
Footnotes
This work was supported in part by Deutsche Forschungsgemeinschaft Grant Ko 1064. We thank Pfizer, Inc. for the gift of CP-101,606, Annette Herold for technical assistance, Drs. Rolf Sprengel, Pavel Osten, and Michael Müller, and members of the Sakmann (Heidelberg, Germany) and D'Angelo (Pavia, Italy) laboratories for discussions and for critical reading of this manuscript.
Correspondence should be addressed to Dr. Georg Köhr, Department of Molecular Neurobiology, Max-Planck-Institute for Medical Research, Jahnstrasse 29, D-69120 Heidelberg, Germany. E-mail:kohr@mpimf-heidelberg.mpg.de.
REFERENCES
- 1.Bergles DE, Jahr CE. Glial contribution to glutamate uptake at Schaffer collateral-commissural synapses in the hippocampus. J Neurosci. 1998;18:7709–7716. doi: 10.1523/JNEUROSCI.18-19-07709.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 3.Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- 4.Burgard EC, Hablitz JJ. NMDA receptor-mediated components of miniature excitatory synaptic currents in developing rat neocortex. J Neurophysiol. 1993;70:1841–1852. doi: 10.1152/jn.1993.70.5.1841. [DOI] [PubMed] [Google Scholar]
- 5.Carlin RK, Grab DJ, Cohen RS, Siekevitz P. Isolation and characterization of postsynaptic densities from various brain regions: enrichment of different types of postsynaptic densities. J Cell Biol. 1980;86:831–845. doi: 10.1083/jcb.86.3.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen N, Luo T, Raymond LA. Subtype-dependence of NMDA receptor channel open probability. J Neurosci. 1999;19:6844–6854. doi: 10.1523/JNEUROSCI.19-16-06844.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 8.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 9.Kennedy MB. The postsynaptic density at glutamatergic synapses. Trends Neurosci. 1997;20:264–268. doi: 10.1016/s0166-2236(96)01033-8. [DOI] [PubMed] [Google Scholar]
- 10.Kennedy MB. Signal transduction molecules at the glutamatergic postsynaptic membrane. Brain Res Brain Res Rev. 1998;26:243–257. doi: 10.1016/s0165-0173(97)00043-x. [DOI] [PubMed] [Google Scholar]
- 11.Kim JH, Huganir RL. Organization and regulation of proteins at synapses. Curr Opin Cell Biol. 1999;11:248–254. doi: 10.1016/s0955-0674(99)80033-7. [DOI] [PubMed] [Google Scholar]
- 12.Kirson ED, Yaari Y. Synaptic NMDA receptors in developing mouse hippocampal neurones: functional properties and sensitivity to ifenprodil. J Physiol (Lond) 1996;497:437–455. doi: 10.1113/jphysiol.1996.sp021779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Köhr G, Seeburg PH. Subtype-specific regulation of recombinant NMDA receptor-channels by protein tyrosine kinases of the src family. J Physiol (Lond) 1996;492:445–452. doi: 10.1113/jphysiol.1996.sp021320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- 15.Kornau HC, Seeburg PH, Kennedy MB. Interaction of ion channels and receptors with PDZ domain proteins. Curr Opin Neurobiol. 1997;7:368–373. doi: 10.1016/s0959-4388(97)80064-5. [DOI] [PubMed] [Google Scholar]
- 16.Leonard AS, Hell JW. Cyclic AMP-dependent protein kinase and protein kinase C phosphorylate N-methyl-d-aspartate receptors at different sites. J Biol Chem. 1997;272:12107–12115. doi: 10.1074/jbc.272.18.12107. [DOI] [PubMed] [Google Scholar]
- 17.Lester RA, Jahr CE. NMDA channel behavior depends on agonist affinity. J Neurosci. 1992;12:635–643. doi: 10.1523/JNEUROSCI.12-02-00635.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li JH, Wang YH, Wolfe BB, Krueger KE, Corsi L, Stocca G, Vicini S. Developmental changes in localization of NMDA receptor subunits in primary cultures of cortical neurons. Eur J Neurosci. 1998;10:1704–1715. doi: 10.1046/j.1460-9568.1998.00169.x. [DOI] [PubMed] [Google Scholar]
- 19.Mainen ZF, Malinow R, Svoboda K. Synaptic calcium transients in single spines indicate that NMDA receptors are not saturated. Nature. 1999;399:151–155. doi: 10.1038/20187. [DOI] [PubMed] [Google Scholar]
- 20.McBain C, Dingledine R. Dual-component miniature excitatory synaptic currents in rat hippocampal CA3 pyramidal neurons. J Neurophysiol. 1992;68:16–27. doi: 10.1152/jn.1992.68.1.16. [DOI] [PubMed] [Google Scholar]
- 21.McBain CJ, Mayer ML. N-methyl-d-aspartic acid receptor structure and function. Physiol Rev. 1994;74:723–760. doi: 10.1152/physrev.1994.74.3.723. [DOI] [PubMed] [Google Scholar]
- 22.Moon IS, Apperson ML, Kennedy MB. The major tyrosine-phosphorylated protein in the postsynaptic density fraction is N-methyl-d-aspartate receptor subunit 2B. Proc Natl Acad Sci USA. 1994;91:3954–3958. doi: 10.1073/pnas.91.9.3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mori H, Manabe T, Watanabe M, Satoh Y, Suzuki N, Toki S, Nakamura K, Yagi T, Kushiya E, Takahashi T, Inoue Y, Sakimura K, Mishina M. Role of the carboxy-terminal region of the GluR epsilon2 subunit in synaptic localization of the NMDA receptor channel. Neuron. 1998;21:571–580. doi: 10.1016/s0896-6273(00)80567-x. [DOI] [PubMed] [Google Scholar]
- 24.Murthy VN, Sejnowski TJ, Stevens CF. Dynamics of dendritic calcium transients evoked by quantal release at excitatory hippocampal synapses. Proc Natl Acad Sci USA. 2000;97:901–906. doi: 10.1073/pnas.97.2.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Omkumar RV, Kiely MJ, Rosenstein AJ, Min KT, Kennedy MB. Identification of a phosphorylation site for calcium/calmodulin-dependent protein kinase II in the NR2B subunit of the N-methyl-d-aspartate receptor. J Biol Chem. 1996;271:31670–31678. doi: 10.1074/jbc.271.49.31670. [DOI] [PubMed] [Google Scholar]
- 26.Sans N, Petralia RS, Wang YX, Blahos J, 2nd, Hell JW, Wenthold RJ. A developmental change in NMDA receptor-associated proteins at hippocampal synapses. J Neurosci. 2000;20:1260–1271. doi: 10.1523/JNEUROSCI.20-03-01260.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sather W, Dieudonne S, MacDonald JF, Ascher P. Activation and desensitization of N-methyl-d-aspartate receptors in nucleated outside-out patches from mouse neurones. J Physiol (Lond) 1992;450:643–672. doi: 10.1113/jphysiol.1992.sp019148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sattler R, Xiong Z, Lu WY, MacDonald JF, Tymianski M. Distinct roles of synaptic and extrasynaptic NMDA receptors in excitotoxicity. J Neurosci. 2000;20:22–33. doi: 10.1523/JNEUROSCI.20-01-00022.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sheng M. PDZs and receptor/channel clustering: rounding up the latest suspects. Neuron. 1996;17:575–578. doi: 10.1016/s0896-6273(00)80190-7. [DOI] [PubMed] [Google Scholar]
- 30.Sprengel R, Suchanek B, Amico C, Brusa R, Burnashev N, Rozov A, Hvalby O, Jensen V, Paulsen O, Andersen P, Kim JJ, Thompson RF, Sun W, Webster LC, Grant SG, Eilers J, Konnerth A, Li J, McNamara JO, Seeburg PH. Importance of the intracellular domain of NR2 subunits for NMDA receptor function in vivo. Cell. 1998;92:279–289. doi: 10.1016/s0092-8674(00)80921-6. [DOI] [PubMed] [Google Scholar]
- 31.Stocca G, Vicini S. Increased contribution of NR2A subunit to synaptic NMDA receptors in developing rat cortical neurons. J Physiol (Lond) 1998;507:13–24. doi: 10.1111/j.1469-7793.1998.013bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stuart GJ, Dodt HU, Sakmann B. Patch-clamp recordings from the soma and dendrites of neurons in brain slices using infrared video microscopy. Pflügers Arch. 1993;423:511–518. doi: 10.1007/BF00374949. [DOI] [PubMed] [Google Scholar]
- 33.Taleb O, Betz H. Expression of the human glycine receptor alpha 1 subunit in Xenopus oocytes: apparent affinities of agonists increase at high receptor density. EMBO J. 1994;13:1318–1324. doi: 10.1002/j.1460-2075.1994.tb06384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tingley WG, Ehlers MD, Kameyama K, Doherty C, Ptak JB, Riley CT, Huganir RL. Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-d-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J Biol Chem. 1997;272:5157–5166. doi: 10.1074/jbc.272.8.5157. [DOI] [PubMed] [Google Scholar]
- 35.Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang YT, Salter MW. Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature. 1994;369:233–235. doi: 10.1038/369233a0. [DOI] [PubMed] [Google Scholar]
- 37.Williams K. Ifenprodil discriminates subtypes of the N-methyl-d-aspartate receptor: selectivity and mechanisms at recombinant heteromeric receptors. Mol Pharmacol. 1993;44:851–859. [PubMed] [Google Scholar]
- 38.Yuste R, Denk W. Dendritic spines as basic functional units of neuronal integration. Nature. 1995;375:682–684. doi: 10.1038/375682a0. [DOI] [PubMed] [Google Scholar]
- 39.Zheng X, Zhang L, Wang AP, Bennett MV, Zukin RS. Protein kinase C potentiation of N-methyl-d-aspartate receptor activity is not mediated by phosphorylation of N-methyl-d-aspartate receptor subunits. Proc Natl Acad Sci USA. 1999;96:15262–15267. doi: 10.1073/pnas.96.26.15262. [DOI] [PMC free article] [PubMed] [Google Scholar]