

Abstract
Neuregulins have been implicated in a number of events in cells in the oligodendrocyte lineage, including enhanced survival, mitosis, migration, and differentiation. At least two signaling pathways have been shown to be involved in neuregulin signaling: the phosphatidylinositol (PI)-3 kinase and the mitogen-activated protein kinase pathways. In the present studies, we examined the signaling pathway involved in the survival function of heregulin, focusing on heregulin-induced changes in Akt activity in cultured glial cells, and the consequences of Akt activation in cells in the oligodendrocyte lineage. Heregulin binds erbB receptors, and in our studies, primary cultures of both oligodendrocyte progenitor cells and differentiating oligodendrocytes expressed erbB2, erbB3, and erbB4 receptors. In C6 glioma cells and primary cultures of oligodendrocytes, heregulin induced time- and dose-dependent Akt phosphorylation at Ser473 in a wortmannin-sensitive manner. To investigate further the signaling pathway for heregulin in glial cells, BAD was overexpressed in C6 glioma cells. In these cells, heregulin induced phosphorylation of BAD at Ser136. Apoptosis of oligodendrocyte progenitor cells induced by growth factor deprivation was effectively blocked by heregulin in a wortmannin-sensitive manner. Overexpression of dominant negative Akt but not of wild-type Akt by adenoviral gene transfer in primary cultures of both oligodendrocytes and their progenitors induced significant apoptosis through activation of the caspase cascade. The present data suggest that the survival function of heregulin is mediated through the PI-3 kinase/Akt pathway in cells in the oligodendrocyte lineage and that the Akt pathway may be quite important for survival of cells in this lineage.
Keywords: oligodendrocyte, survival, Akt, neuregulin, multiple sclerosis, apoptosis
Oligodendrocyte loss and demyelination in the CNS have been proposed to result from apoptotic cell death during normal development (Barres et al., 1992), after growth factor withdrawal in vitro (Yasuda et al., 1995), and in pathological conditions such as multiple sclerosis (MS), Theiler's murine encephalomyelitis virus (TMEV) infection, and experimental allergic encephalitis (EAE; Raine and Scheinberg, 1988; Ozawa et al., 1994;Tsunoda and Fujinami, 1996; Hisahara et al., 2000). Growth factors and receptors can protect cells from apoptosis by blocking cell death signals (Raff et al., 1993). The neuregulin/erbB receptor system seems an ideal candidate to provide survival signals for oligodendrocytes in vivo. In the CNS, neurons, oligodendrocytes, and astrocytes both produce neuregulins and express neuregulin receptors (Chen et al., 1994; Meyer and Birchmeier, 1994;Raabe et al., 1997a; Francis et al., 1999). Furthermore, neuregulins present on the surface of axons play a role in the maintenance of oligodendrocytes and their progenitors (Canoll et al., 1996; Vartanian et al., 1997).
The neuregulins (NRGs) are a class of epidermal growth factor-like molecules that arise by alternative splicing from a single gene (NRG-1). Recently, three other neuregulin-like genes have been described: NRG-2 (Carraway et al., 1997; Chang et al., 1997), NRG-3 (Zhang et al., 1997), and NRG-4 (Harari et al., 1999). Targeted disruptions of the NRG-1 gene as well as the neuregulin receptors demonstrate that neuregulins are essential for the formation of the heart and nervous system (Meyer and Birchmeier, 1995; Kramer et al., 1996; Erickson et al., 1997;Riethmacher et al., 1997; Vartanian et al., 1999; Golding et al., 2000; Lin et al., 2000; Wolpowitz et al., 2000).
The NRG-1/neuregulin family includes heregulin, acetylcholine receptor inducing activity, neu differentiation factor, glial growth factor (GGF), and sensorimotor-derived factor (Burden and Yarden, 1997; Meyer et al., 1997). The heregulin (HRG) members contain two isoforms differing in their EGF-like domain (isoforms: α, β). The α- and β-variants bind differentially to receptors and elicit different biological responses (Marikovsky et al., 1995; Baek and Kim, 1998;Crovello et al., 1998). NRG-1 proteins mediate their action through erbB receptor tyrosine kinases, including erbB2, erbB3 and erbB4.
Although they are clearly involved in cell survival, neuregulins have other effects in the oligodendrocyte lineage, including proliferation or blockage of differentiation (Lemke, 1996;Burden and Yarden, 1997). Also, they have been demonstrated to signal through both the phosphatidylinositol (PI)-3 kinase and the MAP kinase pathways (Canoll et al., 1999). The precise signaling pathways through which neuregulins induce survival in oligodendrocyte lineage cells remain to be established. Thus, understanding which pathway is primarily involved in their survival function for oligodendrocytes and their progenitors is an important question. Because activation of PI-3 kinase/Akt pathway plays a major role in survival signals for multiple growth factors (Dudek et al., 1997; Franke et al., 1995,1997; Kennedy et al., 1997), we have focused this study on whether neuregulin induces cell survival in different stages of the oligodendrocyte lineage through the PI-3 kinase/Akt pathway.
MATERIALS AND METHODS
Reagents. Recombinant heregulin (HRG β1) was obtained from NeoMarkers (Fremont, CA). ErbB3 and erbB4 receptor polyclonal antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA), and erbB2 polyclonal antibody was obtained from NeoMarkers. Anti-total Akt, anti-phospho Akt (Ser473), anti-phospho BAD (Ser112), anti-phospho BAD (Ser136) and anti-total BAD were obtained from New England Biolabs (Beverly, MA). Monoclonal anti-A2B5 antibody was obtained from Roche Molecular Biochemicals (Indianapolis, IN). Polyclonal anti-NG2 antibody was provided by W. B. Stallcup (The Burnham Institute, La Jolla Cancer Research Center, La Jolla, CA). Wortmannin, PD98059, Rapamycin, and SB203580 were obtained from Calbiochem. l-NAME and d-NAME were purchased from Sigma (St. Louis, MO). C6 glioma cells were obtained from American Type Culture Collection (Manassas, VA).
Cell culture, transfections, and stimulation protocols. The C6 glioma cells were grown in DMEM containing 10% fetal bovine serum (Life Technologies, Gaithersburg, MD), plus penicillin and streptomycin at 37°C in 5% CO2 environment. For stimulation with HRG β1, 2 × 106 cells in a 100 mm plate were cultured in serum-free DMEM for 20–24 hr and stimulated as indicated in the figure legends with HRG β1, insulin-like growth factor-1 (IGF-1), or 0.1 mm pervanadate prepared as indicated in Andjelkovic et al. (1997). Cells were exposed to inhibitors for 30 min before HRG β1 addition.
For transient transfections, cells seeded at 1 × 106/100 mm dishes were transfected with 4 μg of plasmid using the Lipofectamine plus reagent (Life Technologies) according to the manufacturer's instructions. Sixteen hours after transfection, the cells were serum-starved for 20–24 hr before HRG β1 stimulation.
Isolation of oligodendrocytes. Primary cultures of rat oligodendrocyte progenitors were prepared with slight modifications to earlier studies (Fuss et al., 1997). Briefly, rat pups (postnatal days 2–3) were decapitated, and meninges were removed. Cortices were dissociated by incubation in 0.06% trypsin, 0.006% pancreatin, and subsequent trituration. Oligodendrocyte precursors were purified by immunopanning using the anti-A2B5 antibody (Eisenbarth et al., 1979). The purified oligodendrocyte precursors were collected by vigorous pipetting and plated at 1 × 106 cells/60 mm dishes coated with 30 μg/ml poly-l-lysine. The cells were cultured overnight in DMEM plus 10% FBS, and the medium was replaced with DMEM containing N2-supplement (Life Technologies) and platelet-derived growth factor (PDGF) (10 ng/ml) and basic fibroblast growth factor (bFGF) (10 ng/ml) for oligodendrocyte progenitors or PDGF (10 ng/ml) and T3 (50 ng/ml) for immature oligodendrocytes. Cells were maintained in that medium for 2–3 d. Mature oligodendrocytes were obtained after a further incubation in CNTF (10 ng/ml) and T3 (50 ng/ml). Before HRG β1 stimulation, the oligodendrocyte progenitors were cultured in DMEM plus 0.5% FBS for an additional 20–24 hr. Cells were exposed to HRG β1 for 30 min.
Adenovirus infection. Adenovirus infection was performed on oligodendrocyte progenitors or immature oligodendrocytes 24 hr after incubation in PDGF/bFGF or PDGF/T3, respectively, with 10 or 50 multiplicity of infection (MOI) as described previously (Kitamura et al., 1998). In experiments on growth factor deprivation, the cells were cultured in growth factors as described above for 24 or 48 hr after infection and then incubated without growth factors or N2 supplements in DMEM plus 0.5% FBS for a further 20–24 hr before the TUNEL or caspase assays. In experiments on HRG β1 effects on oligodendrocytes, cells were removed from growth factors and N2 supplement and incubated in 150 ng/ml HRG β1 during and after viral infection, followed by caspase assay.
Western blot analysis. Cells were lysed in NP-40 lysis buffer (1% NP-40, 10% glycerol, 137 mm NaCl, 20 mm Tris-HCl, pH 7.4) containing 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 mm PMSF, 20 mm NaF, 1 mm Na PPI, and 1 mmNa3VO4. For Western blot analysis, proteins were separated by SDS-PAGE and transferred onto Immobilon-P membranes (Millipore, Bedford, MA). Incubation with the primary antibodies was performed overnight at 4°C. For detection, HRP-conjugated secondary antibodies were used (1:10,000) followed by ECL chemiluminescence development (Amersham, Piscataway, NJ).
Akt phosphorylation. Phosphorylation of Akt was analyzed by Western blots, using a rabbit polyclonal phosphospecific Akt (Ser473) antibody. Total Akt was detected by a rabbit polyclonal Akt antibody.
BAD phosphorylation. BAD-phosphorylation was examined using phospho BAD (Ser136) and phospho BAD (Ser112) antibodies after transient transfection of C6 glioma cells with a BAD eukaryotic expression plasmid (pEBG-MBAD; New England Biolabs). Transiently transfected cells were stimulated with HRG β1. BAD antibody was used for detection of total BAD.
Induction of apoptosis and TUNEL assay.Oligodendrocyte progenitor cell death was induced by growth factor deprivation for 12 or 24 hr in the absence of N2 supplement. Cell death was quantified with the APO-BRDU kit (Pharmingen, San Diego, CA) according to manufacturer's instructions. Bromolated nucleotides were detected by anti-BrdU conjugated to fluorescein, and the positive cells were determined by fluorescence-activated cell sorting (FACS) analysis.
In situ DNA fragmentation detection was performed with the ApopTag in situ apoptosis detection kit (Intergen, Purchase, NY) according to the manufacturer's instructions. Briefly, after infection, immature oligodendrocytes were stained with the monoclonal anti-O4 antibody (1:200) for 30 min at room temperature before fixation. The cells were fixed in 4% paraformaldehyde for 1 hr at room temperature. Digoxigenin nucleotides were catalytically added to the DNA by terminal deoxynucleotidyl transferase (TdT) and detected by anti-digoxigenin antibody conjugated to fluorescein. The number of positive nuclei was determined by counting at least 15 random fields. The results were expressed as percentage of the total O4+ cells.
Caspase activity assay. Caspase activation was measured by CaspACE FITC-VAD-FMK in situ marker (Promega, Madison, WI) according to the manufacturer's instructions. Briefly, live oligodendrocyte progenitor cells infected with adenovirus as described above were treated with the FITC conjugate of the permeable irreversible caspase inhibitor (VAD-FMK) for 20 min to allow binding to activated caspase. Cells were either live-stained with a monoclonal anti-A2B5 antibody (1:100) before fixation or fixed and stained with NG2 antibody (1:200). TR-labeled secondary antibody was used at 1:200. The total number of cells was determined by DAPI staining. The number of positive cells was determined by counting at least 1000 cells per well. The results were expressed as percentage of the total cells ± SEM.
Immunocytochemistry. C6 glioma cells were plated at 80,000 cells/well in four-chamber slide (Becton Dickinson, Franklin Lakes, NJ). After fixation in 4% paraformaldehyde, cells were permeabilized in 0.3% Triton X-100 in PBS for 30 min at room temperature and stained with anti-phospho Akt antibody (1:100) overnight at 4°C. FITC-labeled secondary antibodies were used at 1:200.
Oligodendrocyte progenitors were plated at 0.25 × 106 cells/well in four-chamber slide. After incubation with anti-A2B5 antibody (1:200), the cells were fixed, permeabilized, and stained using anti-erbB2 (1:100), anti-erbB3 (1:200), or anti-erbB4 (1:200) overnight at 4°C. Cells were visualized using FITC- or TR-labeled secondary antibodies at 1:200.
RESULTS
HRG β1 activates Akt in glioma cells
Studies have demonstrated that neuregulins function through both a wortmannin-sensitive PI-3 kinase pathway and the MAP kinase pathway in cells in the oligodendrocyte lineage (Canoll et al., 1999), and that they can have several different effects on these cells. Thus, it is important to establish which pathways are involved with specific effects of the neuregulins. We therefore tested the hypothesis that neuregulins act as survival factors for cells in the oligodendrocyte lineage through activation of Akt.
To determine whether HRG β1 activates Akt in glial cells, we initially studied C6 glioma cells. As shown in Figure1A, after exposure to HRG β1, increased phosphorylation of Akt was detected on immunoblots using phospho-Ser473 specific Akt antibody. Densitometric analysis revealed a threefold to fourfold increase in Akt phosphorylation over basal level. The capacity for HRG β1 to phosphorylate Akt was comparable to that of IGF-I and pervanadate, which are well established activators of Akt, indicating that HRG β1 is a potent activator of Akt in this cell line. HRG β1-induced activation of Akt in C6 cells was dose-dependent. The effect of 100 ng/ml HRG β1 on Akt phosphorylation was comparable to that of 50 ng/ml IGF-I, and the effect was time-dependent (Fig.1B). Heregulin-induced phosphorylation of Akt at Ser473 was robust within 5 min after HRG β1 addition and it was sustained for 30 min, although it was somewhat reduced by 60 min (Fig. 1B).
Fig. 1.

Effect of HRG β1 on Akt in C6 glioma cells. A, C6 glioma cells were treated with HRG β1, IGF-1, and pervanadate for 30 min. Cell lysates were subjected to immunoblot with anti-phospho Akt (Ser473) (top panel) or anti-total Akt (bottom panel). HRG β1 dose-dependently promoted phosphorylation of Akt.B, C6 glioma cells were treated with 100 ng/ml HRG β1 at different times, and Akt phosphorylation was assessed by immunoblot with anti-phospho Akt (Ser473) antibody (top panel) or anti-total Akt (bottom panel). HRG β1 promoted Akt phosphorylation in a time-dependent manner.C, C6 glioma cells were treated with HRG β1, IGF-1, or pervanadate for 30 min and subjected to immunocytochemistry with anti-phospho Akt (Ser473) antibody. HRG β1 induced phosphorylation of Akt in a similar distribution and intensity in cells to that of IGF-1. Pervanadate induced more intense phosphorylation, which was localized in a rather discrete region inside the cells.D, GST-BAD cDNA was transfected into C6 glioma cells, and 48 hr later the cells were treated with 100 ng/ml HRG β1 for 30 min. Cells were subjected to immunoblot with anti-phospho-Ser473 specific Akt (top panel), anti-phospho-Ser136 specific BAD (middle panel), anti-BAD antibodies (bottom panel). HRG β1 promoted BAD phosphorylation at Ser136.
We next investigated the effect of inhibitors of various intracellular signaling mediators on HRG β1-induced Akt phosphorylation. We found that wortmannin (PI-3 kinase inhibitor), but not other kinase inhibitors PD98059 (MAP kinase inhibitor), or rapamycin (p70 S6k inhibitor), blocked the effect of HRG β1-induced Akt activation in C6 glioma cells (data not shown). Thus, in C6 cells, HRG β1 does not appear to function through MAP kinase to activate Akt. In addition, SB20358 (p38 inhibitor) or NOS inhibitors (d-NAME,l-NAME) had no effect on Akt activation, which is consistent with results that we and others (Franke et al., 1995; Kennedy et al., 1997; Reif et al., 1997) have obtained in nonglial cells (data not shown). Akt phosphorylation in C6 cells was also assessed immunocytochemically. As shown in Figure 1C, in cells with no treatment, phosphorylated Akt was barely detectable, but after treatment with HRG β1, diffuse cytoplasmic and nuclear staining of phospho-Akt in C6 glioma cells was seen. The intensity and distribution of phospho-Akt were similar in cells treated with IGF-I. In cells treated with pervanadate, phospho-Akt staining was more intense, and its distribution appeared confined to intracellular organelles. Thus, HRG β1 activated Akt in a similar manner to that of IGF-I, a physiological activator of Akt.
BAD, a cell death-executing molecule, has been reported to be a downstream substrate for Akt involved in survival. Thus, one of the mechanisms by which Akt protects cells from apoptosis is through BAD phosphorylation (Datta et al., 1997; del Peso et al., 1997). We determined that HRG β1 functions in C6 glioma cell survival through phosphorylation of BAD, by transfecting a GST-BAD cDNA into C6 cells, a standard technique for assessing the role of BAD, because it is normally at very low levels in cells (Datta et al., 1997; del Peso et al., 1997). We investigated the effect of HRG β1 on BAD phosphorylation at Ser136 using an antibody specific for this phosphoserine residue. As shown in Figure 1D, HRG β1 promoted BAD phosphorylation at Ser136, a known phosphorylation substrate for Akt, as well as Akt phosphorylation at Ser473. Total BAD protein amounts were similar among transfections with or without treatment with heregulin. As determined by immunoblot with anti-phospho-Ser112 BAD antibody, HRG β1 did not induce BAD phosphorylation at Ser112, a phosphorylation site not normally used by Akt (data not shown). Thus, it appears that in C6 glioma cells, HRG β1 activates Akt, leading to BAD phosphorylation at Ser136, as it does in other cells (Datta et al., 1997; del Peso et al., 1997; Basu et al., 1998). Taken together, these studies suggest that heregulin, acting through erbB receptors, activates Akt and induces phosphorylation of BAD, which would be expected to enhance cell survival.
HRG β1 activates Akt in the oligodendrocyte cell lineage
To assess whether HRG β1 activates Akt in the oligodendrocyte cell lineage directly, we next examined the induction of Akt activation by HRG β1 in primary cultures of oligodendrocyte progenitors. A2B5-positive oligodendrocyte progenitors were purified by immunopanning, and immunostaining with either NG2 or GFAP after 2–3 d in culture showed that they were 93–95% pure (data not shown). As in C6 cells, HRG β1 induced phosphorylation of Akt in a dose-dependent manner in oligodendrocyte progenitors (Fig.2A). Because the total amount of Akt was comparable among the cell preparations treated with different concentrations of HRG β1, the changes in Akt phosphorylation induced by HRG β1 resulted from altered phosphorylation, not different amounts of Akt substrate. The stimulatory effect of HRG β1 on Akt phosphorylation was blocked by pretreatment of cells with wortmannin but not with PD98059 or rapamycin. HRG β1 induced a time-dependent phosphorylation of Akt within 5 min but was reduced by 60 min (Fig. 2B). Thus, it appears that HRG β1 activates Akt in oligodendrocyte progenitors through PI-3 kinase activation, as it does in C6 glioma cells. Essentially all of the oligodendrocyte progenitor cells expressed erbB2, erbB3, and erbB4 receptors (Fig. 2C). ErbB receptors were located in both the plasma membrane and the cytoplasm in oligodendrocyte progenitors (note green stain interspersed with A2B5 stain on processes), which is consistent with results in other cell types (Kumar et al., 1991; Prigent et al., 1992; Francis et al., 1999). Thus, any of these receptors may be involved in the signaling of HRG β1 in oligodendrocyte progenitors.
Fig. 2.

Effect of HRG β1 on Akt in primary cultures of oligodendrocyte progenitors. A, Primary cultures of oligodendrocyte progenitor cells were treated with different concentrations of HRG β1 for 30 min or they were pretreated with different kinase inhibitors (20 μm PD098059, 200 nm wortmannin, and 20 ng/ml rapamycin), and then stimulated for an additional 30 min with 100 ng/ml HRG β1. Cell lysates were subjected to immunoblot with anti-phospho Akt (Ser473) (top panel) and anti-total Akt (bottom panel). HRG β1 dose-dependently promoted phosphorylation of Akt in primary cultures of oligodendrocytes progenitors, which was selectively blocked by wortmannin. B, Oligodendrocytes progenitors were treated with 100 ng/ml HRG β1 at different times, and Akt phosphorylation was assessed by immunoblot with anti-phospho Akt (Ser473) antibody (top panel) or anti-total Akt (bottom panel). HRG β1 promoted Akt phosphorylation in a time-dependent manner. C, Immunofluorescence microscopy of oligodendrocyte progenitor cultures double-stained with anti-A2B5 (red) and anti-erbB2, anti-erbB3, or anti-erbB4 antibodies (green) is shown. Oligodendrocytes progenitors express erbB2, erbB3, and erbB4 receptors.
We next investigated the effect of HRG β1 on Akt phosphorylation in primary cultures of immature and mature oligodendrocytes. HRG β1 induced phosphorylation of Akt in a dose-dependent manner in both immature and mature oligodendrocytes (Fig. 3A,C). As with our other studies, this phosphorylation was only inhibited by wortmannin but not by PD98059 or rapamycin (Fig. 3B, C). Additionally, HRG β1-induced Akt activation was time-dependent in mature oligodendrocytes. Thus, maximal activation was attained by 15 min, and it was sustained for at least 60 min (Fig. 3D). This contrasts with the effect of neuregulin in C6 cells and oligodendrocyte progenitor cells, in which the activation of Akt is attenuated by 60 min. These results suggest that HRG β1 activates Akt in multiple cell types within the oligodendrocyte lineage, although it may have more prolonged effects in mature oligodendrocytes, relative to other cells in the lineage. As seen in oligodendrocyte progenitors, mature oligodendrocytes also expressed erbB2, erbB3, and erbB4 receptors, in both cell bodies and processes (Fig.4).
Fig. 3.

Effect of HRG β1 on Akt in primary cultures of immature and mature oligodendrocytes. A, Primary cultures of immature oligodendrocytes were treated with different concentrations of HRG β1 or vanadate for 30 min. Cell lysates were subjected to immunoblot with anti-phospho Akt (Ser473) (top panel) or anti-total Akt (bottom panel). HRG β1 dose-dependently promoted phosphorylation of Akt.B, Primary cultures of immature oligodendrocytes were pretreated with various protein kinase inhibitors (20 μmPD098059, 200 nm wortmannin, 20 ng/ml rapamycin) for 30 min and then stimulated with 50 ng/ml HRG β1 for an additional 30 min. Cells lysates were subjected to immunoblot with anti-phospho Akt (Ser473) (top panel) or anti-total Akt (bottom panel). HRG β1-induced Akt phosphorylation was selectively blocked by wortmannin. C, Mature oligodendrocytes were treated with different concentrations of HRG β1 for 30 min or pretreated with different kinase inhibitors (20 μm PD098059, 200 nm wortmannin) and then stimulated with 100 ng/ml HRG β1 for an additional 30 min.D, Mature oligodendrocytes were treated with 100 ng/ml HRG β1 at different times, and Akt phosphorylation was assessed by immunoblot with anti-phospho Akt (Ser473) antibody (top panel) or anti-total Akt (bottom panel). HRG β1 promoted Akt phosphorylation in a time-dependent manner in mature oligodendrocytes.
Fig. 4.
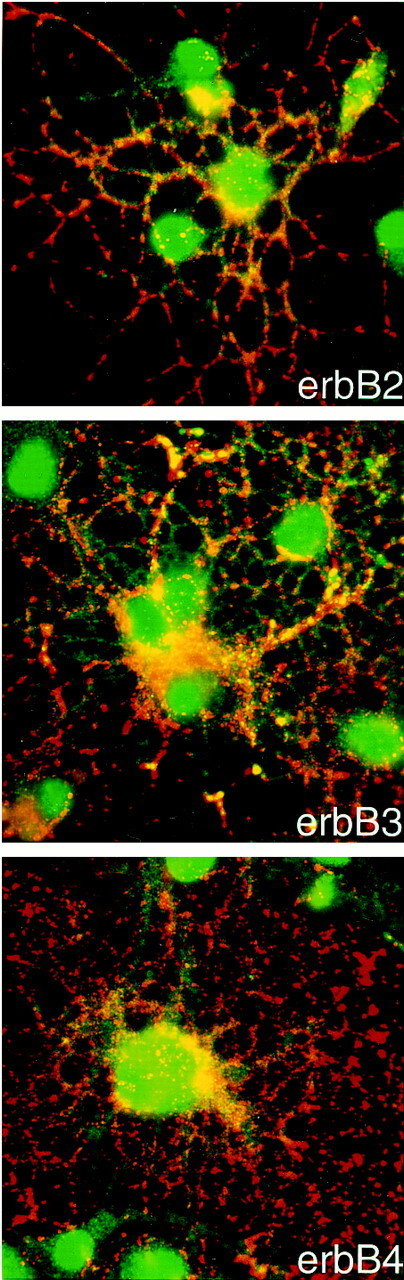
Mature oligodendrocytes express erbB2, erbB3, and erbB4 receptors. Mature oligodendrocytes were obtained after incubation in CNTF/T3 as described in Materials and Methods. Immunofluorescence microscopy of mature oligodendrocyte cultures double-stained with anti-O4 (red) and anti-erbB2, anti-erbB3, or anti-erbB4 antibodies (green) is shown. Note stain for erbB receptors on processes of oligodendrocytes, in addition to cell body stain.
HRG β1 acts as a survival factor in the oligodendrocyte cell lineage
The cellular consequence of heregulin treatment of oligodendrocyte progenitor cells was next examined. Cells were grown in the presence of PDGF/bFGF for 2 d after which they were transferred to medium without growth factors or N2 supplement for 12 or 24 hr. The number of terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL)-positive cells was quantified for cells in the absence of growth factors, for cells supplemented with HRG β1, or for cells supplemented with HRG β1 after 30 min pretreatment with wortmannin. The absolute percentage of dying cells in control samples varied in different experiments, but significant survival was consistently induced in cells supplemented with HRG β1. To standardize data from multiple experiments, the percentage of dying cells in control cultures was defined as 100%. Numbers of dying cells in experimental cultures were expressed as a percentage of that control value. In five experiments, HRG β1 reduced cell death by ∼50% within 12 hr, and this survival effect was blocked by wortmannin (Fig.5). These data clearly indicate the potential for HRG β1 to function as a survival factor inhibiting a cell death pathway in oligodendrocyte progenitor cells. Furthermore, its ability to function as a survival factor in these cells is dependent on an active PI-3 kinase/Akt pathway.
Fig. 5.

HRG β1 maintains cell survival of oligodendrocyte progenitors. Immunopanned oligodendrocyte progenitors were plated in PDGF/bFGF for 2 d and switched to DMEM without any growth factors for 12 or 24 hr (control), with 150 ng/ml HRG β1 with or without a 30 min pretreatment with wortmannin. Apoptotic cells were detected by TUNEL staining and FACS analysis. In each experiment, the number of TUNEL-positive cells was determined for each condition. The absolute number of TUNEL-positive cells varied in different experiments, reaching as much as 50% in some experiments. To combine data from multiple experiments, the number of TUNEL-positive cells in control cultures was therefore defined as 100%. The number of TUNEL-positive oligodendrocyte progenitors in each experimental condition was then expressed as a percentage of control values. Each bar represents the mean ± SEM of five different experiments (*p < 0.0267; **p < 0.0198).
Dominant-negative Akt induces cell death in the oligodendrocyte cell lineage
To test directly whether the Akt pathway regulates the balance between apoptosis and survival of cells in the oligodendrocyte lineage, we expressed a dominant-negative form of Akt, in which both phosphorylation sites (Thr308 and Ser473) were replaced by alanine (AA-AKT;Kitamura et al., 1998) in primary cultures of O4+ immature oligodendrocytes (Fig. 6) or progenitors (Figs. 7,8) by adenovirus-mediated gene transfer. We found adenovirus gene transfer to be an excellent means of introducing foreign genes into primary cultures of oligodendrocytes, because green fluorescent protein was expressed at easily detectable levels in >80% of cells after 48 hr infection (data not shown). Under the same infection conditions, hemagglutinin (HA) epitope-tagged wild-type Akt (wt-Akt, 50 MOI) and dominant-negative Akt (AA-Akt, 10 MOI) expression levels were similar 48 hr after virus infection (Fig.6B), and under these conditions, wt-Akt and AA-Akt were expressed in >90% of the oligodendrocytes, as seen after staining with anti-HA antibody (data not shown). After growth factor withdrawal, dominant-negative Akt effectively induced apoptosis, as assessed by TUNEL analysis (Fig. 6A). O4 staining clearly indicated that immature oligodendrocytes undergo apoptosis under these conditions, whereas only a minor population of cells was apoptotic in wild-type Akt virus infection (Fig. 6A). Infection with control virus (Ad.EGFP) had no effect in cell survival, as shown previously (Matsui et al., 1999). We selected 20 random fields and counted TUNEL-positive cells among the O4-positive cells. O4-positive cells were 88% apoptotic in cultures infected with the dominant-negative Akt virus but only 10% in those infected with the wild-type Akt virus (Fig. 6C). These data clearly indicate a significant role for the Akt pathway in apoptosis in oligodendrocyte progenitors.
Fig. 6.

Dominant-negative Akt induced apoptosis in primary cultures of immature oligodendrocytes. Adenoviruses encoding wild-type Akt or a dominant-negative Akt were used to express recombinant proteins in primary cultures of immature oligodendrocytes. Adenovirus infection was performed on oligodendrocyte precursors 24 hr after incubation in PDGF/T3. A, Cells were subjected to O4 staining and TUNEL analysis 72 hr after infection. The wild-type Akt infection did not induce apoptosis (bottom panel), but dominant-negative Akt infection did induce apoptosis (top and middle panels). B, Expression of wild-type Akt and dominant-negative Akt was assessed by immunoblot with anti-HA antibody; 50 MOI for wild-type Akt and 10 MOI for dominant-negative Akt showed similar levels of Akt expression. Thus, 50 MOI of wild-type Akt and 10 MOI of dominant-negative Akt were used for TUNEL experiments in A. Lysate of cells infected with adenovirus encoding green fluorescent protein was used for control (C). C, O4-positive cells were examined randomly in 20 different fields. In total, 241 cells were examined for dominant-negative Akt virus infection and 171 cells for wild-type Akt virus infection. TUNEL positivity is shown as a percentage of the total number of O4-positive cells. Dominant-negative Akt infection caused apoptosis in O4-positive oligodendrocytes at a significantly higher rate compared to wild-type Akt infection (*p < 0.001).
Fig. 7.
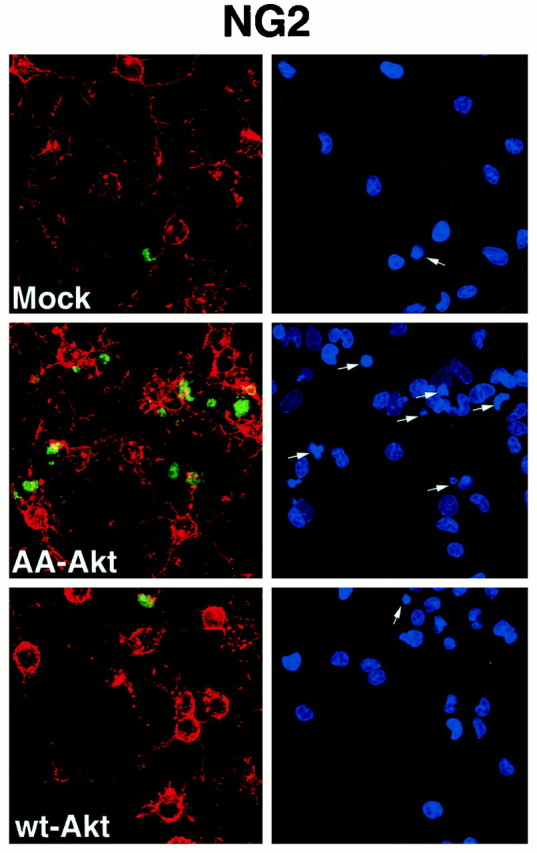
Dominant-negative Akt induced caspase activation in primary cultures of NG2-positive oligodendrocyte progenitors. Oligodendrocyte progenitors were mock-infected or infected with adenovirus expressing wt-Akt (50 MOI) or AA-Akt (10 MOI) for 24 hr as described in Materials and Methods. After infection, cells were subjected to caspase activation analysis (green) and NG2 staining (red). Condensed nuclei were detected by DAPI staining (indicated by arrows). Representative data from one of three independent experiments is shown.
Fig. 8.
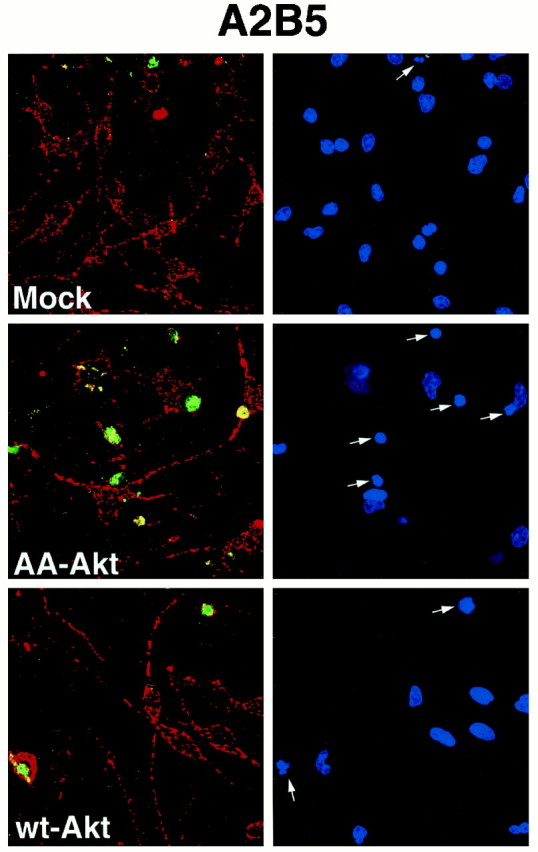
Dominant-negative Akt induced caspase activation in primary cultures of A2B5-positive oligodendrocyte progenitors. Oligodendrocyte progenitors were mock-infected or infected with adenovirus expressing wt-Akt (50 MOI) or AA-Akt (10 MOI) for 24 hr as described in Materials and Methods. After infection, cells were subjected to caspase activation analysis (green) and A2B5 staining (red). Condensed nuclei were detected by DAPI staining (indicated by arrows). Representative data from one of three independent experiments is shown.
Dominant-negative Akt induces apoptosis through caspase activation
The caspase cascade has been shown to play an important role in apoptosis (Thornberry and Lazebnick, 1998). Therefore, to assess the mechanism by which dominant negative Akt induces apoptosis in the oligodendrocyte lineage, we studied caspase activation in infected cultures. Oligodendrocyte progenitors were infected for 24 or 48 hr and then transferred to medium with 0.5% FBS for an additional 20–24 hr. Mock-infected cultures contained >95% of NG2+ or A2B5+ oligodendrocyte progenitors, with only a small number of O4+ cells even after 48 hr infection (data not shown). The number of live progenitor cells seen by phase-contrast microscopy was dramatically reduced in the dominant-negative-expressing cells as compared with mock- or wt-infected cells, although as expected some reduction in cells was noted in the wt-infected plates as well, presumably resulting from the infection itself (data not shown). The number of caspase-positive oligodendrocyte progenitors was significantly increased in the dominant-negative Akt virus infection, in both 24 or 48 hr infection (Table 1). Essentially all caspase-positive oligodendrocyte progenitors (NG2+ cells, Fig. 7; A2B5+ cells, Fig. 8) showed condensed 4′,6-diamidino-2-phenylindole (DAPI)-stained nuclei, suggesting that AA-Akt overexpression induces apoptosis in oligodendrocyte progenitors, which is mediated by activated caspase. Interestingly, apoptotic oligodendrocyte progenitors overexpressing AA-Akt lose cellular processes and/or downregulate NG2 and A2B5 immunoreactivity, because they lose their NG2 or A2B5 immunoreactivity (note cells highlighted with arrows).
Table 1.
Caspase-positive oligodendrocyte progenitors infected with AA-Akt or wt-Akt
| % Caspase-positive cells | ||
|---|---|---|
| 24 hr | 48 hr | |
| Mock-infect | 8.9 ± 1.1 | 10.4 ± 1.0 |
| AA-Akt | 50.1 ± 0.41-160 | 55.8 ± 1.01-160 |
| wt-Akt | 10.5 ± 1.7 | 20.0 ± 1.5* |
Oligodendrocyte progenitor cells were mock-infected or infected with adenovirus expressing AA-Akt or wt-Akt. After a 24 or 48 hr infection, the virus was removed, and cells were cultured in DMEM plus 0.5% FBS for an additional 20–24 hr before caspase inhibitor stain. Caspase-positive cells were counted, and data are expressed as a percentage of total number of cells visualized by DAPI staining. Data are shown as mean ± SEM (n = 3).
F1-160: p < 0.0001;
p< 0.0060.
Dominant-negative Akt blocks the survival function of HRG β1 in the oligodendrocyte cell lineage
To demonstrate a direct link between HRG β1 signaling and Akt, oligodendrocyte progenitors were infected with adenovirus encoding AA-Akt for 48 hr in the presence of 150 ng/ml HRG β1, which maintains cell survival of oligodendrocyte progenitors (Fig. 5). After the infection, the cells were transferred to medium with HRG β1 for an additional 20–24 hr. The number of caspase-positive cells was quantified and compared to mock-infected or control virus-infected cells. As can be seen in Figure 9, after mock infection or infection with a control virus, a small amount of cell death was noted in cells grown in the presence of HRG β1 and the absence of other growth factors. However, when the cells were infected with virus expressing the dominant-negative form of Akt, >70% of the cells were caspase-positive. Furthermore, in the cultures expressing the dominant-negative form of Akt, it is likely that many cells had already died before quantification, because close to twice as many fields had to be analyzed to include 1000 cells, relative to the mock-infected or control-infected cultures. These data further support the conclusion that the survival activity of heregulin acts through Akt in cells in the oligodendrocyte lineage.
Fig. 9.

Dominant-negative Akt induced caspase activation in primary cultures of immature oligodendrocytes in the presence of HRG β1. Oligodendrocyte progenitor cells were mock-infected or infected with 10 MOI of a control adenovirus (Ad.RSV.alphos) or an adenovirus expressing AA-Akt the presence of 150 ng/ml HRG β1. Oligodendrocyte precursors were grown in the presence of PDGF/bFGF for 24 hr; growth factors were then removed, HRG β1 was added to the media, and adenovirus infection was performed. After 48 hr infection in the presence of HRG β1, the virus was removed, and cells were cultured in DMEM plus HRG β1 for an additional 20–24 hr before caspase inhibitor staining. A total of 1000 cells were counted, and the caspase-positive cells were expressed as a percentage of total number of cells visualized by DAPI staining. Data are the mean of a representative experiment made in duplicate. Data are shown as mean ± SEM (n = 2); *p < 0.05.
The data presented here suggest that neuregulins function through ErbB receptors on both oligodendrocytes and their progenitors to activate Akt via PI-3 kinase and that inhibition of Akt function induces apoptosis via caspase activation. Thus, inactivation of the caspase cascade may be one of several mechanisms used by Akt to promote survival in the oligodendrocyte lineage.
DISCUSSION
Significant data indicate that neuregulins are important for oligodendrocyte development. They have been reported to have multiple effects in the oligodendrocyte lineage, including induction of survival, proliferation, or blockage of differentiation (Lemke, 1996; Burden and Yarden, 1997). Our studies are consistent with earlier studies demonstrating that GGF, one of the members of the neuregulin family, promotes survival of oligodendrocytes progenitors under serum-free conditions (Canoll et al., 1996). Additionally, neuregulin has been shown to be a mitogen for cultured mature oligodendrocytes and to inhibit the differentiation and lineage commitment of oligodendrocyte progenitors (Canoll et al., 1999). In addition to these studies in cultured oligodendrocytes, there is clear evidence that the neuregulins are important for in vivo development of oligodendrocytes. For example, in E9 spinal cord explants from neuregulin knock-out mice, oligodendrocytes fail to survive and differentiate, whereas neuron and astrocyte development appears normal (Vartanian et al., 1999).
The neuregulins bind multiple erbB receptors (erbB2, erbB3, and erbB4), all of which are expressed on oligodendrocyte progenitors, and the different erbB receptors recruit multiple docking molecules (Burden and Yarden, 1997). Although neuregulin-1 and neuregulin-2 are both ligands for the erbB3 and erbB4 receptors, a recent study has shown that they exhibit distinct biological activities depending on cellular context (Crovello et al., 1998). These different activities might result from distinct roles of neuregulin-1 and -2 in activation of specific intracellular targets including Akt. Because the different cellular responses to the neuregulins that have been reported could be controlled through a number of different pathways, it is of significant importance to understand which pathways are involved in these different activities in oligodendrocytes. Thus, the current studies focus on the signaling pathways through which heregulin (HRG β1), one of the members of the neuregulin-1 family, induces cellular responses in oligodendrocyte lineage cells. In particular we demonstrate that, in cells of the oligodendroglial lineage, heregulin activates Akt kinase and thereby plays a major role in the survival of these cells. This is consistent with a study in breast cancer cells demonstrating heregulin activation of Akt kinase (Liu et al., 1999). Thus, we demonstrate that (1) in response to growth factor deprivation, heregulin reduces oligodendrocyte progenitor cell death in a wortmannin-sensitive manner; (2) heregulin dose- and time-dependently activates Akt in a wortmannin-sensitive manner in primary cultures of oligodendrocytes as well as their progenitors and C6 glioma cells; (3) in C6 cells, heregulin induces phosphorylation of BAD at Ser136, thereby inhibiting its potential to enhance cell death, as it does in other cells (Datta et al., 1997; del Peso et al., 1997); and (4) dominant-negative Akt induces apoptosis in primary cultures of oligodendrocyte progenitors and immature oligodendrocytes, through activation of the caspase cascade, thereby blocking the survival effect of heregulin. Thus, the present data suggest that the primary pathway through which heregulin mediates survival of oligodendrocyte lineage is the PI-3 kinase/Akt pathway. Interestingly, Canoll et al., (1999) demonstrated that GGF functions through both a PI-3 kinase pathway and a MAP kinase pathway. Our data would suggest that the PI-3 kinase pathway is primarily involved in the survival function of the neuregulins. Other studies indicate that the PI-3 kinase signaling pathway is important for survival of oligodendrocytes and their precursors, because inhibition of this pathway leads to oligodendrocyte and precursor cell death (Vemuri and McMorris, 1996), further supporting the concept that factors such as the neuregulins that act through activation of the PI-3 kinase/Akt signaling pathway are important for oligodendrocyte survival.
Akt activity may function to control cell death by controlling caspase activity. In nonglial cells, caspase activity is inhibited by Akt-induced phosphorylation, thereby reducing apoptosis (Cardone et al., 1998). In vitro experiments show that various stimuli induce apoptosis in mature oligodendrocytes through caspase activation (Hisahara et al., 1997; Gu et al., 1999). In the current studies, dominant-negative Akt expression induced apoptosis in immature oligodendrocytes and oligodendrocyte progenitor cells, perhaps by preventing caspase inactivation by endogenous Akt. Thus, activation of caspases is likely involved in the execution of growth factor deprivation-induced oligodendrocyte cell death, and it appears that this cell death can be prevented by heregulin-dependent Akt activation, acting through BAD phosphorylation.
Differences in the effects of neuregulins could result from activation of specific erbB receptors. A recent knock-out study in mice clearly demonstrated the involvement of erbB3 receptor signaling in the survival and differentiation of Schwann cells in vivo(Riethmacher et al., 1997), which are the myelinating counterparts of oligodendrocytes in the peripheral nervous system. Our preparations of oligodendrocyte progenitors express all three erbB2, erbB3, and erbB4 receptors, which is consistent with some published studies (Canoll et al., 1996; Raabe et al., 1997b). However others have found that oligodendrocytes only express erbB2 and erbB4 but not erbB3 (Vartanian et al., 1997). These differences could result from differences in culture conditions or cell preparation, such as the developmental stage at which the cells are prepared, which could be important in defining the expression of specific erbB receptor proteins. Such differences in receptor expression should be monitored, because they may result in different cellular responses.
Recently PTEN, a member of a phosphatase gene family (Li et al., 1997), was shown to have a key role as a negative upstream regulator of Akt kinase (Maehama and Dixon, 1998; Stambolic et al., 1998). PTEN is frequently deleted in gliomas including oligodendrogliomas (Duerr et al., 1998; Lin et al., 1998), and indeed gliomas that lack PTEN have higher amounts of Akt (Haas-Kogan et al., 1998). Also, recent data indicate that oligodendrocyte progenitors may be the source of oligodendrogliomas (Shoshan et al., 1999). These data suggest that the regulation of the PI-3 kinase/Akt pathway may be critical for oligodendrocyte progenitor survival and/or cellular transformation. Thus, in differentiated oligodendrocytes, it is possible that negative regulators of this pathway, which include PTEN, are upregulated or that positive regulators, such as Akt, are downregulated.
Whereas significant oligodendrocyte cell death occurs during normal brain development, oligodendrocyte cell death is also important in several pathologies of both the adult and the developing nervous system. For example, in addition to oligodendrocyte loss in MS, oligodendrocyte cell death is an element in stroke (McDonald et al., 1998). It is also an important feature of periventricular white matter injury, which is thought to underlie spastic motor and cognitive deficits frequently seen in premature infants (Dambska et al., 1989). The current data suggest that factors that activate Akt, including the neuregulin family members, may be useful in preventing oligodendrocyte death in such pathological conditions. Furthermore, the possibility exists that if neuregulin expression is reduced in some of these pathological conditions, oligodendrocyte loss may be directly triggered.
Understanding how, and under what conditions, oligodendrocytes and their progenitors respond to survival factors is important for therapeutic interventions in these pathologies. MS is the most obvious adult demyelinating pathology in which oligodendrocytes appear to die. Chronic MS lesions contain an important population of cells with the phenotypic morphology of oligodendrocyte precursors (Wolswijk, 1998). Injection of neuregulins in acute or chronic relapsing EAE has been shown to be effective in reducing disease progression (Cannella et al., 1998; Marchionni et al., 1999) and in enhancing remyelination in EAE mice (Marchionni et al., 1999). These results support the hypothesis that neuregulins elicit survival signals for oligodendrocytes in vivo. Other factors have been proposed to enhance survival of the oligodendrocyte lineage. For example CNTF has been shown to be effective in vitro (Louis et al., 1993; Mayer et al., 1994;Dell'Albani et al., 1998) and in MS patients (Webster, 1997; Massaro et al., 1997,1998). IGF-1 was initially thought to be effective in the delay of disease progression in EAE (Yao et al., 1995). However, it has been recently shown that IGF-1 has an adverse effect, because it also augments inflammatory cell survival (Lovett-Racke et al., 1998). Our data and those of others would suggest that neuregulins may be an additional therapeutic avenue for MS treatment and/or for promoting survival of transplanted oligodendrocyte progenitors allowing differentiation and remyelination.
Footnotes
This study is supported by National Institutes of Health (NIH) Grant NS25304 (W.B.M.), Multiple Sclerosis Pilot Grant PP0649 (T.O.), NIH Grant R29-MH56036 (T.O.), Prentiss Foundation (T.O.), and a Postdoctoral Fellowship from the National Multiple Sclerosis Society (A.I.F.).
Correspondence should be addressed to Wendy B. Macklin, Department of Neurosciences NC30, Cleveland Clinic Foundation, 9500 Euclid Avenue, Cleveland, OH 44195. E-mail: mackliw@ccf.org.
Dr. Okamoto's present address: Laboratory for Neurodegeneration and Signaling, Brain Science Institute, The Institute of Physical and Chemical Research (RIKEN), Wako-shi.Saitama 351-0198, Japan.
REFERENCES
- 1.Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJC, Frech M, Cron P, Cohen P, Lucocq JM, Hemmings BA. Role of translocation in the activation and function of protein kinase B. J Biol Chem. 1997;272:31515–31524. doi: 10.1074/jbc.272.50.31515. [DOI] [PubMed] [Google Scholar]
- 2.Baek SY, Kim SU. Proliferation of human Schwann cells induced by neu differentiation factor isoforms. Dev Neurosci. 1998;20:512–517. doi: 10.1159/000017351. [DOI] [PubMed] [Google Scholar]
- 3.Barres BA, Hart IK, Coles HSR, Burne JF, Voyvodic JT, Richardson WD, Raff MC. Cell death and control of cell survival in the oligodendrocyte lineage. Cell. 1992;70:31–46. doi: 10.1016/0092-8674(92)90531-g. [DOI] [PubMed] [Google Scholar]
- 4.Basu S, Bayoumy S, Zhang Y, Lozano J, Kolesnick R. BAD enables ceramide to signal apoptosis via Ras and Raf-1. J Biol Chem. 1998;273:30419–30426. doi: 10.1074/jbc.273.46.30419. [DOI] [PubMed] [Google Scholar]
- 5.Burden S, Yarden Y. Neuregulins and their receptors: a versatile signaling module in organogenesis and oncogenesis. Neuron. 1997;18:847–855. doi: 10.1016/s0896-6273(00)80324-4. [DOI] [PubMed] [Google Scholar]
- 6.Cannella B, Hoban CJ, Gao YL, Garcia-Arenas R, Lawson D, Marchionni M, Gwynne D, Raine CS. The neuregulin, glial growth factor 2, diminishes autoimmune demyelination and enhances remyelination in a chronic relapsing model for multiple sclerosis. Proc Natl Acad Sci USA. 1998;95:10100–10105. doi: 10.1073/pnas.95.17.10100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Canoll PD, Musacchio JM, Hardy R, Reynolds R, Marchionni MA, Salzer JL. GGF/neuregulin is a neuronal signal that promotes the proliferation and survival and inhibits the differentiation of oligodendrocyte progenitors. Neuron. 1996;17:229–243. doi: 10.1016/s0896-6273(00)80155-5. [DOI] [PubMed] [Google Scholar]
- 8.Canoll PD, Kraemer R, Teng KK, Marchionni MA, Salzer JL. GGF/Neuregulin induces a phenotypic reversion of oligodendrocytes. Mol Cell Neurosci. 1999;13:79–94. doi: 10.1006/mcne.1998.0733. [DOI] [PubMed] [Google Scholar]
- 9.Cardone MH, Roy N, Stennicke HR, Salvessen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 10.Carraway KL, III, Weber JL, Unger MJ, Ledesma J, Yu N, Gassmann M, Lai C. Neuregulin-2, a new ligand of ErbB3/ErbB4-receptor tyrosine kinases. Nature. 1997;387:512–516. doi: 10.1038/387512a0. [DOI] [PubMed] [Google Scholar]
- 11.Chang H, Riese DJ, Gilbert W, 2nd, Stern DF, McMahan UJ. Ligands for ErbB-family receptors encoded by a neuregulin-like gene. Nature. 1997;387:509–512. doi: 10.1038/387509a0. [DOI] [PubMed] [Google Scholar]
- 12.Chen MS, Berminghan-McDonogh O, Danehy FT, Nolan C, Jr, Scherer SS, Lucas J, Gwynne D, Marchionni MA. Expression of multiple neuregulin transcripts in postnatal rat brains. J Comp Neurol. 1994;349:389–400. doi: 10.1002/cne.903490306. [DOI] [PubMed] [Google Scholar]
- 13.Crovello CS, Lai C, Cantley LC, Carraway KL., III Differential signaling by the epidermal growth factor-like growth factors neuregulin-1 and neuregulin-2. J Biol Chem. 1998;273:26954–26961. doi: 10.1074/jbc.273.41.26954. [DOI] [PubMed] [Google Scholar]
- 14.Dambska M, Laure-Kamionowska M, Schmidt-Sidor B. Early and late neuropathological changes in perinatal white matter damage. J Child Neurol. 1989;4:291–298. doi: 10.1177/088307388900400408. [DOI] [PubMed] [Google Scholar]
- 15.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 16.Dell'Albani P, Kahn MA, Cole R, Condorelli DF, Giuffrida-Stella AM, de Vellis J. Oligodendroglial survival factors, PDGF-AA, and CNTF, activate similar JAK/STAT signaling pathways. J Neurosci Res. 1998;54:191–205. doi: 10.1002/(SICI)1097-4547(19981015)54:2<191::AID-JNR7>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 17.Del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 18.Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 19.Duerr EM, Rollbrocker B, Hayashi Y, Peters N, Meyer-Puttlitz B, Louis DN, Schramm J, Wiestler OD, Parsons R, Eng C, von Deimling A. PTEN mutations in gliomas and glioneuronal tumors. Oncogene. 1998;16:2259–2264. doi: 10.1038/sj.onc.1201756. [DOI] [PubMed] [Google Scholar]
- 20.Eisenbarth GS, Walsh FS, Nirenberg M. Monoclonal antibody to a plasma membrane antigen of neurons. Proc Natl Acad Sci USA. 1979;76:4913–4917. doi: 10.1073/pnas.76.10.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erickson SL, O'Shea KS, Ghaboosi N, Loverro L, Frants G, Baner M, Lu LH, Moore MW. ErbB3 is required for normal cerebellar and cardiac development: a comparison with ErbB2- and heregulin-deficient mice. Development. 1997;124:4999–5011. doi: 10.1242/dev.124.24.4999. [DOI] [PubMed] [Google Scholar]
- 22.Francis A, Raabe TD, Wen D, DeVries GH. Neuregulins and erbB receptors in cultured neonatal astrocytes. J Neurosci Res. 1999;57:487–494. [PubMed] [Google Scholar]
- 23.Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- 24.Franke TF, Kaplan DR, Cantley LC. PI3K: downstream AKTion blocks apoptosis. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 25.Fuss B, Baba H, Phan T, Tuohy VK, Macklin WB. Phosphodiesterase I, a novel adhesion molecule and/or cytokine involved in oligodendrocyte function. J Neurosci. 1997;17:9095–9103. doi: 10.1523/JNEUROSCI.17-23-09095.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Golding JP, Trainor P, Krumlauf R, Gassmann M. Defects in pathfinding by cranial neural crest cells in mice lacking the neuregulin receptor ErbB4. Nat Cell Biol. 2000;2:103–109. doi: 10.1038/35000058. [DOI] [PubMed] [Google Scholar]
- 27.Gu C, Casaccia-Bonnefil P, Srinivasan A, Chao MV. Oligodendrocyte apoptosis mediated by caspase activation. J Neurosci. 1999;19:3043–3049. doi: 10.1523/JNEUROSCI.19-08-03043.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haas-Kogan D, Shalev N, Wong M, Mills G, Yount G, Stokoe D. Protein kinase B (PKB/Akt) activity is elevated in glioblastoma cells due to mutation of the tumor suppressor PTEN/MMAC. Curr Biol. 1998;8:1195–1198. doi: 10.1016/s0960-9822(07)00493-9. [DOI] [PubMed] [Google Scholar]
- 29.Harari D, Tzahar E, Romano J, Shelly M, Pierce JH, Andrews GC, Yarden Y. Neuregulin-4: a novel growth factor that acts through the ErbB-4 receptor tyrosine kinase. Oncogene. 1999;18:2681–2689. doi: 10.1038/sj.onc.1202631. [DOI] [PubMed] [Google Scholar]
- 30.Hisahara S, Shoji S, Okano H, Miura M. ICE/CED-3 family executes oligodendrocyte apoptosis by tumor necrosis factor. J Neurochem. 1997;69:10–20. doi: 10.1046/j.1471-4159.1997.69010010.x. [DOI] [PubMed] [Google Scholar]
- 31.Hisahara S, Araki T, Sugiyama F, Yagami K, Suzuki M, Abe K, Yamamura K, Miyazaki J, Momoi T, Saruta T, Bernard CCA, Okano H, Miura M. Targeted expression of baculovirus p35 caspase inhibitor in oligodendrocytes protects mice against autoimmune-mediated demyelination. EMBO J. 2000;19:341–348. doi: 10.1093/emboj/19.3.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kennedy SG, Wagner AJ, Cozen SD, Jordan J, Bellacosa A, Tsichlis PN, Hay N. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 1997;11:701–713. doi: 10.1101/gad.11.6.701. [DOI] [PubMed] [Google Scholar]
- 33.Kitamura T, Ogawa W, Sakaue H, Hino Y, Kuroda S, Takata M, Matsumoto M, Maeda T, Konishi H, Kikkawa U, Kasuga M. Requirement for activation of the serine-threonine kinase Akt (protein kinase B) in insulin stimulation of protein synthesis but not of glucose transport. Mol Cell Biol. 1998;18:3708–3717. doi: 10.1128/mcb.18.7.3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kramer R, Bucay N, Kane DJ, Martin LE, Tarpley JE, Theill LE. Neuregulins with an Ig-like domain are essential for mouse myocardial and neuronal development. Proc Natl Acad Sci USA. 1996;93:4833–4838. doi: 10.1073/pnas.93.10.4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumar R, Shepard HM, Mendelson J. Regulation of phosphorylation of the c-erbB-2/HER2 gene product by a monoclonal antibody and serum growth factor(s) in human mammary carcinoma cells. Mol Cell Biol. 1991;11:979–986. doi: 10.1128/mcb.11.2.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lemke G. Neuregulins in development. Mol Cell Neurosci. 1996;7:247–262. doi: 10.1006/mcne.1996.0019. [DOI] [PubMed] [Google Scholar]
- 37.Li L, Ernsting BR, Wishart MJ, Lohse DL, Dixon JE. A family of putative tumor suppressors is structurally and functionally conserved in humans and yeast. J Biol Chem. 1997;272:29403–29406. doi: 10.1074/jbc.272.47.29403. [DOI] [PubMed] [Google Scholar]
- 38.Lin H, Bondy ML, Langford LA, Hess KR, Delclos GL, Wu X, Chan W, Pershouse MA, Yung WK, Steck PA. Allelic deletion analyses of MMAC/PTEN and DMBT1 loci in gliomas: relationship to prognostic significance. Clin Cancer Res. 1998;4:2447–2454. [PubMed] [Google Scholar]
- 39.Lin W, Sanchez HB, Deerinck T, Morris JK, Ellisman M, Lee KF. Aberrant development of motor axons and neuromuscular synapses in erbB2-deficient mice. Proc Natl Acad Sci USA. 2000;97:1299–1304. doi: 10.1073/pnas.97.3.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu W, Li J, Roth RA. Heregulin regulation of Akt/protein kinase B in breast cancer cells. Biochem Biophys Res Commun. 1999;261:897–903. doi: 10.1006/bbrc.1999.1144. [DOI] [PubMed] [Google Scholar]
- 41.Louis JC, Magal E, Takayama S, Varon S. CNTF protection of oligodendrocytes against natural and tumor necrosis factor-induced death. Science. 1993;259:689–692. doi: 10.1126/science.8430320. [DOI] [PubMed] [Google Scholar]
- 42.Lovett-Racke AE, Bittner P, Cross AH, Carlino JA, Racke MK. Regulation of experimental autoimmune encephalomyelitis with insulin-like growth factor (IGF-1) and IGF-1/IGF-binding protein-3 complex (IGF-1/IGFBP3). J Clin Invest. 1998;101:1797–1804. doi: 10.1172/JCI1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 44.Marchionni MA, Canella B, Hoban C, Gao YL, Garcia-Arenas R, Lawson D, Happel E, Noel F, Tofilon P, Gwynne D, Raine CS. Neuregulin in neuron/glial interactions in the central nervous system. GGF2 diminishes autoimmune demyelination, promotes oligodendrocyte progenitor expansion and enhances remyelination. Adv Exp Med Biol. 1999;468:283–295. [PubMed] [Google Scholar]
- 45.Marikovsky M, Lavi S, Pinkas-Kramaski R, Karunagaran D, Liu N, Wen D, Yarden Y. ErbB-3 mediated differential mitogenic effects of NDF/heregulin isoforms on mouse keratinocytes. Oncogene. 1995;10:1403–1411. [PubMed] [Google Scholar]
- 46.Massaro AR. Are there indicators of remyelination in blood or CSF of multiple sclerosis patients? Mult Scler. 1998;4:228–231. doi: 10.1177/135245859800400326. [DOI] [PubMed] [Google Scholar]
- 47.Massaro AR, Soranzo C, Carnevale A. Cerebrospinal-fluid ciliary neurotrophic factor in neurological patients. Eur Neurol. 1997;37:243–246. doi: 10.1159/000117451. [DOI] [PubMed] [Google Scholar]
- 48.Matsui T, Li L, del Monte F, Fukui Y, Franke TF, Hajjar RJ, Rosenzweig A. Adenoviral gene transfer of activated phosphatidylinositol 3′-kinase and Akt inhibits apoptosis of hypoxic cardiomyocytes in vitro. Circulation. 1999;100:2373–2379. doi: 10.1161/01.cir.100.23.2373. [DOI] [PubMed] [Google Scholar]
- 49.Mayer M, Bhakoo K, Noble M. Ciliary neurotrophic factor and leukemia inhibitory factor promote the generation, maturation and survival of oligodendrocytes in vitro. Development. 1994;120:143–153. doi: 10.1242/dev.120.1.143. [DOI] [PubMed] [Google Scholar]
- 50.McDonald JW, Althomsons SP, Hyrc KL, Choi DW, Goldberg MP. Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nat Med. 1998;4:291–297. doi: 10.1038/nm0398-291. [DOI] [PubMed] [Google Scholar]
- 51.Meyer D, Birchmeier D. Distinct isoforms of neuregulin are expressed in mesenchymal and neuronal cells during mouse development. Proc Natl Acad Sci USA. 1994;91:1064–1068. doi: 10.1073/pnas.91.3.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meyer D, Birchmeier D. Multiple essential functions of neuregulin in development. Nature. 1995;378:386–390. doi: 10.1038/378386a0. [DOI] [PubMed] [Google Scholar]
- 53.Meyer D, Yamaai T, Garrat A, Riethmacher-Sonnenberg E, Kane D, Theill LE, Birchmeier C. Isoform-specific expression and function of neuregulin. Development. 1997;124:3575–3586. doi: 10.1242/dev.124.18.3575. [DOI] [PubMed] [Google Scholar]
- 54.Ozawa K, Suchanek G, Breitschopf H, Bruck W, Budka H, Jellinger K, Lassmann H. Patterns of oligodendrocyte pathology in multiple sclerosis. Brain. 1994;117:1311–1322. doi: 10.1093/brain/117.6.1311. [DOI] [PubMed] [Google Scholar]
- 55.Prigent SA, Lemoine NR, Hughes CM, Plowman GD, Selden C, Gullick WJ. Expression of the c-erbB-3 protein in normal human adult and fetal tissues. Oncogene. 1992;7:1273–1278. [PubMed] [Google Scholar]
- 56.Raabe TD, Clive DR, Wen D, DeVries GH. Neonatal oligodendrocytes contain and secrete neuregulins in vitro. J Neurochem. 1997a;69:1859–1863. doi: 10.1046/j.1471-4159.1997.69051859.x. [DOI] [PubMed] [Google Scholar]
- 57.Raabe TD, Suy S, Welcher A, DeVries GH. Effect of neu differentiation factor isoforms on neonatal oligodendrocyte function. J Neurosci Res. 1997b;50:755–768. doi: 10.1002/(SICI)1097-4547(19971201)50:5<755::AID-JNR12>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 58.Raff MC, Barres BA, Burne JF, Cole HS, Ishizaki Y, Jacobson MD. Programmed cell death and the control of cell survival: lessons from the nervous system. Science. 1993;262:695–700. doi: 10.1126/science.8235590. [DOI] [PubMed] [Google Scholar]
- 59.Raine CS, Scheinberg LC. On the immunopathology of plaque development and repair in multiple sclerosis. J Neuroimmunol. 1988;20:189–201. doi: 10.1016/0165-5728(88)90160-9. [DOI] [PubMed] [Google Scholar]
- 60.Reif K, Burgering BMT, Cantrell DA. Phosphatidylinositol 3-kinase links the interleukin-2 receptor to protein kinase B and p70 S6 kinase. J Biol Chem. 1997;272:14426–14433. doi: 10.1074/jbc.272.22.14426. [DOI] [PubMed] [Google Scholar]
- 61.Riethmacher D, Sonnenberg-Riethmacher E, Brinkmann V, Yamaai T, Lewin GR, Birchmeier C. Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature. 1997;389:725–730. doi: 10.1038/39593. [DOI] [PubMed] [Google Scholar]
- 62.Shoshan Y, Nishiyama A, Chang A, Mork S, Barnnet GH, Cowell JK, Trapp BD, Staugaitis SM. Expression of oligodendrocyte progenitor cell antigens by gliomas: implications for the histogenesis of brain tumors. Proc Natl Acad Sci USA. 1999;96:10361–10366. doi: 10.1073/pnas.96.18.10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 64.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 65.Tsunoda I, Fujinami RS. Two models for multiple sclerosis: experimental allergic encephalomyelitis and Theiler's murine encephalomyelitis virus. J Neuropathol Exp Neurol. 1996;55:673–686. doi: 10.1097/00005072-199606000-00001. [DOI] [PubMed] [Google Scholar]
- 66.Vartanian T, Goodearl A, Viehover A, Fischbach G. Axonal neuregulin signals cells of the oligodendrocyte lineage through activation of HER4 and Schwann cells through HER2 and HER3. J Cell Biol. 1997;137:211–220. doi: 10.1083/jcb.137.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vartanian T, Fischbach G, Miller R. Failure of spinal cord oligodendrocyte development in mice lacking neuregulin. Proc Natl Acad Sci USA. 1999;96:731–735. doi: 10.1073/pnas.96.2.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vemuri GS, McMorris FA. Oligodendrocytes and their precursors require phosphatidylinositol 3-kinase signaling for survival. Development. 1996;122:2529–2537. doi: 10.1242/dev.122.8.2529. [DOI] [PubMed] [Google Scholar]
- 69.Webster HD. Growth factors and myelin regeneration in multiple sclerosis. Mult Scler. 1997;3:113–120. doi: 10.1177/135245859700300210. [DOI] [PubMed] [Google Scholar]
- 70.Wolpowitz D, Masin TBA, Dietrich P, Mendelson M, Talmage DA, Role LW. Cysteine-rich domain isoforms of the neuregulin-1 gene are required for maintenance of peripheral synapses. Neuron. 2000;25:79–91. doi: 10.1016/s0896-6273(00)80873-9. [DOI] [PubMed] [Google Scholar]
- 71.Wolswijk G. Chronic stage multiple sclerosis lesions contain a relatively quiescent population of oligodendrocyte precursor cells. J Neurosci. 1998;18:601–609. doi: 10.1523/JNEUROSCI.18-02-00601.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yao DL, Liu X, Hudson LD, Webster HD. Insulin-like growth factor I treatment reduces demyelination and up-regulates gene expression of myelin-related proteins in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 1995;92:6190–6194. doi: 10.1073/pnas.92.13.6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yasuda T, Grinspan J, Stern J, Franceschini B, Bannerman P, Pleasur D. Apoptosis occurs in the oligodendroglial lineage, and in prevented by basic fibroblast growth factor. J Neurosci Res. 1995;40:306–317. doi: 10.1002/jnr.490400304. [DOI] [PubMed] [Google Scholar]
- 74.Zhang D, Sliwkowski MX, Mark M, Frantz G, Akita R, Sun Y, Hillan K, Crowley C, Brush J, Godowski PJ. Neuregulin-3 (NRG3): a novel neural tissue-enriched protein that binds and activates ErbB4. Proc Natl Acad Sci USA. 1997;94:9562–9567. doi: 10.1073/pnas.94.18.9562. [DOI] [PMC free article] [PubMed] [Google Scholar]