

Abstract
The protein kinase C family of enzymes has been implicated in synaptic plasticity and memory in a wide range of animal species, but to date little information has been available concerning specific roles for individual isoforms of this category of kinases. To investigate the role of the β isoform of PKC in mammalian learning, we characterized mice deficient in the PKCβ gene using anatomical, biochemical, physiological, and behavioral approaches. In our studies we observed that PKCβ was predominantly expressed in the neocortex, in area CA1 of the hippocampus, and in the basolateral nucleus of the amygdala. Mice deficient in PKCβ showed normal brain anatomy and normal hippocampal synaptic transmission, paired pulse facilitation, and long-term potentiation and normal sensory and motor responses. The PKCβ knock-out animals exhibited a loss of learning, however; they suffered deficits in both cued and contextual fear conditioning. The PKC expression pattern and behavioral phenotype in the PKCβ knock-out animals indicate a critical role for the β isoform of PKC in learning-related signal transduction mechanisms, potentially in the basolateral nucleus of the amygdala.
Keywords: PKC, Pavlovian fear conditioning, hippocampus, amygdala, knock-out, mice
The calcium- and phospholipid-dependent protein kinases (PKCs) are pluripotent regulators of synaptic transmission and neuronal function. This family of enzymes regulates neurotransmitter release (Malenka et al., 1986;Nicholls, 1998; Stevens and Sullivan, 1998), controls membrane electrical properties (Hoffman and Johnston, 1998; Manseau et al., 1998), modulates neurotransmitter receptor function (Roche et al., 1996; Macek et al., 1998; Suen et al., 1998), and regulates gene expression in mature neurons. (Roberson et al., 1999). Regulation of PKC activity has been implicated in synaptic plasticity and learning and memory in Aplysia (Sacktor et al., 1988; Byrne and Kandel, 1996; Manseau et al., 1998), Hermissenda (Farley and Schuman, 1991; Yamoah and Crow, 1996), Drosophila (Choi et al., 1991; Mihalek et al., 1997), and honeybee (Muller, 1999) and in imprinting in the chick (Sheu et al., 1993). In addition, PKCs have been implicated in learning and memory in mammalian model systems in studies of eye blink conditioning (Van der Zee et al., 1997), spatial learning (Paylor et al., 1991, 1992; Colombo et al., 1997), conditioned taste aversion (Yasoshima and Yamamoto, 1997), and conditioned avoidance (Jerusalinsky et al., 1994).
In mammals the PKC enzyme family is quite heterogeneous and comprises 11 known isozymes divided into three major subsets: conventional [α, βI, βII, and γ (Coussens et al., 1986; Parker et al., 1986)], novel [δ, ε, η, and θ (Ono et al., 1987; Osada et al., 1990,1992; Saido et al., 1992)] and atypical [λ and ζ (Ogita et al., 1992)]. Each isoform is encoded by a separate gene, with the exception of the βI and βII isoforms, which are splice variants. Various isoforms exhibit cell- and tissue-specific expression (Huang et al., 1987; Oehrlein et al., 1998), and each isoform subset is subject to distinct control mechanisms (Conn and Sweatt, 1993; Dekker and Parker, 1994). The conventional isoforms are regulated by calcium in concert with diacylglycerol and membrane phospholipid, whereas the novel and atypical classes are structurally homologous but can be regulated independent of calcium.
Although a role for the PKC enzyme family is broadly established for various forms of mammalian learning, the specific contributions of PKC subtypes to learning and memory are not well known. A knock-out of the brain-specific, γ isoform of PKC resulted in modest effects on memory (Abeliovich et al., 1993), suggesting that other PKC isoforms are involved in mammalian learning and memory. In the present studies we sought to increase our understanding of the roles of PKCβ in synaptic plasticity and memory by investigating a mouse line, generated by homologous recombination, in which the gene coding for the two β isoforms of PKC was replaced by β-galactosidase. We observed pronounced expression of PKCβ, specifically PKCβII, in area CA1 of hippocampus and the basolateral nucleus of the amygdala. Surprisingly PKC-deficient animals did not exhibit deficits in hippocampal synaptic transmission or long-term potentiation (LTP). However, deletion of the PKCβ gene resulted in defects in two amygdala-dependent learning tasks, cued and contextual fear conditioning. Thus, our data suggest an important role for the βII isoform of PKC in the synaptic plasticity underlying amygdala-dependent associative learning.
MATERIALS AND METHODS
Production of PKCβ knock-outs and controls.Production of PKCβ-deficient mice and genotypic determination were performed as described previously (Leitges et al., 1996). Initially, mice heterozygous for the PKCβ gene deletion were bred to C57BL/6 wild-type (wt) animals. Heterozygotes were then backcrossed either 8 or 10 generations with C57BL/6 wt, and our experiments were performed using two different groups of animals. In the first series of experiments, electrophysiology, biochemical, and behavioral analyses were performed with homozygote PKCβ knock-outs and littermate controls obtained from PKCβ heterozygote breeding (8 backcrosses). In the second set of experiments we replicated our fear conditioning results using homozygote knock-out animals obtained from 10 heterozygote backcrosses by comparing them with age-matched controls from the C57BL/6 wild-type line used for the backcrosses. The data obtained from the two different sets of animals were indistinguishable, and the results were pooled for some experiments. Overall, controls used in the two-trial fear conditioning consisted of 17 wild-type littermate controls and 6 age-matched wild-type controls. The analysis of fear conditioning data revealed no statistically significant differences between littermate and age-matched controls. Control experiments involving overtraining (five-trial fear conditioning) and subsequent cue and context trial sessions used knock-outs from 10 backcrosses and age-matched controls exclusively.
Immunohistochemistry. Adult mice were anesthetized intraperitoneally with ketamine and xylazine and perfused transcardially with 10 ml of 0.9% NaCl followed by 50 ml of 3% paraformaldehyde and 1% gluteraldehyde in PBS, pH 7.4. The brains were then cryoprotected in 30% sucrose for 24–48 hr at 4°C, followed by freezing and mounting for cryostat sectioning. Sections (20 μm) were cut and immediately thaw mounted on Plus slides. For β-gal staining, sections were incubated in a solution containing 1 mg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal), 4 mm potassium ferrocyanide, 4 mm potassium ferricyanide, and 2 mm magnesium chloride in PBS overnight in a humidified chamber at 4°C. For isoform-specific antibody immunohistochemistry, sections were incubated in 0.3% H2O2 in methanol for 30 min at room temperature, washed in PBS containing 0.2% Triton X-100 (PBST), and blocked in PBST containing 5% normal goat serum. Blocking media were blotted from the slides, and sections were then incubated with a 1:1000 dilution of primary antibody overnight in a humidified chamber at 4° or 37°C. Sections were washed again in PBST and incubated with a 1:200 dilution of goat anti-rabbit biotinylated secondary antibody for 30 min at room temperature (ImmunoPure ABC peroxidase rabbit IgG staining kit; Pierce, Rockford, IL). Sections were washed again with PBST and then incubated with a 1:50 dilution of ABC reagent for 30 min at room temperature. Sections were again washed in PBST, and staining was revealed with metal-enhanced DAB. Sections were then rinsed, cleared, and coverslipped using a xylene-based medium.
Western blotting. The animals were killed by decapitation. The brains were immediately removed and perfused in ice-cold saline (in mm: 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 d-glucose, 2 CaCl2, and 1 MgCl2, saturated with 95% O2 and 5% CO2, pH 7.4). Hippocampi were dissected and then homogenized in 2–3 ml of buffer (20 mm Tris-HCl, pH 7.5, 1 mm EGTA, 1 mm EDTA, 25 μg/ml aprotinin, 25 μg/ml leupeptin, 1 mmNa4P2O7, 500 μm phenylmethylsulfonyl fluoride, 4 mmpara-nitrophenylphosphate, and 1 mmsodium orthovanadate). Sample buffer was immediately added to the homogenate, and the samples were boiled at 100°C for 10–15 min. Samples were electrophoresed on a 10% SDS-polyacrylamide gel, blotted electrophoretically to Immobilon-P, and blocked in TTBS buffer (50 mm Tris-HCl, pH 7.5, 150 mmNaCl, and 0.05% Tween 20). Because the amygdala is not ideal for this form of biochemical analysis because of its cellular heterogeneity, lack of well circumscribed anatomy, and difficulty of rapid dissection, we chose to assay the hippocampus for our studies to investigate possible compensatory biochemical changes in PKCβ-deficient mice.
Western blots were blocked in TTBS with 3% BSA or 5% dried milk and 1 μm microcysteine. Blots for total PKC amounts were performed as described previously (Chen et al., 1997). All blots were incubated with a secondary antibody conjugated to horseradish peroxidase and developed using the enhanced chemiluminescence method (Amersham Pharmacia Biotech, Arlington Heights, IL). Total protein amounts for each kinase were determined by densitometry and normalized relative to a Lowry assay for each homogenate (Lowry et al., 1951). The density of the bands was quantified by a StudioScan desktop scanner using NIH Image software. Western blots were developed to be linear in the range used for densitometry.
Fear conditioning. Mice were housed on a 12 hr light/dark schedule. All experiments were performed in compliance with the Baylor College of Medicine Institutional Animal Care and Use Committee and national regulations and policies. For cue and contextual fear conditioning, animals were placed in the fear-conditioning apparatus for 2 min, and then a 30 sec acoustic conditioned stimulus (CS; white noise) was delivered. During the last 2 sec of the tone, a 0.5 mA shock [unconditioned stimulus (US)] was applied to the floor grid. This protocol was repeated twice with 2 min between pairings. The stimulus strength and number of training pairs were chosen on the basis of pilot experiments to optimize learning. To assess for contextual learning, the animals were placed back into the training context 24 hr after training and scored for freezing for 5 min. To assess for cue learning, the animals were placed in a different context (novel odor, cage floor, and visual cues) 24 hr after training. Baseline behavior was measured for 3 min in the novel context, and then the acoustic CS was presented for 3 min. Learning was assessed by measuring freezing behavior (i.e., motionless position) every 5 sec. The scorer of the behavioral experiments was blind in reference to animal genotype. One day after the tests for contextual and cue learning, a subset of the knock-out mice and control mice was retrained using a paradigm of five CS–US pairings. To assess short-term cue learning, the animals were placed in a different context 1–2 hr after the completion of the retraining session, and freezing was assessed in response to representation of the acoustic CS. Long-term contextual and cue learning were tested 24 hr later as described above.
Behavioral assessments. General activity levels were measured with an open field task as described previously (Paylor et al., 1998). Animals were placed in the open field (40 × 40 × 30 cm) chamber for 30 min in standard room-lighting conditions. Activity in the open field was monitored by 16 photoreceptor beams on each side of the chamber and analyzed by a computer-operated Digiscan optical animal activity system. The accelerating rotarod test was used to assess overall balance and motor coordination. Passive avoidance was chosen as a control paradigm because of its similarity to fear conditioning in that both have the ability to produce robust associative learning using an aversive stimulus. Nociception was assayed by placing the animals on a 55°C hotplate, and latency to lick the hind paw was measured.
Hippocampal slice preparation. Hippocampal slice preparation and electrophysiology were performed as described previously (Roberson and Sweatt, 1996). The Schaffer collateral pathway was stimulated, and field recordings were made in stratum radiatum of the CA1 region. The initial slope of the population EPSP (pEPSP) was measured. Test stimulation (0.05 Hz) was delivered at a stimulus intensity 30–40% of the maximum pEPSP. Tetanic stimulation consisted of two 100 Hz, 1-sec-long tetani, separated by 20 sec, at test stimulation intensity.
Data analysis. Statistical analysis was conducted by one-way ANOVA followed by paired comparisons using the Tukey test or Student'st test. All values are mean ± SEM; *p< 0.05; **p < 0.01; and ***p < 0.001.
RESULTS
PKCβ expression patterns
Insights into the functional roles of specific signal-transducing proteins can often be obtained through examination of their expression patterns in the CNS (Huang et al., 1987; Hosoda et al., 1989; Saito et al., 1989; Xia et al., 1991). To facilitate this type of analysis we used homologous recombination methods to construct a mouse line wherein the second exon of the PKCβ gene was substituted with a β-galactosidase (β-gal) gene (Leitges et al., 1996). This manipulation simultaneously eliminated PKCβ expression and provided a distinct molecular marker to allow determination of the expression pattern coded for by the regulatory elements upstream of the PKCβ gene. β-gal expression can be tracked by provision of the substrate X-gal, which causes production of a blue marker at sites of β-gal expression in the CNS of our mouse line.
Incubation in X-gal of brains obtained from our PKCβ mouse line revealed an interesting expression pattern (Fig.1A,B). β-Gal expression was pronounced in several brain areas known to be involved in learning and memory, including the neocortex, cerebellum, area CA1 of hippocampus, and basolateral nucleus of the amygdala. This gene expression pattern is somewhat reminiscent of that of the α subunit of Ca2+ calmodulin-dependent protein kinase II (CaMKII), the upstream promoter region of which has been used to selectively eliminate gene expression in cortical areas (Tsien et al., 1996). Overall, the expression pattern suggests the hypothesis that PKCβ is involved in spatial or associative learning in rodents.
Fig. 1.
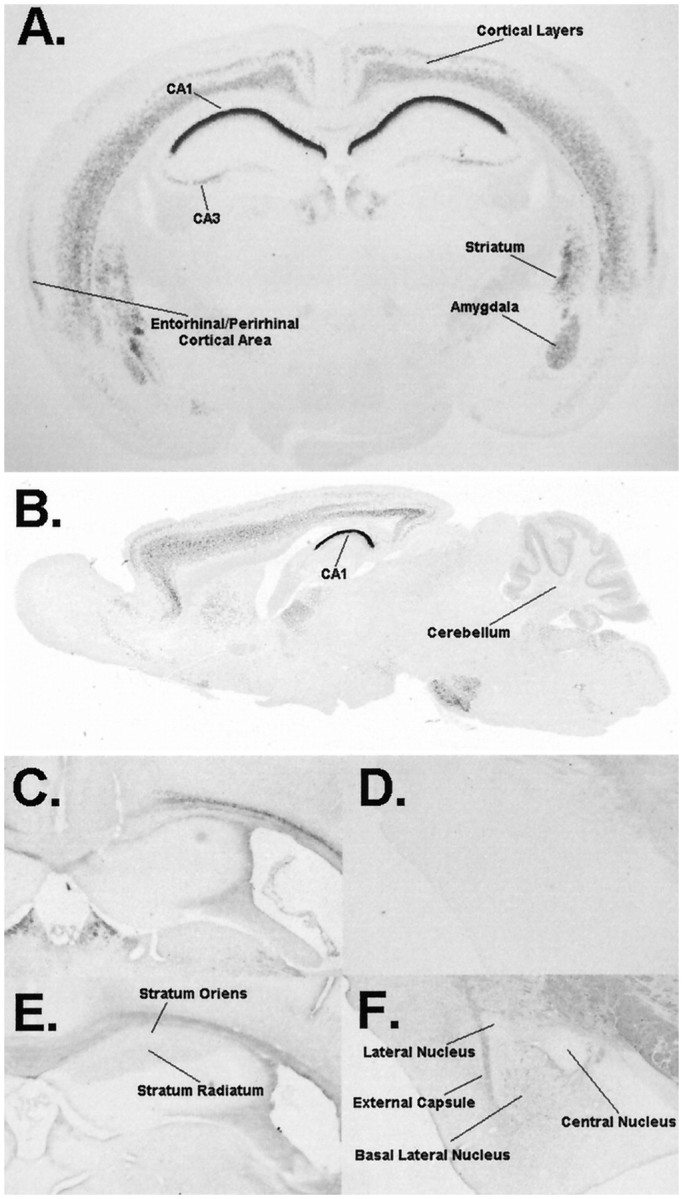
PKCβ distribution in the brain. X-gal staining (A, B) shows prominent staining in CA1 of the hippocampus with mild staining in CA3. Moderate staining is seen in both the lateral and basolateral nuclei of the amygdala, striatum, somatosensory cortex, and cerebellar and entorhinal/perirhinal cortical areas. Immunohistochemistry using antibodies against the βI isoform of PKC (C, D) shows essentially no staining in the hippocampus (C) or amygdala (D). Immunohistochemistry using antibodies against the βII isoform (E, F) shows staining in the stratum oriens and stratum radiatum of CA1 of the hippocampus but not in the stratum pyramidali (E). The amygdala (F) shows staining of fibers in the basolateral nucleus. The structures represented in D–Fwere obtained from a single mouse and processed in parallel.
We infer from the β-gal expression pattern in our mice that the PKCβ gene is normally expressed in the brain areas highlighted by β-gal staining, but this promoter-directed approach has two limitations. First, the β-gal expression pattern may not accurately reflect the expression pattern of native PKC expressed from the intact PKCβ-encoding gene. Second, the β-gal expression pattern does not distinguish between expression of the βI versus βII splice variations, which may have distinct regulatory mechanisms. To address these two limitations, we used immunohistochemistry procedures to localize PKCβI and βII in the CNS of the parent mouse line C57BL/6. Our immunohistochemistry results were quite consistent with the β-gal expression pattern in our knock-out animals, demonstrating a similar pattern of PKCβ expression in the cortex, hippocampus, and amygdala (Fig. 1). Interestingly, the PKCβII splice variant appears to be exclusively expressed in the hippocampus and amygdala (Fig.1E,F) compared with the PKCβI splice variant, which shows essentially no immunoreactivity in these regions (Fig. 1C,D). This observation indicates a high level of control of the splice variant production machinery in these cell types and suggests that any hippocampus- or amygdala-dependent defects exhibited by PKCβ-deficient mice are attributable to loss of the βII isoform of PKC.
Having observed an interesting pattern of PKCβ expression in the mouse CNS, we sought to determine the effects of loss of PKCβ through characterizing our PKCβ-deficient mouse line. The results shown in Figure 1, along with additional histological staining (data not shown), indicated that loss of PKCβ did not result in abnormal gross anatomy of the CNS. These results suggest that PKCβ is not necessary for the normal gross anatomical development of the CNS.
We therefore sought to determine whether loss of PKCβ resulted in more subtle, functional deficits that could not be observed at the anatomical level. Toward this end, we undertook characterization of the synaptic physiology of PKCβ-deficient mice. We chose to undertake these studies using the hippocampal slice preparation in vitro, because this is the best-characterized system for studying synaptic function in the mouse. For these experiments we used extracellular recording of the Schaffer collateral inputs into area CA1, not only because this is a standard technique for studies of this sort but also because of the pronounced expression of PKCβ in the postsynaptic pyramidal neurons in this locus (see Fig. 1). In addition, there is modest but detectable expression of β-galactosidase in the presynaptic CA3 pyramidal neurons of this synapse in our transgenic mouse line. Thus this synapse seemed a likely site to detect derangements of synaptic function if they occurred with knock-out of the PKCβ gene.
Electrophysiological assessments
Loss of the β isoform of PKC appeared to have no deleterious effect on baseline synaptic transmission at Schaffer collateral synapses, because input–output functions for stimulation of area CA1 were not different in control and knock-out mice (Fig.2A). If anything, in these studies the PKCβ knock-outs exhibited a slight increase in the fiber volley amplitude–EPSP slope relationship, suggesting a slight augmentation of synaptic transmission; however, this effect was not statistically significant (p = 0.33). Paired pulse facilitation (PPF) is a form of short-term synaptic plasticity that is commonly held to be caused by residual calcium augmenting neurotransmitter release presynaptically. Because PKCβ is a calcium-dependent form of PKC, we determined whether PPF was attenuated in the PKCβ knock-out mice. As with baseline synaptic transmission, PPF was normal in the knock-outs, indicating that the β isoform of PKC is not a component of the machinery underlying PPF at Schaffer collateral synapses (Fig. 2B).
Fig. 2.

Electrophysiological responses at Schaffer collateral synapses in area CA1 of hippocampus. A, Loss of PKCβ had no effect on baseline synaptic transmission in stratum radiatum of the CA1 region of the hippocampus measured at 25°C in PKCβ-deficient mice (○) or wild-type mice (●).Inset, Representative traces (mean of 6 successive field EPSPs) of previous baseline synaptic transmission. Calibration: 2 mV, 4 msec. B, Paired pulse facilitation was also unaffected in PKCβ-deficient mice.
A large number of studies using a variety of biochemical and pharmacological approaches have demonstrated a necessity for PKC activation in the induction of NMDA receptor-dependent LTP in area CA1 (Malinow et al., 1988; Reymann et al., 1988; Malinow et al., 1989; Wang and Feng, 1992; Hvalby et al., 1994). In particular, elegant studies using microelectrode techniques have shown both sufficiency and necessity of postsynaptic PKC in the induction of LTP at Schaffer collateral synapses (Hu et al., 1987; Malinow et al., 1989; Wang and Feng, 1992; Hvalby et al., 1994). Given the prominent expression of PKCβ in these cells (Fig. 1), we sought to determine whether the β isoform of PKC contributed to LTP induction or expression in area CA1.
In our first series of experiments LTP was induced with two 1 sec, 100 Hz tetani separated by 20 sec, and synaptic efficacy was monitored for 90 min. This LTP induction protocol gives a fairly modest LTP that is useful as a sensitive indicator of differences in the capacity for LTP in area CA1. Furthermore, using this procedure we can monitor the transient post-tetanic potentiation (PTP) produced immediately after high-frequency tetanus (Fig.3A). We observed no difference in either PTP or LTP in the PKCβ-deficient mice using this tetanic stimulation protocol. Furthermore, when the LTP-inducing protocol was followed immediately by a depotentiation protocol (5 Hz stimulation for 5 min; Fig. 3B) that reduces subsequent potentiation, PKCβ-deficient mice were indistinguishable from controls.
Fig. 3.
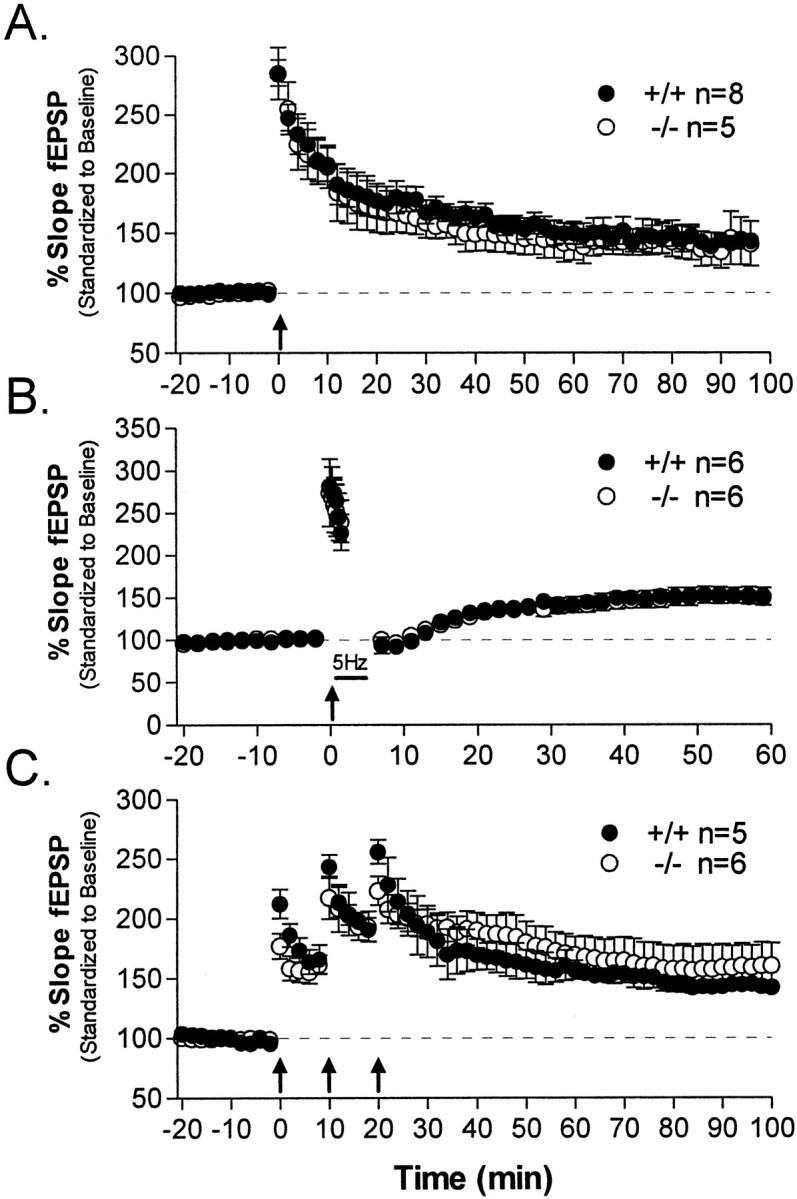
Hippocampal LTP. In the following experiments, hippocampal slices obtained from PKCβ-deficient mice (○) or wild-type mice (●) were given an LTP-inducing stimulus (arrows) delivered after stable baseline responses were recorded for 20 min. Each set of tetani consisted of two trains of 100 Hz stimulation for 1 sec, separated by 20 sec. A, Mutant hippocampal slices showed normal LTP compared with wild types after a modest LTP-inducing protocol consisting of a single set of tetani while maintaining slices at 25°C. B, The extent of depotentiation in PKCβ-deficient mice was equal to that of wild types determined from low-frequency 5 Hz stimulation (5 min) after a single set of tetani at 25°C. C, No differences in PKCβ mutant LTP were observed in experiments using a more robust LTP-inducing protocol consisting of three sets of 100 Hz tetani delivered 10 min apart while maintaining slices at an elevated temperature of 32°C.
A tetanic stimulation protocol using repetitive trains of tetanic stimulation at higher temperatures gives a more robust and longer-lasting LTP that uses different signal transduction mechanisms for its induction (Chetkovich et al., 1993). We also determined whether this stimulus protocol revealed a PKCβ-requiring component of synaptic potentiation and found that, similar to the results above, PTP and LTP were not significantly different in PKCβ knock-out mice (Fig.3C).
Overall, our data indicate that LTP was unimpaired in hippocampal slices from PKCβ-deficient animals. Post-tetanic potentiation was normal, as was short-term potentiation; these findings suggest that both short-term presynaptic plasticity and NMDA receptor-mediated potentiation of synaptic transmission in area CA1 are unaffected by loss of PKCβ. More specifically, our results indicate that the widely reported dependency of hippocampal LTP on PKC is not a manifestation of block of the activity of PKCβ. An additional implication of these results is that any behavioral effects of a knock-out of PKCβ cannot be attributed to nonspecific effects on baseline synaptic transmission or NMDA receptor function in hippocampal area CA1.
One caveat to our conclusion that PKCβ is not required for LTP at Schaffer collateral synapses is that loss of PKCβ might have elicited acute or developmental compensatory changes that alleviate the need for PKCβ in LTP. We obtained two independent lines of evidence that suggest that this alternative scenario is not the case. First, in Western blotting studies of PKCβ-deficient mice we found no alteration in the level of expression of other members of the PKC enzyme family (Fig.4A). Thus compensatory up-regulation of other classical isoforms of PKC did not occur in the PKCβ-deficient mouse line. Moreover, the PKCβ knock-out mice do exhibit a deficiency in PKC-mediated synaptic modulation at Schaffer collateral synapses. We observed that phorbol ester-induced synaptic potentiation in area CA1 is significantly diminished in our PKCβ-deficient mice (Fig. 4B). This control experiment demonstrates that loss of PKCβ does cause detectable alterations in physiological modulation at Schaffer collateral synapses, which have not been compensated for in the PKCβ-deficient mice. Overall these biochemical and physiological control experiments solidify our conclusion that the β isoform of PKC is not necessary for tetanus-evoked LTP using standard LTP-inducing protocols.
Fig. 4.

Lack of compensatory changes in PKC in PKCβ-deficient mice. A, Top, Representative Western analysis of PKCβ, PKCα, and PKCγ protein from hippocampal homogenates of PKCβ knock-outs, control mice, or purified PKCβII protein. Bottom, The percent change in protein kinase C expression in PKCβ knock-out mice versus control mice is shown for each of the Ca2+-dependent protein kinase C isoforms. All PKC densitometric measurements were normalized to corresponding total protein amounts obtained from whole hippocampal homogenates. No statistically significant changes in PKC expression levels were observed for PKCα (n = 4) or PKCγ (n = 4). N/D, Not detectable. B, Phorbol ester-induced potentiation of synaptic transmission was reduced in PKCβ-deficient mice. Results shown are the percent increase in baseline synaptic transmission (relative to predrug application at time 0) produced by a 25 min application of 5 μm phorbol 12, 13 diacetate (PDA) in control (+/+; n = 4) and PKCβ-deficient (−/−; n = 7) hippocampal slices. The difference in control versus knock-out mice is statistically significant (p < 0.01) at all times from 25 min after drug application and is consistent with the biochemical data indicating a lack of other PKC isoform compensatory changes in PKCβ-deficient mice.
Behavioral assessments
Our histochemical studies indicate that the β isoform of PKC exhibits an intriguing pattern of expression in the adult CNS; does loss of PKCβ lead to an interesting behavioral phenotype? In our next series of experiments we sought to determine whether loss of PKCβ led to altered behavior in our knock-out mouse line, with a particular emphasis on behavioral models of learning and memory. Much progress has been made recently in developing standardized procedures for behavioral screening in mice (Crawley and Paylor, 1997). A fairly typical behavioral screen includes evaluation of open-field behavior, rotarod testing, prepulse inhibition, passive avoidance, and cued and contextual fear conditioning. This battery of tests allows evaluation of a variety of sensory responses, including hearing and vision, general activity, reflexes and motor coordination, motor learning, and associative learning.
Contextual fear conditioning and cue learning
We first investigated fear conditioning in our knock-out mice because this conditioning paradigm elicits robust associative learning that involves the hippocampus and amygdala, brain areas we identified as expressing the βII isoform of PKC at high levels. For these experiments, an aversive stimulus (in this case, a mild foot shock; US) was paired two times with an auditory CS (white noise) within a novel environment. When tested 24 hr after training, control mice exhibited marked fear, measured by freezing behavior, in response to representation of either the context (contextual fear conditioning) or the acoustic CS delivered in a different context (cued fear conditioning; Fig. 5). Interestingly, in two separate experiments mice deficient in the β isoform of PKC exhibited significant deficits in fear conditioning in both the cued and contextual variants. This is especially intriguing because both contextual and cued fear conditioning are thought to be dependent on plastic changes in the basolateral nucleus of the amygdala, whereas contextual fear conditioning also involves the hippocampus (Hargreaves and Cain, 1992; Bordi et al., 1996; Favata et al., 1998). Thus, in light of normal synaptic plasticity in the hippocampus of PKCβ-deficient mice, these observations are suggestive of a necessity for PKCβ in normal function of the amygdala and are strongly suggestive of a role for PKCβ in the synaptic plasticity underlying amygdala-dependent learning.
Fig. 5.

Fear conditioning. Left, Our initial findings comparing PKCβ knock-out mice (○) and littermate wild-type mice (●). Right, Replications of these experiments using a set of naïve PKCβ-deficient mice and age-matched C57BL/6 control mice. A, Freezing behavior on the day of training for PKCβ-deficient or wild-type mice. Wild-type mice displayed significantly higher freezing in response to the shock (p < 0.05) than did PKCβ knock-out mice in the initial results, but this difference was not significant when the experiment was replicated. The acoustic CS is presented for the two periods underlined. Foot shocks are presented at the arrowheads. B, In the initial results, PKCβ-deficient mice showed significantly less freezing in response to replacement in the training context (p < 0.01) compared with wild-type mice. This effect was replicated in the second experiment (p < 0.01). C, For these experiments the animals are in a different context than that in which they were trained. Acoustic CS presentation is indicated by theline. PKCβ knock-out mice were impaired in freezing in response to CS presentation 24 hr after training in both the initial experiment (p < 0.01) and the replication experiment (p < 0.001). For all graphs, freezing was scored every 5 sec and averaged over 1 min epochs.
We performed an extensive series of behavioral control experiments to bolster our conclusion that PKCβ-deficient mice are deficient in learning and memory versus having derangements of normal sensory or motor function. First, we monitored animals during the training phase of fear conditioning. We assessed the freezing of the animal in response to foot shock. PKCβ-deficient mice exhibit a freezing response to foot shock presentation, which indicates that they were able to sense the foot shock we delivered. We also undertook an additional series of three control experiments using sensitive tests for normal sensory signal transduction in our PKCβ knock-out mice. First we assessed passive avoidance. In this test, animals learn to suppress their normal dark-seeking reflex because their entry into a dark chamber is paired with a foot shock. This control is particularly appealing because it uses the identical aversive sensory stimulus (0.5 mA foot shock) as cued fear conditioning. In these experiments PKCβ knock-out animals exhibited conditioned avoidance indistinguishable from controls (Fig.6A). This was a surprising result, because it is believed that similar mechanisms exist for both fear-conditioned and passive avoidance learning. However, our results indicate a possible mechanistic difference between the two paradigms, perhaps because of a difference in learning the association between the shock experience and the constellation of environmental cues that form the fear-conditioning context compared with passive avoidance in which only a light or dark area needs to be remembered and recognized. In addition, the hotplate and shock threshold tests were used to test for foot sensitivity to noxious stimuli. PKCβ-deficient animals exhibited normal sensory responsiveness to a 55°C hotplate (p = 0.304; Fig. 6A), nor were any differences seen in the responsiveness of mutants to an increase in foot shock as assessed by recording the amount of flinching, jumping, or vocalization to increasing stimulus intensities (Fig.6A). Finally, the cued variant of fear conditioning is dependent on normal hearing. As a control for this sensory modality and to test for sensorimotor gating, we used prepulse inhibition, a test wherein delivery of a modest volume tone suppresses the animals' subsequent startle response to a loud tone. PKCβ knock-out animals exhibited normal prepulse inhibition for this test (Fig.6A), suggesting that they have normal sensorimotor gating and normal hearing. Overall, these data strongly suggest that the cued and contextual fear conditioning deficits we observed in PKCβ knock-out mice were attributable to a bona fide learning or memory deficit versus simply being attributable to sensory deficits.
Fig. 6.

A, Passive avoidance, hotplate, shock threshold, and prepulse inhibition. Passive avoidance was tested for step-through latencies from a lighted compartment to a dark compartment. This test uses the natural tendency for mice to retreat from a lighted area to darker area during the training session. On entering the dark area a mild foot shock was given, and learning was assessed as the avoidance of the dark area after the training session. Results are shown as step-through latency during trial sessions 1–3 d after training for PKCβ-deficient mice (■) or wild-type mice (▪); mean ± SEM. A hotplate test was used to compare mutant (■) versus wild-type (▪) sensitivity to a noxious stimuli. Thermal nociception was measured on a 55°C hotplate as the latency to hindpaw lick. As an additional control to the hotplate test, the shock threshold test was used to compare sensitivity to foot shock measured by the extent of flinching, jumping, or vocalization to increasing foot shock intensities. Mice normally exhibit a startle response to loud noise. Interestingly, if a modest noise is presented immediately preceding the loud noise, the startle response is significantly attenuated, a phenomenon referred to as prepulse inhibition. Prepulse inhibition is a very sensitive test to evaluate sensorimotor gating as well as hearing, because animals reliably give quantitatively different responses to prepulses varying by only a few decibels. The effect of a pretone (sound intensity given in decibels) to diminish the magnitude of acoustic startle is shown in the bottom graph. Results are given as percent diminution of the force of the subsequent 120 dB startle response for PKCβ-deficient mice (■) or wild- type mice (●). B, Open field behavior. As a test of general activity levels and to test for anxiety, animals were monitored using the open-field test. With this test general activity levels are evaluated by measurements of horizontal activity, vertical activity, and total distance traveled during a 10 min test session in an open box in a lighted room. The open field test also measures anxiety levels, as assessed by the center distance to total distance ratio. Results shown are for PKCβ-deficient mice (○) or wild-type mice (●); mean ± SEM.Top graph, Total distance traveled per minute for a 30 min period. Bottom graph. Ratio of center distance to total distance traveled for each minute over a 30 min period. There were no differences in the total distance traveled or the center to total distance ratio between groups. A small decrease in the general vertical activity of mutants was seen (data not shown).C, Rotarod behavior. We analyzed coordination and motor skill acquisition using the rotarod test. The amount of time an animal can stay on a rotating rod is an index of its general level of coordination. Mice also improve their performance with training, which is an indicator of motor learning. Results shown are total times the animals remain on the rotating rod per training period. Three training trials were given on a single day. The increase in time the animal remained on the rod is taken as an index of motor learning. Results shown are for PKCβ-deficient mice (○) or wild-type mice (●); mean ± SEM.
Finally, we tested PKCβ knock-out animals in two tests for motor behavioral responsiveness, the open-field and rotarod assessments. These serve as general controls for an apparent fear-conditioning phenotype being attributable to hyperactivity or abnormal motor coordination. Also, because PKCβ is expressed in striatum and cerebellar granule cells (Fig. 1A,B), the rotarod test serves as an indication of whether PKCβ is necessary for execution of this cerebellum-dependent task. In these tasks PKCβ-deficient mice again exhibited responses indistinguishable from controls (Fig. 6B,C).
As an additional control, we undertook a series of retraining experiments in a subset of animals. Our goals in these experiments were threefold. First, we sought a control for the potential confounding factor that an apparent fear-conditioning phenotype was simply attributable to an inability of PKCβ knock-out animals to exhibit the freezing we quantitate as an index of learning. A second goal was to determine whether PKCβ knock-out mice exhibited a selective loss of long- versus short-term fear conditioning. Finally, in light of recent work by Maren (1998) showing that overtraining does not overcome deficits in the acquisition or expression of fear conditioning caused by lesion of the basal lateral amygdala in rats, we sought to determine whether overtraining could overcome the fear-conditioning deficit we observed.
Retraining of the animals proceeded as follows. On day 3, control and PKCβ knock-out mice were retrained in the same context as day 1 but using a fear-conditioning protocol consisting of five pairings of acoustic CS and foot shock (Fig.7A). Consistent with the deficit in contextual freezing observed on day 2, mice deficient in PKCβ displayed lower baseline freezing (minutes 1–2) than controls when replaced in the context. Similarly PKCβ knock-out mice showed lower freezing rates than wild-type mice over the first two CS–US pairings (minutes 3–4), which is also consistent with the day 1 training results. However, this freezing deficit disappeared with additional training, and PKCβ-deficient mice displayed freezing levels indistinguishable from those of wild-type mice after three, four, or five CS–US pairings (Fig. 7A). These results indicate that PKCβ knock-out mice are indeed capable of normal freezing behavior.
Fig. 7.

Retraining of PKCβ-deficient and control mice on day 3. A, Freezing behavior on retraining of PKCβ-deficient mice (○) or control mice (●). One day after cue and contextual testing (day 3), these mice were retrained with five pairings of acoustic CS (dark bar) and foot shock (gray arrowhead). Compared with control mice, PKCβ knock-out mice displayed significantly lower freezing during baseline measurements (minutes 1–2; p < 0.05). This freezing deficit disappeared with overtraining (minutes 3–7).B, Freezing in response to CS presentation 1–2 hr after training on day 3. There was no significant difference in freezing levels in response to the CS between PKCβ knock-out mice and controls. C, Freezing in response to representation of the training context on day 4. PKCβ knock-out mice were impaired in terms of freezing behavior in response to the training context (p < 0.05). D, Freezing in response to CS presentation on day 4. PKCβ-deficient mice also showed significantly less freezing in response to the CS than did controls (p < 0.01). In all experiments, freezing is scored every 5 sec and averaged over 1 min epochs.
In addition to the freezing control, we were also interested in the effect of the supplementary training on both short- and long-term emotional memory. When tested 1–2 hr after training, PKCβ-deficient mice displayed normal freezing in response to representation of the acoustic CS compared with controls (Fig. 7B). This is a time point often used for testing short-term memory in fear-conditioning tasks (Abel et al., 1997). Long-term memory retention was assessed 24 hr later. Despite showing normal freezing levels during retraining and when tested 1–2 hr later, PKCβ knock-out mice once again displayed freezing rates significantly lower than those of wild-type controls in response to both the context (Fig. 7C) and the acoustic CS delivered in a different context (Fig. 7D) when tested on day 4. It is unknown whether the differences we have observed in PKC-deficient mice indicate a learning deficit or a deficit in memory retention or retrieval; however, these results add support to our conclusion that the β isoform of PKC plays a role in fear conditioning, an amygdala-dependent form of long-term memory.
Overall, we draw three interesting conclusions from the data we obtained in these retraining studies. First, PKCβ knock-out mice clearly can exhibit freezing behavior indistinguishable from controls. A normal extent of freezing was observed in both the retraining period and in the short-term cue test. Therefore, our observation of altered freezing behavior in the long-term variant of fear conditioning does not appear to be an artifact of an altered freezing response per se. Second, short-term cue testing (2 hr) revealed a robust freezing response in PKCβ knock-out animals, suggesting that loss of PKCβ does not lead to short-term memory deficits. Finally, the long-term fear-conditioning deficit in PKCβ-deficient mice is quite profound; even overtraining the animals was not able to overcome the deficits in either contextual or cued fear conditioning.
DISCUSSION
Tracking the expression of PKCβ
Obtaining information on the cellular distribution of signal transmission proteins is important for obtaining functional insights into their roles in the CNS. In this context the gene substitution method we have used in the present studies provides a valuable tool for studying specific subcellular localization patterns in the CNS. The validity of the gene substitution approach that we used is confirmed by our observations that the cellular labeling pattern obtained with this approach is consistent with our (Fig. 1) and others' (Hosoda et al., 1989; Saito et al., 1989) immunohistochemical studies in the mouse and rat. In addition to the use of the approach that we have described here, the approach is also applicable to studying developmental expression patterns, tracing anatomical circuits, and studying anatomical plasticity with various physiological and pathological stimuli. In general, promoter tracking such as we have done here provides a valuable strategy not only for obtaining expression patterns for specific molecules but also for cell type-specific labeling in the CNS.
PKCβ in synaptic plasticity
On the basis of previous studies, a very convincing case can be made that activation of PKC is necessary for induction of NMDA receptor-dependent LTP in area CA1 of hippocampus. Thus, biochemical studies have shown NMDA receptor-dependent activation of various isoforms of PKC in LTP (Malinow et al., 1988; Klann et al., 1991;Sacktor et al., 1993; Powell et al., 1994), that various pharmacological inhibitors of PKC block LTP (Reymann et al., 1988;Malinow et al., 1989; Wang and Feng, 1992), and that postsynaptic injection of specific PKC-blocking peptides blocks LTP induction (Malinow et al., 1988, 1989; Wang and Feng, 1992; Hvalby et al., 1994). The present studies indicate that, in and of itself, loss of the function of PKCβ does not lead to a loss of LTP. Only one other PKC isoform-specific knock-out has been characterized previously, the PKCγ knock-out. This mutant animal has a pronounced LTP deficit in area CA1 (Abeliovich et al., 1993), and the intriguing model has been proposed that a loss of phosphorylation of the postsynaptic PKC substrate neurogranin contributes to this phenotype (Ramakers et al., 1999). Our understanding of the molecular basis for the dependence of LTP on PKCγ is far from complete at this time, however, because LTP can be recovered in PKCγ-deficient mice by delivery of an LTD-inducing stimulus before LTP-inducing tetanic stimulation (Abeliovich et al., 1993). An intriguing possibility is that LTD-inducing stimulation recruits the capacity of another PKC isoform to compensate for the lack of PKCγ. In this context it will be interesting to evaluate double knock-outs of PKCγ and PKCβ at some future time.
The present studies provide strong evidence that the β isoforms of PKC are not necessary for tetanus-induced, NMDA receptor-dependent LTP induction or early maintenance in area CA1 of hippocampus. These observations do not, of course, preclude the involvement of PKCβ in other forms of LTP in area CA1 and other brain regions. There is, moreover, a potential role for PKCβ in synaptic plasticity in area CA1, because we observed an attenuation of phorbol ester-induced potentiation of synaptic transmission in this region. Interestingly, this finding contrasts with mice deficient in the γ isoform of PKC, which have no loss of phorbol ester-induced synaptic facilitation (Goda et al., 1996). These several observations, when taken together, suggest the possible specific involvement of the β isoforms of PKC in neuromodulation in area CA1.
Behavioral roles of PKCβ
The last decade yielded phenomenal growth in our understanding of the biochemical mechanisms of learning and memory. One theme that has become clear is that protein kinases have a prominent role in the synaptic and cellular plasticity underlying learning and memory. To date, four protein kinases have achieved prominence in learning and memory: PKA, CaMKII, PKC, and MAPK (for review, see Sweatt, 1999). As our understanding of the importance of these protein kinases increases, it is critical to bear in mind that each “protein kinase” is itself a family of enzymes. Thus, just as each protein kinase family plays unique roles in synaptic plasticity and memory, the individual isoforms that make up each protein kinase family are also likely to play specific roles in learning and memory. In identifying specific roles for specific protein kinase isoforms, gene-targeted knock-outs are powerful weapons in the neurobiological experimental armamentarium.
Although particularly powerful in the context of determining specific roles of specific protein isoforms, the use of knock-out animals has a number of well recognized limitations. Prominent among these are developmental side effects, possible compensatory mechanisms at the cellular level, and lack of reversibility. Regulated genetic deletion holds great promise for helping address these limitations, but even these approaches do not constitute a complete solution because of the relatively slow times of onset and reversal when generating loss of a protein through deleting its encoding gene. Thus, it is critical to combine genetic deletion approaches with traditional pharmacological approaches to have a more complete picture of the signal transduction mechanisms operating in learning and memory.
In this context, we reference numerous previous studies using pharmacological approaches that implicate PKC in mammalian learning and memory. An impressive array of observations implicate PKC in eye blink conditioning, spatial learning in the Morris water maze and other tasks, and conditioned taste aversion (Paylor et al., 1991, 1992;Colombo et al., 1997; Van der Zee et al., 1997; Yasoshima and Yamamoto, 1997). Roles for PKC in amygdala-dependent fear conditioning have received relatively little attention to date, however, and one of the few available references also described the use of gene deletion (Abeliovich et al., 1993). Thus, mice deficient in the γ isoform of PKC have modest deficits in contextual fear conditioning but no deficits in cued fear conditioning. The present studies are therefore unique in demonstrating a necessity for PKCβ for amygdala-dependent cued and contextual fear conditioning.
We do not know the mechanisms whereby PKCβ contributes to amygdala-dependent learning. Given the pluripotence of PKC in cellular regulation, the list of candidate effector mechanisms is extensive. Appealing possibilities are regulation of gene expression, regulation of local protein synthesis, control of receptor and ion channel function, and modulation of neurotransmitter release. The paucity of biochemical studies of protein kinase signal transduction in the amygdala makes it difficult to formulate specific hypotheses at this time concerning the mechanisms whereby loss of PKCβ leads to amygdala-dependent learning deficits. This area of pursuit represents an important, interesting, and likely very fruitful line of future investigation.
Footnotes
This work was supported by National Institutes of Health Grants MH 57014 and NS 37444 (J.D.S.), the Mental Retardation Research Center at Baylor College of Medicine, and a National Alliance for Research on Schizophrenia and Depression Independent Investigator Award (J.D.S.).
M.L. and J.D.S. contributed equally to this work.
Correspondence should be addressed to J. David Sweatt, Division of Neuroscience, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030. E-mail: jsweatt@bcm.tmc.edu.
Dr. Atkins's present address: Oregon Health Sciences Center, Vollum Institute, Portland, OR 97201.
REFERENCES
- 1.Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:615–626. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- 2.Abeliovich A, Paylor R, Chen C, Kim JJ, Wehner JM, Tonegawa S. PKC gamma mutant mice exhibit mild deficits in spatial and contextual learning. Cell. 1993;75:1263–1271. doi: 10.1016/0092-8674(93)90614-v. [DOI] [PubMed] [Google Scholar]
- 3.Bordi F, Marcon C, Chiamulera C, Reggiani A. Effects of the metabotropic glutamate receptor antagonist MCPG on spatial and context-specific learning. Neuropharmacology. 1996;35:1557–1565. doi: 10.1016/s0028-3908(96)00101-3. [DOI] [PubMed] [Google Scholar]
- 4.Byrne JH, Kandel ER. Presynaptic facilitation revisited: state and time dependence. J Neurosci. 1996;16:425–435. doi: 10.1523/JNEUROSCI.16-02-00425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen SJ, Sweatt JD, Klann E. Enhanced phosphorylation of the postsynaptic protein kinase C substrate RC3/neurogranin during long-term potentiation. Brain Res. 1997;749:181–187. doi: 10.1016/s0006-8993(96)01159-6. [DOI] [PubMed] [Google Scholar]
- 6.Chetkovich DM, Klann E, Sweatt JD. Nitric oxide synthase-independent long-term potentiation in area CA1 of hippocampus. NeuroReport. 1993;4:919–922. doi: 10.1097/00001756-199307000-00020. [DOI] [PubMed] [Google Scholar]
- 7.Choi KW, Smith RF, Buratowski RM, Quinn WG. Deficient protein kinase C activity in turnip, a Drosophila learning mutant. J Biol Chem. 1991;266:15999–15606. [PubMed] [Google Scholar]
- 8.Colombo PJ, Wetsel WC, Gallagher M. Spatial memory is related to hippocampal subcellular concentrations of calcium-dependent protein kinase C isoforms in young and aged rats. Proc Natl Acad Sci USA. 1997;94:14195–14199. doi: 10.1073/pnas.94.25.14195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conn PJ, Sweatt JD. Protein kinase C in the nervous system. In: Kuo JF, editor. Protein kinase C. Oxford UP; Oxford: 1993. pp. 199–235. [Google Scholar]
- 10.Coussens L, Parker PJ, Rhee L, Yang-Feng TL, Chen E, Waterfield MD, Francke U, Ullrich A. Multiple, distinct forms of bovine and human protein kinase C suggest diversity in cellular signaling pathways. Science. 1986;233:859–866. doi: 10.1126/science.3755548. [DOI] [PubMed] [Google Scholar]
- 11.Crawley JN, Paylor R. A proposed test battery and constellations of specific behavioral paradigms to investigate the behavioral phenotypes of transgenic and knockout mice. Horm Behav. 1997;31:197–211. doi: 10.1006/hbeh.1997.1382. [DOI] [PubMed] [Google Scholar]
- 12.Dekker LV, Parker PJ. Protein kinase C—a question of specificity. Trends Biochem Sci. 1994;19:73–77. doi: 10.1016/0968-0004(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 13.Farley J, Schuman E. Protein kinase C inhibitors prevent induction and continued expression of cell memory in Hermissenda type B photoreceptors. Proc Natl Acad Sci USA. 1991;88:2016–2020. doi: 10.1073/pnas.88.5.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 15.Goda Y, Stevens CF, Tonegawa S. Phorbol ester effects at hippocampal synapses act independently of the gamma isoform of PKC. Learn Mem. 1996;3:182–187. doi: 10.1101/lm.3.2-3.182. [DOI] [PubMed] [Google Scholar]
- 16.Hargreaves EL, Cain DP. Hyperactivity, hyper-reactivity, and sensorimotor deficits induced by low doses of the N-methyl-d-aspartate non-competitive channel blocker MK801. Behav Brain Res. 1992;47:23–33. doi: 10.1016/s0166-4328(05)80249-9. [DOI] [PubMed] [Google Scholar]
- 17.Hoffman DA, Johnston D. Downregulation of transient K+ channels in dendrites of hippocampal CA1 pyramidal neurons by activation of PKA and PKC. J Neurosci. 1998;18:3521–3528. doi: 10.1523/JNEUROSCI.18-10-03521.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hosoda K, Saito N, Kose A, Ito A, Tsujino T, Ogita K, Kikkawa U, Ono Y, Igarashi K, Nishizuka Y, Tanaka C. Immunocytochemical localization of the beta I subspecies of protein kinase C in rat brain. Proc Natl Acad Sci USA. 1989;86:1393–1397. doi: 10.1073/pnas.86.4.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu GY, Hvalby O, Walaas SI, Albert KA, Skjeflo P, Andersen P, Greengard P. Protein kinase C injection into hippocampal pyramidal cells elicits features of long term potentiation. Nature. 1987;328:426–429. doi: 10.1038/328426a0. [DOI] [PubMed] [Google Scholar]
- 20.Huang FL, Yoshida Y, Nakabayashi H, Huang KP. Differential distribution of protein kinase C isozymes in the various regions of brain. J Biol Chem. 1987;262:15714–15720. [PubMed] [Google Scholar]
- 21.Hvalby O, Hemmings HC, Jr, Paulsen O, Czernik AJ, Nairn AC, Godfraind JM, Jensen V, Raastad M, Storm JF, Andersen P, Greengard P. Specificity of protein kinase inhibitor peptides and induction of long- term potentiation. Proc Natl Acad Sci USA. 1994;91:4761–4765. doi: 10.1073/pnas.91.11.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jerusalinsky D, Quillfeldt JA, Walz R, Da Silva RC, Medina JH, Izquierdo I. Post-training intrahippocampal infusion of protein kinase C inhibitors causes amnesia in rats. Behav Neural Biol. 1994;61:107–109. doi: 10.1016/s0163-1047(05)80063-9. [DOI] [PubMed] [Google Scholar]
- 23.Klann E, Chen SJ, Sweatt JD. Persistent protein kinase activation in the maintenance phase of long- term potentiation. J Biol Chem. 1991;266:24253–24256. [PubMed] [Google Scholar]
- 24.Leitges M, Schmedt C, Guinamard R, Davoust J, Schaal S, Stabel S, Tarakhovsky A. Immunodeficiency in protein kinase cbeta-deficient mice. Science. 1996;273:788–791. doi: 10.1126/science.273.5276.788. [DOI] [PubMed] [Google Scholar]
- 25.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 26.Macek TA, Schaffhauser H, Conn PJ. Protein kinase C and A3 adenosine receptor activation inhibit presynaptic metabotropic glutamate receptor (mGluR) function and uncouple mGluRs from GTP-binding proteins. J Neurosci. 1998;18:6138–6146. doi: 10.1523/JNEUROSCI.18-16-06138.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malenka RC, Madison DV, Nicoll RA. Potentiation of synaptic transmission in the hippocampus by phorbol esters. Nature. 1986;321:175–177. doi: 10.1038/321175a0. [DOI] [PubMed] [Google Scholar]
- 28.Malinow R, Madison DV, Tsien RW. Persistent protein kinase activity underlying long-term potentiation. Nature. 1988;335:820–824. doi: 10.1038/335820a0. [DOI] [PubMed] [Google Scholar]
- 29.Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- 30.Manseau F, Sossin WS, Castellucci VF. Long-term changes in excitability induced by protein kinase C activation in Aplysia sensory neurons. J Neurophysiol. 1998;79:1210–1218. doi: 10.1152/jn.1998.79.3.1210. [DOI] [PubMed] [Google Scholar]
- 31.Maren S. Overtraining does not mitigate contextual fear conditioning deficits produced by neurotoxic lesions of the basolateral amygdala. J Neurosci. 1998;18:3088–3097. doi: 10.1523/JNEUROSCI.18-08-03088.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mihalek RM, Jones CJ, Tully T. The Drosophila mutation turnip has pleiotropic behavioral effects and does not specifically affect learning. Learn Mem. 1997;3:425–444. doi: 10.1101/lm.3.5.425. [DOI] [PubMed] [Google Scholar]
- 33.Muller U. Second messenger pathways in the honeybee brain: immunohistochemistry of protein kinase A and protein kinase C. Microsc Res Tech. 1999;45:165–173. doi: 10.1002/(sici)1097-0029(19990501)45:3<165::aid-jemt4>3.3.co;2-q. [DOI] [PubMed] [Google Scholar]
- 34.Nicholls DG. Presynaptic modulation of glutamate release. Prog Brain Res. 1998;116:15–22. doi: 10.1016/s0079-6123(08)60427-6. [DOI] [PubMed] [Google Scholar]
- 35.Oehrlein SA, Maelicke A, Herget T. Expression of protein kinase C gene family members is temporally and spatially regulated during neural development in vitro. Eur J Cell Biol. 1998;77:323–337. doi: 10.1016/S0171-9335(98)80091-5. [DOI] [PubMed] [Google Scholar]
- 36.Ogita K, Miyamoto S, Yamaguchi K, Koide H, Fujisawa N, Kikkawa U, Sahara S, Fukami Y, Nishizuka Y. Isolation and characterization of delta-subspecies of protein kinase C from rat brain. Proc Natl Acad Sci USA. 1992;89:1592–1596. doi: 10.1073/pnas.89.5.1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ono Y, Fujii T, Ogita K, Kikkawa U, Igarashi K, Nishizuka Y. Identification of three additional members of rat protein kinase C family: delta-, epsilon- and zeta-subspecies. FEBS Lett. 1987;226:125–128. doi: 10.1016/0014-5793(87)80564-1. [DOI] [PubMed] [Google Scholar]
- 38.Osada S, Mizuno K, Saido TC, Akita Y, Suzuki K, Kuroki T, Ohno S. A phorbol ester receptor/protein kinase, nPKC eta, a new member of the protein kinase C family predominantly expressed in lung and skin. J Biol Chem. 1990;265:22434–22440. [PubMed] [Google Scholar]
- 39.Osada S, Mizuno K, Saido TC, Suzuki K, Kuroki T, Ohno S. A new member of the protein kinase C family, nPKC theta, predominantly expressed in skeletal muscle. Mol Cell Biol. 1992;12:3930–3938. doi: 10.1128/mcb.12.9.3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parker PJ, Coussens L, Totty N, Rhee L, Young S, Chen E, Stabel S, Waterfield MD, Ullrich A. The complete primary structure of protein kinase C–the major phorbol ester receptor. Science. 1986;233:853–859. doi: 10.1126/science.3755547. [DOI] [PubMed] [Google Scholar]
- 41.Paylor R, Rudy JW, Wehner JM. Acute phorbol ester treatment improves spatial learning performance in rats. Behav Brain Res. 1991;45:189–193. doi: 10.1016/s0166-4328(05)80085-3. [DOI] [PubMed] [Google Scholar]
- 42.Paylor R, Morrison SK, Rudy JW, Waltrip LT, Wehner JM. Brief exposure to an enriched environment improves performance on the Morris water task and increases hippocampal cytosolic protein kinase C activity in young rats. Behav Brain Res. 1992;52:49–59. doi: 10.1016/s0166-4328(05)80324-9. [DOI] [PubMed] [Google Scholar]
- 43.Paylor R, Nguyen M, Crawley JN, Patrick J, Beaudet A, Orr-Urtreger A. Alpha7 nicotinic receptor subunits are not necessary for hippocampal- dependent learning or sensorimotor gating: a behavioral characterization of Acra7-deficient mice. Learn Mem. 1998;5:302–316. [PMC free article] [PubMed] [Google Scholar]
- 44.Powell CM, Johnston D, Sweatt JD. Autonomously active protein kinase C in the maintenance phase of N-methyl-d-aspartate receptor-independent long term potentiation. J Biol Chem. 1994;269:27958–27963. [PMC free article] [PubMed] [Google Scholar]
- 45.Ramakers GM, Gerendasy DD, de Graan PN. Substrate phosphorylation in the protein kinase Cgamma knockout mouse. J Biol Chem. 1999;274:1873–1874. doi: 10.1074/jbc.274.4.1873. [DOI] [PubMed] [Google Scholar]
- 46.Reymann KG, Brodemann R, Kase H, Matthies H. Inhibitors of calmodulin and protein kinase C block different phases of hippocampal long-term potentiation. Brain Res. 1988;461:388–392. doi: 10.1016/0006-8993(88)90274-0. [DOI] [PubMed] [Google Scholar]
- 47.Roberson ED, Sweatt JD. Transient activation of cyclic AMP-dependent protein kinase during hippocampal long-term potentiation. J Biol Chem. 1996;271:30436–30441. doi: 10.1074/jbc.271.48.30436. [DOI] [PubMed] [Google Scholar]
- 48.Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci. 1999;19:4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16:1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- 50.Sacktor TC, Kruger KE, Schwartz JH. Activation of protein kinase C by serotonin: biochemical evidence that it participates in the mechanisms underlying facilitation in Aplysia. J Physiol (Lond) 1988;83:224–231. [PubMed] [Google Scholar]
- 51.Sacktor TC, Osten P, Valsamis H, Jiang X, Naik MU, Sublette E. Persistent activation of the zeta isoform of protein kinase C in the maintenance of long-term potentiation (see comments). Proc Natl Acad Sci USA. 1993;90:8342–8346. doi: 10.1073/pnas.90.18.8342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saido TC, Mizuno K, Konno Y, Osada S, Ohno S, Suzuki K. Purification and characterization of protein kinase C epsilon from rabbit brain. Biochemistry. 1992;31:482–490. doi: 10.1021/bi00117a026. [DOI] [PubMed] [Google Scholar]
- 53.Saito N, Kose A, Ito A, Hosoda K, Mori M, Hirata M, Ogita K, Kikkawa U, Ono Y, Igarashi K, Nishizaka Y, Tanaka C. Immunocytochemical localization of beta II subspecies of protein kinase C in rat brain. Proc Natl Acad Sci USA. 1989;86:3409–3413. doi: 10.1073/pnas.86.9.3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sheu FS, McCabe BJ, Horn G, Routtenberg A. Learning selectively increases protein kinase C substrate phosphorylation in specific regions of the chick brain. Proc Natl Acad Sci USA. 1993;90:2705–2709. doi: 10.1073/pnas.90.7.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stevens CF, Sullivan JM. Regulation of the readily releasable vesicle pool by protein kinase C. Neuron. 1998;21:885–893. doi: 10.1016/s0896-6273(00)80603-0. [DOI] [PubMed] [Google Scholar]
- 56.Suen PC, Wu K, Xu JL, Lin SY, Levine ES, Black IB. NMDA receptor subunits in the postsynaptic density of rat brain: expression and phosphorylation by endogenous protein kinases. Brain Res Mol Brain Res. 1998;59:215–228. doi: 10.1016/s0169-328x(98)00157-0. [DOI] [PubMed] [Google Scholar]
- 57.Sweatt JD. Toward a molecular explanation for long-term potentiation. Learn Mem. 1999;6:399–416. doi: 10.1101/lm.6.5.399. [DOI] [PubMed] [Google Scholar]
- 58.Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain (see comments). Cell. 1996;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- 59.Van der Zee EA, Kronforst-Collins MA, Maizels ET, Hunzicker-Dunn M, Disterhoft JF. gamma isoform-selective changes in PKC immunoreactivity after trace eyeblink conditioning in the rabbit hippocampus. Hippocampus. 1997;7:271–285. doi: 10.1002/(SICI)1098-1063(1997)7:3<271::AID-HIPO3>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 60.Wang JH, Feng DP. Postsynaptic protein kinase C essential to induction and maintenance of long-term potentiation in the hippocampal CA1 region. Proc Natl Acad Sci USA. 1992;89:2576–2580. doi: 10.1073/pnas.89.7.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xia ZG, Refsdal CD, Merchant KM, Dorsa DM, Storm DR. Distribution of mRNA for the calmodulin-sensitive adenylate cyclase in rat brain: expression in areas associated with learning and memory. Neuron. 1991;6:431–443. doi: 10.1016/0896-6273(91)90251-t. [DOI] [PubMed] [Google Scholar]
- 62.Yamoah EN, Crow T. Protein kinase and G-protein regulation of Ca2+ currents in Hermissenda photoreceptors by 5-HT and GABA. J Neurosci. 1996;16:4799–4809. doi: 10.1523/JNEUROSCI.16-15-04799.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yasoshima Y, Yamamoto T. Rat gustatory memory requires protein kinase C activity in the amygdala and cortical gustatory area. NeuroReport. 1997;8:1363–1367. doi: 10.1097/00001756-199704140-00009. [DOI] [PubMed] [Google Scholar]