

Abstract
Neuroactive steroids are synthesized de novo in brain, yet their physiological significance remains elusive. We provide biochemical, electrophysiological, and behavioral evidence that several specific actions of alcohol (ethanol) are mediated by the neurosteroid 3α-hydroxy-5α-pregnan-20-one (3α,5α-THP; allopregnanolone). Systemic alcohol administration elevates 3α,5α-THP levels in the cerebral cortex to pharmacologically relevant concentrations. The elevation of 3α,5α-THP is dose- and time-dependent. Furthermore, there is a significant correlation between 3α,5α-THP levels in cerebral cortex and the hypnotic effect of ethanol. Blockade ofde novo biosynthesis of 5α-reduced steroids using the 5α-reductase inhibitor finasteride prevents several effects of ethanol. Pretreatment with finasteride causes no changes in baseline bicuculline-induced seizure threshold but reverses the anticonvulsant effect of ethanol. Finasteride pretreatment also reverses ethanol inhibition of spontaneous neural activity in medial septal/diagonal band of Broca neurons while having no direct effect on spontaneous firing rates. Thus, elevation of 3α,5α-THP levels by acute ethanol administration represents a novel mechanism of ethanol action as well as an important modulatory role for neurosteroids in the CNS.
Keywords: neuroactive steroids, ethanol, allopregnanolone, 3α-hydroxy-5α-pregnan-20-one, finasteride, nongenomic steroid actions
Steroid hormones have long been recognized as endocrine signals regulating essential functions such as metabolism, catabolism, and reproduction. Classically, steroids are recognized for their genomic action as transcriptional regulators, however they have been more recently characterized in a nongenomic role of direct interaction with membrane-bound receptors (for review, seeRupprecht and Holsboer, 1999). Recent research has indicated that certain of these steroids can be synthesized in brain without peripheral precursors and have nongenomic effects that produce both excitatory and inhibitory actions at various receptor classes in brain (Paul and Purdy, 1992; Robel and Baulieu, 1995). 3α,5α-THP is an endogenous pregnane steroid and potent agonist modulator of the inhibitory neurotransmitter GABAA receptor subtype. 3α,5α-THP is increased in brain in many stress paradigms (Purdy et al., 1991; Barbaccia et al., 1996; Barbaccia et al., 1997), as well as during naturally occurring hormonal fluctuations such as the estrus cycle (Ichikawa et al., 1974; Paul and Purdy, 1992; Finn and Gee, 1993) and pregnancy (Concas et al., 1998). A wealth of evidence demonstrates that the anxiolytic, sedative/hypnotic, and anticonvulsant effects attributed to 3α,5α-THP are mediated by enhancement of chloride ion conductance through GABAA receptors in brain (Morrow, 1995).
Behavioral and neurochemical evidence suggests that alcohol exerts pharmacological effects remarkably similar to GABAA receptor modulators, including 3α,5α-THP (Suzdak et al., 1986b; Grobin et al., 1998). Several ethanol effects are enhanced by the positive GABAA receptor modulators benzodiazepines and barbiturates, but are blocked by GABAA receptor antagonists and inverse agonists (Suzdak et al., 1986a; Nutt and Lister, 1989; Givens and Breese, 1990; Liu and Deitrich, 1998). For example, sedative/hypnotic effects produced by high doses of ethanol are mediated by GABAA receptors and can be enhanced by GABA agonists and blocked by GABA receptor antagonists (Martz et al., 1983). However, the conclusion that ethanol has direct effects on GABAA receptors is controversial. Although ethanol potentiation of GABAAreceptor-mediated responses are observed in Cl− flux studies, there is a lack of consistent electrophysiological evidence that ethanol interacts directly with GABAA receptors in vivoand in vitro. For example, ethanol increases GABA-mediated inhibition in some brain regions, but not others (Givens and Breese, 1990; Simson et al., 1991). Whole-cell patch-clamp recording techniques have also provided mixed results concerning the ability of ethanol to enhance GABA inhibition (Mancillas et al., 1986; Frye et al., 1994; Marszalec et al., 1998; Sapp and Yeh, 1998).
Because ethanol is known to have effects on the GABAA receptor, and 3α,5α-THP is such a potent modulator of the same receptor, we became interested in a possible interaction between these two agents. Ethanol activates the hypothalamic–pituitary axis and has been shown to cause increases in plasma levels of several steroids, including corticosterone, testosterone, and progesterone (Rivier et al., 1984; Korneyev et al., 1993b; Schuckit and Smith, 1996). In this study, we investigated whether ethanol alters brain levels of the GABAergic neurosteroid 3α,5α-THP and if steroid biosynthetic enzyme inhibitors prevent this effect. We also investigated the effect of blockade of steroid biosynthesis on anticonvulsant and electrophysiological effects of ethanol, as well as the temporal relationship between 3α,5α-THP elevation and these effects of ethanol. Our results suggest that 3α,5α-THP plays a modulatory role in ethanol action at the GABAA receptor and contributes to certain electrophysiological and behavioral effects in vivo.
MATERIALS AND METHODS
All experiments were conducted in accordance with National Institutes of Health Guidelines under Institution Animal Care and Use Committee-approved protocols. Male Sprague Dawley rats (200–240 gm) were purchased from Harlan (Indianapolis, IN) and were group-housed in controlled conditions (lights on, 6:00 A.M. to 6:00 P.M.) with ad libitum access to rat chow and water. All animals were handled and habituated to vehicle injections for 5 d before experiments. All experiments were performed at the beginning of the light cycle to minimize diurnal steroid fluctuations.
Radioimmunoassays. RIAs were performed as described previously (Janis et al., 1998). Steroids were extracted from individual cerebral cortical hemispheres that were rapidly dissected in ice-cold saline after euthanasia. Cortices were then frozen at −80°C until use. Recovery was monitored by the incorporation of 4000 dpm of [3H]3α,5α-THP. Samples were digested in 0.3 N NaOH by a sonic dismembrator and extracted three times in 3 ml aliquots of 10% (v/v) ethyl acetate in heptane. The aliquots were combined and diluted with 4 ml of heptane. The extracts were applied to solid phase silica columns (Burdick & Jackson, Muskegon, MI), washed with pentane, and steroids of similar polarity to 3α,5α-THP were eluted off of the column by the addition of 25% (v/v) acetone in pentane. The eluant was then dried under N2 and steroids were redissolved in 20% (v/v) isopropanol RIA buffer (0.1 mNaH2PO4, 0.9 mNaCl, 0.1% w/v BSA, pH 7.0). Extraction efficiency was determined in 50 μl of the redissolved extract by liquid scintillation spectroscopy. Extraction efficiency ranged between 95 and 100%. The remaining 200 μl was used in the determination of 3α,5α-THP by radioimmunoassay.
Reconstituted sample extracts (75 μl) and 3α,5α-THP standards (5–40,000 pg in 6.25% v/v ethanol, 31% v/v isopropyl alcohol in RIA buffer) were assayed in duplicate by the addition of 725 μl of RIA buffer, 100 μl of [3H]3α,5α-THP (20,000 dpm), and 100 μl of anti-3α,5α-THP antibody (1:500; gift of CoCensys, Irvine, CA). Total binding was determined in the absence of unlabeled 3α,5α-THP, and nonspecific binding was determined in the absence of antibody. The antibody-binding reaction was allowed to equilibrate for 120 min at room temperature and was terminated by cooling the mixture to 4°C. Bound 3α,5α-THP was separated from unbound 3α,5α-THP by incubation with 300 μl of cold dextran-coated charcoal (DCC; 0.04% dextran, 0.4% powdered charcoal in double-distilled H2O) for 20 min. DCC was removed by centrifugation at 2000 × g for 10 min. Bound radioactivity in the supernatant was determined by liquid scintillation spectroscopy. Sample values were compared to a concurrently run 3α,5α-THP standard curve (0, 0.005, 0.01, 0.02, 0.04, 0.08, 0.16, 0.31, 0.62, 1.25, 2.5, 5.0, 10.0, 20.0, and 40.0 ng/tube) produced using a one-site competition model (Prism2; GraphPad, San Diego, CA). Sample values were adjusted to account for the previously determined extraction efficiency. The sensitivity of the assay was 15–20 pg/tube, and the interassay coefficient of variation was 9.1%.
Administration of biosynthesis inhibitors. Finasteride (Sigma, St. Louis, MO), a 5α-reductase inhibitor, was administered 4 and 1.5 hr before ethanol injection. It was administered subcutaneously in a 2 ml/kg suspension of finasteride in 20% (w/v) 2-hydroxypropyl-β-cyclodextrin (HCD) (Research Biochemicals, Natick, MA) at either a 25 or 50 mg/kg dose. Doses of finasteride were determined by review of finasteride use in previous behavioral and biochemical experiments in rodents (Lephart et al., 1996; Concas et al., 1998; Kokate et al., 1999). Finasteride (50 mg/kg, s.c.) alters open field behavior in pregnant females rats and decreased enzyme activity by 60–80% (Lephart et al., 1996).
Indomethacin (ICN Biomedicals, Aurora, OH), a 3α-HSD inhibitor, was administered 20 min before ethanol injection. Indomethacin was administered in a 1 ml/kg solution of 20% HCD, intraperitoneally, at a dose of 0.1 mg/kg. This dose has been reported to block 3α,5α-THP formation in a pseudopregnant rat model (Smith et al., 1998a).
Trilostane (Sanofi, Malvern, PA), a 3β-HSD inhibitor, was administered 2 hr before ethanol injection. Trilostane was administered in a 2 ml/kg solution of 20% HCD, intraperitoneally at a dose of 30 mg/kg. This dose has been reported to significantly decrease conversion of pregnenolone to progesterone (Potts et al., 1978; Korneyev et al., 1993b).
Loss of righting reflex. Ethanol-induced sleep time was determined by the duration of the loss of righting reflex, defined as the inability to right three times in 1 min after placement in the supine position. Ethanol (3.5 gm/kg; 20% v/v in saline) was administered by intraperitoneal injection; rats were replaced in their cage until righting reflex was lost and then placed in a V-shaped support in the supine position until recovery. Rats were administered finasteride (25 or 50 mg/kg) or vehicle [20% (w/v) 2-hydroxypropyl-β-cyclodextrin; Research Biochemicals] subcutaneously, 4 and 1.5 hr before ethanol administration.
Seizure thresholds. Rats were administered finasteride (25 or 50 mg/kg) or vehicle [20% (w/v) 2-hydroxypropyl-β-cyclodextrin] subcutaneously, 4 and 1.5 hr before intraperitoneal ethanol administration. Seizure threshold was measured either 10 or 40 min after ethanol (2.0 gm/kg) administration. Rats were gently restrained for insertion of a 25 ga butterfly needle into the lateral tail vein, then held lightly by the tip of the tail while allowed free movement. Bicuculline (0.05 mg/ml) was infused at a rate of 1.6 ml/min until the first myoclonic twitch of the face and/or neck. Seizure threshold (milligrams per kilogram) was calculated as the duration of infusion × dose of bicuculline/body weight.
Single-cell electrophysiological recordings. Rats were anesthetized with urethane (1.5 gm/kg) and placed in a stereotaxic frame. An incision was made in the skin, the skull surface was cleaned, and a burr hole was drilled through the skull 0.3 mm anterior and 1.5 mm lateral of bregma. Single-barrel glass micropipettes were pulled (using Model PE-2; Narishige, Tokyo, Japan) and the tip was broken back to ∼1.0 μm and filled with a 0.9 m NaCl solution saturated with Chicago sky-blue dye. The electrode was lowered into the medial septum/diagonal band of Broca (MS/DB) via a hydraulic microdrive (Trent Wells, South Gate, CA). Extracellular action potentials were amplified, filtered (300 Hz and 8 kHz; Fintronics, Orange, CT), and were monitored with a Tektronix oscilloscope and audiomonitor. Individual action potentials isolated from background activity with at least 3:1 signal-to-noise ratios, constant duration/configuration, and rhythmically bursting patterns were defined as single MS/DB neurons using previously published criteria (Givens and Breese, 1990). Individual action potentials were digitized by a window discriminator (Fintronics), and the pulse was fed into an IBM personal computer that generated 10 sec time bin ratemeter histograms.
After isolation of a single MS/DB neuron, at least a 5 min baseline of spontaneous neural activity was collected, after which rats received a 1.5 gm/kg ethanol (10% w/v) intraperitoneal injection; thereafter, an additional 60 min of spontaneous activity was recorded. Care was taken to monitor the waveform of the action potential, and the electrode location was micromanipulated to prevent waveform alterations. After completion of the recording, a current was passed through the electrode, thereby depositing dye and marking its location. The brain was then removed from the animal, frozen, and sliced to verify electrode location. All neurons analyzed were located in the MS/DB (data not shown).
Statistics. Statistical analysis was done with Statview (Abacus Concepts, Berkeley, CA) and GraphPad Prism (GraphPad Software).
RESULTS
Ethanol induction of 3α,5α-THP in cerebral cortex
Levels of 3α,5α-THP in cerebral cortex were dramatically increased 60 min after acute ethanol administration (1.3–4.0 gm/kg, i.p.; 20% v/v in saline) to male Sprague Dawley rats (Fig.1A). Changes in cerebral cortical levels of 3α,5α-THP induced by ethanol were dose-dependent (Fig. 1A). There was a smaller 3α,5α-THP response at 4.0 gm/kg ethanol, but all doses measured beyond 1 gm/kg were significantly elevated above saline controls (one-way ANOVA, F = 13.20, p < 0.0001; Dunnett's post hoc, p < 0.05). Doses of ethanol from 2–3 g/kg caused increases in 3α,5α-THP levels in cortex from 2.7 ng/gm to 10–12 ng/gm. The effect of ethanol (2 gm/kg, i.p.) was observed 20 min after ethanol injection and peaked between 40 and 80 min (Fig. 1B).
Fig. 1.
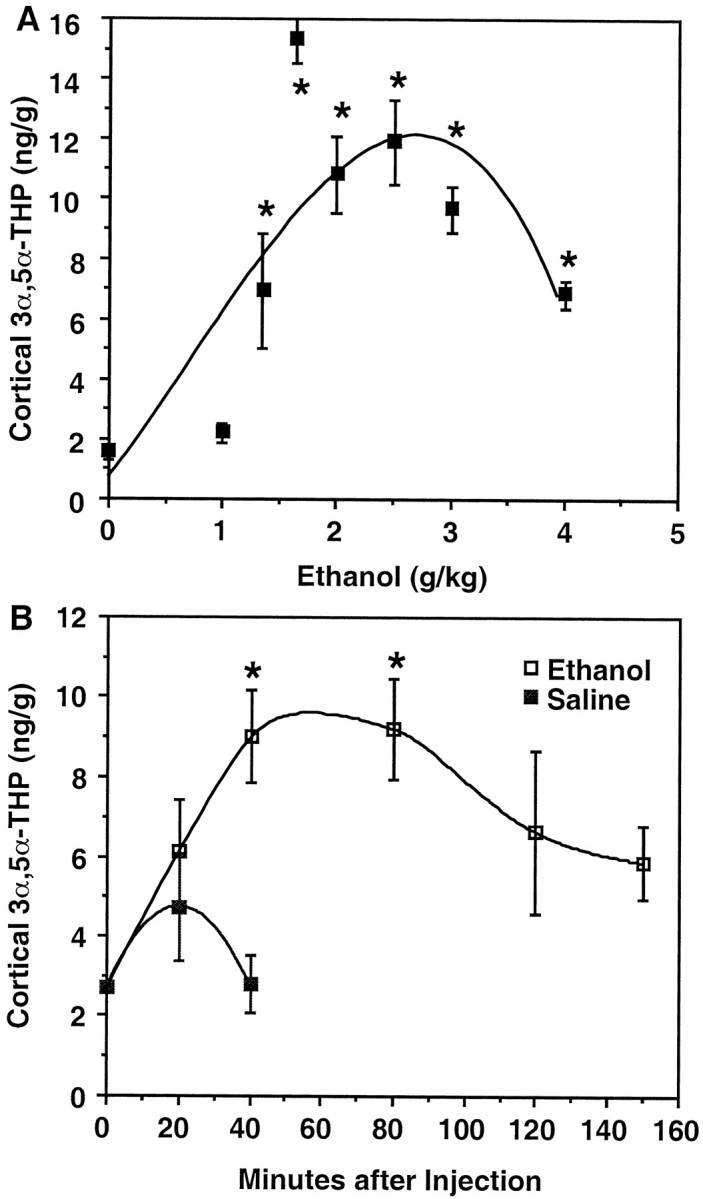
Cerebral cortical 3α,5α-THP levels are elevated after acute, systemic ethanol administration.A, Cerebral cortical 3α,5α-THP levels exhibit a biphasic response to increasing ethanol concentrations. Ethanol increased 3α,5α-THP levels to pharmacologically active concentrations at doses between 1.35 and 4.0 gm/kg (*p < 0.0001, ANOVA; p < 0.05, Dunnett's post hoc) measured 60 min after ethanol administration. Data represent mean ± SEM of duplicate determinations in 6–10 rats per dose from two independent experiments. Further analysis of trend shows a significant match to a quadratic equation (F(1,32) = 17.49;p < 0.05) using random samples of five rats per dose. B, Cerebral cortical 3α,5α-THP levels peak between 40 and 80 min after an injection of ethanol (2 gm/kg, i.p.) followed by a gradual decrease. Data shown are the mean ± SEM of a representative experiment repeated twice with similar results (n = 6/time point; *p < 0.05 Dunnett's post hoc).
3α,5α-THP levels in cerebral cortex of saline-injected rats were only mildly elevated at 20 min after injection and returned to baseline conditions by 40 min (Fig. 1B). Hence, the stress associated with injection did not significantly contribute to cerebral cortical levels of 3α,5α-THP in ethanol-treated rats. Plasma levels of progesterone and corticosterone were positively correlated after ethanol administration (r = 0.829;p < 0.0001); however, these levels did not correlate with ethanol-induced increases in cerebral cortical 3α,5α-THP levels (r = 0, p = 0.964;r = 0.179, p = 0.371, respectively; see Table 2). Moreover, cerebral cortical 3α,5α-THP levels did not correlate with plasma 3α,5α-THP levels measured between 0 and 120 min (r = 0.269, p = 0.074; see Table2).
Table 2.
Effects of steroid biosynthetic enzyme inhibitors
| Treatment groups | Cortical 3α, 5α, THP (ng/gm) | Plasma Progesterone (ng/ml) | Plasma Corticosterone (ng/ml) | n |
|---|---|---|---|---|
| Saline | 1.93 ± 0.34 | 3.40 ± 0.90 | 98.5 ± 23 | 14 |
| Ethanol | 10.5 ± 0.87 | 87.6 ± 7.3 | 476 ± 29 | 18 |
| Ethanol + trilostane | 10.4 ± 0.92 | 37.3 ± 7.3* | 7 | |
| Ethanol + finasteride | 5.9 ± 1.0* | 66.7 ± 8.9 | 202 ± 1.0* | 6 |
| Ethanol + indomethacin | 14.9 ± 89* | 72.3 ± 13 | 32.4 ± 13* | 7 |
*Significantly different from ethanol alone p < 0.01.
Steroid biosynthetic enzyme inhibitors differentially alter ethanol effects on cerebral cortical 3α,5α-THP levels. Ethanol (2 gm/kg, i.p.) was administered, and 3α,5α-THP levels were measured in cerebral cortex 1 hr after ethanol administration. Trilostane was administered 30 mg/kg, i.p. 1 hr before ethanol. The 5α-reductase inhibitor finasteride was administered 25 mg/kg, s.c., 4 and 1.5 hr before ethanol. The 3α-hydroxysteroid oxidoreductase inhibitor indomethacin was administered 0.1 mg/kg, i.p., 20 min before ethanol (Morrow et al., 1998). Cortical allopregnanolone levels were significantly decreased with finasteride pretreatment and significantly increased with indomethacin pretreatment, one-way ANOVA,F = 30.99, df = 4,47, p < 0.0001 with Dunnett's multiple comparison post hoc(p < 0.05 for both finasteride and indomethacin). Pretreatment with trilostane increased plasma progesterone levels, one-way ANOVA, F = 26.05, df = 4,47, p < 0.0001 with Dunnett's multiple comparisonpost hoc (p < 0.01). Pretreatment with finasteride and indomethacin decreased plasma corticosterone levels, one-way ANOVA, F = 34.29, df = 3.41,p < 0.0001 with Dunnett's multiple comparisonpost hoc (p < 0.01 for both finasteride and indomethacin).
Ethanol-induced elevations of 3α,5α-THP levels could occur via several mechanisms, including modulation of de novobiosynthesis, catabolism, or endogenous steroid release. To determine if 3α,5α-THP levels are increased after ethanol exposure as a result of de novo biosynthesis, the effects of steroid biosynthesis inhibitors were investigated. Three of these inhibitors were used, the 3β-hydroyxsteroidoxidoreductase/isomerase inhibitor trilostane, the 5α-reductase inhibitor finasteride, and the 3α-hydroxysteroidoxidoreductase inhibitor indomethacin (Potts et al., 1978; Penning et al., 1985; Steiner, 1996). The irreversible steroid 5α-reductase type II inhibitor finasteride was the only inhibitor that reduced the effect of ethanol on cerebral cortical 3α,5α-THP and plasma corticosterone levels (Table1). As previously reported, indomethacin paradoxically increased 3α,5α-THP levels over those of ethanol-induced 3α,5α-THP in cortex (Morrow et al., 1998), while still decreasing corticosterone levels (Table2). Trilostane reduced plasma progesterone levels, but had no effect on ethanol-induced changes in cortical 3α,5α-THP or plasma corticosterone.
Table 1.
Levels of plasma steroids after ethanol administration
| Steroid | 0 min | 20 min | 40 min | 80 min | 120 min | 150 min | 200 min |
|---|---|---|---|---|---|---|---|
| Allopregnanolone (ng/ml) | 0.068 ± 0.016 | 1.35 ± 0.18 | 1.35 ± 0.092 | 1.43 ± 0.088 | 1.78 ± 0.54 | 0.74 ± 0.22 | 0.455 ± 0.18 |
| Progesterone (ng/ml) | 2.02 ± 0.78 | 93.3 ± 8.2 | 95.3 ± 6.0 | 106 ± 3.7 | 67.2 ± 16 | 44.2 ± 11 | 15.2 ± 5.3 |
| Corticosterone (ng/ml) | 54.3 ± 15 | 386 ± 99 | 517 ± 35 | 597 ± 82 | 481 ± 83 | 391 ± 43 | 174 ± 45 |
Ethanol was administered (2 gm/kg, i.p., 20% ethanol in saline w/v), and animals were euthanized 0, 20, 40, 80, 120, 150, or 200 min after injection. Animals euthanized at 0 min were killed immediately after injection. Each group represents a minimum of six animals, and all measurements were made from aliquots of the same plasma.
3α,5α-THP modulation of ethanol-mediated behaviors
Having established that acute ethanol administration increases cerebral cortical 3α,5α-THP levels, we investigated the hypothesis that 3α,5α-THP contributes to the behavioral effects of ethanol. Because the sedative–hypnotic effects of ethanol involve the activation of GABAA receptors, we investigated whether ethanol-induced 3α,5α-THP levels in the cerebral cortex were correlated with the hypnotic effect of ethanol. Male Sprague Dawley rats were injected with 3.5 gm/kg ethanol [20% (w/v) in saline], and sleep time was measured from loss of righting reflex until righting reflex recovered. Cerebral cortical 3α,5α-THP levels were clearly correlated (r = 0.59; p < 0.0001) with ethanol-induced sleep time (Fig.2).
Fig. 2.

Elevations in cerebral cortical 3α,5α-THP are related to the hypnotic effect of ethanol. Alcohol (3.5 gm/kg, i.p.) was administered to male rats. The duration of the loss of righting reflex (sleep time) was correlated (r = 0.59;p < 0.0001; n = 37) with cerebral cortical 3α,5α-THP levels measured after awakening.
Acute ethanol administration has been shown to increase bicuculline-induced seizure threshold in rats (Rastogi et al., 1986;Glowa et al., 1988; Nutt and Lister, 1989). Ethanol administration (2 gm/kg) increased bicuculline seizure threshold 40 min, but not 10 min after ethanol administration (two-way ANOVA, effect of drug,p < 0.02; Tukey's multiple comparison, *p< 0.02) (Fig. 3A). This time course of anticonvulsant action correlates with the time course of ethanol induction of cerebral cortical 3α,5α-THP levels (Fig.1B). Pretreatment with finasteride (50 mg/kg, s.c.) under the same experimental conditions completely prevented the anticonvulsant effect of ethanol on bicuculline seizure threshold (ANOVA, F = 5.476, p < 0.01; Tukey's multiple comparison, †p< 0.05) (Fig. 3B).
Fig. 3.
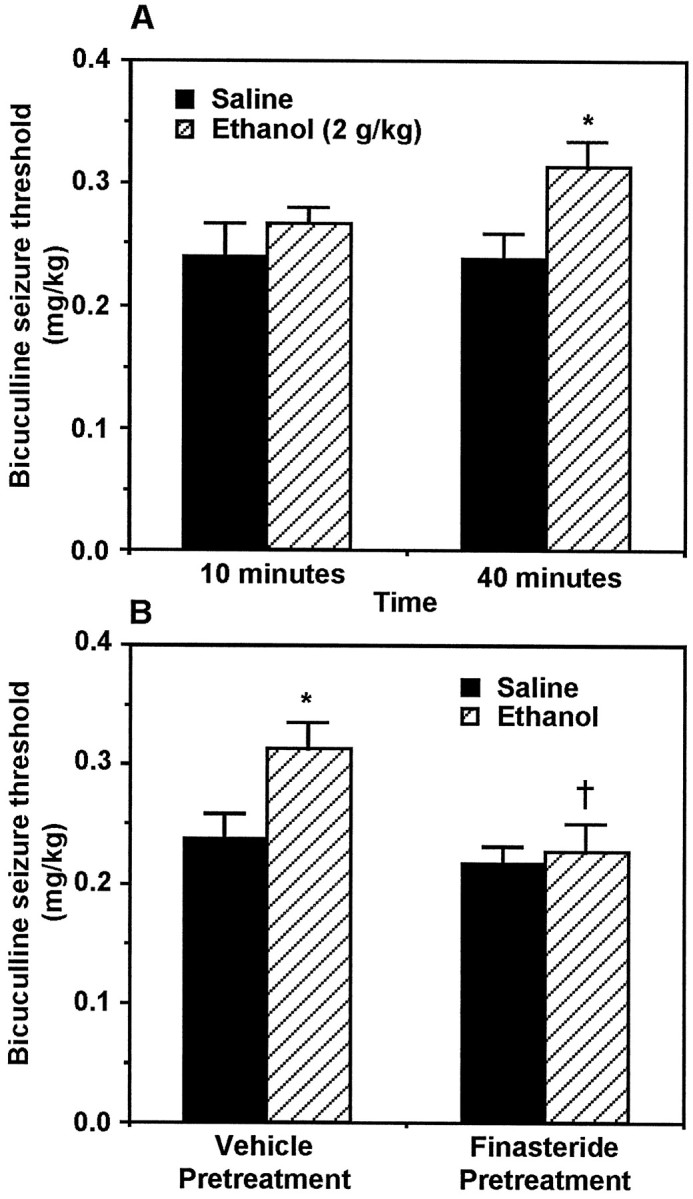
Anticonvulsant effect of ethanol on bicuculline seizure threshold is reversed by finasteride pretreatment.A, Ethanol (2 gm/kg, i.p.) increases bicuculline-induced seizure threshold at 40 min, but not 10 min, after administration (two-way ANOVA, effect of drug, F = 5.96,p < 0.02; Tukey's multiple comparison, *p < 0.02; n = 11–13).B, Finasteride pretreatment selectively reverses (ANOVA,F = 5.476, p < 0.01; Tukey's multiple comparison, †p < 0.05;n = 9–11) the ethanol-induced increase of seizure threshold.
3α,5α-THP modulates electrophysiological actions of ethanol
The ability of finasteride to modulate ethanol-induced effects on neural activity in the brain was investigated by monitoring spontaneous firing rates of MS/DB neurons. MS/DB cells show a high rate of spontaneous firing (Givens and Breese, 1990) that was inhibited by a maximum of 38.0 ± 1.52%, 20–40 min after systemic injection (Fig. 4A). Although maximal inhibition was between 20 and 40 min, spontaneous firing was inhibited from baseline throughout the recording period. Previous work has shown that complete recovery is not attained until 90 min (Givens and Breese, 1990). Finasteride pretreatment reversed the inhibition of spontaneously active MS/DB neurons produced by systemic ethanol administration (p < 0.0001, two-way ANOVA with repeated measures; main effect of finasteride,F(2,152) = 34.77; Fig.4A). Pretreatment with finasteride (25 mg/kg) prevented ethanol-induced inhibition of spontaneously active MS/DB neurons from 20–29 min [Tukey's post hoc test,q = 6.213, p < 0.01 (20–24 min time bin); q = 4.99, p < 0.01 (25–29 min time bin)] but not after 30 min. In contrast, the higher finasteride dose (50 mg/kg) prevented ethanol-induced inhibition throughout the 60 min recording period [Tukey's post hoc test,q = 6.839, p < 0.001 (20–24 min time bin); q = 5.529; p < 0.01 (25–29 min time bin); q = 4.626; p < 0.05 (30–34 min time bin); q = 4.907, p < 0.05 (35–39 min time bin); q = 4.649, p < 0.05 (45–49 min time bin)]. Baseline spontaneous neural activity was not altered by finasteride pretreatment (one-way ANOVA;F(2,13) = 2.13; p > 0.05; Fig. 4B). Baseline firing rates for the ethanol only group ranged from 12.5–59.1 Hz, the ethanol + 25 mg/kg finasteride group ranged from 14.2–31.6 Hz, and the ethanol + 50 mg/kg finasteride group ranged from 10.9–23.3 Hz. Moreover, the time course of the onset of ethanol inhibition of spontaneous neural activity in the MS/DB is very similar to the time course of ethanol-induced 3α,5α-THP elevation in cerebral cortex. However, the time course of the recovery of MS/DB spontaneous neural activity precedes the time course of return to baseline 3α,5α-THP levels in cortex.
Fig. 4.

Finasteride pretreatment prevents ethanol-induced inhibition of spontaneous neural activity in MS/DB neurons.A, Systemic ethanol injection (1.5 gm/kg, i.p.) reduced spontaneous neural activity by a maximum of 38.00 ± 1.52%, 20–40 min after injection. Finasteride pretreatment reversed the inhibition of spontaneously active MS/DB neurons produced by systemic ethanol administration (p < 0.0001, two-way ANOVA with repeated measures; main effect of finasteride,F(2,152) = 34.77). Complete blockade of ethanol inhibition was observed between 16 and 25 min after the ethanol injection at both doses of finasteride. However, only the high dose of finasteride completely blocked ethanol inhibition throughout the recording session. Inset is a representative oscilloscope trace of medial septum/diagonal band of Broca neuron.B, Baseline firing rate (5 min before ethanol); *p < 0.05; †p < 0.07.B, A grand mean of ethanol-induced inhibition for each neuron was calculated by collapsing the data across the 12, 5 min time bins. Acute ethanol administration inhibited the spontaneous activity of MS/DB neurons compared to preinjection baseline (pairedt test, T = 3.12; *p < 0.05). Pretreatment with 25 mg/kg finasteride did not alter the effect of ethanol on spontaneous firing rate. However, pretreatment with 50 mg/kg finasteride completely prevented ethanol-induced inhibition of spontaneous MS/DB neural activity (one-way ANOVA, F(2,13) = 5.45,p < 0.02; Tukey's post hoc test,q = 4.593, †p < 0.05).C, Representative MS/DB neural traces from an animal treated with ethanol only and an animal treated with ethanol + 50 mg/kg finasteride. Each neuron was recorded for 60 min after a 1.5 gm/kg ethanol injection (vertical arrow). The neural trace preceding the vertical arrow is the baseline recording session. Note, ethanol injection resulted in an ∼32% inhibition in the MS/DB neuron from the control rat, whereas the ethanol-induced inhibition was completely blocked by pretreatment with 50 mg/kg finasteride.
DISCUSSION
These data reveal an important interaction between ethanol and the endogenous pregnane neurosteroid 3α,5α-THP. Ethanol induces a dose- and time-dependent increase in cerebral cortical 3α,5α-THP levels to physiologically relevant concentrations. The time course of the induction of this potent GABA modulator at a moderate ethanol dose correlates with the appearance of specific behavioral and electrophysiological effects of ethanol. These effects are reversed when 3α,5α-THP induction is reduced by finasteride, indicating that 3α,5α-THP mediates or modulates some GABAergic effects of ethanol.
Ethanol induces an increase in cortical 3α,5α-THP levels sufficient to potentiate GABAA receptor-mediated inhibition in brain (Harrison et al., 1987; Morrow et al., 1987). The maximal induction of cortical 3α,5α-THP by ethanol is greater than the induction of 3α,5α-THP triggered by various stressors (Purdy et al., 1991; Barbaccia et al., 1996). It is interesting to note that moderate doses of ethanol induced the largest 3α,5α-THP responses at the 60 min time point. This could be reflective of a varying time course in the elevation of 3α,5α-THP levels. Alternatively, this may indicate ethanol effects on 3α,5α-THP biosynthetic enzyme kinetics or desensitization to ethanol. Similar biphasic effects of ethanol and barbiturates on GABAAreceptor-mediated Cl− flux (Schwartz et al., 1986) have previously been observed.
The observation that brain 3α,5α-THP levels do not correlate with plasma corticosterone (a peripheral indicator of HPA axis activity), progesterone (a precursor of 3α,5α-THP), or plasma 3α,5α-THP itself suggests that brain steroid levels may be differentially regulated from circulating steroids. The absence of a correlation between central and circulating 3α,5α-THP levels could be attributable to different time courses for steroid production or metabolism between the periphery and brain. Although it is premature to conclude that brain levels are independent of peripheral steroid sources, evidence suggests that neurosteroid biosynthesis in brain may include novel nonenzymatic pathways (Lieberman and Prasad, 1990; Prasad et al., 1994; Cascio et al., 1998).
The corroboration of single-cell recording data demonstrates the specific physiological role of ethanol-induced 3α,5α-THP in vivo. Ethanol suppression of MS/DB spontaneous activity is temporally correlated with the increase in cortical levels of 3α,5α-THP and reversed by finasteride pretreatment, demonstrating that 3α,5α-THP contributes to this response. The time course of the effects of ethanol on spontaneous activity and 3α,5α-THP levels are very similar, despite differences in the brain regions and the dose of ethanol. Furthermore, single-cell recordings of the neighboring lateral septum, a brain region in which ethanol does not alter the spontaneous firing rate of neurons, show no effect of either ethanol or finasteride (data not shown). The enzymes responsible for 3α,5α-THP production in the brain are regionally expressed (Roselli and Snipes, 1984;Korneyev et al., 1993a; Martini et al., 1996), highlighting the possible importance of studying the regional modulation of these compounds by ethanol.
The correlation of ethanol-induced loss of righting reflex and brain 3α,5α-THP levels suggests that 3α,5α-THP contributes to the hypnotic effect of ethanol, likely via GABAAreceptor activation. The ability of finasteride to block the anticonvulsant effect of ethanol on bicuculline seizure thresholds further substantiates the role of 3α,5α-THP in ethanol-induced behavior. The effects of finasteride on ethanol action provide significant evidence that neurosteroid intermediaries contribute to ethanol action; however, because these steroids share many of the same biosynthetic enzymes, the possible involvement of other 5α-reduced steroids cannot be ruled out.
In fact, it is known that ethanol increases plasma levels of several steroids, including corticosterone, testosterone, and progesterone (Rivier et al., 1984; Korneyev et al., 1993b; Schuckit and Smith, 1996). Further research is needed to determine if similar effects are found with other GABAergic neurosteroids such as 3α,21-dihydroxy-5α-pregnan-20-one or 3α-hydroxy-5β-pregnane-20-one. Recent studies suggest that 3α,5α-THP also modulates other ion channels, including the nicotinic acetylcholine receptor and the serotonin 5-HT3 receptor, but these effects require much greater concentrations (10 μm) than observed in response to ethanol (1–4 gm/kg) (Rupprecht and Holsboer, 1999).
The fact that finasteride inhibits some of the GABAergic effects of ethanol suggests that ethanol may alter the biosynthesis of 3α,5α-THP. Because finasteride is selective for 5α-reductase type 2 activity in vivo (Bramson et al., 1997), inhibition of type 1 enzyme activity may be required to obtain complete inhibition of ethanol effects. However, it appeared that the activity of steroid biosynthesis inhibitors was altered in the presence of ethanol, compared to previously published reports (Korneyev et al., 1993b; Costa et al., 1995; Smith et al., 1998b; Griffin and Mellon, 1999). For example, trilostane significantly decreased plasma progesterone levels, but did not change cortical 3α,5α-THP levels. This is surprising because previous reports suggest that this dose blocks formation of 3α,5α-THP (Concas et al., 1998). This raises the possibility that ethanol increases the activity of the 3α-HSD enzyme, as recently suggested by Griffin and Mellon (1999) in their study of 3α-HSD kinetics. Further studies are needed to distinguish if ethanol has direct effects on the neurosteroid biosynthetic enzymes or the efficacy of the enzyme inhibitors.
Alternatively, the lack of effect of biosynthesis inhibitors on ethanol-induced 3α,5α-THP levels may indicate that the elevation in 3α,5α-THP is independent of de novo biosynthesis of 3α,5α-THP. This could occur if ethanol stimulates the release of 3α,5α-THP from stores in adrenals or glial cells. Finasteride pretreatment may have depleted stores of 3α,5α-THP, because it was administered 4 and 1.5 hr before ethanol, and thereby partially inhibited the effects of ethanol. Further studies are required to investigate this possibility.
Despite the observation that finasteride had a partial effect on cortical 3α,5α-THP levels, it is not surprising that finasteride did not have an effect on baseline seizure threshold and electrophysiological measurements. Endogenous levels of 3α,5α-THP in naïve male rats are very low, certainly below physiologically relevant concentrations (Purdy et al., 1991). Furthermore, the dose of finasteride used (50 mg/kg) is below the EC50 for blockade of the anticonvulsant effects of progesterone in mice (Kokate et al., 1999).
Previous studies demonstrated that brain 3α,5α-THP levels are elevated by the specific serotonin reuptake inhibitors (SSRIs) fluoxetine and paroxetine (Uzunov et al., 1996; Uzunova et al., 1998). Because elevations in endogenous 3α,5α-THP levels enhance the hypnotic effects of pentobarbital (Matsumoto et al., 1999), elevations in 3α,5α-THP may also contribute to hypnotic properties of SSRIs or ethanol. The overlapping effects of ethanol and SSRIs, such as anxiolytic, sedative, and stress-reducing properties, may relate to the ability of these drugs to elevate 3α,5α-THP levels in brain. However, alcohol and SSRIs each have other independent neurochemical actions that likely account for other diverse effects of these drugs. The modulation of 3α,5α-THP levels by SSRIs and ethanol further support the important role of 3α,5α-THP in the regulation of CNS function.
It is important to note that ethanol affects several ion channels (Crews et al., 1996). Some effects of ethanol can be observed minutes after administration. The effects that we measured are observed at later time points. Anticonvulsant effects of ethanol and the depression of MS/DB spontaneous activity occur between 15 and 80 min after ethanol administration and correlate very well with maximal 3α,5α-THP elevation. Thus, our data speak directly to 3α,5α-THP modulation of these GABAergic functions and do not preclude direct ethanol activity at GABAA receptors or other ion channels.
Other investigators have demonstrated important roles for 3α,5α-THP in ethanol action. In a discriminative stimulus paradigm, Bowen et al. (1999) showed that GABAergic neurosteroids substitute for ethanol in both primates and rats. Exogenous administration of 3α,5α-THP altered ethanol reinforced operant responding (Janak et al., 1998). Because neurosteroids are such potent modulators of the GABAergic pathways involved not only in alcohol action, but also in anxiety, seizure, and movement disorders, there are possible clinical ramifications of these data. A specific antagonist of GABAergic neurosteroid action may be useful in treating alcoholism because of the role we have shown for 3α,5α-THP in modulating ethanol actions as well as the roles for 3α,5α-THP in alcohol reinforcement, reward, and perhaps tolerance (Janak et al., 1998; Bowen et al., 1999).
The identification of a neuroactive steroid as an intermediary of ethanol actions at GABAA receptors provides an important example of functional interactions between the genomic and nongenomic actions of neurosteroids. GABAAreceptor function and mRNA expression are modulated by changes in neurosteroid levels during pregnancy, and this modulation can be reversed by finasteride (Concas et al., 1998). In our studies, chronic ethanol administration reduced basal levels of 3α,5α-THP in cerebral cortices of male rats (Janis et al., 1998). We have also found that ethanol-dependent rats are sensitized to the anticonvulsant effect of 3α,5α-THP during ethanol withdrawal (Devaud et al., 1996). Moreover, chronic ethanol exposure and withdrawal (Devaud et al., 1995,1997; Matthews et al., 1998) have been shown to cause changes in GABAA receptor α4 subunit mRNA and peptide expression that mirror the effects of withdrawal from progesterone and 3α,5α-THP (Smith et al., 1998a). Taken together, these data demonstrate that neuroactive steroids such as 3α,5α-THP can elicit synergistic actions involving both nongenomic and genomic effects on GABAA receptors that enable long-term adaptations in receptor function and expression.
Footnotes
This work was supported by United States Public Health Service National Institutes of Health Grants AA10564 and AA11605 to A.L.M., National Science Foundation predoctoral fellowship to M.J.V., and Individual National Research Service Award Grant AA05519 to D.B.M. We thank Drs. Steven Paul, Robert Purdy, and Hugh Criswell for helpful suggestions and discussion. We thank Drs. Fulton Crews and George Koob for review of this manuscript and Tejas Patel for excellent technical assistance.
Correspondence should be addressed to A. Leslie Morrow, Department of Psychiatry, Pharmacology and Center for Alcohol Studies, University of North Carolina School of Medicine, CB #7178, Chapel Hill, NC 27599-7178. E-mail: morrow@med.unc.edu.
Dr. Janis's present address: Department of Pharmacology, Boston University Medical Center, Boston, MA 02118.
Dr. Devaud's present address: Department of Pharmaceutical Sciences, Idaho State University, Pocatello, ID 83209.
Dr. Matthews' present address: Department of Psychology, University of Memphis, Memphis, TN 38152.
REFERENCES
- 1.Barbaccia ML, Roscetti G, Trabucchi M, Mostallino MC, Purdy RH, Biggio G. Time-dependent changes in rat brain neuroactive steroid concentrations and GABAA receptor function after acute stress. Neuroendocrinology. 1996;63:166–172. doi: 10.1159/000126953. [DOI] [PubMed] [Google Scholar]
- 2.Barbaccia ML, Roscetti G, Trabucchi M, Purdy RH, Mostallino MC, Concas A, Biggio G. The effects of inhibitors of GABAergic transmission and stress on brain and plasma allopregnanolone concentrations. Br J Pharmacol. 1997;120:1582–1588. doi: 10.1038/sj.bjp.0701046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bowen CA, Purdy RH, Grant KA. An investigation of endogenous neuroactive steroid-induced modulation of ethanol's discriminative stimulus effects. Behav Pharmacol. 1999;10:297–311. doi: 10.1097/00008877-199905000-00006. [DOI] [PubMed] [Google Scholar]
- 4.Bramson HN, Hermann D, Batchelor KW, Lee FW, James MK, Frye SV. Unique preclinical characteristics of GC745, a potent dual inhibitor of 5AR. J Pharmacol Exp Ther. 1997;282:1496–1502. [PubMed] [Google Scholar]
- 5.Cascio C, Prasad VVK, Lin YY, Lieberman S, Papadopoulos V. Detection of P450c17-independent pathways for dehydroepiandrosterone (DHEA) biosynthesis in brain glial tumor cells. Proc Natl Acad Sci USA. 1998;95:2862–2867. doi: 10.1073/pnas.95.6.2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Concas A, Mostallino MC, Porcu P, Follesa P, Barbaccia ML, Trabucchi M, Purdy RH, Grisenti P, Biggio G. Role of brain allopregnanolone in the plasticity of gamma-aminobutyric acid type A receptor in rat brain during pregnancy and after delivery. Proc Natl Acad Sci USA. 1998;95:13284–13289. doi: 10.1073/pnas.95.22.13284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Costa A-M, Spence KT, Smith SS, ffrench-Mullen J. Withdrawal from the endogenous steroid progesterone results in GABAA currents insensitive to benzodiazepine modulation in rat CA1 hippocampus. J Neurophysiol. 1995;74:464–469. doi: 10.1152/jn.1995.74.1.464. [DOI] [PubMed] [Google Scholar]
- 8.Crews F, Morrow AL, Criswell H, Breese G. Effects of ethanol on ion channels. In: Bradley RJ, Harris RA, editors. International review of neurobiology, Vol 39. Academic; New York: 1996. pp. 283–367. [DOI] [PubMed] [Google Scholar]
- 9.Devaud LL, Smith FD, Grayson DR, Morrow AL. Chronic ethanol consumption differentially alters the expression of γ-aminobutyric acidA receptor subunit mRNAs in rat cerebral cortex: competitive, quantitative reverse transcriptase-polymerase chain reaction analysis. Mol Pharmacol. 1995;48:861–868. [PubMed] [Google Scholar]
- 10.Devaud LL, Purdy RH, Finn DA, Morrow AL. Sensitization of γ-aminobutyric acidA receptors to neuroactive steroids in rats during ethanol withdrawal. J Pharmacol Exp Ther. 1996;278:510–517. [PubMed] [Google Scholar]
- 11.Devaud LL, Fritschy J-M, Sieghart W, Morrow AL. Bidirectional alterations of GABAA receptor subunit peptide levels in rat cortex during chronic ethanol consumption and withdrawal. J Neurochem. 1997;69:126–130. doi: 10.1046/j.1471-4159.1997.69010126.x. [DOI] [PubMed] [Google Scholar]
- 12.Finn DA, Gee KW. The influence of estrus cycle on neurosteroid potency at the gamma-aminobutyric acidA receptor complex. J Pharmacol Exp Ther. 1993;265:1374–1379. [PubMed] [Google Scholar]
- 13.Frye GD, Fincher AS, Grover CA, Griffith WH. Interaction of ethanol and allosteric modulators with GABAA-activated currents in adult medial septum/diagonal band neurons. Brain Res. 1994;635:283–292. doi: 10.1016/0006-8993(94)91449-4. [DOI] [PubMed] [Google Scholar]
- 14.Givens BS, Breese GR. Site-specific enhancement of γ-aminobutyric acid-mediated inhibition of neural activity by ethanol in the rat medial septum. J Pharmacol Exp Ther. 1990;254:528–538. [PMC free article] [PubMed] [Google Scholar]
- 15.Glowa JR, Crawley J, Suzdak PD, Paul SM. Ethanol and the GABA receptor complex: studies with the partial inverse benzodiazepine receptor agonist Ro 15–4513. Pharmacol Biochem Behav. 1988;31:767–772. doi: 10.1016/0091-3057(88)90263-8. [DOI] [PubMed] [Google Scholar]
- 16.Griffin LD, Mellon SH. Selective serotonin reuptake inhibitors directly alter activity of neurosteroidogenic enzymes. Proc Natl Acad Sci USA. 1999;965:13512–13517. doi: 10.1073/pnas.96.23.13512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grobin AC, Matthews DB, Devaud LL, Morrow AL. The role of GABAA receptors in the acute and chronic effects of ethanol. Psychopharmacology. 1998;139:2–19. doi: 10.1007/s002130050685. [DOI] [PubMed] [Google Scholar]
- 18.Harrison NL, Majewska MD, Harrington JW, Barker JL. Structure-activity relationships for steroid interaction with the gamma-aminobutyric acid-A receptor complex. J Pharmacol Exp Ther. 1987;241:346–353. [PubMed] [Google Scholar]
- 19.Ichikawa S, Sawada T, Nakamura Y, Morioka H. Ovarian secretion of pregnane compounds during the estrus cycle and pregnancy in rats. Endocrinology. 1974;94:1615–1620. doi: 10.1210/endo-94-6-1615. [DOI] [PubMed] [Google Scholar]
- 20.Janak PH, Redfern JEM, Samson HH. The reinforcing effects of ethanol are altered by the endogenous neurosteroid, allopregnanolone. Alcohol Clin Exp Res. 1998;22:1106–1112. [PubMed] [Google Scholar]
- 21.Janis GC, Devaud LL, Mitsuyama H, Morrow AL. Effects of chronic ethanol consumption and withdrawal on the neuroactive steroid 3α-hydroxy-5α-pregnan-20-one in male and female rats. Alcohol Clin Exp Res. 1998;22:2055–2061. [PubMed] [Google Scholar]
- 22.Kokate TG, Banks MK, Magoo T, Yamaguchi S-I, Rogawski MA. Finasteride, a 5α-reductase inhibitor, blocks the anticonvulsant activity of progesterone in mice. J Pharmacol Exp Ther. 1999;288:679–684. [PubMed] [Google Scholar]
- 23.Korneyev A, Guidotti A, Costa E. Regional and interspecies differences in brain progesterone metabolism. J Neurochem. 1993a;61:2041–2047. doi: 10.1111/j.1471-4159.1993.tb07440.x. [DOI] [PubMed] [Google Scholar]
- 24.Korneyev AY, Costa E, Guidotti A. During anesthetic-induced activation of hypothalamic pituitary adrenal axis, blood borne steroids fail to contribute to the anesthetic effect. Neuroendocrinology. 1993b;57:559–565. doi: 10.1159/000126405. [DOI] [PubMed] [Google Scholar]
- 25.Lephart ED, Ladle DR, Jacobson NA, Rhees RW. Inhibition of brain 5α-reductase in pregnant rats: Effects on enzymatic and behavioral activity. Brain Res. 1996;739:356–360. doi: 10.1016/s0006-8993(96)01068-2. [DOI] [PubMed] [Google Scholar]
- 26.Lieberman S, Prasad VVK. Heterodox notions of pathways of steroidogenesis. Endocr Rev. 1990;11:469–493. doi: 10.1210/edrv-11-4-469. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Deitrich RA. Role of GABA in the actions of ethanol in rats selectively bred for ethanol sensitivity. Pharmacol Biochem Behav. 1998;60:793–801. doi: 10.1016/s0091-3057(97)00479-6. [DOI] [PubMed] [Google Scholar]
- 28.Mancillas JR, Siggins JR, Bloom FE. Systemic ethanol: enhancement of responses to acetylcholine and somatostatin in hippocampus. Science. 1986;231:161–163. doi: 10.1126/science.2867600. [DOI] [PubMed] [Google Scholar]
- 29.Marszalec W, Aistrup GL, Narahashi T. Ethanol modulation of excitatory and inhibitory synaptic interactions in cultured cortical neurons. Alcohol Clin Exp Res. 1998;22:1516–1524. [PubMed] [Google Scholar]
- 30.Martini L, Celotti F, Melcangi RC. Testosterone and progesterone metabolism in the central nervous system: cellular localization and mechanism of control of the enzymes involved. Cell Mol Biol. 1996;16:271–282. doi: 10.1007/BF02088095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martz A, Dietrich RA, Harris RA. Behavioral evidence for the involvement of γ-aminobutyric acid in the actions of ethanol. Eur J Pharmacol. 1983;89:53–62. doi: 10.1016/0014-2999(83)90607-6. [DOI] [PubMed] [Google Scholar]
- 32.Matsumoto K, Uzunova V, Pinna G, Taki K, Uzunov DP, Watanabe H, Mienville JM, Guidotti A, Costa E. Permissive role of brain allopregnanolone content in the regulation of pentobarbital-induced righting reflex loss. Neuropharmacology. 1999;38:955–963. doi: 10.1016/s0028-3908(99)00018-0. [DOI] [PubMed] [Google Scholar]
- 33.Matthews DB, Devaud LL, Fritschy J-M, Sieghart W, Morrow AL. Differential regulation of GABAA receptor gene expression by ethanol in the rat hippocampus vs. cerebral cortex. J Neurochem. 1998;70:1160–1166. doi: 10.1046/j.1471-4159.1998.70031160.x. [DOI] [PubMed] [Google Scholar]
- 34.Morrow AL. Regulation of GABAA receptor function and gene expression in the central nervous system. In: Bradley RJ, Harris RA, editors. International review of neurobiology, Vol 38. Academic; San Diego: 1995. pp. 1–41. [DOI] [PubMed] [Google Scholar]
- 35.Morrow AL, Suzdak PD, Paul SM. Steroid hormone metabolites potentiate GABA receptor-mediated chloride ion flux with nanomolar potency. Eur J Pharmacol. 1987;142:483–485. doi: 10.1016/0014-2999(87)90094-x. [DOI] [PubMed] [Google Scholar]
- 36.Morrow AL, VanDoren MJ, Devaud LL. Effects of progesterone or neuroactive steroid? Nature. 1998;395:652–653. doi: 10.1038/27106. [DOI] [PubMed] [Google Scholar]
- 37.Nutt DJ, Lister RG. Antagonizing the anticonvulsant effect of ethanol using drugs acting at the benzodiazepine/GABA receptor complex. Pharmacol Biochem Behav. 1989;31:751–755. doi: 10.1016/0091-3057(88)90260-2. [DOI] [PubMed] [Google Scholar]
- 38.Paul SM, Purdy RH. Neuroactive steroids. FASEB J. 1992;6:2311–2322. [PubMed] [Google Scholar]
- 39.Penning TM, Sharp RB, Kriegers NR. Purification and properties of 3α-hydroxysteroid dehydrogenase from rat brain cytosol. J Biol Chem. 1985;260:15266–15282. [PubMed] [Google Scholar]
- 40.Potts GO, Creange JW, Harding HR, Schane HP. Trilostane, an orally active inhibitor of steroid biosynthesis. Steroids. 1978;32:257–267. doi: 10.1016/0039-128x(78)90010-7. [DOI] [PubMed] [Google Scholar]
- 41.Prasad VVK, Vegesna SR, Welch M, Lieberman S. Precursors of the neurosteroids. Proc Natl Acad Sci USA. 1994;91:3220–3223. doi: 10.1073/pnas.91.8.3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Purdy RH, Morrow AL, Moore PH, Jr, Paul SM. Stress-induced elevations of gamma-aminobutyric acid type A receptor-active steroids in the rat brain. Proc Natl Acad Sci USA. 1991;88:4553–4557. doi: 10.1073/pnas.88.10.4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rastogi SK, Thyagarajan R, Clothier J, Ticku MK. Effect of chronic treatment of ethanol on benzodiazepine and picrotoxin sites on the GABA receptor complex in regions of the brain of the rat. Neuropharmacology. 1986;25:1179–1184. doi: 10.1016/0028-3908(86)90167-x. [DOI] [PubMed] [Google Scholar]
- 44.Rivier C, Bruhn T, Vale W. Effect of ethanol on the hypothalamic-pituitary-adrenal axis in the rat: role of corticotropin-releasing factor (CRF). J Pharmacol Exp Ther. 1984;229:127–131. [PubMed] [Google Scholar]
- 45.Robel P, Baulieu EE. Neurosteroids: biosynthesis and function. Crit Rev Neurobiol. 1995;9:383–395. [PubMed] [Google Scholar]
- 46.Roselli CE, Snipes CA. Progesterone 5α-reductase in mouse brain. Brain Res. 1984;305:197–202. doi: 10.1016/0006-8993(84)90425-6. [DOI] [PubMed] [Google Scholar]
- 47.Rupprecht R, Holsboer F. Neuroactive steroids: mechanisms of action and neuropsychopharmacological perspectives. Trends Neurosci. 1999;22:410–416. doi: 10.1016/s0166-2236(99)01399-5. [DOI] [PubMed] [Google Scholar]
- 48.Sapp DW, Yeh HH. Ethanol-GABAA receptor interactions: A comparison between cell lines and cerebellar Purkinje cells. J Pharmacol Exp Ther. 1998;284:768–776. [PubMed] [Google Scholar]
- 49.Schuckit MA, Smith TL. An 8-year follow-up of 450 sons of alcoholic and control subjects. Arch Gen Psychiatry. 1996;53:202–210. doi: 10.1001/archpsyc.1996.01830030020005. [DOI] [PubMed] [Google Scholar]
- 50.Schwartz RD, Suzdak PD, Paul SM. γ-aminobutyric acid (GABA) and barbiturate receptor-mediated 36Cl- uptake in rat brain synaptoneurosomes: evidence for rapid desensitization of the GABA receptor-coupled chloride ion channel. Mol Pharmacol. 1986;30:419–426. [PubMed] [Google Scholar]
- 51.Simson PE, Criswell HE, Breese GR. Ethanol potentiates γ-aminobutyric acid-mediated inhibition in the inferior colliculus: evidence for local ethanol/γ-aminobutyric acid interactions. J Pharmacol Exp Ther. 1991;259:1288–1293. [PubMed] [Google Scholar]
- 52.Smith SS, Gong QH, Hsu F-C, Markowitz RS, ffrench-Mullen JMH, Li X. GABAA receptor α4 subunit suppression prevents withdrawal properties of an endogenous steroid. Nature. 1998a;392:926–930. doi: 10.1038/31948. [DOI] [PubMed] [Google Scholar]
- 53.Smith SS, Gong QH, Li X, Moran MH, Bitran D, Frye CA, Hsu F. Withdrawal from 3α-OH-5α-pregnan-20-one using a pseudopregnancy model alters the kinetics of hippocampal GABAA-gated current and increases the GABAA receptor α4 subunit in association with increased anxiety. J Neurosci. 1998b;18:5275–5284. doi: 10.1523/JNEUROSCI.18-14-05275.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Steiner JF. Clinical pharmacokinetics and pharmacodynamics of finasteride. Clin Pharmacokinet. 1996;30:16–27. doi: 10.2165/00003088-199630010-00002. [DOI] [PubMed] [Google Scholar]
- 55.Suzdak PD, Glowa JR, Crawley JN, Schwartz RD, Skolnick P, Paul SM. A selective imidazobenzodiazepine antagonist of ethanol in the rat. Science. 1986a;234:1243–1247. doi: 10.1126/science.3022383. [DOI] [PubMed] [Google Scholar]
- 56.Suzdak PD, Schwartz RD, Skolnick P, Paul SM. Ethanol stimulates γ-aminobutyric acid receptor-mediated chloride transport in rat brain synaptoneurosomes. Proc Natl Acad Sci USA. 1986b;83:4071–4075. doi: 10.1073/pnas.83.11.4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Uzunov DP, Cooper TB, Costa E, Guidotti A. Fluoxetine-elicited changes in brain neurosteroid content measured by negative ion mass fragmentography. Proc Natl Acad Sci USA. 1996;93:12599–12604. doi: 10.1073/pnas.93.22.12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Uzunova V, Sheline Y, Davis JM, Rasmusson A, Uzunov DP, Costa E, Guidotti A. Increase in the cerebrospinal fluid content of neurosteroids in patients with unipolar major depression who are receiving fluoxetine or fluvoxamine. Proc Natl Acad Sci USA. 1998;95:3239–3244. doi: 10.1073/pnas.95.6.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]