

Abstract
We describe a new mutation, flathead(fh), that arose spontaneously in an inbred colony of Wistar rats. The mutation is autosomal recessive, and the behavioral phenotype of fh/fh rats includes spontaneous seizures, tremor, impaired coordination, and premature death. A striking feature of the fh mutation is a dramatic reduction in brain size (40% of normal at birth). In contrast, no abnormalities are evident in the peripheral nervous system or in other tissues outside of the CNS. Although bromodeoxyuridine incorporation assays indicate that the rate of cell proliferation in thefh/fh cortex is similar to that of unaffected animals,in situ terminal deoxynucleotidyl transferase-mediated dUTP-biotin end-labeling assays reveal a dramatic increase in apoptotic cell death beginning after embryonic day 16 (E16). At E18 there is a 20-fold increase in cell death in the ventricular zone offh/fh neocortex, and at postnatal day 1 (P1), the number of apoptotic cells is still two times that of normal. However, by P8 the extent of cell death in fh/fh is comparable to that of unaffected littermates, indicating that the reduction in brain growth is caused by abnormally high apoptosis during a discrete developmental period. Late-developing structures such as the cerebellum, neocortex, hippocampus, and retina are most severely affected by the fh mutation. Within these structures, later-generated neuronal populations are selectively depleted. Together, these results suggest that the flathead gene is essential for a developmental event required for the generation and maturation of late-born cell populations in the brain.
Keywords: proliferation, seizures, neural development, neurological mutant, apoptosis, autosomal recessive
The development of the mammalian brain depends on a complex and highly regulated sequence of events organized into distinguishable phases. Cell division typically occurs within proliferative zones lining the inner surface of the neural tube. During later stages of embryonic neurodevelopment, proliferation of these neuroepithelial cells ceases at specific times while differentiation begins leading to the formation of all the neurons and macroglia of the adult brain. In many brain regions, such as the cerebral cortex or the cerebellum, postmitotic neurons migrate considerable distances before extending axons that then make highly precise synaptic connections. Mutations that affect the ability of neural cells to traverse these orderly and precisely paced steps of neurodevelopment result in developmental arrest, often leading to early death of the affected cell populations. Indeed, the degeneration seen in many rodent neurological mutants is known to result from defects in a variety of processes including neural tube development (Gunther et al., 1994), proliferation of neural progenitors (Herrup and Busser, 1995; Lee et al., 1998), differentiation (Mullen et al., 1976;Sibilia et al., 1998), neuronal migration (Rakic and Sidman, 1973;Herrup and Mullen, 1978; Goldowitz et al., 1997), axon formation (Dahme et al., 1997; Cohen et al., 1998), synaptogenesis (Landis et al., 1975;Roffler-Tarlov et al., 1984), or cellular organization (Ross et al., 1990; Goldowitz et al., 1997). It is generally accepted that similar developmental defects may underlie human genetic neurological disorders. In addition to being highly reproducible genetic tools for examining the events governing proper brain development, analyses of rodent mutants are likely to shed insight into human neuropathologies.
In this report, we describe a new rat mutation, flathead(fh), that appears to be brain specific. Rats homozygous for the flathead mutation display a massive reduction in brain size and dysgenesis of neocortex, hippocampus, cerebellum, and retina. Mutant rats suffer from severe ataxia, tremors, and spontaneous seizures, and die within 4 weeks after birth. The reduced brain size inflathead appears to result from a burst of apoptotic cell death that occurs during late neurogenesis. In contrast to most other neurological mutants in which selected cell populations are affected, the flathead mutation kills neuronal populations throughout the brain. Thus, flathead is a “temporal mutation” that kills diverse neuronal populations during a specific time window of brain development. Because the effects of the flathead mutation are evident only in the later stages of neural development, this mutant represents a valuable tool to further our understanding of the cellular and molecular events necessary for the generation and maturation of the late-born cell populations.
MATERIALS AND METHODS
Animals. All fh/fh mutants and unaffected littermates were generated from a breeding colony maintained at the University of Connecticut. For embryonic studies, the detection of a vaginal plug 3–4 hr after breeding pairs were put together was considered embryonic day (E0). All animal care procedures were in compliance with animal welfare guidelines.
Perfusion and histology. Rats were anesthetized with halothane (Sigma, St. Louis, MO) and perfused through the heart first with saline followed by a phosphate-buffered (PB, 0.1m) fixative containing 4% paraformaldehyde. Brains were removed from the skull and fixed with 4% paraformaldehyde and processed for either paraffin or cryostat sectioning. Nuclei were stained by incubating slides in 4,6-diamidino-2-phenylindole (DAPI, 2 μg/ml, Sigma) for 15 min at room temperature and viewed under ultraviolet light (260 nm). Non-CNS tissue and retina were fixed in 10% neutral-buffered formalin; gastrointestinal tissue was flushed with 10% formalin. Paraffin sections were cut at 10 μm and stained with hematoxylin and eosin. Stained sections were coverslipped with permount and viewed with a Nikon Optiphot-2 microscope. For the whole-brain histology shown in Figure 2, brains were dissected from animals perfused with saline and then 4% paraformaldehyde, rinsed in PBS, embedded in agar, and serially sectioned on a vibratome at 50 μm. Horizontal sections were mounted on gel-coated slides, dehydrated in ethanol and xylenes, and stained with cresyl violet.
Fig. 2.
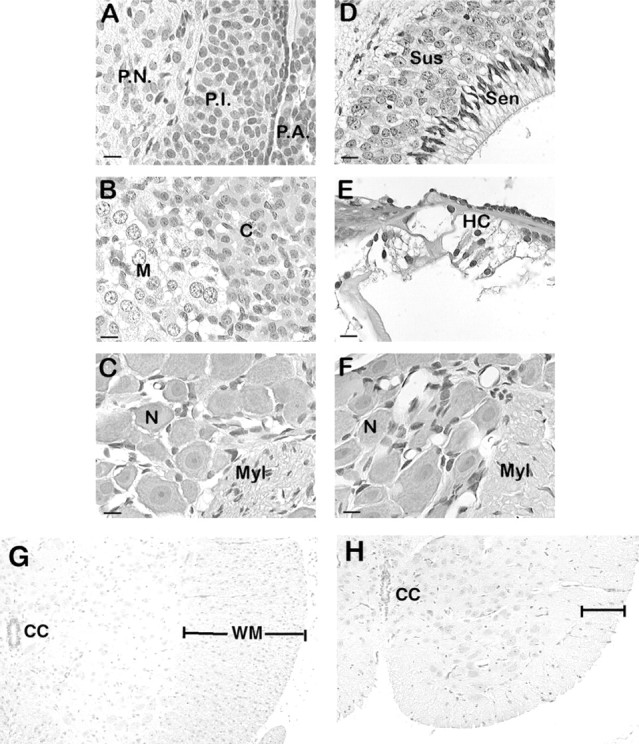
Light microscopic comparison of body tissues from mutant and unaffected animals did not reveal any differences.fh/fh and unaffected littermates of ages 1, 7, 14, and 21 d were perfused, fixed, and sectioned as described in Materials and Methods. Sections were stained with hematoxylin and eosin and examined by light microscopy. Images from representativefh/fh tissues are shown. A, Pituitary at P14. B, Adrenal gland at P14. C, Dorsal root ganglion at P21. D, Olfactory epithelium at P21.E, Cochlea at P14. F, Superior cervical ganglion at P21. G and H show thoracic spinal cord from normal and fh/fh animals at P14.P.N., Pars nervosa; P.I., pars intermedia; P.A., pars anterior; C, adrenal cortex; M, adrenal medulla; N, neuron; Myl, myelinated nerve fibers;Sus, suscentacular cells; Sen, sensory cells; HC, hair cells; CN, central canal;WM, white matter.
For cytochrome oxidase staining, 50 μm vibratome sections were incubated in DAB (Vector Laboratories, Burlingame, CA; 75 mg/ml) and cytochrome c (Sigma; 35 mg/ml) in PBS for 2 hr at 37°C.
Immunohistochemistry. Perfused brains were fixed in 4% paraformaldehyde in 0.1 m PB, pH 7.4, at 4°C overnight. Tissue was cryoprotected in a solution of 30% sucrose in PBS at 4°C for 24 hr, quick-frozen, and sectioned at 10 μm on a cryostat. Sections were first incubated in 5% BSA for 45 min and then in PBS containing calbindin D-28K polyclonal rabbit antibody (1:5000; Calbiochem, San Diego, CA) for 1 hr at room temperature. The sections were washed three times in PBS and incubated with the secondary antibody goat anti-rabbit IgG conjugated with Texas Red (Jackson ImmunoResearch, West Grove, PA) at a 1:200 dilution for 45 min. After incubation with the secondary antibody, the sections were washed in PBS. During one of the three 10 min washes, DAPI (2 μg/ml) was added to stain cell nuclei. Digital images of immunostained tissue were acquired using a SPOT-cooled CCD camera (Diagnostics Instruments) attached to an Olympus fluorescence upright microscope. After visualization using the SPOT software the images were transferred to Adobe Photoshop software (Adobe Systems, Mountain View, CA).
Bromodeoxyuridine labeling and birth-dating. Timed-pregnant females were injected with 60 mg/kg bromodeoxyuridine (BrdU, Sigma) 1–2 hr before they were killed by halothane inhalation. Embryos were rapidly removed and placed in cold HBSS (Life Technologies, Grand Island, NY). Whole brains were dissected and fixed in 4% paraformaldehyde in 0.1 m PB, pH 7.4, at 4°C overnight. Tissue was cryoprotected in a solution of 30% sucrose in PBS at 4°C for 24 hr, quick-frozen, sectioned at 10 μm on a cryostat, and processed for immunohistochemical identification of BrdU using the avidin–biotin–horseradish peroxidase technique. Tissue was incubated in PBS at 65°C for 10 min, cooled to room temperature, and incubated in 0.1 mg/ml pepsin in 0.1N HCl. Slides were then rinsed in PBS and incubated in 2N HCl for 10 min at 37°C. After several rinses in PBS, slides were blocked with 5% goat serum/0.2% Triton X-100 for 45 min and incubated for 1 hr in mouse anti-BrdU (Novocastra) diluted 1:200 in PBS/1% normal goat serum/0.3% Triton X-100. Sections were visualized using the indirect avidin–biotin–horseradish peroxidase technique and followed by nickel-intensified diaminobenzidine. After visualization, sections were lightly counterstained with 1% pararosanaline, dehydrated, cleared, and coverslipped in Permount. To quantify the number of cells positive for BrdU in the neocortical ventricular and subventricular zones and external granule layer (EGL) of cerebellum, cell counts were made from parasagittal sections obtained from the same relative position in fh/fh and unaffected littermates. The area of VZ that was quantified was at the mid anterior–posterior region of dorsal telencephalon.
For birth-dating experiments, timed pregnant rats were injected with BrdU at either 15 or 18 d of gestation. Two weeks after birth,fh/fh animals and unaffected littermates were perfused and processed for BrdU immunocytochemistry. Images of sections were digitized and analyzed with NIH Image 1.59. To quantify the migration pattern in neocortex, labeled cells in equivalent areas of somatosensory cortex of fh/fh and unaffected littermates were analyzed. The shortest radial distance from the pia was then determined for each of 200–300 BrdU cells for each injection time. Because of the difference in cortical thickness between mutant and unaffected brains, the migration distance was expressed as a percentage of the thickness of neocortex.
Terminal deoxynucleotidyl transferase-mediated dUTP-biotin end-labeling assay. Detection of apoptotic cells in brain slices was performed using the apoptosis detection system from Promega (Madison, WI), which is based on the terminal deoxynucleotidyl transferase-mediated dUTP-biotin end-labeling (TUNEL) method described by Gavrieli et al. (1992). Briefly, 10 μm cryosections were washed for 5 min in PBS, dehydrated through an ascending series of ethanol and xylene, and rehydrated through descending ethanols. Next, sections were washed in 0.85% NaCl for 5 min and in PBS for 5 min, and fixed in 4% formaldehyde for 15 min. After two 5 min rinses in PBS, sections were incubated in 20 μg/ml proteinase K for 8–10 min. Sections were washed in PBS for 5 min, post-fixed in 4% formaldehyde for 5 min, and washed in PBS for 5 min. Sections were equilibrated in the provided equilibration buffer for at least 10 min before incubation in a mixture of terminal transferase/florescein-conjugated dNTP for 1 hr at 37°C in a humidified chamber. The reaction was terminated by rinsing in 2× SSC for 15 min, rinsed twice in PBS for 5 min, and counterstained in 40 ng/ml propidium iodide (Sigma) for 15 min. Sections were rinsed three times for 5 min in deionized H2O and coverslipped in antifade reagent (Molecular Probes, Eugene, OR). Sections were viewed with a Nikon Optiphot-2 microscope with an episcopic fluorescence attachment (EFD-3, Nikon.) To quantify the number of TUNEL-positive cells in neocortical ventricular and subventricular zones, and the EGL of cerebellum, cell counts were made from parasagittal sections obtained from the same relative position infh/fh and unaffected littermates.
Blunt-end ligation-mediated PCR. Genomic DNA was analyzed by blunt-end ligation-mediated PCR (LM-PCR) as described by Blaschke et al. (1996). This technique is based on the attachment of partially overlapping primers to the ends of genomic DNA that is then subjected to PCR amplification. Because the DNA from apoptotic cells is fragmented, it serves as a better substrate for primer ligation and amplification than the uncleaved DNA from healthy cultures. The “ladder-like” pattern of amplified DNA indicates nonrandom DNA cleavage, a characteristic feature of apoptosis. Briefly, 1 nm each of 24- and 12-mer oligonucleotides (24 bp: 5′-AGCACTCTCGAGCCTCTCACGGCA-3′; 12 bp: 5′-TGCGGTGAGAGG-3′) was annealed by heating the mixture to 55°C followed by slow cooling to 10°C. Annealed primers were ligated to 2.5 μg of genomic DNA isolated from E19 fh/fh and unaffected brains. The mixture was diluted to 25 μg/ml, and 5 μl of this was used with 125 pmol of the 24-mer primer in a 30-cycle, two-step (94°C for 1 min, 72°C for 3 min) PCR amplification reaction. Amplified DNA was electrophoresed through a 1.5% agarose gel and stained with ethidium bromide.
RESULTS
Origin of the flathead mutation
After ∼1.5 years of inbreeding in a colony of Wistar rats generated from approximately 10 original founders, several litters were observed to contain animals with the fh/fh phenotype. Three breeding pairs that gave rise to fh/fh offspring were isolated and used to generate a separate colony (WUC1). Inquiries to Charles River Laboratories, the source of the original animals in the colony, indicated that they have not observed animals in their Wistar colony that display the fh/fh phenotype. The mutation, therefore, appears to have arisen spontaneously in the breeding colony at the University of Connecticut animal facility. Because breeding records were not maintained in the years before the appearance of the first fh/fh animals, it is not possible to determine when the mutation arose. However, considering that there were at least threefh/+ breeding pairs in a colony that contained approximately 20 breeding pairs, the mutation probably arose within 1 year of the colony's origin.
General appearance of fh mutant
Affected rats can be distinguished at birth by a flattened skull and resting tremor. The behavioral phenotype also includes a lack of balance, ataxia, and progressive dragging of the hindlimbs. Closer observation of affected rats revealed certain features of behavioral seizures (Sarkisian et al., 1999), such as periodic tail flexion (strub tail) and tonic limb extension. The deficiency in motor control worsens with age; by 3 weeks of age, affected animals often drag their hindlimbs and fall to one side when walking. However, normal righting reflexes are observed, which suggests that the lack of balance is attributable to a deficiency in motor control and not to a vestibular defect. Affected rats die between 25 and 30 d of age.
fh is an autosomal recessive mutation
To determine the inheritance pattern of the new mutation, we established breeding colonies starting from three different breeding pairs, selected on the basis of their ability to produce offspring with the fh phenotype. These animals were designated as the WUC1 strain of rats. To determine whether the defect in this strain segregated as a single gene, we performed a series of intercrosses and backcrosses. As shown in Table 1, in 26 of 26 WUC1 breedings, approximately one-fourth (26%) of the offspring born had the fh phenotype. This is consistent with the expected distribution of a recessive mutation, although a reduced penetrance dominant mutation was also conceivable. However, because crosses into wild-type colony animals from vendor did not produce litters with the fh phenotype, that possibility was ruled out. To test the hypothesis that this mutation segregates as a single gene, a backcross was performed between 17 unaffected F1 progeny from a WUC1 × WUC1 intercross. As shown in Table 1, approximately two-thirds of these matings gave rise to offspring with fhphenotype (12 of the 17 backcrosses produced litters containing affected animals). In addition, the phenotype was not associated with a particular gender. Of the flathead animals in which gender was determined, 54% were female and 46% were male. Taken together, these results indicate that fh is an autosomal recessive mutation.
Table 1.
Backcross and F1 intercross experiments with animals from the WUC1 strain
| Cross | Number of crosses | Number of litters w/affected | Number of litters w/o affected | Number of offspring/litter | % affected | |
|---|---|---|---|---|---|---|
| In affected | In unaffected | |||||
| WUC1 × WUC1 | 26 | 26 | 0 | 9.3 ± 2.94 | 26.34 (n = 186) | |
| WUC1 × F1 (backcross) | 17 | 12 | 5 | 9.9 ± 2.91 | 8.8 ± 3.42 | 26.89 (n = 119) |
WUC1 is the strain defined by those breeding pairs that generate offspring with the flathead phenotype. In WUC1 breedings, 26% of the offspring are affected. In addition there appears to be no embryonic lethality because the litter sizes from these breedings or other breedings that do not generateflathead animals are similar. The backcross experiments were breedings between unaffected littermates from litters that contained affected animals and their parents. In these breedings, approximately two-thirds of the litters contained affected animals. Together, this is consistent with a single autosomal recessive gene with 100% penetrance segregating at 1:2:1.
The effects of the fh/fh mutation are restricted to the CNS
As a first step toward characterization of the fh/fhmutation, we compared brain and body weights of fh/fh rats and unaffected littermates at various ages. As shown in Figure1A,B, although fh/fh rats often weigh slightly less than their unaffected littermates prenatally and in the first week of life, their body weights fall within the range of expected variations through this period. After the first week, however, the difference in body size increases progressively until the death of the animal, at which time affected animals weigh less than one-half that of their unaffected littermates (data not shown). Histological examinations were therefore performed to determine whether any non-neural tissues and organs were affected. Examination of several structures of the peripheral nervous system and other organ and tissue types revealed no difference betweenfh/fh and unaffected animals (Table2, Fig. 2). Given that no abnormality is evident outside the CNS, it is likely that the progressive decrease in body weight is an indirect effect, possibly caused by poor feeding by fh/fh animals after birth.
Fig. 1.

The flathead mutation preferentially affects the brain. A, The bodies of unaffected (left) and flathead(right) littermates on P10 are similar in size. Also note the slightly flattened curvature of the flatheadskull (arrow). B, A graph of body weights of flathead (striped bars) and unaffected rats (black bars) reveals no significant difference in body weights from E19 until the end of the first postnatal week. C, A flathead brain (right) is significantly smaller than an unaffected (left) littermate on P14. Note that although the entire brain is smaller, the cortex and cerebellum are especially reduced in size. D, A graph of brain weights from E19 to P7 shows that a flathead brain weighs approximately one-half that of a normal brain. Four to eight animals were used for each point. *p < 0.05.
Table 2.
Organ and tissues found to be normal infh/fh
| Organ system | Tissue/cells |
|---|---|
| Peripheral nervous | Peripheral nerves, dorsal root ganglia, superior cervical ganglia, trigeminal ganglia, and cochlear and vestibular ganglia |
| Musculoskeletal | Connective tissue, bone, adipose, and skeletal muscle |
| Gastrointestinal | Tongue, teeth, salivary glands, esophagus, stomach, small intestine, liver, and exocrine pancreas |
| Urogenital | Glomeruli, tubules, juxtamedullary apparatus, and urinary bladder |
| Respiratory | Respiratory epithelium, trachea, and lung parenchyma |
| Cardiovascular | Heart, Purkinje fibers, aorta, arteries, veins, capillaries, and lymphatics |
| Hematopoietic | Spleen, thymus, lymph nodes and loose lymphoid tissues, and bone marrow |
| Endocrine | Pituitary, adrenal gland, thyroid, parathyroid, and islets of Langerhans |
| Skin | Epidermis, dermis, hair, and sebaceous and apocrine glands |
Gross and light microscopic examinations were performed on hematoxylin and eosin-stained sections obtained from 1-, 7-, and 14-d-old unaffected and fh/fh rats. Shown is a list of organs and tissues found to be indistinguishable from unaffected littermates.
In contrast, the brain is much smaller in fh/fh rats than in their unaffected littermates (Fig. 1C,D). Although no difference was detected at E16 in several litters from WUC1 breedings (data not shown), at E18 the fh/fh brain weighed ∼26% less than their unaffected littermates (data not shown), and by E19 the discrepancy in brain size increased to 51%. After this point, the fh/fh brain remained approximately one-half the size of a normal brain throughout the 3–4 week life-span. Reduction in the size of the brain is not limited to a specific area but affects many regions, at least to some extent (Figs. 1C,3). Most dramatic was the reduction in the size of the cortex and cerebellum, whereas the tectum was less severely affected (Figs. 1C, 3). An examination of thefh/fh spinal cord showed reduced cross-sectional area characterized by an overall decrease in white matter with normal gray matter (Fig. 2G,H). It is possible that this is the result of the reduced number of axonal processes exiting the brain.
Fig. 3.
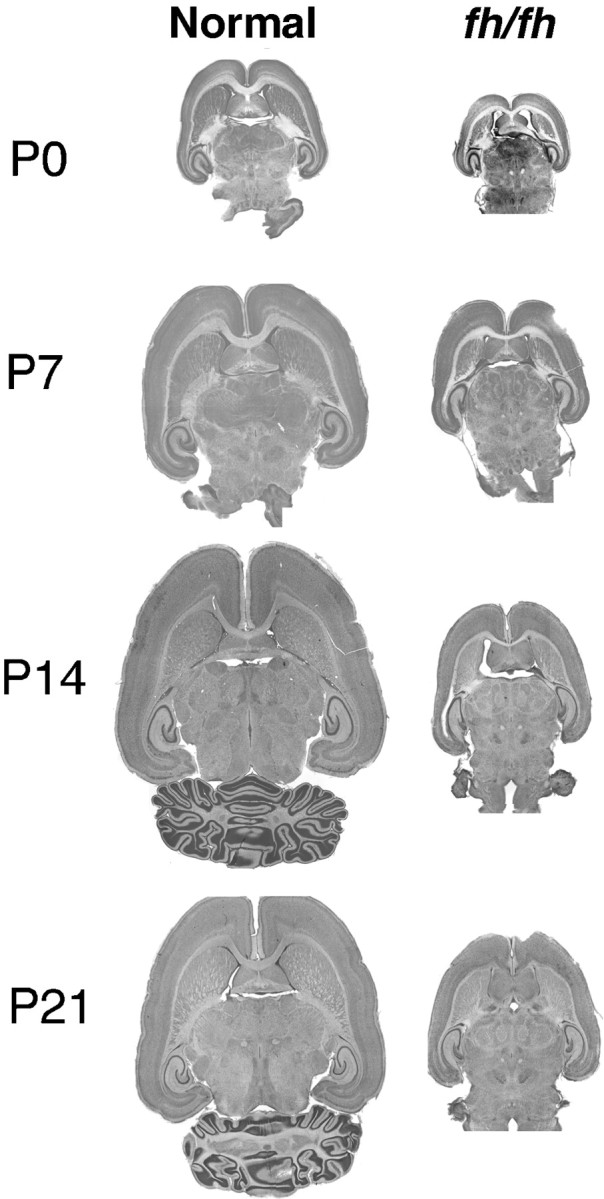
Light microscopic comparison of horizontal brain sections of fh/fh and unaffected littermates at P0, P7, P14, and P21. Halothane-anesthetized animals were intracardially perfused and then fixed and sectioned on a vibratome at 50 μm. Horizontal sections were mounted on gel-coated slides and stained with cresyl violet. The fh/fh brain is significantly smaller at all ages. Although all brain regions are reduced in size, the cerebral cortex and cerebellum are particularly affected infh/fh.
Structures containing postnatally generated cells are most affected in fh/fh rats
To examine the fh/fh brain in more detail, we focused on four areas: the neocortex, the hippocampus, the cerebellum, and the retina. These areas were chosen because of their organized structures and defined cell populations.
Neocortex
The neocortex of the fh/fh rat is reduced in overall size and thickness, flattened on the dorsal surface, and shortened at the caudal aspect (Fig. 3). During normal cortical development, neuronal precursors migrate radially outward to populate the cerebral isocortex after their terminal mitosis in the ventricular zone between days 14 and 20 of gestation in the rat (Berry and Rogers, 1965). The earliest born neuronal precursors form the deepest layers of the isocortex and later born precursors migrate through the existing layers to form the superficial layers (Angevine and Sidman, 1961; Luskin and Schatz, 1985).
Examination of the neocortex by light microscopy suggested that the overall pattern of lamination in fh/fh is generally normal, with a clear, cell-sparse layer I under the pia and a thin, dense layer VIb overlying the white matter. Relatively minor alterations in specific layers can be clearly observed, however, in the mutant cortex. In particular, layer 2/3 appears thinned. To investigate this further we used two markers, one for layer IV in somatosensory cortex (cytochrome oxidase staining) and another for layer 2/3 pyramidal cells (calbindin). As shown in Figure4A, the cytochrome oxidase stain in the mutant is both thinner and transposed toward the pial surface relative to the staining in normal cortex. In addition, calbindin antibody, which in addition to labeling non-pyramidal cells in all layers, labels pyramidal cells in layer 2/3, shows a clear difference between mutants and wild type. The staining infh/fh for calbindin-positive pyramidal cells is confined to a narrow band under layer I, whereas in controls, layer 2/3 is approximately one-third the cortical thickness (Fig.4B). This result indicates selective depletion of later-generated neurons in the fh/fh neocortex.
Fig. 4.
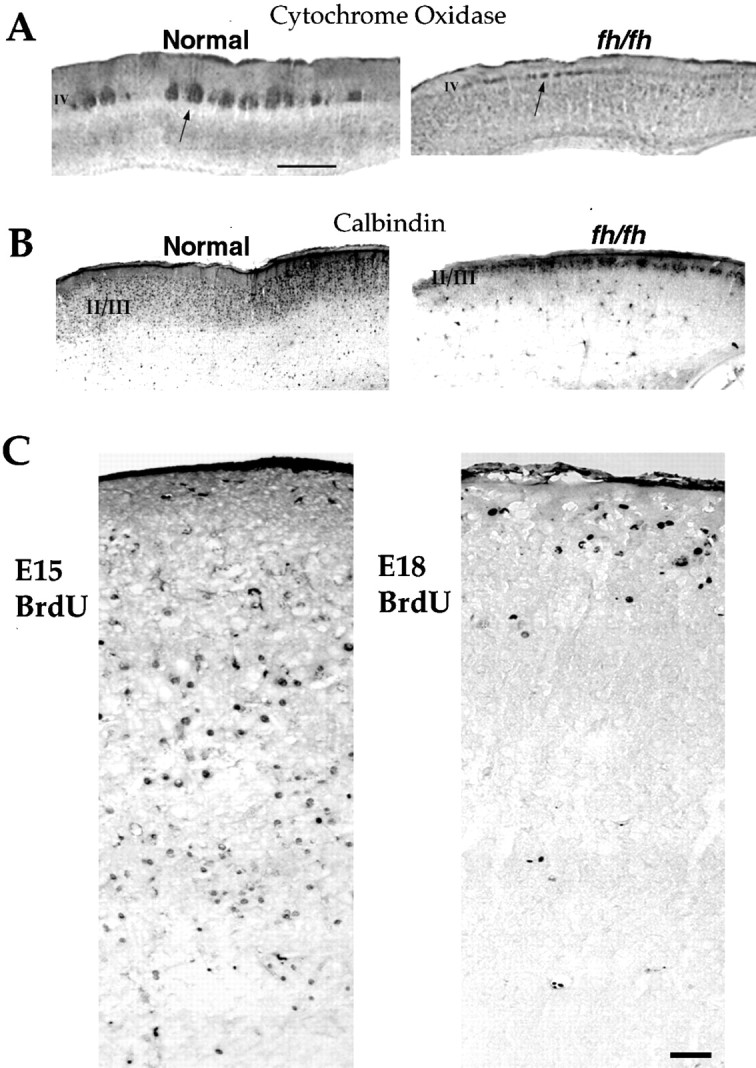
fh/fh mutants have reduced thickness of superficial layers but display a normal inside/out pattern of migration in neocortex. A, Cytochrome oxidase stain of somatosensory cortex. Note the thinning and transposition of layer IV toward the pial surface. Scale bar, 1 mm. B, Calbindin immunostaining in fh/fh labels a thin upper layer, which is wider in wild type. C, BrdU immunocytochemistry of a P14 fh/fh animal that was pulsed with BrdU at E15 (left) and another P14fh/fh that was pulsed with BrdU at E18 (right). Labeling was examined in sections 2 weeks after birth. Most of the cells labeled by an E18 injection are localized to upper layers of somatosensory cortex, whereas the E15 injection resulted in staining throughout lower layers of neocortex. Scale bar, 50 μm.
To determine whether reduced thickness of the upper layers in the flathead mutant is caused by inappropriate migration and whether the normal “inside-out” of cortical migration occurs infh/fh, we performed BrdU birth-dating experiments in which premigratory proliferating neurons were labeled and their position examined postnatally. Figure 4C shows BrdU staining of the somatosensory cortex of P14 fh/fh animals that were pulsed with BrdU at E15 and E18. In fh/fh animals pulsed at E15 and examined 2 weeks after birth, BrdU-positive cells were localized throughout the neocortex with a higher proportion found in the lower layers. When pulsed with BrdU at E18, however, the majority of labeled cells in both fh/fh and littermates were localized to the upper cortical layers (Fig. 4B). These results indicate that despite the overall reduction in the thickness of neocortex and specifically of the upper layers in fh/fh, an inside-out pattern of migration is still preserved.
Hippocampus
The dentate gyrus, the last structure of the hippocampus to form, is greatly reduced in fh/fh mutants (Figs. 3,5A,B). In fact, the inferior blade of the dentate gyrus is completely missing. The dentate gyrus is composed of three layers: the cells of the hilus and molecular layers of the dentate gyrus are born between E16 and E18 in the ventricular neuroepithelium, and the dentate granule cells arise from a secondary proliferative zone during the first postnatal week (Schlessinger et al., 1975; Bayer, 1980). In contrast, the hippocampus proper (Ammon's horn), which grows most rapidly between embryonic days 16 and 17, is less affected in the fh/fh brain (Fig.5A,B).
Fig. 5.
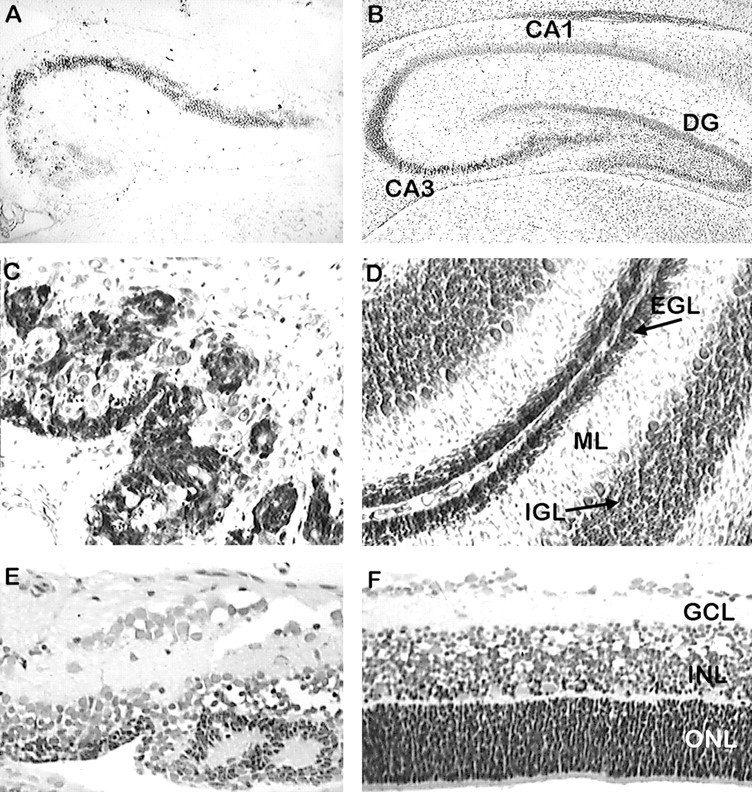
Thionin staining reveals depletion of neuronal populations born late in neurogenesis and disruption of normal laminar structure. Paraffin-embedded sections (10 μm) of fh/fh (A) and unaffected (B) hippocampus at P21 were stained with thionin and analyzed by light microscopy. The figure shows that the dentate gyrus (DG) is virtually absent in the mutant, and the CA3 region of Ammon's horn is shortened and cell sparse. Sagittal sections of P14 fh/fh(C) and unaffected (D) cerebellum show that some external granule cell layer (EGL) and scattered PCs are present in thefh/fh, but the internal granule cell layer (IGL) is absent. Sections of the P12 retina fromfh/fh (E) and unaffected (F) show that the photoreceptors in the outer nuclear layer (ONL) are severely depleted. Although the laminar structure is disrupted, the inner nuclear layer (INL) and ganglion cell layer (GCL) are less affected.
Cerebellum
The cerebellum of the fh/fh mutant is extremely hypoplasic. Although the characteristic cortical foliations are discernible in fh/fh, they are rudimentary, and the depths of the folds are markedly reduced (Fig. 3). Light microscopic studies have revealed a severe dysgenesis of the lamination pattern. The normal cerebellum has four layers: the external granule cell layer, molecular layer, Purkinje cell layer, and internal granule cell layer (Fig.5D). Purkinje neurons are born in the ventricular neuroepithelium and exit this germinal zone between E11 and E13 (Altman and Bayer, 1985a,b; Goldowitz and Hamre, 1998). Cerebellar granule cells arise between E13 and E15 from a more caudal germinal zone adjacent to the rhombic lip (Adler et al., 1996; Altman and Bayer, 1985a,b). In contrast to the Purkinje cells, however, granule cell precursors continue to proliferate over the next few days as they migrate to form the EGL (Rakic, 1985; Hatten, 1990; Adler et al., 1996;Goldowitz and Hamre, 1998). Postnatally, the cells in the expanding EGL begin a secondary migration inward on Bergmann glia to form the internal granule layer (Rakic, 1985; Hatten, 1990; Goldowitz and Hamre, 1998). Therefore, although cells giving rise to granule neurons arise prenatally, they reach their final numbers and exit the cell cycle only after birth. In the fh/fh mutant, distinct cell layers are not observed (Fig. 5C). Large Purkinje neurons are scattered throughout the cerebellar cortex, and only a thin external granule layer is seen. The late developing internal granule cells are virtually absent (Fig. 5C).
Retina
In the fully developed normal retina (Fig. 5F), there are three distinct layers: the ganglion cell layer, the inner nuclear layer (containing bipolar and amacrine cells), and the outer nuclear layer (containing photoreceptors). The bipolar and ganglion cell precursors are generated by E17, and although the genesis of cone precursors is completed by E16, the peak of genesis of the more abundant rod photoreceptors is reached on the day of birth and extends to P5 (Braekevelt and Hollenberg, 1970). In contrast to the normal retina, in fh/fh only two layers are discernible: the ganglion cell layer and a nuclear cell layer that appears to be a fusion of the inner nuclear layer and photoreceptor layer (Fig.5E).
Our observations suggest that cell populations generated late during brain development (postnatally) are more affected in fh/fhthan those that are generated relatively early. To further investigate this issue, we subjected sections of the 7-d-old cerebellum from normal and fh/fh animals to immunohistochemical analysis using a calbindin antibody. Within the cerebellum, calbindin stains Purkinje neurons specifically. As expected in normal cerebellum, calbindin staining is restricted to the Purkinje cell layer, which is situated above the nascent IGL layer (Fig. 6). Infh/fh, however, the cerebellum appears to be devoid of mature granule cells, and with the exception of the narrow EGL, most of the cells within the cerebellum stain positively for calbindin (Fig.6). The late-developing granule cells are therefore affected much more severely than Purkinje neurons, which are generated before the period at which cell death is rampant in the fh/fh brain.
Fig. 6.

Calbindin immunostaining reveals thatfh/fh cerebellum contains mostly Purkinje neurons. Frozen sagittal sections of 7-d-old normal and fh/fhbrains were processed for immunohistological detection of calbindin, a protein expressed specifically by Purkinje neurons in the cerebellum.Red represents calbindin staining, whereasblue represents nuclei stained with DAPI.
Reduced brain size in fh/fh is caused by an increase in apoptosis, rather than decreased cell proliferation
During development, brain growth is regulated by two opposing processes: cell proliferation and cell death. The fh/fhbrain grows at the same rate as a normal brain until approximately E18, when a 26% reduction in brain size is observed. The reducedfh/fh brain size could be caused by a decreased rate of cell proliferation, an increase in the amount of cell death, or a combination of the two processes. To determine the cause of the reducedfh/fh brain size, we quantified the proliferating population using BrdU incorporation assays. To determine the percentage of cells in S-phase of the cell cycle in embryonic cerebral cortex and postnatal cerebellum, animals were injected with BrdU at E19 and P1, and the percentage of BrdU-positive cells in the periventricular epithelium (PVE), ventricular and subventricular zones, and EGL was determined. These ages were chosen for analysis because reduction in the sizes of cerebral cortex and cerebellum are clearly discernible. As shown in Figure 7, the percentage of proliferating cells in the PVE of fh/fh cerebral cortex was similar to that of unaffected animals. Analysis of P1 cerebellar sections also showed similar proportions of BrdU-positive staining in the EGL offh/fh and unaffected rats (Fig. 7).
Fig. 7.

BrdU pulse labeling reveals a comparable rate of proliferation in fh/fh and unaffected cortex. Animals were injected with 60 μg/g of BrdU 1 hr before they were killed. Paraffin sections were processed for immunohistological detection of BrdU incorporation as described in Materials and Methods. BrdU labeling in the E19 unaffected (left) and fh/fh(right) cortex and P1 cerebellum is shown. The percentage of total BrdU-positive cells in the PVE of the cerebral cortex and EGL of the cerebellum has been quantified (n = 4) and shown graphically. No significant difference in the proportion of BrdU-positive cells was observed in cortical or cerebellar sections between unaffected andfh/fh animals.
Because the rate of proliferation in two different proliferative zones was not altered significantly, we proceeded to determine whetherfh/fh brains displayed a higher incidence of cell death. As a first step, we used the DNA-binding nuclear stain DAPI. As shown in Figure 8A, an examination of DAPI staining in the cortex of normal brains at E18 reveals only a very small percentage of dying neurons as judged by the characteristic pyknotic or fragmented nuclei. In contrast, nuclear staining of fh/fh brains of the same age revealed a large number of dying cells scattered throughout the cerebral cortex (Fig.8A). Although condensed nuclei are most characteristic of cells in late stages of apoptosis, nuclei may also appear smaller in necrotic cells and during certain stages of mitosis. To determine whether these pyknotic nuclei indicate increased apoptotic cell death, we looked at the extent of DNA fragmentation in the unaffected and fh/fh brain at E19 using the LM-PCR method described by Blashke et al. (1996), which is known to be significantly more sensitive than the standard electrophoresis of soluble DNA. Using this technique, a ladder-like pattern of nucleosomal-length DNA fragments, a hallmark of apoptotic cells, is detectable in DNA extracted from the fh/fh brain but not from the normal brain at this age (Fig. 8B). This supports the results obtained from DAPI staining and also confirms that the increased cell death at E19 is caused by apoptosis. This was further verified using the TUNEL method, a technique capable of specifically labeling apoptotic nuclei in situ. At E18 we found 20-fold more TUNEL-positive cells in the fh/fh cortex than in the unaffected cortex (Fig. 9). Quantification of TUNEL-positive cells (Table3) indicated that although fewer than 1 in 400 cells was apoptotic at a given time in the normal cerebral cortex, approximately 1 in 20 cells was undergoing apoptosis in thefh/fh cortex. Interestingly, although dying cells were scattered throughout the cortex, proliferative areas displayed the highest density of TUNEL staining, a pattern also seen during normal brain development, albeit to a much smaller extent (Blaschke et al., 1996; Thomaidou et al., 1997). Together, these data suggest that the decreased brain size in fh/fh is not caused by decreased cell proliferation but by a dramatic increase in the amount of cell death.
Fig. 8.

Increased cell death in fh/fhcortex. Cell death was examined by nuclear staining (A) and LM-PCR amplification of DNA (B). DAPI staining reveals a greater number of pyknotic nuclei in the fh/fh cortex (arrows) than in the unaffected cortex at E18. To determine whether these pyknotic nuclei might be apoptotic (rather than necrotic or mitotic), we used the LM-PCR technique to preferentially amplify fragmented DNA from E18 fh/fh and normal brains. After amplification, DNA from unaffected and fh/fhanimals was analyzed by electrophoresis, which revealed a laddering pattern in DNA from the fh/fh but not the unaffected brain. Numbers on the left indicate molecular sizes in kilobases.
Fig. 9.
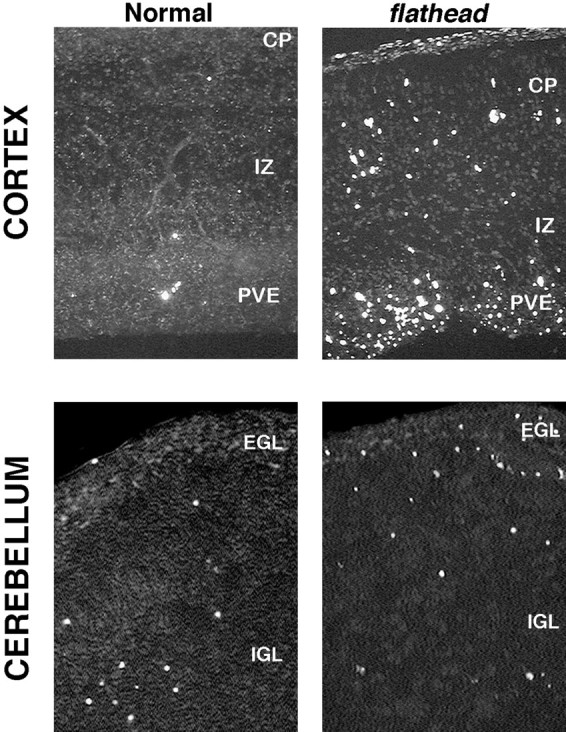
TUNEL-labeling reveals an increase of apoptotic cell death in the fh/fh cortex and cerebellum. Frozen tissue was sectioned at 10 μm, permeabilized with proteinase K, and incubated with florescein-conjugated terminal transferase for 1 hr to label fragmented DNA. Top panels, TUNEL labeling in the E18 unaffected (left) and fh/fh(right) cortex. Although the number of dying cells is dramatically increased in all areas of the cortex at E18, the pattern of cell death in the fh/fh cortex mirrors the pattern observed in the normal cortex at this age. Specifically, the areas of highest cell death occur in the periventricular epithelium (PVE), which contains mostly proliferating cells, whereas the cortical plate (CP) and intermediate zone (IZ) have less cell death. Bottom panels, TUNEL labeling in the unaffected (left) andfh/fh (right) cerebellum at P1. The external granule layer (EGL) of fh/fhdisplays substantially more TUNEL-positive cells than that of an unaffected littermate. Although a distinct internal granule layer (IGL) is lacking in the fh/fh cerebellar, the proportion of TUNEL-positive cells in the region internal to the EGL was comparable to that of the normal IGL.
Table 3.
Cell death as determined in situ by the TUNEL method
| PVE | IZ | CP | |
|---|---|---|---|
| E16 | 0.65 ± 0.53 | 0.90 ± 0.85 | 0.40 ± 0.42 |
| n = 2750 | n = 791 | n = 1980 | |
| E18 | |||
| fh | 18.61 ± 5.68 | 6.45 ± 3.70 | 9.83 ± 3.43 |
| n = 1837 | n = 305 | n = 644 | |
| Normal | 0.36 ± 0.63 | 0.63 ± 0.80 | 0.12 ± 0.27 |
| n = 1459 | n = 480 | n = 835 | |
| P1 | |||
| fh | * | 10.32 ± 4.25 | 3.84 ± 2.63 |
| n = 163 | n = 426 | ||
| Normal | * | 0.86 ± 0.89 | 0.21 ± 0.48 |
| n = 479 | n = 470 | ||
| P10 | |||
| fh | * | ** | 2.21 ± 2.78 |
| n = 215 | |||
| Normal | * | ** | 1.91 ± 3.27 |
| n = 450 |
The percentage of cell death was compared in the periventricular epithelium (PVE), intermediate zone (IZ), and cortical plate (CP). Counts of TUNEL-positive cells were taken from two to three sections from a representative brain of fh/fh and unaffected littermates at E18, P1, and P10. Further experiments supported these results. No difference in cell death was observed in a litter at E16, and cell death from representative brain is shown. nindicates the total number of cells counted. *By P1, the PVE has disappeared. **By P10, the IZ has disappeared.
Examination of the fh/fh cerebellum also shows a marked increase in the proportion of apoptotic cells compared with normal (Fig. 9). As in the cerebral cortex, the difference in the extent of cell death is particularly dramatic in the proliferative zone (EGL) of the cerebellum (sixfold higher in fh/fh). Although a distinct IGL is not discernible in fh/fh, the extent of cell death in the region internal to the EGL was relatively similar to that seen in unaffected animals.
Cell death in fh/fh occurs in a narrow time window beginning around E18
The TUNEL method was used to determine the location and extent of apoptosis in the developing brain, the TUNEL method was used to label fragmented DNA ends in situ. Because the cerebral isocortex has a well defined structure and is dramatically reduced in size in mutants, we concentrated our efforts on this region. A time course study of cell death in the fh/fh isocortex using the TUNEL technique was performed. At E16, before the reduction infh/fh brain size is apparent, no significant difference in the pattern or amount of cell death could be detected. By E18, the average fh/fh brain weighs 26% less than an unaffected brain, and in the proliferative zone, more than one of every six cells is apoptotic (Table 3). The number of TUNEL-positive cells infh/fh is still greater than unaffected at P1 (Table 3, Fig.10), but the rate of death is roughly one-half of what it was at E18. By P8, the percentage of TUNEL-positive cells is similar between unaffected and fh/fh brains (Fig. 10).
Fig. 10.
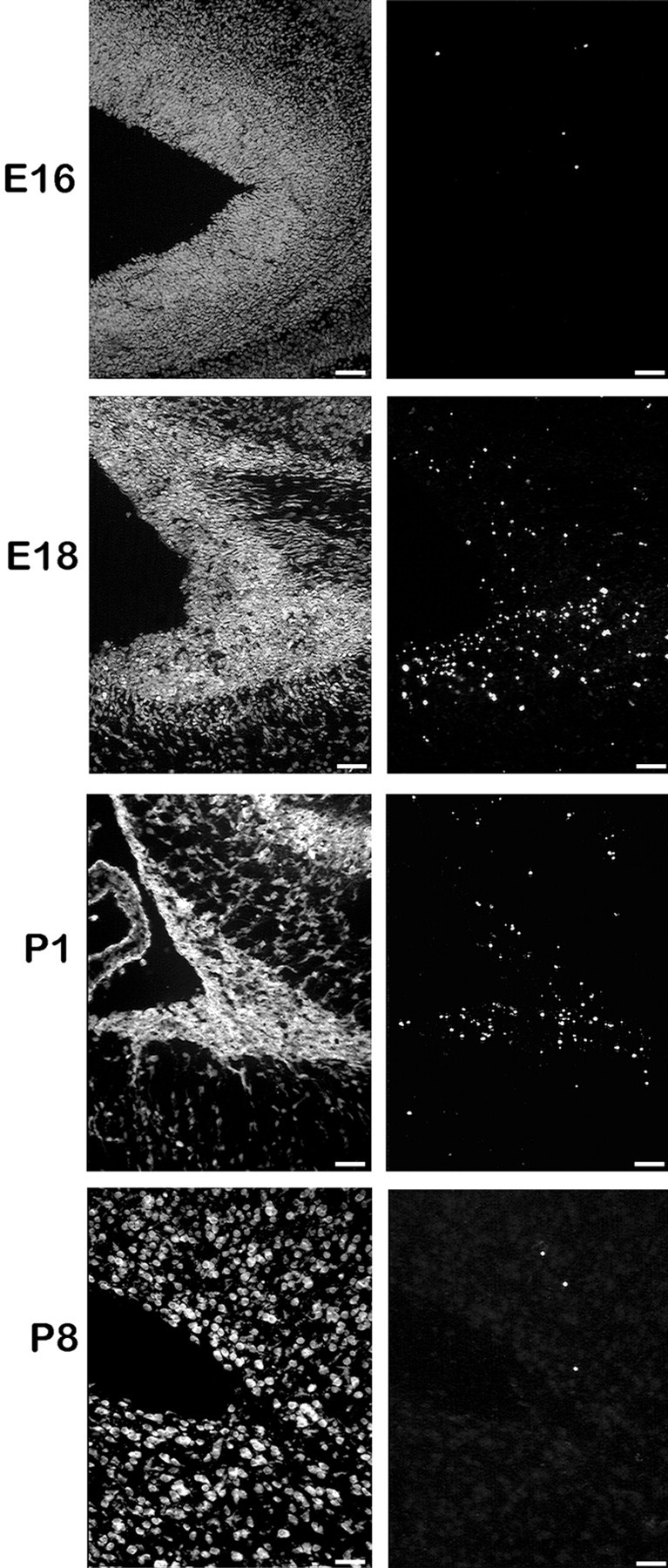
TUNEL labeling of the ventricular region of the cerebral cortex at E16, E18, P1, and P8 reveals that cell death occurs within a narrow time window during development of thefh/fh cortex. Frozen tissue was sectioned at 10 μm, rinsed in xylenes to remove lipids, permeabilized with proteinase K, and incubated with florescein-conjugated terminal transferase to label fragmented DNA. Sections were counterstained with propidium iodide (seepanels at left). Our results indicate that the onset and peak of cell death occurs around E18. Although thefh/fh cortex has a higher rate of cell death than a normal brain at P1, the frequency of cell death is approximately one-half of what it was at E18. By P8, only a few scattered cells are TUNEL positive in the fh/fh, which is similar to the pattern observed in a normal brain (data not shown).
fh/fh animals display recurrent spontaneous seizures
As described previously, fh/fh animals display certain features of behavioral seizures such as periodic tail flexing and tonic limb extension. To confirm that these behaviors are indicators of spontaneous seizures, electroencephalogram (EEG) recordings were performed. Figure 11 shows an EEG of a spontaneously occurring seizure in a P14 fh/fh animal. Recordings from the surface of neocortex in several sites indicate that the seizures are generalized across the entire cortex and synchronously involve both the left and right hemispheres. Such electroencephalographic seizures occur in mutants at a frequency as high as one seizure every 8–12 min from P7 to P18. After P19, seizures become far less frequent but more severe in their behavioral manifestations, often including loud vocalizations leading to complete tonus. A more complete characterization of seizures in fh/fhhas just been published (Sarkisian et al., 1999).
Fig. 11.

Representative EEG recording from afh/fh rat at P14 before (top), during (middle), and after (bottom) a spontaneous convulsive seizure. Such seizure activity has been recorded in 72 animals, with seizures occurring at a rate of four to six per hour from P7 to P18.
DISCUSSION
We describe a novel neurological rat mutant that displays reduced brain growth, ataxia, spontaneous seizures, and premature death. We have named this mutant flathead based on the first outward phenotypic trait that we noticed in affected animals: reduced cranial curvature. Our results indicate that fh is an autosomal recessive mutation with complete penetrance. While the entire brain and the spinal cord are reduced in size, certain areas such as the cerebral cortex and cerebellum are disproportionately smaller. Although a decrease in fh/fh body weight is also measurable after the first week, this is likely to be a consequence of poor feeding rather than a direct effect of the mutation. Indeed, histological analysis of sections from various parts of the fh/fh peripheral nervous system and non-neural tissues failed to reveal any abnormalities. Histological examination indicates that even within the brain, the mutation appears to be without any discernible effect until E16. Thefh/fh mutation therefore exerts its effects during later stages of brain development.
Because the smaller brain size in fh/fh could result from a reduction in cell proliferation, we used BrdU pulse-labeling to assess the proportion of proliferating cells in fh/fh cerebral cortex at E19, the time at which the size difference between mutant and unaffected brains becomes evident. This assay indicates that the rate of proliferation in the fh/fh brain is comparable to that of unaffected rats. In contrast, we discovered that there is a dramatic increase in cell death within the fh/fh brain at the time at which reduction of brain size becomes evident, indicating that increased cell death rather than reduced cell production is responsible for the smaller fh/fh brain. Based on the condensed nuclear morphology, positive TUNEL staining, and the detection of nonrandom DNA fragmentation, we conclude that the aberrant cell death infh/fh animals is caused by apoptosis rather than necrosis. Although much of our attention has focused on the cerebral cortex, it deserves mentioning that the increased cell death in fh/fhoccurs throughout the brain (M. Roberts and S. R. D'Mello, unpublished observations).
An interesting feature of the aberrant cell death in fh/fhis that it occurs in a narrow time window late in neurodevelopment, a period during which proliferation of neuronal progenitors in many brain regions, including the neocortex, is ceasing and differentiation is beginning. Although proliferative zones display a higher amount of cell death, cells that undergo apoptosis may include both differentiated neurons and proliferating progenitors. The depletion of progenitors would be expected to severely affect the neuronal populations that are derived from them. Consistent with such a possibility is the observation that neuronal populations generated later during brain development are more severely affected by the late burst of cell death in fh/fh. For example, granule neurons within the cerebellum, most of which are produced postnatally within the EGL, are severely depleted. In comparison, Purkinje neurons that are generated by E13 appear to be spared. In the absence of the internal granule layer, however, these neurons are scattered throughout the cerebellar cortex rather than being organized into a single layer. In the hippocampus, the dentate gyrus is missing almost entirely, whereas the cells of the Ammon's horn, which are generated mainly between E16 and E17, are relatively unaffected. Within the retina, photoreceptor cells that are generated postnatally are significantly reduced in number, whereas the deeper layers containing earlier born bipolar and ganglion cells appear less affected (V. Leung and J. J. LoTurco, unpublished data). Finally, within the neocortex, layers 2/3, which are composed of neurons generated late during cortical development, are severely reduced in thickness, whereas the deep layers that contain earlier-born neurons are relatively unaffected.
Despite extensive cell loss, the inside-out pattern of neocortical lamination is generally unaffected in fh/fh, indicating that migration per se is not defective in fh/fh. Failure of neurons to accurately migrate during development has been described in other mutants such as weaver, reeler, andscrambler (Rakic and Sidman, 1973; Pinto-Lord et al., 1982;Goldowitz et al., 1997; Gonzales et al., 1997). Although the dysgenesis in the fh/fh cerebellum is reminiscent of these mutants, the relatively normal neocortical layering suggests that the increased cell death is not attributable to defective migration but to the failure of granule cells (which make up much of the cerebellar cortex) to form. Similarly, disruption of lamination in the retina likely results from the failure of photoreceptor cells to form. It is possible, however, that although the fh mutation does not affect the ability of migrating neurons to find their appropriate location, it may prevent the initiation of migration (for example, a process such as interaction with radial glia) of neurons that are generated after E18. The higher incidence of cell death in the proliferative zones of the cerebral cortex and the cerebellum would be consistent with such a notion. Moreover, a careful examination of the cerebellar EGL offh/fh shows that most of the dying cells are located along the internal edge of the EGL. Failure of late-born cells that have exited the cell cycle to differentiate appropriately could also be possible. Taken together with the finding that BrdU incorporation infh/fh cortex and cerebellum is normal, our results raise the possibility that the fh/fh mutation affects a step between cell proliferation and neuronal migration that leads to an arrest in normal brain development. Supporting the idea that the fh/fhmutation causes developmental arrest is the observation that (Figs.1C, 3) the P14 fh/fh brain resembles that of a P0 wild type. Therefore, although it is possible that aberrant induction of apoptosis is the primary cause of the reduced brain size, it is more likely that cell death represents an epiphenomenon of a larger developmental arrest involving defective proliferation, cell-cycle exit, differentiation, or initiation of migration.
The precise cause of the demise of mutant animals is not known. Because of their neurological abnormalities, fh/fh rats are unable to compete with their unaffected littermates for food. However, we have observed that the life-span of fh/fh rats is not significantly extended even if nonmutant littermates are separated at birth, suggesting that the inability to compete for food is in itself not responsible for the short life-span. Moreover, autopsy offh/fh reveals milk in the stomach. Another possibility is that the recurrent episodes of seizures is responsible for the premature demise of these animals. EEG recordings indicate that seizures in fh/fh occur at a frequency of four to six episodes per hour for much of the brief life-span of the animal (Sarkisian et al., 1999). Although we have not determined whether the seizures in the days preceding death do in fact cause cell death, such cell loss could kill the animal directly or indirectly by affecting important respiratory, circulatory, or visceral systems.
In summary, we describe a new neurological mutant, fh, that displays extensive and brain-specific cellular degeneration. Degeneration has also been observed in a number of other neurological mutants. Although similar to other mutants in this regard, the pattern of neuronal death in fh/fh is different in two respects. First, this mutation affects diverse populations of cells that appear to share no functional or biochemical relationship, and cell loss occurs all over the brain. Second, cell death occurs in a narrow time window late in neurodevelopment and therefore represents a temporal mutation. Several developmental events such as proliferation, differentiation, and migration, are underway during the period at which cell-death is seen in fh/fh, and it is likely that the mutation affects one of these important processes. Regardless of the actual process that is affected, the unique phenotype displayed byfh renders it a valuable tool for investigating the mechanisms that regulate later stages of normal neurodevelopment and for ultimately identifying the molecules involved. Because virtually every region of the brain is affected, the mutation likely affects a common and fundamental process as opposed to cell or region-specific feature of neurodevelopment. Recently, we mapped fh to a region of rat chromosome 12 ∼2 cm telomeric to Nos-1, and there are no known mouse mutations in the homologous region of mouse chromosome 5 or human chromosome 12 that have a neurological phenotype (Cogswell et al., 1998). Therefore, the fh mutation is likely to involve a novel gene essential to normal perinatal brain development. Because reduced brain size and seizures are also seen in humans with autosomal recessive microcephaly (Holmes and Logan, 1980; Tolmie et al., 1987;Perlman and Argyle, 1992), and aberrant neuronal loss via induction of apoptosis is seen in a number of neurodegenerative diseases, theflathead mutation could shed valuable insight into the genetic and biochemical mechanisms affected in numerous neuropathologies.
Footnotes
Correspondence should be addressed to Santosh R. D'Mello, Department of Molecular and Cell Biology, University of Texas at Dallas, 2601 North Floyd Road, Richardson, TX 75083. E-mail:dmello@utdallas.edu.
REFERENCES
- 1.Alder J, Cho NK, Hatten ME. Embryonic precursor cells from the rhombic lip are specified to a cerebellar granule neuron identity. Neuron. 1996;17:389–399. doi: 10.1016/s0896-6273(00)80172-5. [DOI] [PubMed] [Google Scholar]
- 2.Altman J, Bayer SA. Embryonic development of the rat cerebellum. I. Delineation of the cerebellar primordium and early cell movements. J Comp Neurol. 1985a;231:1–26. doi: 10.1002/cne.902310103. [DOI] [PubMed] [Google Scholar]
- 3.Altman J, Bayer SA. Embryonic development of the rat cerebellum. III. Regional differences in the time of origin, migration, and settling of Purkinje cells. J Comp Neurol. 1985b;231:42–65. doi: 10.1002/cne.902310105. [DOI] [PubMed] [Google Scholar]
- 4.Angevine JB, Sidman RL. Autoradiographic study for cell migration during histogenesis of the cerebral cortex in the mouse. Nature. 1961;192:766–768. doi: 10.1038/192766b0. [DOI] [PubMed] [Google Scholar]
- 5.Bayer SA. Development of the hippocampal region in the rat. II. Morphogenesis during embryonic and early postnatal life. J Comp Neurol. 1980;190:115–134. doi: 10.1002/cne.901900108. [DOI] [PubMed] [Google Scholar]
- 6.Berry M, Rogers AW. Pattern of cell migration during cortical histogenesis. Nature. 1965;203:591–593. doi: 10.1038/203591b0. [DOI] [PubMed] [Google Scholar]
- 7.Blaschke AJ, Staley K, Chun J. Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development. 1996;122:1165–1174. doi: 10.1242/dev.122.4.1165. [DOI] [PubMed] [Google Scholar]
- 8.Braekevelt CR, Hollenberg MJ. The development of the retina of the albino rat. Am J Anat. 1970;127:281–302. doi: 10.1002/aja.1001270305. [DOI] [PubMed] [Google Scholar]
- 9.Cogswell CA, Sarkisian MR, Leung V, Patel R, D'Mello SR, LoTurco JJ. A gene essential to brain growth maps to the distal arm of rat chromosome 12. Neurosci Lett. 1998;251:5–8. doi: 10.1016/s0304-3940(98)00478-9. [DOI] [PubMed] [Google Scholar]
- 10.Cohen NR, Taylor JS, Scott LB, Guillery RW, Soriano P, Furley AJ. Errors in corticospinal axon guidance in mice lacking the neural cell adhesion molecule L1. Curr Biol. 1998;8:26–33. doi: 10.1016/s0960-9822(98)70017-x. [DOI] [PubMed] [Google Scholar]
- 11.Dahme M, Bartsch U, Martini R, Anliker B, Schachner M, Mantei N. Disruption of the mouse L1 gene leads to malformations of the nervous system. Nat Genet. 1997;17:346–349. doi: 10.1038/ng1197-346. [DOI] [PubMed] [Google Scholar]
- 12.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldowitz D, Hamre K. The cells and molecules that make a cerebellum. Trends Neurosci. 1998;21:375–382. doi: 10.1016/s0166-2236(98)01313-7. [DOI] [PubMed] [Google Scholar]
- 14.Goldowitz D, Cushing RC, Laywell E, D'Arcangelo G, Sheldon M, Sweet H, Davisson M, Steindler D, Curran T. Cerebellar disorganization characteristic of reeler in scrambler mutant mice despite presence of Reelin. J Neurosci. 1997;17:8767–8777. doi: 10.1523/JNEUROSCI.17-22-08767.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzales JL, Russo CJ, Goldowitz D, Sweet HO, Davisson MT, Walsh CA. Birthdate and cell marker analysis of scrambler: a novel mutation affecting cortical development with a reeler-like phenotype. J Neurosci. 1997;17:9204–9211. doi: 10.1523/JNEUROSCI.17-23-09204.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gunther T, Struwe M, Aguzzi A, Schughart K. Open brain, a new mouse mutant with severe neural tube defects, shows altered gene expression patterns in the developing spinal cord. Development. 1994;120:3119–3130. doi: 10.1242/dev.120.11.3119. [DOI] [PubMed] [Google Scholar]
- 17.Hatten ME. Mechanisms of glial-guided neuronal migration in vitro and in vivo. Experientia. 1990;46:907–916. doi: 10.1007/BF01939383. [DOI] [PubMed] [Google Scholar]
- 18.Herrup K, Busser JC. The induction of multiple cell cycle events precedes target-related neuronal death. Development. 1995;121:2385–2395. doi: 10.1242/dev.121.8.2385. [DOI] [PubMed] [Google Scholar]
- 19.Herrup K, Mullen RJ. Staggerer chimeras: intrinsic nature of Purkinje cell defects and implications for normal cerebellar development. Brain Res. 1978;178:443–457. doi: 10.1016/0006-8993(79)90705-4. [DOI] [PubMed] [Google Scholar]
- 20.Holmes GL, Logan WJ. A syndrome of infantile CNS degeneration. Am J Dis Child. 1980;134:262–266. doi: 10.1001/archpedi.1980.02130150020006. [DOI] [PubMed] [Google Scholar]
- 21.Landis SC, Shoemaker WJ, Schlumpf M, Bloom FE. Catecholamines in mutant mouse cerebellum: fluorescence microscopic and chemical studies. Brain Res. 1975;93:253–266. doi: 10.1016/0006-8993(75)90349-2. [DOI] [PubMed] [Google Scholar]
- 22.Lee KS, Collins JL, Anzivino MJ, Frankel EA, Schottler F. Heterotopic neurogenesis in a rat with cortical heterotopia. J Neurosci. 1998;18:9365–9375. doi: 10.1523/JNEUROSCI.18-22-09365.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luskin MB, Schatz CJ. Neurogenesis of the cat's primary visual cortex. J Comp Neurol. 1985;242:611–631. doi: 10.1002/cne.902420409. [DOI] [PubMed] [Google Scholar]
- 24.Mullen RJ, Eicher EM, Sidman RL. Purkinje cell degeneration, a new neurological mutation in the mouse. Proc Natl Acad Sci USA. 1976;73:208–212. doi: 10.1073/pnas.73.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perlman JM, Argyle C. Lethal cytomegalovirus infection in preterm infants: clinical, radiological, and neuropathological findings. Ann Neurol. 1992;31:64–68. doi: 10.1002/ana.410310112. [DOI] [PubMed] [Google Scholar]
- 26.Pinto-Lord MC, Evrard P, Caviness VS. Obstructed neuronal migration along radial glial fibers in the neocortex of the reeler mouse: a Golgi-EM analysis. Brain Res. 1982;263:379–393. doi: 10.1016/0165-3806(82)90181-x. [DOI] [PubMed] [Google Scholar]
- 27.Rakic P. Contact regulation of neuronal migration. Wiley; New York: 1985. [Google Scholar]
- 28.Rakic P, Sidman RL. Sequence of developmental abnormalities leading to granule cell deficit in cerebellar cortex of weaver mutant mice. J Comp Neurol. 1973;152:103–132. doi: 10.1002/cne.901520202. [DOI] [PubMed] [Google Scholar]
- 29.Roffler-Tarlov S, Landis SC, Zigmond MJ. Effects of Purkinje cell degeneration on the naradrenergic projection to mouse cerebellar cortex. Brain Res. 1984;298:303–311. doi: 10.1016/0006-8993(84)91429-x. [DOI] [PubMed] [Google Scholar]
- 30.Ross ME, Fletcher C, Mason CA, Hatten ME, Heintz N. Meander tail reveals a discrete developmental unit in the mouse cerebellum. Proc Natl Acad Sci USA. 1990;87:4189–4192. doi: 10.1073/pnas.87.11.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sarkisian MR, Rattan S, D'Mello SR, LoTurco JJ. Characterization of seizures in the flathead rat: a new genetic model of epilepsy in early postnatal development. Epilepsia. 1999;40:394–400. doi: 10.1111/j.1528-1157.1999.tb00732.x. [DOI] [PubMed] [Google Scholar]
- 32.Schlessinger AR, Cowan WM, Gottlieb DI. An autoradiographic study of the time of origin and the pattern of granule cell migration in the dentate gyrus of the rat. J Comp Neurol. 1975;159:149–176. doi: 10.1002/cne.901590202. [DOI] [PubMed] [Google Scholar]
- 33.Sibilia M, Steinbach JP, Stingl L, Aguzzi A, Wagner EF. A strain-independent postnatal neurodegeneration in mice lacking the EGF receptor. EMBO J. 1998;17:719–731. doi: 10.1093/emboj/17.3.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomaidou D, Mione MC, Cavanagh JFR, Parnavelas JG. Apoptosis and its relation to the cell cycle in the developing cerebral cortex. J Neurosci. 1997;17:1075–1085. doi: 10.1523/JNEUROSCI.17-03-01075.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 36.Tolmie JL, McNay M, Stephenson JB, Doyle D, Connor JM. Microcephaly: genetic counselling and antenatal diagnosis after the birth of an affected child. Am J Med Genet. 1987;27:583–594. doi: 10.1002/ajmg.1320270311. [DOI] [PubMed] [Google Scholar]
- 37.Wullner U, Loschman PA, Weller M, Klockgether T. Apoptotic cell death in the cerebellum of mutant weaver and lurcher mice. Neurosci Lett. 1995;200:109–112. doi: 10.1016/0304-3940(95)12090-q. [DOI] [PubMed] [Google Scholar]