

Abstract
Neuronal α1E Ca channel subunits are widely expressed in mammalian brain, where they are thought to form R-type Ca channels. Recent studies have demonstrated that R-type channels contribute to neurosecretion and dendritic Ca influx, but little is known concerning their modulation. Here we show that α1E channels are strongly stimulated, and only weakly inhibited, through M1 muscarinic acetylcholine receptors. Both forms of channel modulation are mediated by pertussis toxin-insensitive G-proteins. Channel stimulation is blocked by regulator of G-protein signaling 2 (RGS2) or the C-terminal region of phospholipase C-β1 (PLCβ1ct), which have been previously shown to function as GTPase-activating proteins for Gαq. In contrast, RGS2 and PLCβ1ct do not block inhibition of α1E through M1 receptors. Inhibition is prevented, however, by the C-terminal region of β-adrenergic receptor kinase 1, which sequesters Gβγ dimers. Thus, stimulation of α1E is mediated by a pertussis toxin-insensitive Gα subunit (e.g., Gαq), whereas inhibition is mediated by Gβγ. The ability of RGS2 and PLCβ1ct to selectively block stimulation indicates these proteins functioned primarily as effector antagonists. In support of this interpretation, RGS2 prevented stimulation of α1E with non-hydrolyzable guanosine 5′-0-(3-thiotriphosphate). We also report strong muscarinic stimulation of rbE-II, a variant α1E Ca channel that is insensitive to voltage-dependent inhibition. Our results predict that Gαq-coupled receptors predominantly stimulate native R-type Ca channels. Receptor-mediated enhancement of R-type Ca currents may have important consequences for neurosecretion, dendritic excitability, gene expression, or other neuronal functions.
Keywords: CaV2.3, R-type calcium channel, α1E, RGS protein, phospholipase C-β1, GAP, effector antagonist
Native R-type Ca channels have been defined by their resistance to selective antagonists of L-, N-, and P/Q-type Ca channels (Randall and Tsien, 1995). Only recently has a selective antagonist of R-type channels been reported (Newcomb et al., 1998). Antisense depletion experiments suggest that neuronal R-type Ca channels are formed by α1E subunits (Piedras-Rentería and Tsien, 1998; Tottene et al., 2000). α1E subunits are widely expressed in mammalian brain (Niidome et al., 1992; Soong et al., 1993; Wakamori et al., 1994; Williams et al., 1994; Yokoyama et al., 1995), and several splice variants have been described (cf. Pereverzev et al., 1998). Although the physiological functions of R-type Ca channels are incompletely known, available evidence indicates that they contribute to dendritic Ca influx (Kavalali et al., 1997) and neurosecretion by some presynaptic terminals (Turner et al., 1995; Wu et al., 1998, 1999;Allen, 1999; Wang et al., 1999).
The G-protein-dependent modulation of N- and P/Q-type Ca channels has been extensively studied (for review, see Hille, 1994; Jones and Elmslie, 1997; Zamponi and Snutch, 1998; Ikeda and Dunlap, 1999; Bean, 2000). In contrast, much less is known concerning the modulation of R-type Ca channels. Previously, we reported that R-type channels formed by rabbit α1E subunits are both inhibited and stimulated through M2 muscarinic acetylcholine receptors (Meza et al., 1999). Our experiments demonstrated that inhibition and stimulation of α1E are separate processes that occur through distinct signaling pathways, both of which couple to M2 receptors. Inhibition occurs through a fast, pertussis toxin (PTX)-sensitive pathway, whereas stimulation occurs through a slower, PTX-insensitive pathway (Meza et al., 1999). It is intriguing that R-type channels can be stimulated through muscarinic receptors, because the closely-related N- and P/Q-type channels are typically inhibited through G-protein-coupled receptors. Receptor-mediated enhancement of R-type Ca currents may have important consequences for dendritic Ca signaling, neurosecretion, or other neuronal functions.
In the present study, we have further examined the muscarinic stimulation of recombinant R-type Ca channels. α1E subunits were expressed in HEK293 cells with M1 muscarinic acetylcholine receptors, which preferentially couple to heterotrimeric G-proteins of the Gαq subfamily (Felder, 1995). We find that M1 receptors predominantly stimulate, rather than inhibit, α1E channels. Additionally, we find that stimulation of α1E is selectively blocked by regulator of G-protein signaling 2 (RGS2) and the C-terminal region of phospholipase C-β1 (PLCβ1ct), two proteins known to function as GTPase-activating proteins (GAPs) for Gαq. Interestingly, the effects of RGS2 and PLCβ1ct in our experiments can only be explained if these proteins functioned primarily as effector antagonists. Altogether, our data suggest that α1E is stimulated through a Gαq-coupled signaling pathway. These observations predict that Gαq-coupled receptors stimulate native R-type Ca channels in vivo. Our results also provide new insight into mechanisms by which RGS proteins can influence the receptor-mediated modulation of voltage-gated Ca channels.
MATERIALS AND METHODS
Cell culture and transfection. Human embryonic kidney (HEK293) cells were obtained from the American Type Culture Collection (Manassas, VA) and propagated in culture medium containing 90% DMEM, 10% fetal bovine serum, and 50 μg/ml gentamycin. The cells were trypsinized weekly and replated onto 60 mm culture dishes at 20% confluence. CaPO4 precipitation was used to transfect these cells within 3–5 d of plating. The transfection mixture contained expression plasmids encoding α1E, α2-δ, and β3 Ca channel subunits at 1.25 μg of each cDNA per dish, plus an expression plasmid encoding the M1 muscarinic acetylcholine receptor at 0.25 μg of cDNA per dish. In selected experiments, the transfection mixture also included expression plasmids encoding RGS2, RGS8, or the C-terminal region (Thr903-Leu1216) of PLCβ1 at 0.625 μg of cDNA per dish. RGS2, RGS8, and PLCβ1ct were expressed as fusions to the C terminus of enhanced green fluorescence protein (EGFP). In specific experiments, an expression plasmid encoding Gly495-Leu689of β-adrenergic receptor kinase 1 (βARK1; denoted βARK1ct) was transfected at 1.25 μg of cDNA per dish. For the experiments illustrated in Figure 7, cells were transfected at 1.25 μg/dish with an expression plasmid encoding a variant α1E subunit (denoted rbE-II) cloned from rat hippocampus by Soong et al. (1993). These cells were cotransfected with the M1 receptor as described above or alternatively with a plasmid encoding the M2 muscarinic acetylcholine receptor at 0.0625 μg/dish. For all transfections that did not include EGFP fusion proteins, a separate plasmid encoding EGFP was included at 0.125 μg/dish. The day after transfection, cells were briefly trypsinized and replated onto 12 mm round glass coverslips. Electrophysiological experiments were performed 16–24 hr later. Successfully transfected cells were visually identified by their green fluorescence under ultraviolet illumination; only green cells were used for experiments.
Fig. 7.

rbE-II Ca channels are strongly stimulated through muscarinic receptors. Left, rbE-II current amplitudes are plotted as a function of time during representative experiments.Right, Whole-cell Ca currents recorded at times indicated in the corresponding plot. A, Stimulation of rbE-II current through M2 receptors (M2R). Ca currents were evoked every 3 sec by step depolarizations from −90 to +30 mV.C = 50 pF; RS = 1.1 MΩ. The application of CCh (50 μm) is indicated by ahorizontal bar; this CCh concentration produces maximal activation of M2 receptors (Melliti et al., 1999; Meza et al., 1999).B, Stimulation of rbE-II current through M1 receptors (M1R). Ca currents were evoked every 5 sec by step depolarizations from −90 to +30 mV. C = 28 pF;RS = 1.5 MΩ. The concentration of applied CCh was 1 mm. C, Average stimulation of rbE-II currents through M1 and M2 receptors.
Expression plasmids. cDNA encoding rabbit α1E (GenBank accession number X67856) was in pcDNA3.1+ (Invitrogen, San Diego, CA). rbE-II (accession number L15453) and rat α2-δ (M86621) were in pMT2 (Genetics Institute, Cambridge, MA). Rabbit β3 (X64300) was in pcDNA3 (Invitrogen). Human M1 muscarinic acetylcholine receptor (X52068) was in pCD. Human M2 muscarinic receptor (X15264) was in pRK5 (Genentech, South San Francisco, CA). Jellyfish enhanced green fluorescent protein (U55763) was in pEGFP (Clontech, Cambridge, UK). Human RGS2 (L13463) and rat RGS8 (AB006013) were in pCI (Promega, Madison, WI). EGFP-RGS2 and EGFP-RGS8 were in pEGFP-C2 and pEGFP-C1 (Clontech), respectively. A deletion mutant of RGS8 (denoted ΔRGS8) was in pEGFP-C1; this construct encodes EGFP fused to an RGS8 protein lacking Phe57-Pro160. cDNA encoding Thr903-Leu1216of rat PLCβ1 (M20636) was in pEGFP-C1 (Clontech). cDNA encoding Gly495-Leu689of βARK1 (M34019) was in pRK5 (Koch et al., 1994).
Patch-clamp recordings. Large-bore patch pipettes were pulled from 100 μl borosilicate glass micropipettes (VWR 53432-921) and filled with a solution containing (in mm): 155 CsCl, 10 Cs2-EGTA, 4 Mg-ATP, 0.32 Li-GTP, and 10 HEPES, pH 7.4, with CsOH. For the experiments illustrated in Figure 6, equimolar guanosine 5′-0-(3-thiotriphosphate) (GTP-γ-S) was substituted for GTP in the pipette solution. Aliquots of the pipette solution were stored at −80°C, kept on ice after thawing, and filtered at 0.22 μm immediately before use. Pipette tips were coated with paraffin to reduce capacitance and then fire-polished. Filled patch pipettes had DC resistances of 1.0–1.5 MΩ. The bath solution contained (in mm): 145 NaCl, 40 CaCl2, 2 KCl, and 10 HEPES, pH 7.4, with NaOH. Ca currents were recorded in the whole-cell configuration. After forming a gigaohm seal in the cell-attached configuration, residual pipette capacitance was compensated using the negative capacitance circuit of the amplifier. The DC resistance of the whole-cell configuration was routinely >1 GΩ. The steady holding potential was −90 mV. No corrections were made for liquid junction potentials. Depolarizations to potentials near the peak of the current–voltage relationship (+30 mV) were delivered every 1–10 sec; the stimulation rate was adjusted for each cell to maximize sampling resolution and to minimize cumulative inactivation. Currents were filtered at 2–10 kHz using the built-in Bessel filter (four-pole low-pass) of an Axopatch 200B amplifier (Axon Instruments, Foster City, CA) and sampled at 10–50 kHz using a Digidata 1200 analog-to-digital board installed in a Gateway Pentium computer. The pCLAMP 7.0 software programs Clampex and Clampfit were used for data acquisition and analysis, respectively. Figures were made using the software program Origin (version 6.0).
Fig. 6.

RGS2 prevents stimulation of α1E by intracellular GTP-γ-S. Whole-cell Ca currents were recorded from HEK293 cells expressing rabbit α1E Ca channels and human M1 receptors; some cells also expressed RGS2. The pipette solution contained 0.32 mm GTP-γ-S in place of GTP; aliquots of this solution were thawed from −80°C, kept on ice during experiments, and discarded within 3 hr of thawing. Recordings from control and RGS2-expressing cells were alternated. The cells were incubated with PTX (200–500 ng/ml) overnight before experiments. Immediately after establishment of the whole–cell configuration (break-in), Ca currents were evoked by 10 msec depolarizations to +30 mV, delivered every 10 sec from a steady holding potential of −90 mV.Top, Representative whole-cell Ca currents recorded at break-in and 5 min later from a control (left) and an RGS2-expressing (right) cell. RS= 2.5 MΩ (control) and 2.4 MΩ (RGS2). Bottom, Average ± SEM Ca current amplitudes at various times after break-in. Current amplitudes are expressed relative to the initial Ca current amplitude recorded in each cell at break-in. For times between 0 and 2 min, symbols represent data from 15 control and 10 RGS2-expressing cells; beyond 2 min, symbolsrepresent data from 8 control and 7 RGS2-coexpressing cells. Initial Ca current densities were 99 ± 17 pA/pF (control) and 124 ± 22 pA/pF (RGS2).
Linear cell capacitance (C) was determined by integrating the area under the whole-cell capacity transient, which was evoked by a voltage-clamp step from −90 to −80 mV; the whole-cell capacitance compensation circuit of the amplifier was turned off during this measurement. The average value of C was 18 ± 1 pF (mean ± SEM; n = 160 cells). To minimize voltage errors, the time constant for decay of the whole-cell capacity transient (t) was reduced as much as possible using the analog series resistance compensation circuit of the amplifier. Series resistance (RS) was calculated ast × (1/C), where t was the time constant for decay of the whole-cell capacity transient. The average values of t and RS, measured before electronic compensation, were 54 ± 2 μsec and 3.2 ± 0.1 MΩ, respectively (n = 160). Maximal Ca current amplitude was 1790 ± 140 pA (n = 160; test potential, +30 mV). After electronic compensation of tand RS, the average maximum voltage error was 3.9 ± 0.3 mV (n = 160).
All currents were corrected for linear capacitance, and leakage currents using −P/6 or −P/4 subtraction. Ca current amplitudes were measured at the time of peak inward current. Comparisons were by ANOVA or by unpaired, two-tailed t tests, withp < 0.05 considered significant. Application of carbachol (CCh) was by bath exchange or local superfusion through macropipette positioning close to the cell under study. CCh was dissolved directly in the bath solution. Temperature (20–24°C) was continuously monitored using a miniature thermocouple placed in the recording chamber.
RESULTS
α1E Ca channels are predominantly stimulated through M1 muscarinic acetylcholine receptors
Figure 1 illustrates Ca currents recorded from an HEK293 cell coexpressing rabbit α1E subunits and human M1 muscarinic acetylcholine receptors. As seen in the plot of current amplitudes versus time (Fig. 1A), application of CCh initially produced a small, rapid decrease (inhibition) of α1E current amplitude (point b). This initial inhibition was soon followed (point c) by a substantial increase in the current amplitude (stimulation). After CCh washout, current amplitude again transiently increased (point d) by an amount comparable with that of the initial inhibition. This secondary increase at washout apparently corresponds to relief of the initial inhibition (see below). The observed modulation of α1E Ca channels was attributable to coexpressed M1 receptors, because it was absent from cells not cotransfected with muscarinic receptors and was completely blocked by atropine (Meza et al., 1999).
Fig. 1.

α1E Ca channels are predominantly stimulated through M1 muscarinic acetylcholine receptors. A, Whole-cell Ca currents were evoked every 5 sec by step depolarizations from −90 to + 30 mV. Ca current amplitudes are plotted as a function of time during a representative experiment. Application of CCh (1 mm) is indicated by a horizontal bar. Linear cell capacitance (C) = 13 pF; series resistance (RS) = 2.1 MΩ.B, Selected Ca currents recorded at the times indicated inA. C, Summary data for inhibition and stimulation of α1E Ca currents through coexpressed M1 receptors. For each cell, inhibition was measured as the difference between the amplitudes of currents a and b, normalized with respect to the amplitude of current a. Stimulation was measured as the difference between the amplitudes of currents band c, normalized with respect to the amplitude of current a. Error bars represent ±SEM.
α1E current amplitudes were inhibited by only 6 ± 2% (n = 17) through M1 receptors. In contrast, the magnitude of stimulation was substantially larger (40 ± 6%;n = 17). Thus, M1 receptors predominantly stimulate α1E Ca channels (Fig. 1C). Previously, we found that α1E currents were inhibited by ∼40% and stimulated by ∼20% through the M2 subtype of muscarinic acetylcholine receptor (Meza et al., 1999). This comparison indicates that M1 receptors produce smaller inhibition and larger stimulation of α1E Ca channels than M2 receptors.
We next examined the dose dependency for stimulation of α1E Ca channels through M1 receptors (Fig. 2). Application of 10 μm CCh produced approximately half-maximal stimulation, and 100 μm CCh generated maximal stimulation. The concentration of CCh used throughout the remainder of this study (1 mm) was therefore clearly saturating. These dose–response data are in general agreement with previously reported agonist binding affinities of cloned M1 receptors (Peralta et al., 1987).
Fig. 2.

Dose–response data for modulation of α1E Ca channels through M1 receptors. Top, Representative Ca currents recorded before the application of CCh and during maximal stimulation by various concentrations of CCh. The control currents are aligned with the horizontal dotted line.Bottom, Average inhibition and stimulation of α1E currents by various CCh concentrations. Inhibition and stimulation are expressed relative to the control Ca current in each cell, recorded immediately before CCh application. Each cell was exposed once to a single concentration of CCh. The voltage protocol is as in Figure 1. In selected experiments, cells were exposed to 100 nmstaurosporine before, during, and after the application of CCh. Staurosporine (STAURO.) was dissolved in DMSO to make a stock solution of 1 mm. The final concentration of DMSO in the bath was 0.01%, which alone had no effects on α1E currents.
In our previous study (Meza et al., 1999), we found that the M2 receptor-mediated stimulation of α1E was prevented by staurosporine, a broad-spectrum inhibitor of serine-threonine kinases. As shown in Figure 2, staurosporine also prevented stimulation of α1E through M1 receptors, suggesting that stimulation results from a pathway that couples to both receptor subtypes. Because M1 receptors produce larger stimulation than M2 receptors, the responsible signaling pathway apparently couples more efficiently to M1 receptors. The effect of staurosporine is consistent with a previous report that α1E Ca channels are stimulated through a protein kinase C-dependent pathway inXenopus oocytes (Stea et al., 1995).
As shown in Figure 2, the average magnitude of inhibition was somewhat larger in staurosporine-treated cells (18 ± 10%) than in control cells (6 ± 2%), although this difference was not statistically significant (p > 0.05). Our impression is that stimulation causes a slight underestimation in the measurement of inhibition; however, this underestimation does not affect the conclusions of this study (see below).
Stimulation of α1E is mediated by a PTX-insensitive Gα subunit
PTX catalyzes the ADP ribosylation of Gαi subfamily proteins at a cysteine residue near the C terminus, thereby decoupling these Gα subunits from receptors (West et al., 1985; Avigan et al., 1992). We used PTX to investigate which G-proteins are responsible for modulation of α1E through M1 receptors. Figure3B shows currents recorded from a cell exposed to PTX (500 ng/ml) overnight, and Figure3A illustrates currents recorded from an untreated control cell. It was clear that PTX had no appreciable effects on inhibition or stimulation of α1E. Averaged results are presented in Figure 5. These data suggest that, in the case of M1 receptors, inhibition and stimulation of α1E are both mediated by PTX-insensitive G-proteins.
Fig. 3.
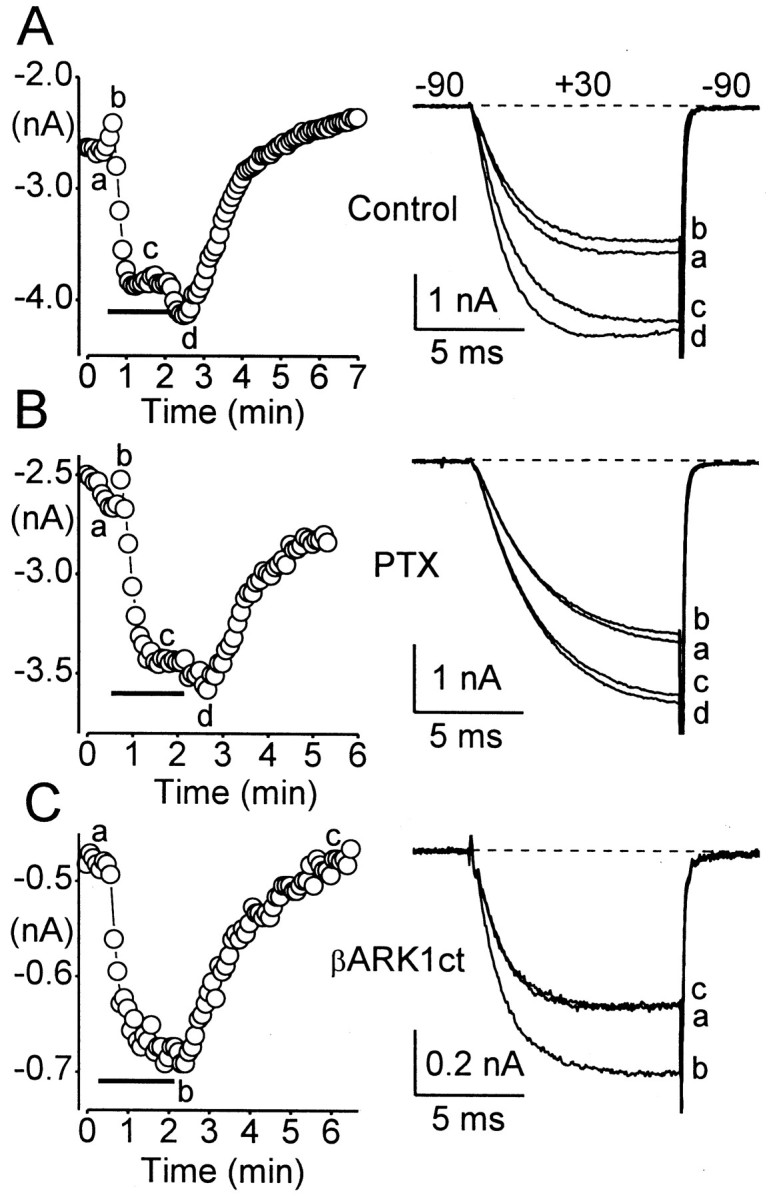
Stimulation of α1E is mediated by a PTX-insensitive Gα subunit. Left, α1E current amplitudes are plotted as a function of time during representative experiments.Right, Whole-cell Ca currents recorded at times indicated in the corresponding plots. A, Modulation of α1E currents by M1 receptors in a control cell (same cell as in Fig.1A). C = 13 pF; RS = 2.1 MΩ. B, PTX does not affect inhibition or stimulation of α1E through M1 receptors. Cells were preincubated with PTX; 200–500 ng/ml) for at least 20 hr.C = 9 pF; RS = 3.6 MΩ. C, Coexpression of βARK1ct selectively blocks inhibition of α1E through M1 receptors. C = 11 pF; RS = 3.2 MΩ. Other details are as in Figure 1.
Fig. 5.

Modulation of α1E Ca currents through M1 receptors. Inhibition and stimulation are expressed relative to the control Ca current in each cell, recorded immediately before CCh application. Each cell was exposed once to a single concentration of CCh. Means were compared using one-way ANOVA or by an unpaired, two-tailed t test. Asterisks indicate significant differences from the control mean (*p< 0.05; **p < 0.01; ***p < 0.001). The average Ca current densities (measured in response to a test pulse to +30 mV) in each group were 106 ± 17 pA/pF (n = 17) in control, 104 ± 23 pA/pF (n = 10) in PTX-treated, 80 ± 21 pA/pF (n = 10) in PLCβ1ct-expressing, 196 ± 88 pA/pF (n = 9) in RGS2-expressing, 68 ± 10 pA/pF (n = 8) in βARK1ct-expressing, 124 ± 19 pA/pF (n = 18) in RGS8-expressing, and 149 ± 38 pA/pF (n = 7) in ΔRGS8-expressing cells.
To determine which subunits (Gα or Gβγ) of the PTX-insensitive G-protein are responsible for modulation of α1E, we expressed βARK1ct. This region (Gly495-Leu689) of βARK1 sequesters Gβγ dimers (Koch et al., 1994), including Gβγ released through activation of Gαq-coupled M3 receptors (Stehno-Bittel et al., 1995). As shown in Figure 3C, coexpression of βARK1ct blocked the initial inhibition of α1E as well as the secondary increase in current amplitude after CCh washout. Overall, α1E currents were inhibited by only 0.7 ± 0.7% (n = 8) in βARK1ct-expressing cells, compared with 6.4 ± 1.8% (n = 17) inhibition in control cells (p < 0.05; see Fig. 5). These results with βARK1ct suggest that inhibition of α1E is mediated by Gβγ dimers released as a consequence of M1 receptor activation.
In contrast, stimulation of α1E was unaffected by βARK1ct (see Fig.5). Thus, α1E currents were stimulated by 40 ± 6% in control cells (n = 17) and by 43 ± 7% in βARK1ct-expressing cells (n = 7). Altogether, these data indicate that stimulation involves signaling by a PTX-insensitive Gα subunit.
PLCβ1ct, RGS2, and RGS8 selectively block stimulation of α1E
M1 receptors preferentially couple to G-proteins of the Gαq subfamily (Felder, 1995), suggesting that Gαq mediates stimulation of α1E. To test this hypothesis, we took advantage of recent work showing that PLCβ1 and RGS2 can modify interactions of Gαq with its downstream effectors. PLCβ1 is the principal effector enzyme of Gαq; interestingly, this phopholipase also functions as a powerful GAP for Gαq (Berstein et al., 1992; Biddlecome et al., 1996). For our purposes, we chose to express only the C terminus of PLCβ1 (Thr903-Leu1216; denoted PLCβ1ct), because this portion of the protein contains intrinsic GAP activity (Paulssen et al., 1996), but it completely lacks phospholipase activity (Wu et al., 1993).
As illustrated in Figure4B, coexpression of PLCβ1ct greatly reduced stimulation of α1E. Thus, stimulation was only ∼4% in cells expressing PLCβ1ct, compared with ∼40% stimulation in control cells (Fig. 5). Surprisingly, PLCβ1ct did not also reduce inhibition of α1E through M1 receptors. Overall, α1E currents were inhibited by 10 ± 2% (n = 10) in PLCβ1ct-expressing cells, compared with 6 ± 2% (n = 17) inhibition in control cells (p > 0.1). The lack of effect on inhibition was unexpected, given the previously demonstrated ability of PLCβ1 to act as a powerful GAP for Gαq (Berstein et al., 1992).
Fig. 4.

Stimulation of α1E is selectively blocked by PLCβ1ct, RGS2, and RGS8. A, Modulation of α1E by M1 receptors in a control cell. C = 16 pF;RS = 2.8 MΩ. B, Modulation of α1E by M1 receptors in a cell coexpressing PLCβ1ct. C = 24 pF; RS = 1.3 MΩ. C, Modulation of α1E by M1 receptors in a cell coexpressing RGS2.C = 11 pF; RS = 5.1 MΩ. D, Modulation of α1E by M1 receptors in a cell coexpressing RGS8. C = 13 pF;RS = 2.9 MΩ. Other details are as in Figure1.
To further test the hypothesis that Gαq mediates stimulation of α1E, we used RGS2, because previous studies have found that RGS2 preferentially interacts with Gαq in vitro (Chen et al., 1997; Heximer et al., 1997; but see Ingi et al., 1998). Stimulation of α1E was also significantly reduced by coexpression of RGS2 (Fig.4C). Altogether, stimulation was 7 ± 4% in RGS2-expressing cells (n = 9), compared with 40 ± 6% (n = 17) stimulation in control cells (p < 0.01). In contrast, inhibition of α1E was unaffected by RGS2 (Fig. 5). Thus, the effects of RGS2 were basically identical to those of PLCβ1ct; that is, both proteins strongly reduced stimulation of α1E without also reducing inhibition. Because PLCβ1ct and RGS2 have been previously demonstrated to interact with Gαq, these findings support the hypothesis that stimulation is mediated through a Gαq-coupled signaling pathway.
For comparison with PLCβ1ct and RGS2, we tested RGS8, which has been previously shown to act as a GAP for Gαi subfamily proteins (Saitoh et al., 1997; Melliti et al., 1999). Coexpression of RGS8 significantly reduced stimulation of α1E (Fig. 4D) but to a lesser extent than either PLCβ1ct or RGS2 (Fig. 5). Inhibition of α1E was unaffected by RGS8 (Fig. 5). The ability of RGS8 to reduce stimulation suggests that it can interact with Gαq, although less effectively than either PLCβ1ct or RGS2.
As a control for expression of PLCβ1ct, RGS2, and RGS8, we used the deletion mutant ΔRGS8 (Melliti et al., 1999). This mutant lacks amino acids Phe57-Pro160, which constitute the major portion of the conserved RGS core domain (Berman and Gilman, 1998). The RGS core domain mediates binding of RGS proteins to the switch regions of Gα (Tesmer et al., 1997). We have previously shown that ΔRGS8 does not act as a GAP to attenuate N-type Ca channel inhibition by Gαi subfamily proteins (Melliti et al., 1999). As summarized in Figure 5, coexpression of ΔRGS8 had no effect on the modulation of α1E through M1 receptors. Thus, the RGS core domain is apparently necessary for the observed effects of RGS2 and RGS8 in our experiments.
In summary, only stimulation of α1E was attenuated by PLCβ1ct, RGS2, and RGS8. Notably, these proteins failed to reduce inhibition of α1E through M1 receptors (Fig. 5). The selective block of stimulation by PLCβ1ct, RGS2, and RGS8 cannot be explained by the GAP activity of these proteins, because simply accelerating GTP hydrolysis should have attenuated signaling by both Gα and Gβγ and should have reduced both stimulation and inhibition of α1E. However, the selective block of stimulation is consistent with the interpretation that PLCβ1ct, RGS2, and RGS8 functioned primarily as effector antagonists in our experiments.
RGS2 prevents GTP-γ-S-mediated stimulation of α1E
Previous studies have found that RGS2 and RGS4 reduce signaling by Gαq activated with GTP-γ-S (Hepler et al., 1997; Heximer et al., 1997). GTP-γ-S is nonhydrolyzable; thus, these effects of RGS2 and RGS4 cannot be attributed to their GAP activities. Rather, it is thought that RGS2 and RGS4 can function as effector antagonists by binding to the switch regions of Gαq, thereby blocking its interactions with downstream effectors. To further examine whether PLCβ1ct, RGS2, and RGS8 could have functioned as effector antagonists in our experiments, we used GTP-γ-S to produce stimulation of α1E. These experiments were performed on cells coexpressing α1E Ca channels and M1 receptors and incubated with PTX (200–500 ng/ml) overnight to inactivate Gαi/o proteins. Equimolar GTP-γ-S was substituted for GTP in the pipette solution, and α1E current amplitudes were monitored as a function of time after establishing the whole-cell configuration (i.e., break-in). As shown in Figure6, intracellular dialysis with GTP-γ-S significantly increased α1E currents in control cells. The magnitude of this increase was comparable (∼40%) with that produced by coexpressed M1 receptors (compare Fig. 1). By contrast, GTP-γ-S failed to stimulate α1E currents in cells coexpressing RGS2 (Fig. 6). Apparently, GTP-γ-S activated the stimulatory pathway in control cells but not in cells coexpressing RGS2. Because GTP-γ-S is nonhydrolyzable, RGS2 cannot have blocked the stimulatory pathway by functioning as a GAP; it must have functioned as an effector antagonist. These results support our hypothesis that PLCβ1ct, RGS2, and RGS8 selectively blocked stimulation of α1E by acting as effector antagonists.
rbE-II is stimulated through muscarinic receptors
Previous studies have demonstrated that rbE-II, an α1E subunit cloned from rat hippocampus (Soong et al., 1993), is insensitive to inhibition through μ-opioid and dopamine receptors (Bourinet et al., 1996; Page et al., 1998). It was recently shown by Page et al. (1998)that an N-terminal domain within α1B and α1E subunits is essential for their voltage-dependent inhibition by Gβγ dimers. rbE-II lacks this domain, which accounts for its insensitivity to voltage-dependent inhibition (Page et al., 1998).
To determine whether rbE-II can undergo muscarinic stimulation, we coexpressed it with M1 or M2 receptors. In agreement with Bourinet et al. (1996) and Page et al. (1998), we found that rbE-II displayed no appreciable voltage-dependent inhibition through either muscarinic receptor. However, rbE-II was prominently stimulated through both receptors. In response to M2 receptors, rbE-II exhibited stimulation that simply reversed after CCh washout (Fig.7A). However, with M1 receptors rbE-II currents often exhibited a secondary increase after CCh washout (Fig. 7B). The mechanism of this secondary increase is presently unclear; further work is being done.
Results obtained with rbE-II Ca channels are summarized in Figure7C. Altogether, rbE-II currents were stimulated by 35 ± 11% (n = 10) through M2 receptors and by 55 ± 8% (n = 13) through M1 receptors (only the primary phase of stimulation is included in the latter measurement). The larger stimulation of rbE-II through the M1 receptor is consistent with the hypothesis that stimulation is mediated by Gαq, because M1 receptors couple preferentially to this Gα subunit (Felder, 1995). Stimulation of rbE-II was statistically indistinguishable (p> 0.1) from stimulation of the rabbit α1E Ca channel [19 ± 3% through M2 receptors (n = 11) and 40 ± 6% through M1 receptors (n = 17)]. In summary, these experiments demonstrate that rbE-II Ca channels are significantly stimulated through M1 and M2 muscarinic receptors. Because rbE-II lacks the N-terminal domain essential for voltage-dependent, Gβγ-mediated inhibition (Page et al., 1998), this domain apparently is not involved in the muscarinic stimulation of α1E Ca channels.
DISCUSSION
We have shown that α1E Ca channels are strongly stimulated and only weakly inhibited through M1 muscarinic acetylcholine receptors. Both forms of channel modulation are insensitive to PTX. Stimulation of α1E is blocked by PLCβ1ct and RGS2, two proteins previously demonstrated to function as GAPs for Gαq (Berstein et al., 1992;Paulssen et al., 1996; Heximer et al., 1997). In contrast, stimulation is unaffected by βARK1ct, which sequesters Gβγ subunits and which blocks inhibition of α1E through M1 receptors (Figs. 4C,5). Together these results indicate that stimulation of α1E involves signaling by a PTX-insensitive Gα subunit. Because M1 receptors preferentially couple to heterotrimeric G-proteins of the Gαq subfamily (Felder, 1995), we hypothesize that stimulation is mediated through Gαq. Our results predict that native R-type Ca channels are predominantly stimulated through endogenous muscarinic and possibly other Gαq-coupled receptors. This possibility is supported by the strong muscarinic stimulation of rbE-II Ca channels (Fig. 7).
The finding that βARK1ct blocks inhibition of α1E suggests that inhibition is mediated by Gβγ dimers (Figs. 3C, 5). Notably, M1 receptors produce relatively weak inhibition of α1E (compare Figs. 1, 5). Thus, even in staurosporine-treated cells where stimulation was prevented and inhibition was consequently not underestimated, α1E currents were inhibited by only ∼18% (Fig. 2). In comparison, the M2 subtype of muscarinic receptor produces ∼40% inhibition of α1E Ca currents under identical experimental conditions (Meza et al., 1999). The weak inhibition of α1E through M1 receptors may reflect the type of Gβγ dimer involved. Previously, Fletcher et al. (1998) found that Gβ5 preferentially associates with Gαq, suggesting that Gαq-coupled receptors such as the M1 receptor will liberate Gβγ dimers containing Gβ5 (assuming that Gβ5 is present in HEK293 cells). Because Gβ5 produces relatively weak voltage-dependent inhibition of native N-type Ca channels (García et al., 1998; Ruiz-Velasco and Ikeda, 2000), it may also produce relatively weak inhibition of R-type Ca channels formed by α1E.
PLCβ1ct, RGS2, and RGS8 functioned primarily as effector antagonists
PLCβ1ct, RGS2, and RGS8 blocked stimulation of α1E without also reducing its inhibition through M1 receptors (Fig. 5). The selective block of stimulation is surprising, given that PLCβ1 and RGS2 have been previously shown to act as powerful GAPs for Gαqin vitro (Berstein et al., 1992; Biddlecome et al., 1996;Ingi et al., 1998). If PLCβ1ct and RGS2 had behaved mainly as GAPs, they would have accelerated conversion of Gα-GTP into Gα-GDP, thereby promoting reassociation of Gα with its Gβγ dimer (Berman and Gilman, 1998). In this event, both inhibition and stimulation of α1E would have been attenuated. That only stimulation was reduced argues that PLCβ1ct, RGS2, and RGS8 functioned primarily as effector antagonists in our experiments. This interpretation is supported by results obtained using GTP-γ-S. We found that GTP-γ-S produced stimulation of α1E in control cells but not in cells coexpressing RGS2 (Fig. 6). Because GTP-γ-S cannot be hydrolyzed by Gα subunits, RGS2 must have functioned exclusively as an effector antagonist to prevent stimulation of α1E in these experiments (Fig. 6). However, these experiments with GTP-γ-S do not preclude the possibility that RGS2, RGS8, and PLCβ1ct also functioned as GAPs, in addition to functioning as effector antagonists, in experiments in which M1 receptors and hydrolyzable GTP produced stimulation of α1E (e.g., Fig. 4).
Previous studies have demonstrated that RGS2 and RGS4 can act as effector antagonists under certain conditions (Hepler et al., 1997;Heximer et al., 1997; Yan et al., 1997). In contrast, effector antagonism by PLCβ1 or RGS8 has not been previously reported. However, Kammermeier and Ikeda (1999) found that PLCβ1ct blocked the voltage-independent, Gαq-mediated component of N-type Ca channel inhibition in superior cervical ganglion neurons but left the voltage-dependent, Gβγ-mediated component of inhibition intact. Their experiments suggest that PLCβ1ct interfered with signaling by Gαq but not with signaling by its Gβγ dimer. These results ofKammermeier and Ikeda (1999) are also consistent with the idea that PLCβ1ct can function as an effector antagonist for Gαq.
Physical interactions between Gα subunits and RGS proteins take place at the switch regions of Gα, which are also involved in binding Gβγ dimers and downstream effectors such as PLCβ1 (Wall et al., 1995; Lambright et al., 1996; Tesmer et al., 1997; Berman and Gilman, 1998). It is therefore unlikely that Gα can associate with Gβγ while an RGS protein (or effector) is bound. Prolonged association between Gα and an RGS protein would be expected to delay heterotrimer formation and would be predicted to block signaling by Gα but to allow continued signaling by its Gβγ dimer. This mechanism was recently proposed by Bünemann and Hosey (1998) to explain the apparently increased availability of Gβγ dimers in cells overexpressing RGS4. Prolonged association between Gα and an RGS protein might result if GTP hydrolysis was relatively slow or if the RGS protein dissociated slowly from Gα after GTP hydrolysis (Berman and Gilman, 1998). Alternatively, an RGS protein might remain associated with Gα over multiple GTPase cycles, as proposed for PLCβ1 and Gαq (Biddlecome et al., 1996). In this latter example, an RGS protein (or PLCβ1) could function simultaneously, or alternately, as an effector antagonist and as a GAP.
Previously, we demonstrated that RGS proteins shift the dose–response curve for Ca channel inhibition to higher agonist concentrations (Melliti et al., 1999). RGS proteins have also been shown to accelerate recovery of Ca channels from inhibition after agonist washout (Jeong and Ikeda, 1998; Melliti et al., 1999). These effects of RGS proteins can be adequately explained by their GAP activity. In these two previous studies, Ca channels were inhibited by PTX-sensitive G-proteins belonging to the Gαi subfamily (Jeong and Ikeda, 1998;Melliti et al., 1999). In the present study, channel modulation was mediated by PTX-insensitive G-proteins (probably Gαq), and the coexpressed RGS proteins (and PLCβ1ct) appeared to function primarily as effector antagonists. Thus, whether Gα-interacting proteins such as RGS and PLCβ1 behave mainly as GAPs or as effector antagonists might depend on the Gα subunit involved. Consistent with this idea, most previous studies have used Gαq to reveal the effector antagonist function of RGS proteins (Hepler et al., 1997; Heximer et al., 1997;Yan et al., 1997; Kammermeier and Ikeda, 1999).
Our present results are the first demonstration that RGS proteins (and PLCβ1ct) can influence receptor-mediated stimulation, as opposed to inhibition, of voltage-gated Ca channels. Our findings contribute to a growing body of evidence that RGS and other Gα-interacting proteins play important roles in ion channel modulation (cf. Doupnik et al., 1997; Saitoh et al., 1997; Bünemann and Hosey, 1998; Jeong and Ikeda, 1998; Herlitze et al., 1999; Kammermeier and Ikeda, 1999;Melliti et al., 1999).
Physiological significance of α1E Ca channel stimulation
Muscarinic receptors are widely expressed in mammalian brain and are prevalent in hippocampus, dentate gyrus, amygdala, and cortex (Buckley et al., 1988). These same regions of the brain also express α1E subunits (Niidome et al., 1992; Soong et al., 1993; Wakamori et al., 1994; Williams et al., 1994; Yokoyama et al., 1995). Additionally, muscarinic receptors and α1E subunits are both found on neuronal somata and dendrites (Hersch et al., 1994; Yokoyama et al., 1995;Westenbroek et al., 1998). Thus, we speculate that native R-type Ca channels are modulated through muscarinic receptors in central neurons.
Only a few studies have examined receptor-mediated modulation of native R-type Ca channels. Jeong and Wurster (1997) observed muscarinic inhibition of R-type currents in intracardiac neurons, and Overholt and Prabhakar (1999) found adrenergic inhibition of R-type currents in carotid body glomus cells. To our knowledge, receptor-mediated stimulation of native R-type Ca channels has not been reported. However, our present results demonstrate that α1E Ca channels are strongly stimulated through Gαq-coupled M1 receptors. It seems likely that native R-type Ca channels are also stimulated through muscarinic and perhaps other Gαq-coupled receptors in vivo. Stimulation of native R-type currents may have important consequences for dendritic Ca signaling, neurosecretion, gene expression, or other neuronal functions.
Footnotes
This work was supported by National Institutes of Health Grant NS34423 to B.A., American Heart Association Established Investigator Award 0040067N to B.A., and Consejo Nacional de Ciencia y Tecnologia Grant 31391-N to U.M. K.M. was the recipient of a fellowship from the Philippe Foundation.
Correspondence should be addressed to Brett Adams, Department of Biology, 5305 Old Main Hill, Utah State University, Logan, UT 84322-5305. E-mail: brett@biology.usu.edu.
REFERENCES
- 1.Allen TGJ. The role of N-, Q- and R-type Ca2+ channels in feedback inhibition of ACh release from rat basal forebrain neurones. J Physiol (Lond) 1999;515:93–107. doi: 10.1111/j.1469-7793.1999.093ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avigan J, Murtagh JJ, Stevens LA, Angus CW, Moss J, Vaughan M. Pertussis toxin-catalyzed ADP-ribosylation of Goα with mutations at the carboxyl terminus. Biochemistry. 1992;31:7736–7740. doi: 10.1021/bi00148a039. [DOI] [PubMed] [Google Scholar]
- 3.Bean BP. Modulating modulation. J Gen Physiol. 2000;115:273–275. doi: 10.1085/jgp.115.3.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berman DM, Gilman AG. Mammalian RGS proteins: barbarians at the gate. J Biol Chem. 1998;273:1269–1272. doi: 10.1074/jbc.273.3.1269. [DOI] [PubMed] [Google Scholar]
- 5.Berstein G, Blank JL, Jhon D-Y, Exton JH, Rhee SG, Ross EM. Phospholipase C-β1 is a GTPase-activating protein for Gq/11, its physiological regulator. Cell. 1992;70:411–418. doi: 10.1016/0092-8674(92)90165-9. [DOI] [PubMed] [Google Scholar]
- 6.Biddlecome GH, Berstein G, Ross EM. Regulation of phospholipase C-β1 by Gq and m1 muscarinic cholinergic receptor. J Biol Chem. 1996;271:7999–8007. doi: 10.1074/jbc.271.14.7999. [DOI] [PubMed] [Google Scholar]
- 7.Bourinet E, Soong TW, Stea A, Snutch TP. Determinants of the G-protein-dependent opioid modulation of neuronal calcium channels. Proc Natl Acad Sci USA. 1996;93:1486–1491. doi: 10.1073/pnas.93.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buckley NJ, Bonner TI, Brann MR. Localization of a family of muscarinic receptor mRNAs in rat brain. J Neurosci. 1988;8:4646–4652. doi: 10.1523/JNEUROSCI.08-12-04646.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bünemann M, Hosey MM. Regulators of G protein signaling (RGS) proteins constitutively activate Gβγ-gated potassium channels. J Biol Chem. 1998;273:31186–31190. doi: 10.1074/jbc.273.47.31186. [DOI] [PubMed] [Google Scholar]
- 10.Chen C, Zheng B, Han J, Lin SC. Characterization of a novel mammalian RGS protein that binds to Gα proteins and inhibits pheromone signaling in yeast. J Biol Chem. 1997;272:8679–8685. doi: 10.1074/jbc.272.13.8679. [DOI] [PubMed] [Google Scholar]
- 11.Doupnik GA, Davidson N, Lester HA, Kofuji P. RGS proteins reconstitute the rapid gating kinetics of Gβγ-activated inwardly rectifying K+ channels. Proc Natl Acad Sci USA. 1997;94:10461–10466. doi: 10.1073/pnas.94.19.10461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Felder CC. Muscarinic acetylcholine receptors: signal transduction through multiple effectors. FASEB J. 1995;9:619–625. [PubMed] [Google Scholar]
- 13.Fletcher JE, Lindorfer MA, DeFilippo JM, Yasuda H, Guilmard M, Garrison JC. The G protein β5 subunit interacts selectively with Gq α subunit. J Biol Chem. 1998;273:636–644. doi: 10.1074/jbc.273.1.636. [DOI] [PubMed] [Google Scholar]
- 14.García DE, Li B, García-Ferreiro RE, Hernández-Ochoa EO, Yang K, Gautam N, Catterall WA, Mackie K, Hille B. G-protein β-subunit specificity in the fast membrane-delimited inhibition of Ca channels. J Neurosci. 1998;18:9163–9170. doi: 10.1523/JNEUROSCI.18-22-09163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hepler JR, Berman DM, Gilman AG, Kozasa T. RGS4 and GAIP are GTPase-activating proteins for Gqα and block activation of phospholipase Cβ by γ-thio-GTP-Gqα. Proc Natl Acad Sci USA. 1997;94:428–432. doi: 10.1073/pnas.94.2.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herlitze S, Ruppersberg JP, Mark MD. New roles for RGS2, 5 and 8 on the ratio-dependent modulation of recombinant GIRK channels expressed in Xenopus oocytes. J Physiol (Lond) 1999;517:341–352. doi: 10.1111/j.1469-7793.1999.0341t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hersch SM, Gutekunst CA, Rees HD, Heilman CJ, Levey AI. Distribution of m1–m4 muscarinic receptor proteins in the rat striatum: light and electron microscopic immunocytochemistry using subtype-specific antibodies. J Neurosci. 1994;14:3351–3363. doi: 10.1523/JNEUROSCI.14-05-03351.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heximer SP, Watson N, Linder ME, Blumer KJ, Hepler JR. RGS2/GOS8 is a selective inhibitor of Gqα function. Proc Natl Acad Sci USA. 1997;94:14389–14393. doi: 10.1073/pnas.94.26.14389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hille B. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 20.Ikeda SR, Dunlap K. Voltage-dependent modulation of N-type calcium channels: role of G protein subunits. Adv Second Messenger Phosphoprotein Res. 1999;33:131–151. doi: 10.1016/s1040-7952(99)80008-1. [DOI] [PubMed] [Google Scholar]
- 21.Ingi T, Krumins AM, Chidiac P, Brothers GM, Chung S, Snow BE, Barnes CA, Lanahan AA, Siderovski DP, Ross EM, Gilman AG, Worley PF. Dynamic regulation of RGS2 suggests a novel mechanism in G-protein signaling and neuronal plasticity. J Neurosci. 1998;18:7178–7188. doi: 10.1523/JNEUROSCI.18-18-07178.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jeong SW, Ikeda SR. G protein α subunit Gαz couples neurotransmitter receptors to ion channels in sympathetic neurons. Neuron. 1998;21:1201–1212. doi: 10.1016/s0896-6273(00)80636-4. [DOI] [PubMed] [Google Scholar]
- 23.Jeong SW, Wurster RD. Muscarinic receptor activation modulates Ca2+ channels in rat intracardiac neurons via a PTX- and voltage-sensitive pathway. J Neurophysiol. 1997;78:1476–1490. doi: 10.1152/jn.1997.78.3.1476. [DOI] [PubMed] [Google Scholar]
- 24.Jones SW, Elmslie KS. Transmitter modulation of neuronal calcium channels. J Membr Biol. 1997;155:1–10. doi: 10.1007/s002329900153. [DOI] [PubMed] [Google Scholar]
- 25.Kammermeier PJ, Ikeda SR. Expression of RGS2 alters the coupling of metabotropic glutamate receptor 1a to M-type K+ and N-type Ca2+ channels. Neuron. 1999;22:819–829. doi: 10.1016/s0896-6273(00)80740-0. [DOI] [PubMed] [Google Scholar]
- 26.Kavalali ET, Zhuo M, Bito H, Tsien RW. Dendritic Ca channels characterized by recordings from isolated hippocampal dendritic segments. Neuron. 1997;18:651–663. doi: 10.1016/s0896-6273(00)80305-0. [DOI] [PubMed] [Google Scholar]
- 27.Koch WJ, Hawes BE, Inglese J, Luttrell LM, Lefkowitz RJ. Cellular expression of the carboxyl terminus of a G protein-coupled receptor kinase attenuates Gβγ-mediated signaling. J Biol Chem. 1994;269:6193–6197. [PubMed] [Google Scholar]
- 28.Lambright DG, Sondek J, Bohm A, Skiba NP, Hamm HE, Sigler PB. The 2.0 A crystal structure of a heterotrimeric G protein. Nature. 1996;379:311–319. doi: 10.1038/379311a0. [DOI] [PubMed] [Google Scholar]
- 29.Melliti K, Meza U, Fisher R, Adams B. Regulators of G protein signaling attenuate the G protein-mediated inhibition of N-type Ca channels. J Gen Physiol. 1999;113:97–109. doi: 10.1085/jgp.113.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meza U, Bannister R, Melliti K, Adams B. Biphasic, opposing modulation of cloned neuronal α1E Ca channels by distinct signaling pathways coupled to M2 muscarinic acetylcholine receptors. J Neurosci. 1999;19:6806–6817. doi: 10.1523/JNEUROSCI.19-16-06806.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Newcomb R, Szoke B, Palma A, Wang G, Chen Xh, Hopkins W, Cong R, Miller J, Urge L, Tarczy-Hornoch K, Loo JA, Dooley DJ, Nadasdi L, Tsien RW, Lemos J, Miljanich G. Selective peptide antagonist of the class E calcium channel from the venom of the tarantula Hysterocrates gigas. Biochemistry. 1998;37:15353–15362. doi: 10.1021/bi981255g. [DOI] [PubMed] [Google Scholar]
- 32.Niidome T, Kim M-S, Friedrich T, Mori Y. Molecular cloning and characterization of a novel calcium channel from rabbit brain. FEBS Lett. 1992;308:7–13. doi: 10.1016/0014-5793(92)81038-n. [DOI] [PubMed] [Google Scholar]
- 33.Overholt JL, Prabhakar NR. Norepinephrine inhibits a toxin resistant Ca2+ current in carotid body glomus cells: evidence for a direct G protein mechanism. J Neurophysiol. 1999;81:225–233. doi: 10.1152/jn.1999.81.1.225. [DOI] [PubMed] [Google Scholar]
- 34.Page KM, Cantí C, Stephens GJ, Berrow NS, Dolphin AC. Identification of the amino terminus of neuronal Ca2+ channel α1 subunits α1B and α1E as an essential determinant of G-protein modulation. J Neurosci. 1998;18:4815–4824. doi: 10.1523/JNEUROSCI.18-13-04815.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paulssen RH, Woodson J, Liu Z, Ross EM. Carboxyl-terminal fragments of phospholipase C-β1 with intrinsic Gq GTPase-activating protein (GAP) activity. J Biol Chem. 1996;271:26622–26629. doi: 10.1074/jbc.271.43.26622. [DOI] [PubMed] [Google Scholar]
- 36.Peralta EG, Ashkenazi A, Winslow JW, Smith DH, Ramachandran J, Capon DJ. Distinct primary structures, ligand-binding properties and tissue-specific expression of four human muscarinic acetylcholine receptors. EMBO J. 1987;6:3923–3929. doi: 10.1002/j.1460-2075.1987.tb02733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pereverzev A, Klöckner U, Henry M, Grabsch H, Vajna R, Olyschläger S, Viatchenko-Karpinski S, Schröder R, Hescheler J, Schneider T. Structural diversity of the voltage-dependent Ca2+ channel α1E-subunit. Eur J Neurosci. 1998;10:916–925. doi: 10.1046/j.1460-9568.1998.00099.x. [DOI] [PubMed] [Google Scholar]
- 38.Piedras-Rentería ES, Tsien RW. Antisense oligonucleotides against α1E reduce R-type calcium currents in cerebellar granule cells. Proc Natl Acad Sci USA. 1998;95:7760–7765. doi: 10.1073/pnas.95.13.7760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Randall A, Tsien RW. Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J Neurosci. 1995;15:2995–3012. doi: 10.1523/JNEUROSCI.15-04-02995.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruiz-Velasco V, Ikeda SR. Multiple G-protein βγ combinations produce voltage-dependent inhibition of N-type calcium channels in rat superior cervical ganglion neurons. J Neurosci. 2000;20:2183–3191. doi: 10.1523/JNEUROSCI.20-06-02183.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saitoh O, Kubo Y, Miyatani Y, Asano T, Nakata H. RGS8 accelerates G-protein-mediated modulation of K+ currents. Nature. 1997;390:525–529. doi: 10.1038/37385. [DOI] [PubMed] [Google Scholar]
- 42.Soong TW, Stea A, Hodson CD, Dubel SJ, Vincent SR, Snutch TP. Structure and functional expression of a member of the low voltage-activated calcium channel family. Science. 1993;260:1133–1136. doi: 10.1126/science.8388125. [DOI] [PubMed] [Google Scholar]
- 43.Stea A, Soong TW, Snutch TP. Determinants of PKC-dependent modulation of a family of neuronal calcium channels. Neuron. 1995;15:929–940. doi: 10.1016/0896-6273(95)90183-3. [DOI] [PubMed] [Google Scholar]
- 44.Stehno-Bittel L, Krapivinski G, Krapivinski L, Perez-Terzic C, Clapham DE. The G protein βγ subunit transduces the muscarinic receptor signal for Ca2+ release in Xenopus oocytes. J Biol Chem. 1995;270:30068–30074. doi: 10.1074/jbc.270.50.30068. [DOI] [PubMed] [Google Scholar]
- 45.Tesmer JJG, Berman DM, Gilman AG, Sprang SR. Structure of RGS4 bound to AlF4--activated Giα1: stabilization of the transition state for GTP hydrolysis. Cell. 1997;89:251–261. doi: 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- 46.Tottene A, Volsen S, Pietrobon D. α1E subunits form the pore of three cerebellar R-type calcium channels with different pharmacological and permeation properties. J Neurosci. 2000;20:171–178. doi: 10.1523/JNEUROSCI.20-01-00171.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Turner TJ, Lampe RA, Dunlap K. Characterization of presynaptic calcium channels with ω-conotoxin MVIIC and ω-grammotoxin SIA: role for a resistant calcium channel type in neurosecretion. Mol Pharmacol. 1995;47:348–353. [PubMed] [Google Scholar]
- 48.Wakamori M, Niidome T, Furutama D, Furuichi T, Mikoshiba K, Fujita Y, Tanaka I, Katayama K, Yatani A, Schwartz A, Mori Y. Distinctive functional properties of the neuronal BII (class E) calcium channel. Receptors Channels. 1994;2:303–314. [PubMed] [Google Scholar]
- 49.Wall MA, Coleman DE, Lee E, Iniguez-Lluhi JA, Posner BA, Gilman AG, Sprang SR. The structure of the G protein heterotrimer Gi alpha 1 beta 1 gamma 2. Cell. 1995;83:1047–1058. doi: 10.1016/0092-8674(95)90220-1. [DOI] [PubMed] [Google Scholar]
- 50.Wang G, Dayanithi G, Newcomb R, Lemos JR. An R-type Ca2+ current in neurohypophysial terminals preferentially regulates oxytocin secretion. J Neurosci. 1999;19:9235–9241. doi: 10.1523/JNEUROSCI.19-21-09235.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.West RE, Moss J, Vaughan M, Liu T, Liu T-Y. Pertussis toxin-catalyzed ADP-ribosylation of transducin: cysteine 347 is the ADP-ribose acceptor site. J Biol Chem. 1985;260:14428–14430. [PubMed] [Google Scholar]
- 52.Westenbroek RE, Hoskins L, Catterall WA. Localization of Ca channel subtypes on rat spinal motor neurons, interneurons, and nerve terminals. J Neurosci. 1998;18:6319–6330. doi: 10.1523/JNEUROSCI.18-16-06319.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Williams ME, Marubio LM, Deal CR, Hans M, Brust PF, Philipson LH, Miller RJ, Johnson EC, Harpold MM, Ellis SB. Structure and functional characterization of neuronal α1E calcium channel subtypes. J Biol Chem. 1994;269:22347–22357. [PubMed] [Google Scholar]
- 54.Wu D, Jiang H, Katz A, Simon MI. Identification of critical regions on phospholipase C-β1 required for activation by G-proteins. J Biol Chem. 1993;268:3704–3709. [PubMed] [Google Scholar]
- 55.Wu L-G, Borst JGG, Sakmann B. R-type Ca2+ currents evoke transmitter release at a rat central synapses. Proc Natl Acad Sci USA. 1998;95:4720–4725. doi: 10.1073/pnas.95.8.4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu L-G, Westenbroek RE, Borst JGG, Catterall WA, Sakmann B. Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx-type synapses. J Neurosci. 1999;19:726–736. doi: 10.1523/JNEUROSCI.19-02-00726.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yan Y, Chi PP, Bourne HR. RGS4 inhibits Gq-mediated activation of mitogen-activated protein kinase and phosphoinositide synthesis. J Biol Chem. 1997;272:11924–11927. doi: 10.1074/jbc.272.18.11924. [DOI] [PubMed] [Google Scholar]
- 58.Yokoyama CT, Westenbroek RE, Hell JW, Soong TW, Snutch TP, Catterall WA. Biochemical properties and subcellular distribution of the neuronal class E calcium channel α1 subunit. J Neurosci. 1995;15:6419–6432. doi: 10.1523/JNEUROSCI.15-10-06419.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zamponi GW, Snutch TP. Modulation of voltage-dependent calcium channels by G proteins. Curr Opin Neurobiol. 1998;8:351–356. doi: 10.1016/s0959-4388(98)80060-3. [DOI] [PubMed] [Google Scholar]