

Abstract
One of the primary physiological roles of group II and group III metabotropic glutamate receptors (mGluRs) is to presynaptically reduce synaptic transmission at glutamatergic synapses. Interestingly, previous studies suggest that presynaptic mGluRs are tightly regulated by protein kinases. cAMP analogs and the adenylyl cyclase activator forskolin inhibit the function of presynaptic group II mGluRs in area CA3 of the hippocampus. We now report that forskolin has a similar inhibitory effect on putative mGluR2-mediated responses at the medial perforant path synapse and that this effect of forskolin is blocked by a selective inhibitor of cAMP-dependent protein kinase (PKA). A series of biochemical and molecular studies was used to determine the precise mechanism by which PKA inhibits mGluR2 function. Our studies reveal that PKA directly phosphorylates mGluR2 at a single serine residue (Ser843) on the C-terminal tail region of the receptor. Site-directed mutagenesis combined with biochemical measures of mGluR2 function reveal that phosphorylation of this site inhibits coupling of mGluR2 from GTP-binding proteins
Keywords: cAMP, metabotropic, dentate gyrus, perforant path, glutamate, phosphorylation, desensitization
Fast synaptic responses at the majority of excitatory synapses in mammalian brain are mediated by activation of a well characterized family of glutamate receptor cation channels referred to as the ionotropic glutamate receptors (Dingledine et al., 1999). In addition, glutamate can modulate cell excitability and synaptic transmission by activation of metabotropic glutamate receptors (mGluRs), which are coupled to effector systems through GTP-binding proteins. To date eight mGluR subtypes have been cloned and classified into three groups on the basis of their primary structures, second messenger coupling, and pharmacology. Group I mGluRs (mGluR1 and mGluR5) couple to Gq and activation of phospholipase C and are selectively activated by 3,5-dihydroxyphenylglycine. Group II (mGluR2 and mGluR3) and group III (mGluR4 and mGluR6–8) mGluRs couple to Gi/Go and are selectively activated by (2S, 2′R, 3′R)-2-(2′, 3′-dicarboxycyclopropyl)-glycine (DCG-IV) andl-2-amino-4-phosphonobutyric acid (L-AP4), respectively (for review, see Conn and Pin, 1997; Pin et al., 1999).
One of the primary functions of mGluRs seen throughout the CNS is their role in presynaptically reducing transmission at glutamatergic synapses (for review, see Anwyl, 1999; Conn and Pin, 1997). Although members of each major group of mGluRs can serve this role, the predominant mGluRs involved in regulating glutamate release belong to group II and group III mGluRs. Interestingly, previous studies reveal that presynaptic mGluRs are tightly regulated. For instance, activation of PKC inhibits the function of multiple presynaptic group II and group III mGluR subtypes at several glutamatergic synapses (Swartz et al., 1993; Tyler and Lovinger, 1995; Kamiya and Yamamoto, 1997; Macek et al., 1998). More recently, Kamiya and Yamamoto (1997) and Maccaferri et al., (1998)reported that the adenylyl cyclase activator forskolin or cAMP analogs reduce the ability of group II mGluRs to inhibit transmission at excitatory synapses in area CA3 of the hippocampus. The finding that cAMP can inhibit presynaptic group II mGluR function is particularly interesting in light of several recent studies suggesting the cAMP plays a critical role in both acute and long-lasting modulation of synaptic transmission at various hippocampal synapses (Nguyen and Kandel, 1996; Kamiya and Yamamoto, 1997). However, it is not yet clear whether this effect is restricted to area CA3 or can be seen at other synapses where group II mGluRs regulate synaptic transmission. Furthermore, the precise molecular mechanism by which an increase in cAMP concentration inhibits group II mGluR function is not known. We now report that forskolin also inhibits the function of group II mGluRs at the medial perforant path (MPP)–dentate granule cell synapse, a synapse where inhibition of synaptic transmission is likely mediated by mGluR2 (Neki et al., 1996; Petralia et al., 1996; Shigemoto et al., 1997). We then used a combination of electrophysiological, biochemical, and molecular approaches to show that this effect is mediated by activation of cAMP-dependent protein kinase (PKA) and that PKA phosphorylates mGluR2 at a single site on the C-terminal tail (Ser843). Phosphorylation of this site inhibits mGluR2-mediated responses by inhibiting coupling of the receptor to GTP-binding proteins.
MATERIALS AND METHODS
Materials. Chinese hamster ovary (CHO) cells were obtained from American Type culture collection (Manassas, VA). pCMV-script vector, site-directed mutagenesis system, BL21 Gold supercompetent cells were obtained from Stratagene (La Jolla, CA). pGEX-6P3 vector was obtained from Pharmacia (Piscataway, NJ). The mGluR2/3 antibody was obtained from Chemicon (Temecula, CA). [P32]-orthophosphate, [35S]-GTPγS, and [32P]-γ-ATP were obtained from NEN-Dupont (Boston, MA). [3H]-myo-inositol was purchased from American Radiolabeled Chemicals (St. Louis, MO). 8-Bromoadenosine-3′, 5′-cyclic monophosphate (8-bromo-cAMP), dibutyryl-cAMP, and adenylate cyclase, 9-(tetrahydro-2-furyl)-adenine (SQ22536) were obtained fromBiomol (Plymouth Meeting, PA). A water-soluble form of forskolin,N-[2-(p-bromocinnamylamino)-ethyl]-5-isoquinoline-sulfonamide 2HCl (H89), and PKA inhibitor 6–22 amide (PKI) were obtained from Calbiochem (San Diego, CA). DCG-IV was obtained from Tocris Cookson (Ballwin, MO). Purified catalytic subunit of PKA and all other materials were obtained from Sigma.
Field potential recording. Hippocampal slices were prepared from 6- to 8-week-old rats, and field potential recordings were performed at MPP–dentate gyrus (MPP–DG) synapses as described previously (Macek et al., 1996). Both stimulating and recording electrodes were placed in the middle third of the molecular layer in the dentate gyrus, and the previously described criteria were used to confirm selective recording from MPP synapses (Kahle and Cotman, 1993;Macek et al., 1996).
Plasmid constructions. The cDNA encoding mGluR2 originally in the pBluescript vector (gift from Dr. S. Nakanishi) was subcloned into the pCMV-script vector or into the retroviral vector pCL2 by standard protocols. pCL2 was constructed from pLNCX (Miller et al., 1993) by removing from the latter the region encoding the Neo gene and internal CMV promoter and replacing it with a linker that forms a new multicloning region with two noncohesive SfiI restriction sites. Point mutations were introduced into the fusion protein or in the full-length mGluR2 using the Stratagene site-directed mutagenesis system as per the manufacturer's instructions. Each mutant was sequenced to verify the changes.
Cell culture and transfection. Rat thoracic aorta smooth muscle cells (VSMC) and Phoenix cells are kept as a continuous cell line as described previously (Wang and Murphy, 1998). For transfection, CHO cells were plated in six-well plates (105 cells per well) the day before. Cells were transfected by the calcium-phosphate method with 2 μg of mGluR2 in combination with 3 μg of Gα15. After an overnight incubation the CaPO4-DNA medium was replaced by fresh medium. The retrovirus production and transfection protocols of VSMC were performed as described previously (Wang and Murphy, 1998).
Culture of rat cerebellar granule cells. Cerebella from postnatal day 4 (P4) rats were removed and washed in ice-cold calcium/magnesium-free HBSS solution. Tissue fragments were triturated using a P1000 pipette, and dissociated cells were centrifuged 5 min at 600 rpm and suspended in culture medium described below and plate in poly-d-lysine-coated six-well plates. Cultures were maintained at 37°C in 5% CO2 for 2 weeks in DMEM supplemented with penicillin and streptomycin and 25 mm KCl.
Glutathione-S-transferase–fusion protein generation and purification. Sense and antisense single-stranded DNA primers were generated for regions of mGluR2 that code for either the predicted first (I1) or second (I2) intracellular loops or the C-terminal tail of the receptor. Primers were designed that incorporated either anEcoRI (sense) or NotI (antisense) restriction site proximal to the 5′ end of the oligomer. Sense and antisense primers were used to amplify by PCR the appropriate regions of mGluR2 (Ho et al., 1989). The resultant PCR products were then digested withEcoRI and NotI and subcloned, in-frame, into the polylinker region of pGEX-6P3 (Pharmacia), a glutathione-S-transferase (GST)-fusion protein bacterial expression vector. Subcloned DNA was then transformed into BL21Escherichia coli (Stratagene) in accordance with the manufacturer's protocols and plated onto LB medium plus ampicillin agar plates. Single colonies were then grown overnight in LB plus ampicillin, and plasmid DNA was purified using Qiagen DNA prep kits. Correct orientation was determined by restriction digest and DNA sequencing. Predicted amino acid sequences were determined by computer translation of the sequenced DNA. GST fusions proteins were purified according to the manufacturer's protocol.
Immunoprecipitation. Rat cerebellar granule cells (15 din vitro) were incubated with [P32]-orthophosphate (1 mCi/ml) for 3 hr in phosphate-free DMEM at 37°C in a tissue-culture incubator. After labeling, the cells were exposed to PKA activator for 30 min. Group II mGluRs were immunoprecipitated using 1 μg mGluR2/3 antibody as described previously (Alagarsamy et al., 1999).
In vitro phosphorylation assay. GST-fusion proteins or hippocampal membranes were incubated in PKA assay buffer containing 20 mm Tris-HCl, pH 7.4, 10 mmMgCl2, 1 mm EGTA, and 30 U purified catalytic subunit of PKA. The reaction was started by the addition of 10 μCi [32P]-γ-ATP at 30°C for 30 min. The reaction was terminated by the addition of sample buffer. The phosphoproteins were separated by electrophoresis on SDS-polyacrylamide gels. The dried gel was exposed to a phosphoscreen or x-ray film at −80°C, and radioactivity in the peptide or the mGluR2/3 bands was quantified with a Molecular Dynamics phosphorImager.
Sequencing analysis. Purified fusion proteins were phosphorylated as described above except that 10 mm cold ATP was added instead of [32P]-γ-ATP. The purified phosphorylated protein [(mass = 6857 average mass unit (amu), expected mass = 6857 amu)] was digested with trypsin, and the peptides were separated by RP-HPLC. The masses of the peptides were determined by electrospray-ionization mass spectrometry (ESI-MS) in a model API300 triple quadrupole mass spectrometer equipped with MicroIon Spray source (PE-Biosystems). Phosphorylated peptides were identified by ESI-MS using the precursor ion scanning technique (Carr et al., 1996) or by matrix-assisted laser desorption-ionization time-of-flight mass spectrometry in a Bruker Daltonics model Reflex-II mass spectrometer before and after alkaline phosphatase treatment to confirm phosphorylation (Zhang et al., 1998). Purified phosphopeptides (10–20 pmol) were sequenced by Edman degradation in a PE-Biosystems model cLC-Procise sequencer. The serine phosphorylation sites in the peptides were unambiguously identified by measuring the relative sequencing [phenylthiohydantoin (PTH)] yields of Ser and products of β-elimination of Ser or Ser(P), or both, in Edman cycles (Meyer et al., 1991; Chambers et al., 1993).
Phosphoinositide hydrolysis. Phosphoinositide (PI) hydrolysis was measured as described by Peavy and Conn (1998) except that CHO cells in six-well plates were labeled overnight in glutamine-free DMEM containing 5 μCi/ml [3H]-myo-inositol. Protein kinase activators or inhibitors were added 15 min before the addition of DCG-IV.
Preparation of hippocampal synaptosomes and vascular smooth muscle cell membranes and measurement of [35S]-GTPγS binding. Hippocampi of 5- to 7-week-old male Sprague Dawley rats were removed and bathed in ice-cold oxygenated Krebs' buffer and cross-sectioned (350 × 350 μm) using a McIlwayne tissue chopper. After incubation for 1 hr in oxygenate Krebs' buffer at 37°C, an equal volume of gravity-packed slices was incubated for 30 min in the absence or presence of 8-bromo-cAMP (1 mm). The slices were diluted 1:10 (w/v) in ice-cold buffer 1 containing 20 mm HEPES, 10 mmEDTA, and 320 mm sucrose, pH 7.4, and homogenized with a Teflon pestle. The resulting homogenate was centrifuged at 900 ×g for 10 min. The pellet was discarded, and the supernatant was recentrifuged at 48,000 × g for 10 min. This pellet was resuspended with a Polytron and washed with buffer 1 without sucrose followed by two washes with buffer 2 containing 20 mm HEPES, 0.1 mm EDTA, pH 7.4. Membranes were stored in aliquots at −80°C. Protein concentration was determined according to the method of Bradford (Pierce BCA reagent, Rockford, Il), with bovine serum albumin as a standard.
Vascular smooth muscle cells were treated with or without 8-bromo-cAMP (1 mm) for 30 min. Membranes were made as described above and stored in aliquot at −80°C. On the day of the experiment, the membranes were thawed and washed once with the assay buffer containing 20 mm HEPES, 100 mm NaCl, 10 mmMgCl2, pH 7.4, and resuspended at 250 μg/ml. The [35S]-GTPγS binding was measured according to Kowal et al. (1998).
RESULTS
Consistent with previous reports, the selective group II mGluR agonist, DCG-IV (0.5 μm), induced a reversible depression field EPSP (fEPSP) at the MPP–dentate gyrus synapse (Fig.1). Previous physiological (Brown and Reymann, 1995; Macek et al., 1996; Dietrich et al., 1997; Huang et al., 1999; Macek et al., 1999) and anatomical (Neki et al., 1996; Petralia et al., 1996; Shigemoto et al., 1997) studies suggest that this response is likely mediated by presynaptically localized mGluR2.
Fig. 1.

cAMP-dependent regulation of group II mGluR function at MPP–DG synapses. fEPSPs were recorded before and after application of DCG-IV (0.5 μm) in the presence and absence of the adenylyl cyclase activator forskolin (50 μm). The mean (±SEM) slope of the fEPSP from five independent experiments was plotted against time in the bottom panel. Bars represent periods of drug application. Top panels show fEPSP traces corresponding to time points designated in the time course graph. Calibration: 0.2 mV, 10 msec.
There was no obvious desensitization of the response to DCG-IV and the magnitude of the response remained unchanged with repeated DCG-IV applications (data not shown). Application of the adenylyl cyclase activator forskolin (50 μm) induced a significant increase in fEPSP slope at the MPP–dentate gyrus synapse, which typically stabilized after 10 min (Fig. 1). After the fEPSP stabilized, the effect of DCG-IV was measured. Consistent with the previous reports at the mossy fiber–CA3 synapse (Kamiya and Yamamoto, 1997) and at excitatory synapses onto CA3 interneurons (Maccaferri et al., 1998), forskolin markedly attenuated the inhibitory effect of DCG-IV on EPSPs (Fig. 1). In other studies, we have also found that forskolin inhibits the effect of group II mGluR agonists on transmission at excitatory synapses in the substantia nigra pars reticulata (our unpublished observations). Taken together, these data suggest that the ability of forskolin to inhibit presynaptic group II mGluR-mediated responses is a widespread phenomenon that occurs at multiple glutamatergic synapses.
At present, the molecular mechanism by which forskolin inhibits this mGluR2-mediated responses is not known. As is common with many other receptors that regulate neurotransmitter release, mGluR2 can couple to Gi and inhibition of adenylyl cyclase (Schoepp et al., 1992, 1995; Conn and Pin, 1997). If inhibition of adenylyl cyclase is the mechanism by which mGluR2 inhibits glutamate release, one possible mechanism by which forskolin and cAMP analogs could inhibit mGluR2-mediated response is by simply overcoming the ability of mGluR agonists to reduce cAMP accumulation. However, this is extremely unlikely given the large body of literature suggesting that multiple Gi-coupled receptors (including mGluRs) inhibit neurotransmitter release by a mechanism that is unrelated to their ability to inhibit adenylyl cyclase (Limbird, 1988; Gereau and Conn, 1994; Herrero et al., 1996; Schoppa and Westbrook, 1997; Rauca et al., 1998). However, to verify that DCG-IV-induced inhibition of transmission at the MPP synapse is not mediated by inhibition of adenylyl cyclase, we determined the effect of the adenylyl cyclase inhibitor SQ22536 and the PKA inhibitor H89 on this response. Consistent with previous studies at the Schaffer collateral synapse (Gereau and Conn, 1994), SQ22536 (300 μm) did not inhibit transmission at the MPP synapse at concentrations that completely inhibit adenylyl cyclase (Dixon and Atwood, 1989; Goldsmith and Abrams, 1991) and completely inhibit cAMP accumulation in hippocampal slices (Gereau and Conn, 1994). In fact, SQ22536 induced an increase on fEPSP slope at the MPP synapse. Furthermore, SQ22536 had no effect on the inhibitory action of DCG-IV (Fig.2A,C). In the presence of SQ22536 (300 μm), the percentage inhibition of fEPSP slope induced by DCG-IV was identical to the response obtained by DCG-IV alone (Fig. 2C). The PKA inhibitor H89 had a similar lack of effect on transmission at the MPP synapse and did not affect the percentage of inhibition induced by DCG-IV alone (Fig. 2C). These data are consistent with previous studies and suggest that DCG-IV-induced inhibition of transmission at the MPP synapse is independent of DCG-IV-induced regulation of adenylyl cyclase and PKA.
Fig. 2.
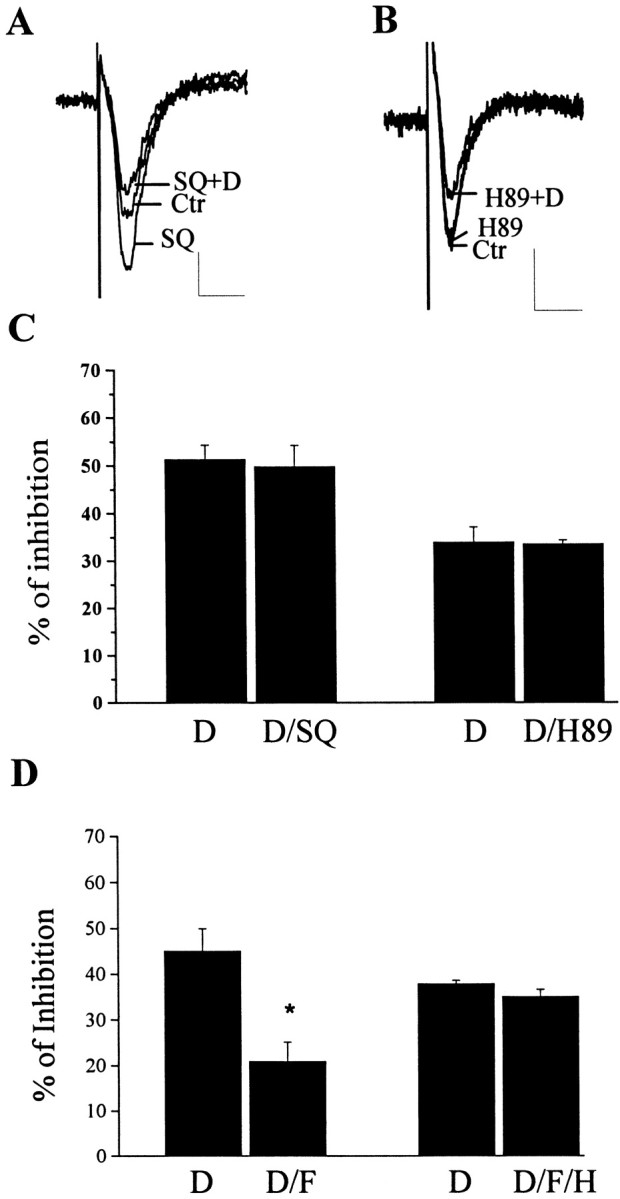
Effects of adenylyl cyclase inhibitor (SQ22536) and PKA inhibitor (H89) on DCG-IV-induced depression of fEPSP.A, Overlaid traces of fEPSP show that SQ22536 (SQ; 300 μm) enhanced fEPSP but had no effect on the inhibitory action of DCG-IV (D). B, Overlaid traces showing that H89 (10 μm) had no effect on basal fEPSP and DCG-IV-induced depression of fEPSP. C, Bar graph summarizing the effects of SQ22536 and H89 on DCG-IV-induced depression of fEPSP (p > 0.05,t test). The results are expressed as percentage of inhibition and represent the mean (±SEM) of five (SQ22536) or six (H89) independent experiments. D, Bar graph summarizing the results in which the effect of DCG-IV was determined in the presence and absence of forskolin (F) or forskolin plus the PKA inhibitor H89 (H) (10 μm). The results are expressed as percentage of inhibition and represent the mean (±SEM) of four (H89) or five (forskolin) independent experiments. Forskolin significantly reduced DCG-IV-induced depression of fEPSP in the absence of H89 (*p < 0.05, paired t test) but was without effect in the presence of H89 (p > 0.05, t test). Calibration: 0.2 mV, 10 msec.
If forskolin-induced inhibition of the response to DCG-IV is mediated by activation of PKA rather than a nonspecific action of the drug, this response should be blocked by the PKA inhibitor. Consistent with this, H89 completely blocked the ability of forskolin to inhibit the response to DCG-IV (Fig. 2D). These data suggest that forskolin inhibits the response to DCG-IV by a mechanism that is dependent on activation of PKA. However, they provide no information as to the substrate of PKA or the mechanism by which PKA exerts this effect. We next performed a series of studies to determine the exact mechanism by which PKA inhibits mGluR2 signaling.
PKA could inhibit mGluR2 function by phosphorylation of any number of proteins, including the receptor, the G-protein, or downstream effector proteins. If PKA acts at the level of the receptor or the G-protein, activation of this enzyme should inhibit coupling of the receptor to GTP binding proteins. In contrast, if PKA acts at a downstream effector, such as an ion channel or a protein involved in the exocytotic process, it may have no effect on G-protein coupling of mGluR2. To determine whether PKA acts at the level of the receptor or G-protein, we determined the effect of PKA on coupling of mGluR2 to G-proteins. To accomplish this, DCG-IV-induced increases in [35S]-GTPγS binding were measured as a direct measure of G-protein coupling in hippocampal synaptosomes and vascular smooth muscle cells infected with a retrovirus containing mGluR2 (Kowal et al., 1998). The retroviral infection system that we used provides high efficiency of protein expression that is typically required for reliable measurement of [35S]-GTPγS binding (Wang and Murphy, 1998). DCG-IV induced a concentration-dependent increase in [35S]-GTPγS binding in both hippocampal synaptosomes and vascular smooth muscle cell membranes (Fig. 3). Interestingly, 8-bromo-cAMP (1 mm) induced a significant reduction in the response to DCG-IV (Fig. 3) in both preparations. A similar inhibition of the response was seen with forskolin (data not shown). These data suggest that PKA-induced inhibition of mGluR2 function is at least partially mediated by inhibition of coupling of the receptor from G-proteins.
Fig. 3.

Effect of PKA activation on DCG-IV-induced increases in [35S]-GTPγS-binding in hippocampal synaptosomes (A) or in vascular smooth muscle cell membranes (B). Vascular smooth muscle cells have been infected with an mGluR2-expressing virus as described in Materials and Methods. Concentration–response curves show the effect of increasing concentrations of DCG-IV on [35S]-GTPγS binding in the absence (▪) or in the presence of 8-bromo-cAMP (1 mm) (●). Results are expressed as percentage of maximal response and are the mean (±SEM) of three independent experiments, each performed in triplicate.
PKA could inhibit G-protein coupling of mGluR2 by phosphorylation of either the receptor or the G-protein. Alternatively, PKA could phosphorylate another protein that in some way regulates coupling of mGluR2 to G-proteins. We next performed a series of experiments to test the hypothesis that PKA directly phosphorylates mGluR2. Previous studies reveal that cerebellar granule cells in primary culture express mGluR2 (Prezeau et al., 1994). This provides a convenient system to determine whether PKA activation leads to phosphorylation of this receptor in a native system. mGluR2/3 antibodies were used to immunoprecipitate group II mGluRs from primary rat cerebellar granule cells that had been incubated with32P-orthophosphate to label endogenous ATP pools. Radioactivity in mGluR2/3 was markedly increased by incubation with 8-bromo-cAMP (1 mm), and the response to 8-bromo-cAMP was significantly attenuated by the PKA inhibitor H89 (30 μm) (Fig.4A). The increase in phosphorylation of group II mGluRs could be seen as an increase in32P incorporation in both the monomer and dimer bands of the receptor. We also used an in vitrophosphorylation assay to determine whether exogenously applied PKA can phosphorylate mGluR2/3 receptors in hippocampal membranes. Membranes prepared from hippocampal slices were incubated with32P-ATP in the presence or absence of the purified catalytic subunit of PKA. Immunoprecipitation of mGluR2/3 from PKA-treated membranes revealed a robust PKA-induced increase in phosphorylation of mGluR2/3 that was inhibited by PKI (1 μm), a highly specific peptide inhibitor of PKA (Fig. 4B).
Fig. 4.

PKA phosphorylates group II mGluRs.A, Autoradiogram showing mGluR2/3 phosphorylation in cerebellar granule cells. Cells were labeled with [32P]-orthophosphate and exposed to vehicle, 8-bromo-cAMP (1 mm), or 8-bromo-cAMP + H89 (30 μm) for 30 min. H89 was added 10 min before 8-bromo-cAMP addition. B, Autoradiogram showing PKA-induced phosphorylation of mGluR2/3 immunoprecipitated from hippocampal membranes. Membranes were incubated with [32P]-γ-ATP and purified PKA in the presence or absence of a selective inhibitor of PKA (PKI).C, Autoradiogram showing PKA-induced phosphorylation of GST-fusion proteins of intracellular domains of mGluR2. Fusion proteins were incubated with [32P]-γ-ATP and purified PKA. In each case, the immunoprecipitated group II mGluRs or fusion proteins were separated by SDS-PAGE and analyzed by autoradiography. Each figure is representative of at least three independent experiments. I1, First intracellular loop;I2, second intracellular loop; CT, C terminal.
The above studies suggest that activation of PKA leads to an increase in phosphorylation of group II mGluRs in two native preparations. However, the antibodies used do not differentiate between mGluR2 and mGluR3, both of which are likely present in these cells. Furthermore, it is possible that PKA does not directly phosphorylate mGluR2/3 but does induce an increase in phosphorylation by activation of another kinase or inhibition of a phosphatase that is present in these nonpurified preparations. To test the hypothesis that PKA can phosphorylate mGluR2, GST-fusion proteins containing the putative intracellular domains of mGluR2 were constructed and used to determine whether PKA phosphorylates mGluR2 sequences in a highly purified preparation. Analysis of the sequence of mGluR2 reveals that potential PKA consensus sites are present on the second intracellular loop (I2) and on the C-terminal tail of the protein. In vitrophosphorylation assays revealed that PKA directly phosphorylates the fusion protein for the C-terminal domain (residues 820–872). In contrast, there was no detectable phosphorylation of the I1 loop (residues 591–604) and only minimal phosphorylation of the I2 loop (residues 656–680) (Fig. 4C). The finding that the C-terminal domain is heavily phosphorylated by PKA is interesting in light of previous studies suggesting that the C-terminal region of mGluRs is important for G-protein coupling (Pin et al., 1994; Prezeau et al., 1996). The C-terminal tail of mGluR2 contains several serine residues but only two putative PKA consensus sites, Ser837 and Ser843. Quantitative phosphopeptide analysis of tryptic peptides revealed that the vast majority of phosphate incorporated into the fusion protein was present in a fragment containing residues 842–861. Sixty percent of the total fusion protein was phosphorylated on this fragment. Sequence analysis of fragment (842–861) unambiguously identified Ser843 as the primary PKA phosphorylation site in the protein (Fig. 5A,B). A second fragment containing residues 835–841 was minimally phosphorylated (at Ser837); however, only 5% of the total fusion protein contained a phosphate at this site.
Fig. 5.

Identification of Ser843 as the major phosphorylation site in the C-terminal tail of mGluR2.A, Deconvoluted ESI-MS spectra of phosphorylated (A1) and dephosphorylated (phosphatase-treated,A2) fragment (842–861) demonstrate a loss of 80 mass units (−PO3H). B, Edman degradation of fragment (842–861): PTH-amino acid yields in cycles 1, 2, 9, and 11 normalized using PTH-Ala842. A 10-fold lower PTH yield of Ser843 for the phosphorylated but not for nonphosphorylated or dephosphorylated peptide demonstrates the presence of modification at this site; similar PTH yields of Ser850 and Ser852 in cycles 9 and 11 for all three peptides demonstrate absence of modification at these two residues.
Mutation analysis confirmed the results of sequence analysis in identifying Ser843 as the major site of phosphorylation by PKA. Thus, mutation of Ser843 to alanine dramatically reduced phosphorylation of the mGluR2 C-terminal fusion protein by PKA (Fig.6A,B). In contrast, mutation of Ser837 to alanine was without effect, as were mutations of Ser833, Ser827, or Thr832 (Fig.6A,B). Mutation analysis was also used to identify the minor site of phosphorylation on the I2 loop. The I2 loop contains only a single residue that is a predicted PKA consensus site (Ser675). Mutation of this site completely abolished the phosphorylation of the I2 fusion protein (data not shown).
Fig. 6.

Identification of Ser843 as phosphorylation site of mGluR2 C terminus fusion protein.A, In vitro phosphorylation of mGluR2 C terminus-fusion protein by PKA GST-fusion proteins of intracellular domains were phosphorylated in vitro by purified PKA for 30 min. The peptides were separated by SDS-PAGE and analyzed by autoradiography. B, The bar graph represents the mean data obtained by the phosphorImager. Results are expressed as percentage of wild type and are the mean (±SEM) of three independent experiments. Statistical significance was defined by using a paired Student's t test; *p < 0.05 versus wild type (WT).
The finding that PKA directly phosphorylates mGluR2 is consistent with the hypothesis that PKA inhibits mGluR2 function by direct phosphorylation of the receptor. Furthermore, the robust phosphorylation of Ser843 raises this site as a likely candidate for mediating this effect of PKA. To test this hypothesis directly, we transiently transfected CHO cells with mGluR2 and Gα15 (Wilkie et al., 1991). Previous studies reveal that mGluR2 can couple to this G-protein and thereby activate phospholipase C and increase PI hydrolysis (Gomeza et al., 1996). This robust mGluR2-mediated response lends itself well to screening of mutant receptors in that it does not require the use of viral constructs and can be readily measured in transiently transfected cells (Gomeza et al., 1996). DCG-IV induced a concentration-dependent increase in PI hydrolysis in CHO cells expressing Gα15 and mGluR2 (Fig.7A). Consistent with the studies presented above, 8-bromo-cAMP (1 mm) inhibited the response to DCG-IV (Fig. 7A). This effect was mimicked by application of forskolin (30 μm) or dibutyryl cAMP (1 mm), another cAMP analog (Fig.7B). Furthermore, the effects of 8-bromo-cAMP and forskolin were completely blocked by the PKA inhibitor H89 (30 μm) (Fig. 7B), suggesting that this response is mediated by activation of PKA.
Fig. 7.

Effect of PKA activation in CHO cells transiently transfected with mGluR2 and Gα15. A, Concentration–response curves show the effect of increasing concentrations of DCG-IV in the absence (▪) or in the presence (●) of 8-bromo-cAMP (1 mm) on PI hydrolysis. B, Effect of different PKA activator on the DCG-IV-induced PI hydrolysis in the presence or absence of H89 (30 μm). Results are expressed as means (±SEM) of three independent experiments performed in triplicate. Statistical significance was defined by using a paired Student's t test; *p < 0.05 versus PKA activators alone.
Site-directed mutagenesis was used to develop several mutant mGluR2 constructs in which potential phosphorylation sites were mutated on the basis of the biochemical studies outlined above. Each construct was coexpressed with Gα15 in CHO cells, and the effect of 8-bromo-cAMP on the PI response was determined. Mutation of Ser843 to alanine (mGluR2S843A) or to glycine (mGluR2S843G) virtually abolished the ability of 8-bromo-cAMP to inhibit the response (Fig.8). In contrast, mutation of several other potential phosphorylation sites had no significant effect on the response to 8-bromo-cAMP. Mutations that were without effect included the minor PKA phosphorylation site on the C-terminal tail identified above (mGluR2S837A), the PKA consensus site on the I2 loop that was slightly phosphorylatedin vitro (mGluR2S675A), and a serine residue on the I1 loop that was not phosphorylated in thein vitro studies (mGluR2S601A). We also determined the effect of a double mutation of Ser843 and Ser675. The response of this mutant to 8-bromo-cAMP was similar to that of mGluR2S843A (Fig. 8A). We next determined the effect of PKA activation on mGluR2S843A coupling to Gi/Go by determining the effect of PKA activation on [35S]-GTPγS binding in vascular smooth muscle cells infected with the mutant receptor. DCG-IV induced a dose-dependent increase in [35S]-GTPγS binding. Consistent with the studies with Gα15 in CHO cells, 8-bromo-cAMP (1 mm) failed to inhibit DCG-IV-induced increases in [35S]-GTPγS binding in membranes from cells expressing the mutant receptor (Fig. 8B).
Fig. 8.

Effect of different mutations on DCG-IV-induced PI hydrolysis (A) or in [35S]-GTPγS-binding (B) in the presence of 8-bromo-cAMP (1 mm). A, CHO cells were transfected with mGluR2 (WT) and mutant receptors in combination with Gα15. Cells were stimulated with DCG-IV (3 μm) in the presence of 8-bromo-cAMP (1 mm). Results are expressed as percentage of DCG-IV (3 μm) and are the mean (±SEM) of three independent experiments performed in triplicate. Statistical significance was defined by using a paired Student's t test; *p < 0.05 versus WT.B, Vascular smooth muscle cells were infected with an mGluR2-S843A-expressing virus as described in Materials and Methods. Concentration–response curves showing the effect of increasing concentrations of DCG-IV on [35S]-GTPγS binding in the absence (●) or in the presence of 8-bromo-cAMP (1 mm) (▪). Results are expressed as percentage of maximal response and are the mean (±SEM) of three independent experiments, each performed in triplicate.
DISCUSSION
The present data add to a growing body of literature suggesting that mGluRs are tightly regulated by protein phosphorylation. As discussed above, previous studies have shown that forskolin also inhibits presynaptic group II mGluRs at the hippocampal mossy fiber synapse (Kamiya and Yamamoto, 1997) and synapses of hippocampal CA3 pyramidal cells onto interneurons (Maccaferri et al., 1998). Although selective compounds that differentiate between mGluR2 and mGluR3 are not available, immunocytochemical studies suggest that mGluR2 is the most likely candidate for the group II mGluR subtype that reduces transmission at these synapses (Petralia et al., 1996; Shigemoto et al., 1997). In addition, activation of protein kinase C inhibits the function of multiple presynaptic mGluR subtypes at a wide variety of glutamatergic synapses. These include group II mGluRs (likely mGluR2) at corticostriatal (Swartz et al., 1993), MPP (Macek et al., 1998), and mossy fiber (Kamiya and Yamamoto, 1997) synapses as well as group III mGluRs at the lateral perforant path synapse (likely mGluR8) and the Schaffer collateral–CA1 synapse (likely mGluR7) (Macek et al., 1998). At the Schaffer collateral–CA1 synapse, activation of A3 adenosine receptors induces a PKC-mediated inhibition of group III mGluR-mediated responses (Macek et al., 1998). Thus PKC participates in a heterologous form of desensitization analogous to that observed here with activation of PKA by forskolin. In addition, PKC is involved in homologous desensitization of mGluR5, a group I mGluR that couples to Gq and activation of phospholipase C (Abe et al., 1992). PKC is also involved in induction of oscillatory calcium responses by mGluR5 (Flint and Connors, 1996; Nakahara et al., 1997).
We have performed a number of studies that strongly suggest that PKA inhibits mGluR2 signaling by direct phosphorylation of a single residue on the receptor and leads to inhibition of mGluR2 coupling to G-proteins. Thus, we clearly demonstrate that mGluR2 is a substrate for PKA and that PKA activation inhibits mGluR2 coupling to G-proteins as assessed by DCG-IV-induced increases [35S]-GTPγS binding in hippocampal synaptosomes and in a heterologous expressing system. A search for PKA consensus sites in the primary structure of mGluR2 (Tanabe et al., 1992) identified three potential PKA phosphorylation sites, one in the second intracellular domain (Ser675) and two (Ser837, Ser843) in the C-terminal tail region of mGluR2. In vitro phosphorylation experiments using GST-fusion proteins from intracellular domains and purified catalytic subunits of PKA revealed that only one of these sites, Ser843, is a major site of PKA phosphorylation. Functional studies of wild-type and mutant forms of mGluR2 clearly identified Ser843 as the only site required for PKA-induced inhibition of mGluR2 signaling.
Few studies have been successful in unambiguously identifying specific phosphorylation sites involved in regulation of mGluR function by protein kinases. However, it is interesting that in each of the cases in which the site of mGluR modulation has been identified, the critical site or sites have been found to reside on the C-terminal intracellular domain of the receptor. For instance, evidence suggests that PKC-induced desensitization of mGluR5 is mediated by phosphorylation of two major sites (Ser881 and Ser890) on the C-terminal tail region (Gereau and Heinemann, 1998). PKC-induced phosphorylation of another site on the C-terminal tail (Thr840) has been implicated in induction of calcium oscillations by mGluR5 (Kawabata et al., 1996). Although the precise molecular mechanism by which PKC inhibits signaling by each of the different presynaptic group II and group III mGluR subtypes is not entirely known, recent studies suggest that PKC inhibits coupling of these receptors to G-proteins in a manner similar to that reported here for PKA effects on mGluR2 (Macek et al., 1998). Furthermore, recent studies with mGluR7 suggest that PKC directly phosphorylates the C-terminal region of the receptor at a calmodulin binding site located immediately after the seventh transmembrane spanning domain (Nakajima et al., 1999). Interestingly, calmodulin binding to this site is required for normal signaling by mGluR7 (O'Connor et al., 1999). Phosphorylation of the receptor by PKC inhibits the interaction with calmodulin (Nakajima et al., 1999), providing a possible mechanism by which PKC could inhibit mGluR7-mediated responses.
It is interesting to note that previous studies suggest that the first 28 N-terminal amino acid residues of the C-terminal domain of mGluRs are critical for coupling of these receptors to G-proteins. The present finding that PKA inhibits mGluR2-mediated responses by phosphorylation of Ser843 provides further evidence that this region of the C-terminal domain is important for G-protein coupling (Pin et al., 1994). Interestingly, sequence alignment of the mGluR C-terminal intracellular domain revealed that, except for mGluR1, all other mGluR subtypes contain a single or two PKA consensus sites, suggesting that these receptors might be regulated by PKA (Fig.9). In particular, mGluR3, which has a high homology with mGluR2, has a similar PKA consensus site at Ser845. It is conceivable that this site on mGluR3 is also phosphorylated by PKA, resulting in uncoupling of the receptor from the G-protein in a manner analogous to that reported here for mGluR2.
Fig. 9.

Sequence alignment of the C-terminal intracellular domains of mGluRs. Serine residues involved in a potential PKA consensus site are boxed in black.
At present the precise physiological roles of PKA-induced inhibition of mGluR2 signaling are not entirely clear. However, it is interesting to note that several previous studies suggest that cAMP and activation of PKA can induce both acute and long-lasting increases in transmission at several glutamatergic synapses in the hippocampus, including the MPP (Nguyen and Kandel, 1996) and mossy fiber (Kamiya and Yamamoto 1997) synapses where PKA-induced inhibition of group II mGluR function has been observed. For instance, activation of adenylyl cyclase is critical for activation of long-term potentiation (LTP) at mossy fiber synapses. Furthermore, β-adrenergic receptor-mediated increases in cAMP induce a long-lasting increase in transmission at perforant path synapses in the dentate gyrus (Stanton and Sarvey, 1987; Dahl and Sarvey, 1990). It is interesting to note that agonists of group II mGluRs acutely depress excitatory transmission at both of these synapses and inhibit induction of LTP at the MPP synapses (Huang et al., 1997; Kilbride et al., 1998). Although we do not know the physiological setting in which mGluR2 is phosphorylated, it is conceivable that PKA-induced inhibition of mGluR2 signaling could play an important role in facilitating transmission and promoting induction of LTP. In future studies, it will be important to determine whether mGluR2 is phosphorylated on Ser843 in response to physiological stimuli that lead to activation of PKA.
Footnotes
This work was supported by National Institutes of Health (NIH)–National Institute of Neurological Diseases and Stroke (P.J.C.), NIH–NCRR (J.P.), NIH–National Heart, Lung, and Blood Institute (T.J.M.), and NARSAD grants (H.S., P.J.C.). We thank N. F. Ciliax and M. Ellington for their technical assistance. We thank also Dr. T. M. Wilkie for providing the original Gα15 construct.
Correspondence should be addressed to P. Jeffrey Conn, Department of Pharmacology, Emory University School of Medicine, 5015 Rollins Research Center, Atlanta, GA 30322-3090. E-mail:pconn@emory.edu.
REFERENCES
- 1.Abe T, Sugihara H, Nawa H, Shigemoto R, Mizuno N, Nakanishi S. Molecular characterization of a novel metabotropic glutamate receptor mGluR5 coupled to inositol phosphate/Ca2+ signal transduction. J Biol Chem. 1992;267:13361–13368. [PubMed] [Google Scholar]
- 2.Alagarsamy S, Marino MJ, Rouse ST, Gereau RW, Heinemann SF, Conn PJ. Activation of NMDA receptors reverses desensitization of mGluR5 in native and recombinant systems. Nat Neurosci. 1999;2:234–240. doi: 10.1038/6338. [DOI] [PubMed] [Google Scholar]
- 3.Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Brain Res Rev. 1999;29:83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- 4.Brown RE, Reymann KG. Metabotropic glutamate receptor agonists reduce paired-pulse depression in the dentate gyrus of the rat in vitro. Neurosci Lett. 1995;196:17–20. doi: 10.1016/0304-3940(95)11825-h. [DOI] [PubMed] [Google Scholar]
- 5.Carr SA, Huddleston MJ, Annan RS. Selective detection and sequencing of phosphopeptides at the femtomole level by mass spectrometry. Anal Biochem. 1996;239:180–192. doi: 10.1006/abio.1996.0313. [DOI] [PubMed] [Google Scholar]
- 6.Chambers TC, Pohl J, Raynor RL, Kuo JF. Identification of specific sites in human P-glycoprotein phosphorylated by protein kinase C. J Biol Chem. 1993;268:4592–4595. [PubMed] [Google Scholar]
- 7.Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 8.Dahl D, Sarvey JM. Beta-adrenergic agonist-induced long-lasting synaptic modifications in hippocampal dentate gyrus require activation of NMDA receptors, but not electrical activation of afferents. Brain Res. 1990;526:347–350. doi: 10.1016/0006-8993(90)91245-c. [DOI] [PubMed] [Google Scholar]
- 9.Dietrich D, Beck H, Kral T, Clusmann H, Elger CE, Schramm J. Metabotropic glutamate receptors modulate synaptic transmission in the perforant path: pharmacology and localization of two distinct receptors. Brain Res. 1997;767:220–227. doi: 10.1016/s0006-8993(97)00579-9. [DOI] [PubMed] [Google Scholar]
- 10.Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 11.Dixon D, Atwood HL. Adenylate cyclase system is essential for long-term facilitation at the crayfish neuromuscular junction. J Neurosci. 1989;9:4246–4252. doi: 10.1523/JNEUROSCI.09-12-04246.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flint AC, Connors BW. Two types of network oscillations in neocortex mediated by distinct glutamate receptor subtypes and neuronal populations. J Neurophysiol. 1996;75:951–957. doi: 10.1152/jn.1996.75.2.951. [DOI] [PubMed] [Google Scholar]
- 13.Gereau RW, Conn PJ. Potentiation of cAMP responses by metabotropic glutamate receptors depresses excitatory synaptic transmission by a kinase-independent mechanism. Neuron. 1994;12:1121–1129. doi: 10.1016/0896-6273(94)90319-0. [DOI] [PubMed] [Google Scholar]
- 14.Gereau RW, Heinemann SF. Role of protein kinase C phosphorylation in rapid desensitization of metabotropic glutamate receptor 5. Neuron. 1998;20:143–151. doi: 10.1016/s0896-6273(00)80442-0. [DOI] [PubMed] [Google Scholar]
- 15.Goldsmith BA, Abrams TW. Reversal of synaptic depression by serotonin at Aplysia sensory neuron synapses involves activation of adenylyl cyclase. Proc Natl Acad Sci USA. 1991;88:9021–9025. doi: 10.1073/pnas.88.20.9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gomeza J, Mary S, Brabet I, Parmentier ML, Restituito S, Bockaert J, Pin JP. Coupling of metabotropic glutamate receptors 2 and 4 to G alpha 15, G alpha 16, and chimeric G alpha q/i proteins: characterization of new antagonists. Mol Pharmacol. 1996;50:923–930. [PubMed] [Google Scholar]
- 17.Herrero I, Vazquez E, Miras-Portugal MT, Sanchez-Prieto J. Decrease in [Ca2+]c but not in cAMP mediates L-AP4 inhibition of glutamate release: PKC-mediated suppression of this inhibitory pathway. Eur J Neurosci. 1996;8:700–709. doi: 10.1111/j.1460-9568.1996.tb01255.x. [DOI] [PubMed] [Google Scholar]
- 18.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 19.Huang L, Killbride J, Rowan MJ, Anwyl R. Activation of mGluRII induces LTD via activation of protein kinase A and protein kinase C in the dentate gyrus of the hippocampus in vitro. Neuropharmacology. 1999;38:73–83. doi: 10.1016/s0028-3908(98)00168-3. [DOI] [PubMed] [Google Scholar]
- 20.Huang LQ, Rowan MJ, Anwyl R. mGluR II agonist inhibition of LTP induction, and mGluR II antagonist inhibition of LTD induction, in the dentate gyrus in vitro. NeuroReport. 1997;8:687–693. doi: 10.1097/00001756-199702100-00022. [DOI] [PubMed] [Google Scholar]
- 21.Kahle JS, Cotman CW. Synaptic reorganization in the hippocampus: an electrophysiological analysis. Ann NY Acad Sci. 1993;702:61–74. doi: 10.1111/j.1749-6632.1993.tb17242.x. [DOI] [PubMed] [Google Scholar]
- 22.Kamiya H, Yamamoto C. Phorbol ester and forskolin suppress the presynaptic inhibitory action of group-II metabotropic glutamate receptor at rat hippocampal mossy fibre synapse. Neuroscience. 1997;80:89–94. doi: 10.1016/s0306-4522(97)00098-5. [DOI] [PubMed] [Google Scholar]
- 23.Kawabata S, Tsutsumi R, Kohara A, Yamaguchi T, Nakanishi S, Okada M. Control of calcium oscillations by phosphorylation of metabotropic glutamate receptors. Nature. 1996;383:89–92. doi: 10.1038/383089a0. [DOI] [PubMed] [Google Scholar]
- 24.Kilbride J, Huang LQ, Rowan MJ, Anwyl R. Presynaptic inhibitory action of the group II metabotropic glutamate receptor agonists, LY354740 and DCG-IV. Eur J Pharmacol. 1998;356:149–157. doi: 10.1016/s0014-2999(98)00526-3. [DOI] [PubMed] [Google Scholar]
- 25.Kowal D, Hsiao CL, Ge A, Wardwell-Swanson J, Ghosh K, Tasse R. A [35S]GTP-gamma-S binding assessment of metabotropic glutamate receptor standards in Chinese hamster ovary cell lines expressing the human metabotropic receptor subtypes 2 and 4. Neuropharmacology. 1998;37:179–187. doi: 10.1016/s0028-3908(98)00011-2. [DOI] [PubMed] [Google Scholar]
- 26.Limbird LE. Receptors linked to inhibition of adenylate cyclase: additional signaling mechanisms. FASEB J. 1988;2:2686–2695. doi: 10.1096/fasebj.2.11.2840317. [DOI] [PubMed] [Google Scholar]
- 27.Maccaferri G, Toth K, McBain CJ. Target-specific expression of presynaptic mossy fiber plasticity. Science. 1998;279:1368–1370. doi: 10.1126/science.279.5355.1368. [DOI] [PubMed] [Google Scholar]
- 28.Macek TA, Winder DG, Gereau RW, Ladd CO, Conn PJ. Differential involvement of group II and group III mGluRs as autoreceptors at lateral and medial perforant path synapses. J Neurophysiol. 1996;76:3798–3806. doi: 10.1152/jn.1996.76.6.3798. [DOI] [PubMed] [Google Scholar]
- 29.Macek TA, Schaffhauser H, Conn PJ. Protein kinase C and A3 adenosine receptor activation inhibit presynaptic metabotropic glutamate receptor (mGluR) function and uncouple mGluRs from GTP-binding proteins. J Neurosci. 1998;18:6138–6146. doi: 10.1523/JNEUROSCI.18-16-06138.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Macek TA, Schaffhauser H, Conn PJ. Activation of PKC disrupts presynaptic inhibition by group II and group III metabotropic glutamate receptors and uncouples the receptor from GTP-binding proteins. Ann NY Acad Sci. 1999;868:554–557. doi: 10.1111/j.1749-6632.1999.tb11327.x. [DOI] [PubMed] [Google Scholar]
- 31.Meyer HE, Hoffmann-Posorske E, Heilmeyer LM. Determination and location of phosphoserine in proteins and peptides by conversion to S-ethylcysteine. Methods Enzymol. 1991;201:169–185. doi: 10.1016/0076-6879(91)01016-u. [DOI] [PubMed] [Google Scholar]
- 32.Miller AD, Miller DG, Garcia JV, Lynch CM. Use of retroviral vectors for gene transfer and expression. Methods Enzymol. 1993;217:581–599. doi: 10.1016/0076-6879(93)17090-r. [DOI] [PubMed] [Google Scholar]
- 33.Nakahara K, Okada M, Nakanishi S. The metabotropic glutamate receptor mGluR5 induces calcium oscillations in cultured astrocytes via protein kinase C phosphorylation. J Neurochem. 1997;69:1467–1475. doi: 10.1046/j.1471-4159.1997.69041467.x. [DOI] [PubMed] [Google Scholar]
- 34.Nakajima Y, Yamamoto T, Nakayama T, Nakanishi S. A relationship between protein kinase C phosphorylation and calmodulin binding to the metabotropic glutamate receptor subtype 7. J Biol Chem. 1999;274:27573–27577. doi: 10.1074/jbc.274.39.27573. [DOI] [PubMed] [Google Scholar]
- 35.Neki A, Ohishi H, Kaneko T, Shigemoto R, Nakanishi S, Mizuno N. Pre- and postsynaptic localization of a metabotropic glutamate receptor, mGluR2, in the rat brain: an immunohistochemical study with a monoclonal antibody. Neurosci Lett. 1996;202:197–200. doi: 10.1016/0304-3940(95)12248-6. [DOI] [PubMed] [Google Scholar]
- 36.Nguyen PV, Kandel ER. A macromolecular synthesis-dependent late phase of long-term potentiation requiring cAMP in the medial perforant pathway of rat hippocampal slices. J Neurosci. 1996;16:3189–3198. doi: 10.1523/JNEUROSCI.16-10-03189.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Connor V, El Far O, Bofill-Cardona E, Nanoff C, Freissmuth M, Karschin A, Airas JM, Betz H, Boehm S. Calmodulin dependence of presynaptic metabotropic glutamate receptor signaling. Science. 1999;286:1180–1184. doi: 10.1126/science.286.5442.1180. [DOI] [PubMed] [Google Scholar]
- 38.Peavy RD, Conn PJ. Phosphorylation of mitogen-activated protein kinase in cultured rat cortical glia by stimulation of metabotropic glutamate receptors. J Neurochem. 1998;71:603–612. doi: 10.1046/j.1471-4159.1998.71020603.x. [DOI] [PubMed] [Google Scholar]
- 39.Petralia RS, Wang YX, Niedzielski AS, Wenthold RJ. The metabotropic glutamate receptors, mGluR2 and mGluR3, show unique postsynaptic, presynaptic and glial localizations. Neuroscience. 1996;71:949–976. doi: 10.1016/0306-4522(95)00533-1. [DOI] [PubMed] [Google Scholar]
- 40.Pin JP, Joly C, Heinemann SF, Bockaert J. Domains involved in the specificity of G protein activation in phospholipase C-coupled metabotropic glutamate receptors. EMBO J. 1994;13:342–348. doi: 10.1002/j.1460-2075.1994.tb06267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pin JP, De Colle C, Bessis AS, Acher F. New perspectives for the development of selective metabotropic glutamate receptor ligands. Eur J Pharmacol. 1999;375:277–294. doi: 10.1016/s0014-2999(99)00258-7. [DOI] [PubMed] [Google Scholar]
- 42.Prezeau L, Carrette J, Helpap B, Curry K, Pin JP, Bockaert J. Pharmacological characterization of metabotropic glutamate receptors in several types of brain cells in primary cultures. Mol Pharmacol. 1994;45:570–577. [PubMed] [Google Scholar]
- 43.Prezeau L, Gomeza J, Ahern S, Mary S, Galvez T, Bockaert J, Pin JP. Changes in the carboxyl-terminal domain of metabotropic glutamate receptor 1 by alternative splicing generate receptors with differing agonist-independent activity. Mol Pharmacol. 1996;49:422–429. [PubMed] [Google Scholar]
- 44.Rauca C, Henrich-Noack P, Schafer K, Hollt V, Reymann KG. (S)-4C3HPG reduces infarct size after focal cerebral ischemia. Neuropharmacology. 1998;37:1649–1652. doi: 10.1016/s0028-3908(98)00146-4. [DOI] [PubMed] [Google Scholar]
- 45.Schoepp DD, Johnson BG, Monn JA. Inhibition of cyclic AMP formation by a selective metabotropic glutamate receptor agonist. J Neurochem. 1992;58:1184–1186. doi: 10.1111/j.1471-4159.1992.tb09381.x. [DOI] [PubMed] [Google Scholar]
- 46.Schoepp DD, Johnson BG, Salhoff CR, Valli MJ, Desai MA, Burnett JP, Mayne NG, Monn JA. Selective inhibition of forskolin-stimulated cyclic AMP formation in rat hippocampus by a novel mGluR agonist, 2R,4R-4-aminopyrrolidine-2,4-dicarboxylate. Neuropharmacology. 1995;34:843–850. doi: 10.1016/0028-3908(95)00061-a. [DOI] [PubMed] [Google Scholar]
- 47.Schoppa NE, Westbrook GL. Modulation of mEPSCs in olfactory bulb mitral cells by metabotropic glutamate receptors. J Neurophysiol. 1997;78:1468–1475. doi: 10.1152/jn.1997.78.3.1468. [DOI] [PubMed] [Google Scholar]
- 48.Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M, Flor PJ, Neki A, Abe T, Nakanishi S, Mizuno N. Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J Neurosci. 1997;17:7503–7522. doi: 10.1523/JNEUROSCI.17-19-07503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stanton PK, Sarvey JM. Norepinephrine regulates long-term potentiation of both the population spike and dendritic EPSP in hippocampal dentate gyrus. Brain Res Bull. 1987;18:115–119. doi: 10.1016/0361-9230(87)90039-6. [DOI] [PubMed] [Google Scholar]
- 50.Swartz KJ, Merritt A, Bean BP, Lovinger DM. Protein kinase C modulates glutamate receptor inhibition of Ca2+ channels and synaptic transmission. Nature. 1993;361:165–168. doi: 10.1038/361165a0. [DOI] [PubMed] [Google Scholar]
- 51.Tanabe Y, Masu M, Ishii T, Shigemoto R, Nakanishi S. A family of metabotropic glutamate receptors. Neuron. 1992;8:169–179. doi: 10.1016/0896-6273(92)90118-w. [DOI] [PubMed] [Google Scholar]
- 52.Tyler EC, Lovinger DM. Metabotropic glutamate receptor modulation of synaptic transmission in corticostriatal co-cultures: role of calcium influx. Neuropharmacology. 1995;34:939–952. doi: 10.1016/0028-3908(95)00066-f. [DOI] [PubMed] [Google Scholar]
- 53.Wang X, Murphy TJ. Inhibition of cyclic AMP-dependent kinase by expression of a protein kinase inhibitor/enhanced green fluorescent fusion protein attenuates angiotensin II-induced type 1 AT1 receptor mRNA down-regulation in vascular smooth muscle cells. Mol Pharmacol. 1998;54:514–524. doi: 10.1124/mol.54.3.514. [DOI] [PubMed] [Google Scholar]
- 54.Wilkie TM, Scherle PA, Strathmann MP, Slepak VZ, Simon MI. Characterization of G-protein alpha subunits in the Gq class: expression in murine tissues and in stromal and hematopoietic cell lines. Proc Natl Acad Sci USA. 1991;88:10049–10053. doi: 10.1073/pnas.88.22.10049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang X, Herring CJ, Romano PR, Szczepanowska J, Brzeska H, Hinnebusch AG, Qin J. Identification of phosphorylation sites in proteins separated by polyacrylamide gel electrophoresis. Anal Chem. 1998;70:2050–2059. doi: 10.1021/ac971207m. [DOI] [PubMed] [Google Scholar]