

Abstract
c-Jun N-terminal protein kinase (JNK) and p38 mitogen-activated protein kinase are activated by stress and are implicated in regulation of apoptosis in several tissues. However, their contribution to stress-induced apoptosis in CNS neurons is not well defined. Here we investigated the role of JNK and p38 in cortical neuron apoptosis caused by sodium arsenite treatment. Sodium arsenite is an environmental toxicant that causes developmental defects in the CNS. Treatment of cortical neurons with sodium arsenite activated p38 and JNK3 but not JNK1 or JNK2. It also induced c-Jun phosphorylation. Furthermore, sodium arsenite induced cortical neuron apoptosis. This apoptosis was attenuated by SB203580, an inhibitor of p38, and by CEP-1347, an inhibitor of JNK activation. Expression of dominant-interfering mutants of the JNK or p38 pathways inhibited apoptosis induced by arsenite, whereas expression of constitutive active mutants for either pathway induced apoptosis. Moreover, the caspase inhibitor zVAD-fluoromethylketone as well as expression of bcl-2 or bcl-xL inhibited cortical neuron apoptosis induced by arsenite or by constitutive activation of JNK or p38. These data indicate that both JNK and p38 contribute to arsenite-induced apoptosis in primary CNS neurons, and this apoptosis requires the bcl-2–caspase pathway. This is the first evidence that a specific JNK isoform is differentially activated by stress and contributes to neuronal apoptosis.
Keywords: apoptosis, signal transduction, CNS, neurons, arsenite, JNK, p38, MAP kinase
Apoptosis plays an important role during neuronal development and in the homeostasis of the adult nervous system (Cowan et al., 1984; Oppenheim, 1991; Raff et al., 1993). Abnormal neuronal apoptosis may contribute to various neurodegenerative diseases (Thompson, 1995; Stefanis et al., 1997). Elucidation of mechanisms that regulate neuronal apoptosis may provide insights concerning the prevention and treatment of these disorders.
Recent studies have implicated the stress-activated protein kinase c-Jun N-terminal protein kinase (JNK) and p38 mitogen-activated protein (MAP) kinase pathways as key regulators of apoptosis (Estus et al., 1994; Ham et al., 1995; Mesner et al., 1995). For example, activation of JNK or p38 contributes to PC12 cell apoptosis caused by nerve growth factor (NGF) withdrawal (Xia et al., 1995). Nevertheless, the contribution of JNK and p38 to neuronal apoptosis is not well defined. Although some studies suggested a role for JNK or p38 in neuronal apoptosis (Kawasaki et al., 1997; Yang et al., 1997; Bazenet et al., 1998; Luo et al., 1998; Behrens et al., 1999; Le Niculescu et al., 1999; Camandola et al., 2000; Kanamoto et al., 2000), other studies failed to implicate these kinases in neuronal apoptosis (Migheli et al., 1997; Gunn-Moore and Tavare, 1998; Watson et al., 1998; Anderson and Tolkovsky, 1999). For example, c-Jun phosphorylation but not JNK activation is required for superior cervical ganglion neuron apoptosis after NGF deprivation (Eilers et al., 1998). Depending on the specific region of the brain examined, there is either an increase or a decrease of apoptosis in the brains from JNK1/2−/−mice during development, suggesting both a proapoptotic and an antiapoptotic role of JNK in brain development (Kuan et al., 1999). More interestingly, activation of p38 is implicated in promoting survival of cerebellar granule cells (CGCs) (Mao et al., 1999).
The objective of this study was to define the role for JNK and p38 MAP kinases in cortical neuron apoptosis and to determine whether both JNK and p38 are required. Another goal was to determine which of the JNK isoforms contribute to neuronal apoptosis. There are three genes encoding JNK, JNK1, JNK2, and JNK3, and mRNA for all three genes is expressed in the brain (Gupta et al., 1996). However, JNK3 is the only neural-specific isoform (Martin et al., 1996). Do the JNK isozymes differentially contribute to stress-induced apoptosis? To address these issues, we induced apoptosis in cortical neurons using sodium arsenite and evaluated the role of p38 and specific isoforms of JNKs in neuron death. Sodium arsenite was chosen because it is an environmental toxicant that causes human cancer and CNS defects (Clarkson et al., 1985; Clarkson, 1987; Snow, 1992; Domingo, 1994; Hartwig, 1995; Shalat et al., 1996). Exposure of mouse embryos in culture to micromolar levels of sodium arsenite results in open neural tubes, probably because of increased cell death (Chaineau et al., 1990; Tabocova et al., 1996). Cortical neurons were chosen because the importance of JNK and p38 for apoptosis in these neurons has not been evaluated, and they are frequently damaged during neurodegenerative diseases. Our data suggest that both p38 and JNK3 contribute to arsenite-induced cortical neuron apoptosis.
MATERIALS AND METHODS
Plasmids. The following plasmids have been described previously: pON260, which encodes β-galactosidase (Cherrington and Mocarski, 1989), the wild-type MKK3, the constitutively active pRc/RSV-Flag-MKK3(Glu), the dominant negative pRc/RSV-Flag-MKK3(Ala), the wild-type p38α (Xia et al., 1995), pcDNA3-Flag-bcl-2 and bcl-xL (del Peso et al., 1997), the constitutive active pCMV5-MEKK1 (Whitmarsh et al., 1995), the kinase dead pCMV5-MEKK1 (Xia et al., 1995), the dominant negative pcDNA3-ASK1 (K709R) (Ichijo et al., 1997), the constitutive active cdc42 (pCMV-cdc42 V12-myc-tagged) (Minden et al., 1995), the dominant negative cdc42 (pCEV-cdc42 N17) (Coso et al., 1995), and the c-Jun dominant negative mutants pcDNA1-Flag-Δ169 (Ham et al., 1995) and pCMV-TAM67 (Δ3–122) (Rapp et al., 1994). The pCaMKII-JBD plasmid was constructed as follows. AHindIII–EcoRI fragment containing a Flag-tagged JNK binding domain (JBD) cDNA fragment (0.5 kb) was isolated from pcDNA3-Flag-JBD plasmid (Dickens et al., 1997), blunt-ended, and inserted into the EcoRV site of pNN265 (a gift from Dr. Mark Mayford, University of California, La Jolla, CA) to give rise to pNN265-Flag-JBD. The NotI fragment containing the SV40 small-t intron and early polyadenylation sequences as well as the Flag-JBD region was isolated from pNN265-Flag-JBD and inserted into pMM403 plasmid, which contains the Ca2+/calmodulin-activated protein kinase (CaM kinase) II-α promoter (pCaMKII) (Mayford et al., 1996). The final plasmid construct pCaMKII-Flag-JBD (13.4 kb) contains a 10.5 kbSfiI DNA fragment including an 8.5 kb CaMKII-α promoter and Flag-JBD cDNA coding sequence.
Primary cortical neuron cultures. Cortical neurons were prepared from newborn Sprague Dawley rats as described (Hetman et al., 1999, 2000). Briefly, dissociated cortical neurons were plated in 60 mm culture dishes for biochemistry experiments or in 35 mm dishes for transfection experiments at a density of 2000–2500 cells/mm2 and cultured in basal medium Eagle (BME) supplemented with 10% heat-inactivated bovine calf serum, 35 mm glucose, 1 mml-glutamine, 100 U/ml penicillin, and 0.1 mg/ml streptomycin and maintained in a humidified incubator with 5% CO2 at 37°C. Plates and glass coverslips were coated with poly-d-lysine and laminin. Cytosine-β-d-arabinofuranoside (2.5 μm; Sigma, St. Louis, MO) was added to cultures on the second day after seeding [day in vitro 2 (DIV 2)] to inhibit the proliferation of non-neuronal cells. Previous studies demonstrated that >90% of the cells in this culture preparation are neurons (Hetman et al., 1999). Cortical neurons were cultured for 6 d (DIV 6) before drug treatment.
Transient transfection of primary cortical neurons. Cortical neurons were transiently transfected at DIV 3 using a calcium phosphate co-precipitation protocol as described (Xia et al., 1996; Hetman et al., 1999). Briefly, the DNA–calcium phosphate precipitates were prepared by mixing 1 volume of DNA in 250 mmCaCl2 with an equal volume of 2× HEPES-buffered saline (2× HBS; 274 mm NaCl, 10 mm KCl, 1.4 mm Na2HPO4, 15 mmd-glucose, and 42 mm HEPES, pH 7.07). The precipitates were allowed to form for 25–30 min at room temperature before addition to the cultures. The conditioned culture media were removed and saved. Cells were washed three times with BME, and 1.5 ml of transfection media was added to each 35 mm dish. The transfection media consisted of BME supplemented with 1 mmsodium kynurenate, 10 mm MgCl2, and 5 mm HEPES. The pH of the transfection media was kept high by incubating BME in a dish at 37°C and 0% CO2for 30 min to “de-gas.” Sixty microliters of the DNA–calcium phosphate precipitates were added drop-wise to each 35 mm dish and mixed gently. Plates were incubated at room temperature and ambient air for 5 min and then in a humidified incubator with 5% CO2 at 37°C for 35–45 min. The incubation was stopped 20–25 min after the layer of precipitate formed on the plates by “shocking” the cells for 2 min with 1× HBS, 1 mmsodium kynurenate, 10 mm MgCl2 in 5 mm HEPES, pH 7.5, and 5% glycerol. Cells were then washed three times with 2 ml of BME. The saved conditioned media were added back to each plate, and cells were returned to the 5% CO2 incubator at 37°C for 24–48 hr before treatment or harvest. In our experience, expression of the transfected genes is quite high by 24 hr and normally peaks at ∼40–48 hr after transfection. Therefore, neurons were treated or harvested 48 hr after transfection for most experiments.
Quantitation of cell death and apoptosis. The effect of sodium arsenite on cortical neuron viability was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) metabolism assay (Hansen et al., 1989; Hetman et al., 1999). Cells were stained with a 2.5 μg/ml concentration of the DNA dye Hoechst 33258 (bis-benzimide) to visualize nuclear morphology (Hetman et al., 1999). Apoptosis was quantitated by scoring the percentage of apoptotic cells in the adherent cell population. Uniformly stained nuclei were scored as healthy, viable neurons. Condensed or fragmented nuclei were scored as apoptotic. To examine DNA cleavage, soluble cytoplasmic DNA was isolated and subjected to agarose gel electrophoresis (Hockenbery et al., 1990). Statistical analysis of the data was performed using one-way ANOVA and Fisher's predicted least square determinationpost hoc test.
Detection of transfected cells. Primary neuron cultures were always cotransfected with an expression vector encoding β-galactosidase (pON260) as a marker for transfected cells (Xia et al., 1995; Hetman et al., 1999, 2000). Neuron cultures were fixed for immunostaining 1–3 d after transfection. Transfected cells were detected by immunostaining with a polyclonal antibody to β-galactosidase (5′→3′, Boulder, CO; 1:500 dilution) and Texas Red-conjugated goat antibody to rabbit IgG. Cells transfected with β-galactosidase stained red. Some of the expression vectors used in this study are epitope-tagged. The expression of these epitope-tagged proteins was directly detected by the corresponding anti-epitope-specific antibodies: M2 monoclonal antibody to Flag (Eastman Kodak, New Haven, CT; 15 μg/ml) and 9E10 monoclonal antibody to c-myc (Oncogene Research Products, Boston, MA; 10 μg/ml).
Assay of apoptosis in transfected cells. Apoptosis in transfected cells was assayed by nuclei fragmentation and condensation after Hoechst staining (Xia et al., 1995; Hetman et al., 1999, 2000). To visualize the nuclei of transfected cells, we included the DNA dye Hoechst 33258 (2.5 μg/ml) in the wash after the secondary antibody incubation. Transfected cells were scored blind for apoptosis under the fluorescence microscope at the single-neuron level. The percentage of apoptotic cells in the total transfected cell population was quantitated.
Western analysis and protein kinase assays. Cell lysates were prepared as described (Dérijard et al., 1994), and 150 μg of proteins was used for each kinase assay. The p38 activity was measured as described using an immune complex kinase assay and glutathione S-transferase (GST)-ATF-2 as substrates (Xia et al., 1995) or by Western analysis using an anti-phospho-p38 antibody (New England Biolabs, Beverly, MA) and an anti-p38 antibody (Santa Cruz Biotechnology, Santa Cruz, CA). To assay for total JNK activity (JNK1–3), a JNK capture assay was performed (Faris et al., 1998). Briefly, cell lysates were incubated with recombinant GST-c-Jun (1–79) bound to glutathione-coupled agarose beads (Sigma), and the complex was washed extensively with lysis buffer. Kinase activity in the complex was assayed by addition of [γ-32P]ATP. The JNK1 and 2 MAP kinase activity was quantitated by an immune complex kinase assay using GST-cJun (1–79) as substrate and a polyclonal antibody to JNK that recognizes both JNK1 and 2 (Dérijard et al., 1994) or a monoclonal antibody that recognizes JNK1 (PharMingen, San Diego, CA) to immune precipitate JNK1 and 2 together or JNK1 alone, respectively (Xia et al., 1995). JNK3 activity was assayed as described (Yang et al., 1997). Briefly, cell lysates were immunoprecipitated with a mixture of a polyclonal antibody that recognizes both JNK1 and 2 (Dérijard et al., 1994) and a monoclonal antibody that recognizes JNK1 (PharMingen) to remove both JNK1 and 2 from the lysates. The remaining JNK3 kinase activity in the supernatant was assayed by the JNK capture assay. To ensure that JNK1 and 2 were completely removed from the supernatant, cell lysates (150 μg) before and after JNK1 and 2 immunodepletion were analyzed by Western blotting using a monoclonal antibody that recognizes both JNK1 and 2 (PharMingen). Relative increases in kinase activity were determined by ImageQuant program of the autoradiographic image. Western analysis for c-Jun phosphorylation was performed using anti-phospho-c-Jun (Ser-73) antibody (New England Biolabs).
RESULTS
Arsenite induces apoptosis in cultured cortical neurons
To examine the toxic effects of sodium arsenite, cortical neurons were treated with varying concentrations of sodium arsenite (0–15 μm) and assayed for cell viability at various times after treatment using the MTT metabolism assay (Fig.1). Sodium arsenite reduced cell viability in a dose- and time-dependent manner. The cytotoxic concentrations of sodium arsenite reported here are comparable with those that are neurotoxic for mouse embryos (Morrissey and Mottet, 1983; Chaineau et al., 1990; Tabocova et al., 1996).
Fig. 1.

Sodium arsenite (NaAsO2) reduces cortical neuron viability. Cortical neuron viability was determined by MTT metabolism assay. Data are mean ± SEM (n = 3).
To determine whether the reduced cell viability is attributable to apoptosis, cortical neurons were stained with the DNA dye Hoechst 33258 to visualize nuclear morphology. Sodium arsenite at concentrations from 2.0 to 10 μm caused morphological changes characteristic of apoptosis, including degeneration of neurites, shrinkage of cell bodies, and fragmentation of nuclei (Fig.2A). Induction of the apoptotic phenotype by sodium arsenite was dependent on the arsenite concentration and the time of incubation (Fig. 2B). Approximately 90% of the cells were apoptotic 48 hr after treatment with 10 μm arsenite. Sodium arsenite also caused DNA cleavage into oligonucleosome fragments manifested as “DNA laddering,” another hallmark of apoptosis (Fig. 2C). The effect of arsenite on cell viability (data not shown) and apoptosis (Fig. 2D) was inhibited by treatment with cycloheximide and actinomycin D. Collectively, these data indicate that sodium arsenite induces apoptosis in cortical neurons, which is dependent on gene expression and protein synthesis.
Fig. 2.

Sodium arsenite induces apoptosis in cortical neurons. A, Representative photomicrograph of Hoechst-stained nuclei of cortical neurons treated with sodium arsenite. Arrowheads indicate apoptotic nuclei. Scale bar, 20 μm. B, Quantitation of the kinetics and dose response of sodium arsenite-induced apoptosis. C, Sodium arsenite induces DNA fragmentation manifested as a “DNA ladder.” Positions of molecular size markers (in kilobases) are indicated on theright. D, Arsenite-induced cortical neuron apoptosis was blocked by cycloheximide (CXM) or actinomycin D (ActD), inhibitors for protein or RNA synthesis, respectively. Cortical neurons were treated with 10 μm sodium arsenite for 24 hr in the presence or absence of 10 μg/ml cycloheximide or 1 μg/ml actinomycin D. Data are mean ± SEM (n = 6). At least 2000 cells were scored for each data point.
Contribution of the bcl-2 and caspase pathways to arsenite-induced apoptosis
Antioxidants including N-acetyl cysteine, glutathione, and taurine inhibit sodium arsenite-induced apoptosis in non-neuronal cells, suggesting a role for reactive oxygen species in arsenite-mediated apoptosis (Watson et al., 1996; Wang et al., 1997). Both bcl-2 and bcl-xL protect against several forms of apoptosis possibly by blocking the generation of reactive oxygen species as one of the mechanisms (Hockenbery et al., 1993; Davies, 1995; Park et al., 1996; Reed, 1997). Therefore, we examined the effect of expression of bcl-2 and bcl-xL on arsenite-induced apoptosis. To transiently express these proteins in cortical neurons, we used a modified calcium phosphate method that has been optimized in this laboratory (for details, see Materials and Methods). Cortical neurons were co-transfected with varying amounts of plasmid DNA encoding Flag-tagged bcl-2 or bcl-xL and an expression vector encoding β-galactosidase as a marker for transfection (Fig.3A). The expression of Flag-tagged bcl-2 or bcl-xL was also directly confirmed by anti-Flag immunostaining (data not shown). More than 90% of the transfected cells co-expressed the Flag epitope and β-galactosidase. Expression of bcl-2 or bcl-xL almost completely protected cortical neurons from arsenite-induced apoptosis in transfected cells (Fig. 3B).
Fig. 3.

Expression of bcl-2 or bcl-xL protects cortical neurons from arsenite-induced apoptosis. Cortical neurons at DIV 3 were transfected with expression vectors for Flag-bcl-2 or Flag-bcl-xL (0, 2, or 4 μg). All cells were also cotransfected with 1 μg of plasmid DNA encoding β-galactosidase as a marker for transfection. Empty vector pcDNA3 was used to supplement the total DNA to 5 μg in each case. Two days after transfection, neurons were treated with 7 μm sodium arsenite for 24 hr. A, Representative photomicrographs of cortical neurons transfected with bcl-2 or a vector control. Transfected cells were identified by positive β-galactosidase staining (red, arrows). Note that after sodium arsenite treatment, although many of the vector control-transfected cells were apoptotic, showing fragmented or condensed nuclei, as indicated by arrowheads, most of the bcl-2-transfected cells were healthy. Scale bar, 25 μm.B, Quantitative effect of bcl-2 or bcl-xL on arsenite-induced apoptosis. At least 800 cells were counted for each data point. Data are mean ± SEM (n = 3).
Cortical neurons were also treated with zVAD-fluoromethylketone (zVAD), a broad-spectrum caspase inhibitor. zVAD almost completely inhibited arsenite-induced apoptosis (Fig.4A). Moreover, zVAD treatment eliminated DNA laddering (Fig. 4B). These results suggest that regulation by bcl-2 family proteins and caspase activation are important for arsenite-mediated apoptosis in cortical neurons.
Fig. 4.

Cortical neuron apoptosis induced by arsenite requires caspase activation. A, Cortical neurons were pretreated with 100 μm caspase inhibitor zVAD or DMSO as vehicle control for 30 min and then stimulated with 10 μm sodium arsenite for 24 or 48 hr. The results are mean ± SEM (n = 6). At least 1000 cells were counted for each data point. B, Treatment with zVAD inhibits arsenite-induced DNA fragmentation manifested as a DNA ladder. Positions of molecular size markers (in kilobases) are indicated on theright.
Activation of p38 is required for arsenite-induced cortical neuron apoptosis
To evaluate the contribution of p38 for arsenite-induced apoptosis in cortical neurons, p38 activity was measured by an immune complex kinase assay at various times after arsenite treatment (Fig.5A). Within 0.5 hr after treatment, p38 was activated by 10 μm sodium arsenite, a concentration that induces apoptosis. The activation of p38 preceded the induction of apoptosis, and p38 activity remained elevated for at least 8 hr. Activation of p38 was confirmed by Western analysis using an anti-phospho-p38 antibody that specifically recognizes phosphorylated and activated p38 (Fig. 5B).
Fig. 5.
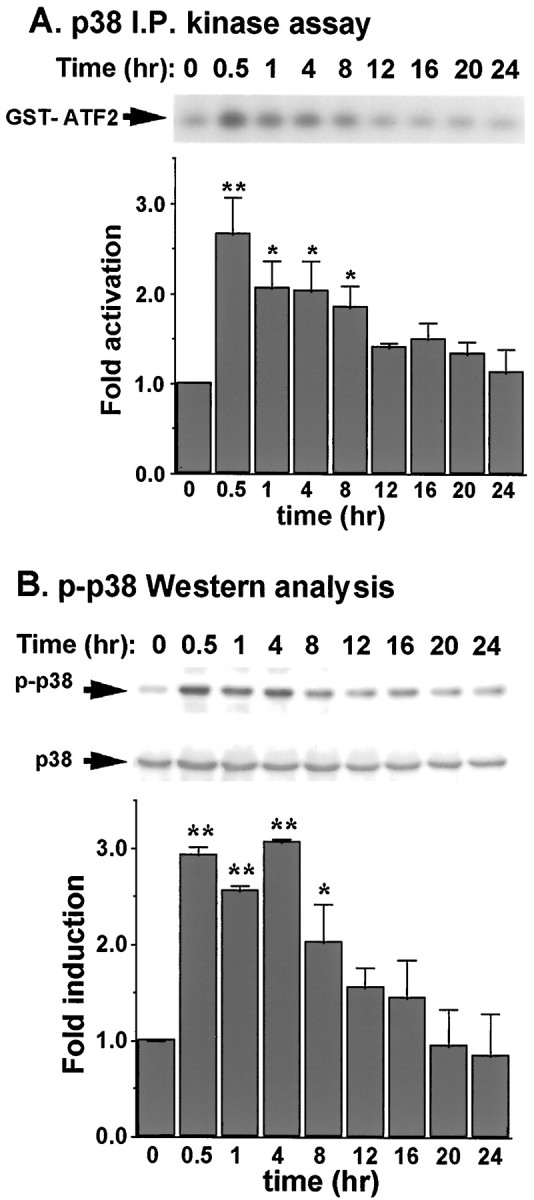
Sodium arsenite activates p38 in cortical neurons. Cortical neurons at DIV 6 were stimulated with 10 μmsodium arsenite for the indicated times. p38 activity was determined by an immune complex kinase assay (A) or by Western analysis using antibodies recognizing either phosphorylated and activated (p-) p38 (top) or unphosphorylated p38 (bottom) (B). Anti-p38 Western was used to confirm an equal amount of protein loading in each gel lane and that changes of p38 activity did not result from changes in protein levels of p38. The intensity of the bands on Western blots was quantitated by scanning the Western blots and analyzed by ImageQuant analysis. The relative phospho-p38 was normalized to the total p38 from anti-p38 Western blots. Results are averages of three independent experiments ± SEM. I.P., Immunoprecipitation.
The importance of p38 activation for induction of apoptosis was investigated by inhibition of p38. Treatment with 10 μmSB203580, a specific inhibitor for p38 (Cuenda et al., 1995;Clifton et al., 1996), partially protected cortical neurons from 5 μm sodium arsenite (Fig.6A). We also transfected cortical neurons with a dominant interfering mutant of MAP kinase kinase 3 (MKK3) to selectively block p38 activation. MKK3 is an upstream kinase that activates and phosphorylates p38 (Ip and Davis, 1998). Expression of the dominant-interfering mutant of MKK3 protected neurons against 5 μm sodium arsenite (Fig.6B). The ineffective inhibition of apoptosis after 10 μm arsenite treatment could be attributable to incomplete inhibition of p38 MAP kinase activity by SB203580 or the dominant negative MKK3. For example, SB203580 does not inhibit all isoforms of p38 MAP kinase (Cuenda et al., 1997). Moreover, other signaling pathways, e.g., the JNK pathway, may also play a key role in apoptosis induced by higher concentrations of arsenite.
Fig. 6.

Inhibition of p38 MAP kinase attenuates arsenite-induced apoptosis in cortical neurons. A, Cortical neurons were pretreated for 30 min with 0 (vehicle control DMSO), 2, or 10 μm SB203580 and then challenged with 0, 5, or 10 μm sodium arsenite for 24 hr. At least 1000 cells were counted for each data point. B, Cortical neurons were transfected with 3 μg of plasmid DNA encoding either a dominant-interfering MKK3(Ala) mutant (d.n. MKK3) or a vector control (V) for 24 hr. Cells were then treated with 0, 5, or 10 μm sodium arsenite for 24 hr. At least 500 transfected cells were counted for each data point. Data are mean ± SEM (n = 3). *p < 0.05; **p < 0.01.
If activation of p38 contributes to arsenite-induced apoptosis in cortical neurons, then direct and selective stimulation of p38 may induce apoptosis in the absence of other stress stimuli. Cortical neurons were transfected with a constitutive active form of MKK3 to activate p38. Expression of the constitutive active MKK3 increased apoptosis to 37%, threefold greater than control cells transfected with the expression vector (Fig.7A). Similarly, co-expression of a constitutive active MKK3 together with a wild-type p38 caused a threefold increase in apoptosis (Fig.7B,C). This apoptosis was inhibited by coexpression of bcl-2 or bcl-xL (Fig. 7B) or by zVAD inhibition of caspase activity (Fig. 7C). Expression of p38 and various forms of MKK3 in cortical neurons after transient transfection was confirmed by Western analysis (J. E. Cavanaugh and Z. Xia, unpublished observation). Furthermore, the constitutive active MKK3 but not the wild-type or the dominant negative MKK3 activated the co-transfected p38 (Cavanaugh and Xia, unpublished observation). Collectively, these data indicate that activation of p38 plays an important role in arsenite-induced apoptosis in cortical neurons.
Fig. 7.
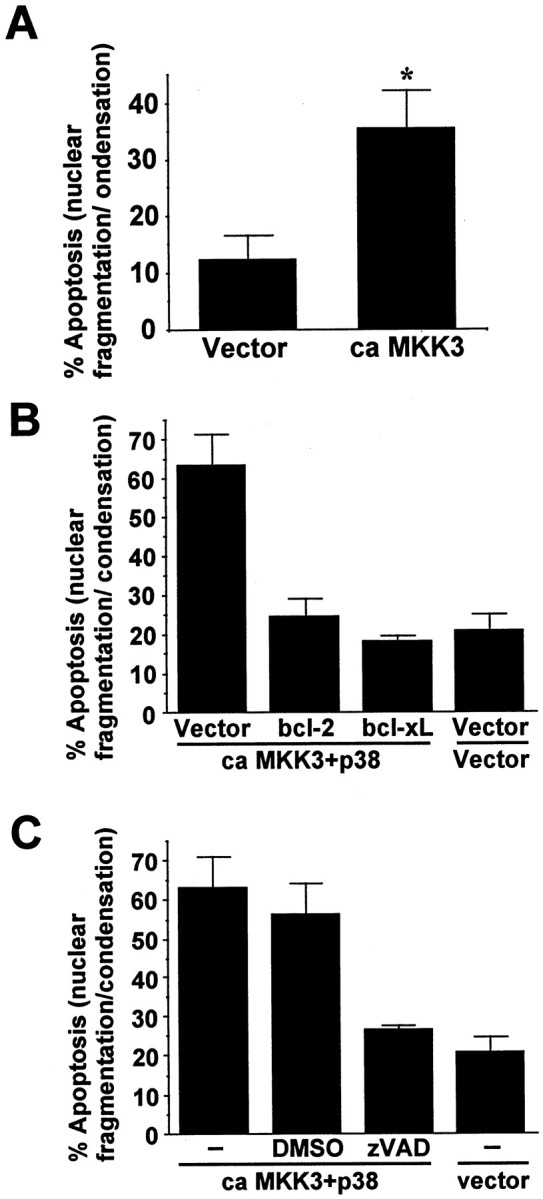
Constitutive and selective activation of p38 in cortical neurons is sufficient to induce apoptosis, which depends on caspase activity. A, Expression of constitutive active MKK3 induces cortical neuron apoptosis. Cortical neurons were transfected with 1 μg of DNA encoding a constitutive active form of MKK3(Glu) (ca MKK3) or vector control. At least 500 transfected cells were counted. B, Expression of bcl-2 or bcl-xL blocks p38-induced apoptosis. Cortical neurons were cotransfected with cDNA encoding constitutive active MKK3 (ca MKK3; 1 μg), wild-type p38α (1 μg), bcl-2 (3 μg), bcl-xL (3 μg), or vector control as indicated.C, p38-induced apoptosis requires caspase activity. Cortical neurons were cotransfected with cDNA encoding constitutive active MKK3 (ca MKK3; 1 μg), wild-type p38 (1 μg), or vector control as indicated. Four hours after transfection, 100 μm zVAD or vehicle control DMSO was added to the medium to inhibit caspase activity. Apoptosis in transfected cells was scored 48 hr after transfection. Data are mean ± SEM (n = 4–6). *p < 0.05.
JNK3 but not JNK1 or 2 is activated by arsenite in cortical neurons
To determine whether JNK contributes to arsenite-induced apoptosis in cortical neurons, JNK activity was assayed after arsenite treatment. There was a twofold increase in total JNK activity when cortical neurons were treated with 10 μm sodium arsenite (Fig.8A). The relative contribution of different isoforms of JNK was evaluated by assaying JNK1, JNK1 and 2, and JNK3 activities separately. JNK1 and 2 showed high basal activity that was not stimulated by arsenite (Fig.8B). The high basal JNK1 and 2 activity is consistent with published observations concerning basal levels of JNK in rat brain (Xu et al., 1997). Similar results were obtained when JNK1 activity alone was measured (Fig. 8C). The antibodies used for monitoring JNK1 or JNK1 and 2 activities have been used in PC12 cells to show activation of JNK1 and 2 (Xia et al., 1995; Xia, unpublished observations). Furthermore, the kinase assays were performed several times using varying amounts of cell lysates in an attempt to detect arsenite activation of JNK1 or 2 (data not shown). These results suggest that the increase in total JNK activity after arsenite treatment is not a result of JNK1 or 2 activation.
Fig. 8.

JNK1 and 2 are not activated by arsenite. Cortical neurons at DIV 6 were stimulated with 10 μm sodium arsenite for the indicated times. A, Total JNK activity.B, JNK1 and 2 activity. C, JNK1 activity. One hundred fifty micrograms of protein extracts were used for each assay. Data are representative of two or three independent experiments.
To test whether JNK3 is activated by arsenite, we first depleted JNK1 and JNK2 from the cell lysates using a mixture of a polyclonal antibody that recognizes both JNK1 and 2 (Dérijard et al., 1994) and a monoclonal antibody that recognizes JNK1 only. This treatment completely removed JNK1 and 2 from cell lysates (Fig.9A). The remaining JNK3 kinase activity in the supernatant was assayed by a capture kinase assay. In contrast to JNK1 and 2, the basal activity of JNK3 was not high, and JNK3 was activated 3.5-fold by 10 μm sodium arsenite (Fig. 9B). The JNK3 activation was apparent 30 min after arsenite treatment and persisted for at least 16 hr. These data indicate that JNK3 is selectively activated by arsenite stimulation in cortical neurons.
Fig. 9.
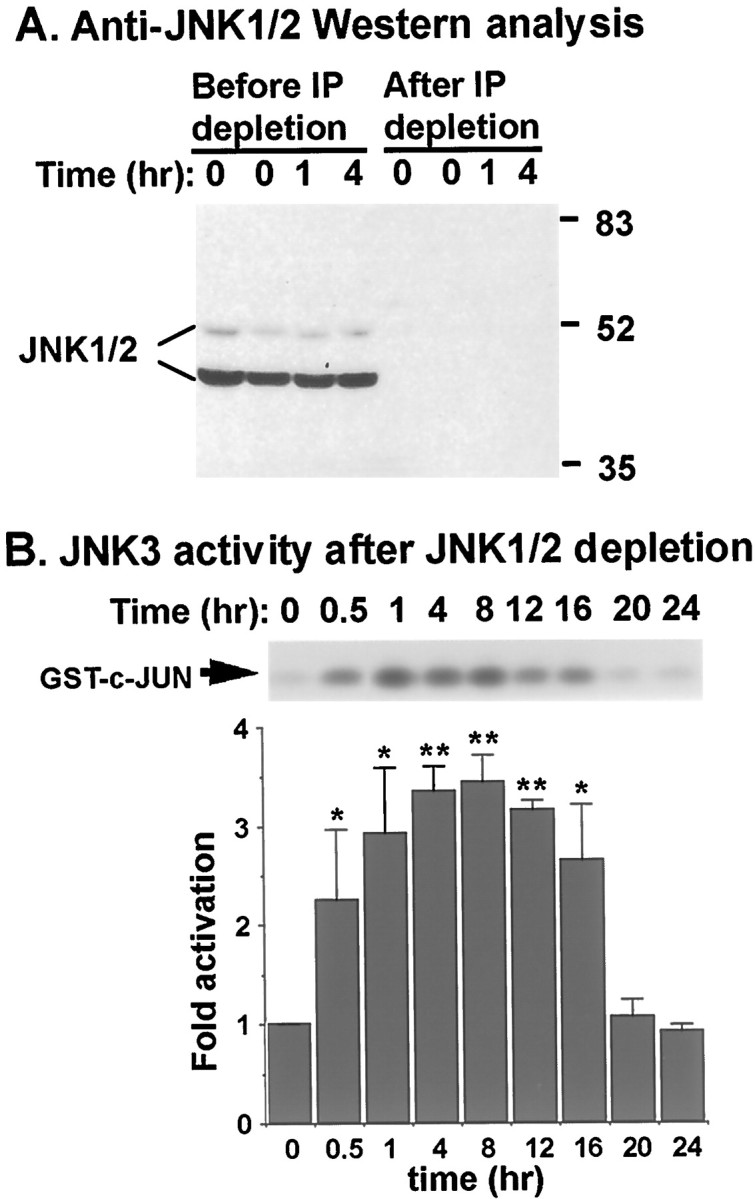
JNK3 is activated by arsenite. Cortical neurons at DIV 6 were stimulated with 10 μm sodium arsenite for the indicated times. JNK1 and 2 in the cell lysates were depleted by immunoprecipitation. A, Western analysis using an antibody that recognizes both JNK1 and 2. One hundred fifty micrograms of protein extracts before and after JNK1 and 2 immunodepletion were used. Positions of molecular size markers (in kilodaltons) are indicated on the right.B, JNK3 activity. JNK1 and 2 in the cell lysates (150 μg) were depleted by immunoprecipitation, and the remaining JNK3 activity in the supernatant was determined by a capture JNK assay. Data are results from four independent experiments ± SEM. *p < 0.05; **p < 0.01.
To determine whether c-Jun, a known JNK substrate, is phosphorylated in cortical neurons exposed to arsenite, we performed Western analysis using an anti-phospho-c-Jun antibody (Fig.10). Treatment of cortical neurons with sodium arsenite induced c-Jun phosphorylation. The kinetics of c-Jun phosphorylation was similar to that of JNK3 activation. Co-treatment of cortical neurons with CEP1347, an inhibitor of the JNK pathway (Maroney et al., 1998), prevented c-Jun phosphorylation. These results suggest that increased c-Jun phosphorylation and transcription by activated JNK3 may be important for arsenite-induced apoptosis in cortical neurons.
Fig. 10.

Sodium arsenite induces c-Jun phosphorylation in cortical neurons. Cortical neurons at DIV 6 were stimulated with 10 μm sodium arsenite for the indicated times. Forty micrograms of protein extracts were used for Western analysis with anti-phospho c-Jun antibody. A, Quantitation of c-Jun phosphorylation. The intensity of the bands on Western blots was quantitated by scanning the Western blots and analyzed by ImageQuant analysis. Results are averages of two independent experiments ± SEM. B, Blockade of arsenite-induced c-Jun phosphorylation by CEP1347 (5 μm). Results are representative of two experiments.
Activation of JNK is critical for arsenite-induced cortical neuron apoptosis
If activation of JNK contributes to arsenite-induced apoptosis, then direct and selective stimulation of JNK may induce apoptosis in the absence of other stress stimuli. To address this issue, cortical neurons were transfected with a constitutive active cdc42, or a constitutive active MAP kinase/extracellular signal-regulated kinase (ERK) kinase kinase (MEKK) 1 to determine whether activation of JNK induces apoptosis. cdc42 is an upstream GTPase that activates the JNK pathway (Coso et al., 1995; Minden et al., 1995), and MEKK1 is a JNK kinase kinase that activates JNK (Minden et al., 1994). Expression of either constitutive active cdc42 or the MEKK1 was sufficient to induce apoptosis in cortical neurons (Fig.11A). We also cotransfected cortical neurons with plasmids encoding a constitutive active MEKK1 and a JBD. JBD is a fragment of the JNK-scaffolding protein JIP, which interacts with cytoplasmic JNK (Dickens et al., 1997; Whitmarsh et al., 1998). JBD prevents JNK translocation into the nucleus, thereby inhibiting JNK stimulation of transcription (Dickens et al., 1997). It does not interfere with the signaling of p38 or ERK (Dickens et al., 1997). The cDNA encoding JBD was placed under the control of a CaMKIIα promoter, which directs neuron-specific expression of target genes (Mayford et al., 1995). MEKK1-induced apoptosis was suppressed by co-expression of JBD but not by its vector control (Fig. 11B), confirming that MEKK1-induced apoptosis is mediated by a JNK pathway. Furthermore, MEKK1-induced apoptosis was inhibited by coexpression of bcl-2 or bcl-xL (Fig. 11C) and by zVAD inhibition of caspase activity (Fig. 11D), suggesting that JNK-induced apoptosis in cortical neurons is caspase-dependent.
Fig. 11.

Activation of the JNK signaling pathway is sufficient to induce caspase-dependent apoptosis in cortical neurons.A, Induction of apoptosis after expression of a constitutive active cdc42 or MEKK1, upstream activators of the JNK signaling pathway. Cortical neurons were transfected with 0, 1, or 2 μg of plasmid DNA for a constitutive active cdc42 (V12) or MEKK1 as indicated. Vector control DNA was supplemented to maintain equal amounts of the total DNA added to each dish. Apoptosis in transfected cells was scored 48 hr later. B, Apoptosis induced by the constitutive active MEKK1 was blocked by co-expression of JBD. Cortical neurons were transfected with 1 μg of DNA for a constitutive active MEKK1 together with 9 μg of DNA for pCaMKII-JBD or vector control. Apoptosis in transfected cells was scored 24 and 48 hr later.C, Expression of bcl-2 or bcl-xL blocks MEKK1-induced apoptosis. Cortical neurons were cotransfected with cDNA encoding a constitutive active MEKK1 (1 μg), bcl-2 (3 μg), bcl-xL (3 μg), or vector control as indicated. Apoptosis in transfected cells was scored 48 hr later. D, MEKK1-induced apoptosis requires caspase activity. Cortical neurons were cotransfected with cDNA encoding a constitutive active MEKK1 (1 μg) or vector control. Four hours after transfection, 100 μm zVAD or vehicle control DMSO was added to the medium to inhibit caspase activity. Apoptosis in transfected cells was scored 48 hr later. Data are mean ± SEM (n = 3–6). *p < 0.05; **p < 0.01. At least 500 transfected cells were counted for each data point.
To assess the importance of JNK activation for arsenite-induced apoptosis, cortical neurons were transfected with two different expression vectors for dominant-interfering c-Jun. c-Jun is an AP-1 transcription factor and a downstream target of JNK. Transient expression of the dominant-interfering c-Jun constructs Δ169 and TAM 67 antagonized arsenite-induced apoptosis in transfected cells (Fig.12A). These data suggest a role for JNK activation in arsenite-induced apoptosis. However, the dominant negative c-Jun could potentially dimerize with other AP-1 proteins, thereby interfering with pathways independent from JNK. Therefore, JNK signaling was also inhibited by other approaches, and the effect on arsenite-induced apoptosis was evaluated. Cortical neurons were transiently transfected with an expression vector for JBD. Treatment of neurons with 7 μm sodium arsenite induced apoptosis in 50% of control transfected cells, which was blocked by expression of JBD (Fig. 12B). Furthermore, expression of dominant negative forms of cdc42 (Fig. 12C) or ASK1 (Fig. 12D), but not a kinase dead form of MEKK1 (Fig. 12E), reduced arsenite-induced cortical neuron apoptosis. ASK1 is a JNK kinase kinase that activates JNK and has been implicated in several forms of apoptosis (Ichijo et al., 1997;Wang et al., 1999; Yamamoto et al., 1999; Kanamoto et al., 2000). These data suggest that Cdc42 and ASK1 may be upstream components of the JNK3 signaling pathway that mediate arsenite-induced apoptosis in cortical neurons. The kinase dead MEKK1 is not a strong dominant negative mutant for blocking JNK activation; this may explain why it exhibted little effect on arsenite-induced apoptosis.
Fig. 12.

Blocking the JNK signaling pathway inhibits arsenite-induced cortical neuron apoptosis. A, Inhibition of apoptosis by expression of dominant-interfering mutants of c-Jun. Cortical neurons were transfected with 3 μg of plasmid DNA encoding the dominant interfering c-Jun mutants Flag-Δ169 and TAM67 or vector control. B, Inhibition of apoptosis by expression of JBD, a strong dominant negative mutant that blocks JNK signaling specifically. Cortical neurons were transfected with 9 μg of plasmid DNA encoding a Flag-tagged JBD directed by the CaMKII-α promoter (pCaMKII-JBD) or a vector control.C–E, Effects of a dominant negative cdc42 (C), a dominant negative ASK (D), or a kinase dead MEKK1 (E) on arsenite-induced apoptosis. Cortical neurons were transfected with cDNA encoding dominant negative forms of cdc42 (3 μg), ASK1 (3 μg), kinase dead MEKK1 (3 μg), or control vectors. For A–E, cells were treated 24 hr after transfection with 7 μm sodium arsenite or vehicle control for 24 hr. F, Cortical neurons were pretreated for 30 min with vehicle control DMSO, 10 μm SB203580, or 5 μm CEP-1347 as indicated and then challenged with 0, 5, or 10 μm sodium arsenite for 24 hr. At least 1000 cells were counted for each data point. Data are mean ± SEM (n = 3–6). *p < 0.05; **p < 0.01.
Cortical neurons were also treated with CEP-1347, a pharmacological inhibitor that prevents activation of JNK but not p38 or ERK (Maroney et al., 1998) (Fig. 12F). Treatment with CEP-1347 caused a statistically significant (p < 0.01) reduction in cortical neuron apoptosis after treatment with 5 or 10 μmsodium arsenite. Cotreatment of cortical neurons with both SB203580 and CEP-1347 provided more protection against cortical neuron apoptosis after 5 μm arsenite treatment than either inhibitor alone. When cortical neurons were challenged with 10 μm sodium arsenite, SB203580 alone was not neuroprotective, consistent with the data in Figure 6. SB203580 also did not potentiate the neuroprotection afforded by CEP-1347 after 10 μm arsenite treatment. Together, these data indicate that activation of one or more JNKs is obligatory for arsenite-induced cortical neuron apoptosis and implicate downstream transcriptional events because of the inhibition of apoptosis by dominant-interfering c-Jun constructs.
DISCUSSION
Most mechanistic studies of apoptosis have focused on proliferating, non-neuronal cells or neurons derived from the peripheral nervous system (PNS). However, the biochemical and cellular properties of mature CNS neurons are quite distinct from those of dividing non-neuronal cells or PNS neurons. Consequently, it is important to define the mechanisms for apoptosis in CNS neurons caused by specific types of cellular stress.
Although JNK and p38 are implicated for apoptosis in PC12 cells and non-neuronal cells, their contribution to apoptosis in primary CNS neurons was not well defined. There has been no clear consensus in the literature concerning the role of JNKs and p38 for regulation of apoptosis in CNS neurons (Kawasaki et al., 1997; Yang et al., 1997;Gunn-Moore and Tavare, 1998; Maroney et al., 1998; Watson et al., 1998;Kuan et al., 1999; Mao et al., 1999). One difficulty in solidifying a role for the JNK and p38 signaling in neuronal apoptosis is attributable to the limitations of research tools used in the previous studies. For instance, pharmacological inhibitor studies have implicated p38 and JNK in several forms of neuronal apoptosis (Kawasaki et al., 1997; Glicksman et al., 1998; Maroney et al., 1998, 1999;Pirvola et al., 2000). However, interpretation of data using inhibitors without complementary transfection studies using dominant interfering or constitutive active constructs is compromised by the uncertainties concerning the specificity of these drugs. Disruption of the JNK genes in mice has also provided clues concerning the importance of JNKs for neuronal apoptosis (Yang et al., 1997; Kuan et al., 1999). Mice deficient in JNK3 do not exhibit hippocampal neuron death after kainate injection, implicating JNK3 in excitotoxicity-induced apoptosis (Yang et al., 1997). However, these mice are more resistant to kainate-induced seizure; therefore, it is not clear whether reduced cell death in JNK3 mutant mice is attributable to decreased seizure activity or the loss of an intrinsic cell death pathway (Yang et al., 1997).
The objective of this study was to investigate the importance of the JNK and the p38 pathways for apoptosis in CNS neurons caused by arsenite, an environmental toxicant. Treatment of cortical neurons with micromolar concentrations of sodium arsenite decreased cell viability and caused cellular changes typical of apoptosis. JNK3 and p38 but not JNK1 or 2 were activated by arsenite at concentrations that induced apoptosis. Furthermore, arsenite induced c-Jun phosphorylation. Using pharmacological inhibitors and expression of dominant-interfering constructs for the kinase pathways, we demonstrated that inhibition of either JNK or p38 signaling protects cortical neurons from arsenite toxicity. In contrast, selective activation of either pathway was sufficient to induce apoptosis. This is the first demonstration that both p38 and JNK pathways are required for apoptosis in primary CNS neurons.
The discovery that different isoforms of JNK are differentially regulated by arsenite provides new insights concerning the function of the JNK signaling pathways during stress-induced apoptosis. mRNAs for all of the JNK genes are expressed in the brain (Gupta et al., 1996), and our data indicate that cortical neurons express JNK1, 2, and 3 kinase activities. Interestingly, arsenite stimulated JNK3 activity but not JNK1 or 2. Because general inhibition of the JNK signaling pathway blocked apoptosis, and only JNK3 was activated by arsenite, these results suggest that JNK3 specifically mediates arsenite-induced apoptosis. This is the first evidence showing differential stimulation of a JNK isoform by a specific form of stress and suggests that these kinases are not simply redundant activities. Although our data do not formally exclude a role for JNK1 and 2, the fact that only JNK3 is activated by arsenite strongly suggests a pivotal role for JNK3 in arsenite-stimulated apoptosis. Our data showing high basal JNK activities and isoform-specific activation of JNKs in response to a specific stress signal illustrate the regulatory diversity of the JNKs and indicate that JNK3 plays a critical role in stress-induced apoptosis.
Cortical neuron apoptosis induced by arsenite or by selective activation of JNK or p38 was also inhibited by a caspase inhibitor, zVAD, as well as by expression of the anti-apoptotic proteins Bcl-2 and Bcl-xL. These data suggest that arsenite activates JNK3 and p38, which then induce apoptosis through a bcl-2–caspase-dependent mechanism. In contrast, camptothecin-induced apoptosis in the same preparations of postnatal cortical neurons was not affected by zVAD (M. Hetman and Z. Xia, unpublished observations). These results are consistent with recent observations that some forms of neuronal cell death are caspase-independent (Johnson et al., 1998, 1999; Stefanis et al., 1999). Interestingly, it was recently reported that p53-dependent caspase activation contributes to embryonic but not postnatal cortical neuron apoptosis (Johnson et al., 1999). Our data showed that postnatal cortical neurons can undergo both caspase-dependent and -independent forms of apoptosis depending on the source of injury. We also report here that arsenite-induced apoptosis requires transcriptional and translational events. It is possible that JNK3/p38 activates the bcl-2–caspase pathway via transcriptional induction of FasL expression, as suggested in trophic withdrawal-induced apoptosis in CGCs (Le Niculescu et al., 1999).
The finding that sodium arsenite induces apoptosis in CNS neurons may explain some of the CNS defects attributed to environmental exposure to arsenite. The ubiquitous presence of arsenicals in the environment arises from natural rock formations and leaching into ground water, as well as from industrial and agricultural uses. For example, inorganic arsenicals were widely used in agricultural products, including pesticides and herbicides. Arsenic is a well established teratogen in rodents (Ferm et al., 1971; Hood and Bishop, 1972; Beaudoin, 1974;Machado et al., 1999). Exposure of mouse embryos in culture to 3–4 μm sodium arsenite results in open neural tubes (Chaineau et al., 1990; Tabocova et al., 1996), and developmental exposure of arsenic to the dam in rodent models causes exencephaly (Morrissey and Mottet, 1983). Data reported here suggest that activation of both JNK and p38, and subsequent stimulation of apoptosis may contribute to arsenite neurotoxicity. Activation of JNK and p38 as well as subsequent induction of apoptosis may also contribute to the neurotoxicity of other environmental toxicants. It has become increasingly clear that many toxicants exert their toxic effects by inducing apoptosis (Bolon et al., 1993; Alison and Sarraf, 1995; Bulleit and Cui, 1998). For example, other heavy metals, including lead, mercury, and lithium, all induce neuronal apoptosis (D'Mello et al., 1994; Kunimoto, 1994;Sarafian et al., 1994; Oberto et al., 1996). Chlorpyrifos, a commonly used organophosphate pesticide, also induces apoptosis in PC12 cells and in the neuroepithelium of cultured rat embryos (Bagchi et al., 1995; Roy et al., 1998). Interestingly, chlorpyrifos also induces apoptosis in primary cultured cortical neurons and activates both JNK and p38 (A. Caughlan and Z. Xia, unpublished observations).
In summary, sodium arsenite induction of apoptosis in cortical neurons depends on the activity of both p38 and JNK. Because JNK3 is neural-specific and is selectively activated during apoptosis, JNK3 may be a useful target for blocking neuronal apoptosis.
Footnotes
This work was supported by Grant NS37359 from the National Institute of Neurological Disorders and Stroke and Grant APP3010 from the Burroughs Wellcome Fund for New Investigator Awards in Toxicology (Z.X.). We thank Drs. L. del Peso and G. Nunez for providing bcl-2 and bcl-xL plasmids, Dr. Gutkind for dominant negative cdc42, Dr. H. Ichijo for dominant negative ASK1, Dr. Mark Mayford for pNN265 and pMM403 plasmids, Dr. R. Davis for pcDNA3-JBD, anti-JNK1/2 polyclonal antibody, and helpful discussions, and Dr. Anna C. Maroney (Cephalon) for CEP-1347.
Correspondence should be addressed to Zhengui Xia, Department of Environmental Health, Box 357234, University of Washington, Seattle, WA 98195. E-mail: zxia@u.washington.edu.
REFERENCES
- 1.Alison MR, Sarraf CE. Apoptosis: regulation and relevance to toxicology. Hum Exp Toxicol. 1995;14:234–247. doi: 10.1177/096032719501400302. [DOI] [PubMed] [Google Scholar]
- 2.Anderson CNG, Tolkovsky AM. A role for MAPK/ERK in sympathetic neuron survival: protection against a p53-dependent, JNK-independent induction of apoptosis by cytosine arabinoside. J Neurosci. 1999;19:664–673. doi: 10.1523/JNEUROSCI.19-02-00664.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bagchi D, Bagchi M, Hassoun EA, Stohs SJ. In vitro and in vivo generation of reactive oxygen species, DNA damage and lactate dehydrogenase leakage by selected pesticides. Toxicology. 1995;104:129–140. doi: 10.1016/0300-483x(95)03156-a. [DOI] [PubMed] [Google Scholar]
- 4.Bazenet CE, Mota MA, Rubin LL. The small GTP-binding protein Cdc42 is required for nerve growth factor withdrawal-induced neuronal death. Proc Natl Acad Sci USA. 1998;95:3984–3989. doi: 10.1073/pnas.95.7.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beaudoin AR. Teratogenicity of sodium arsenate in rats. Teratology. 1974;10:153–157. doi: 10.1002/tera.1420100211. [DOI] [PubMed] [Google Scholar]
- 6.Behrens MM, Strasser U, Koh JY, Gwag BJ, Choi DW. Prevention of neuronal apoptosis by phorbol ester-induced activation of protein kinase C: blockade of p38 mitogen-activated protein kinase. Neuroscience. 1999;94:917–927. doi: 10.1016/s0306-4522(99)00212-2. [DOI] [PubMed] [Google Scholar]
- 7.Bolon B, Dorman DC, Bonnefoi MS, Randall HW, Morgan KT. Histopathologic approaches to chemical toxicity using primary cultures of dissociated neural cells grown in chamber slides. Toxicol Pathol. 1993;21:465–479. doi: 10.1177/019262339302100506. [DOI] [PubMed] [Google Scholar]
- 8.Bulleit RF, Cui H. Methylmercury antagonizes the survival-promoting activity of insulin-like growth factor on developing cerebellar granule neurons. Toxicol Appl Pharmacol. 1998;153:161–168. doi: 10.1006/taap.1998.8561. [DOI] [PubMed] [Google Scholar]
- 9.Camandola S, Poli G, Mattson MP. The lipid peroxidation product 4-hydroxy-2,3-nonenal increases AP-1- binding activity through caspase activation in neurons. J Neurochem. 2000;74:159–168. doi: 10.1046/j.1471-4159.2000.0740159.x. [DOI] [PubMed] [Google Scholar]
- 10.Chaineau E, Binet S, Pol D, Chatellier G, Meininger V. Embryotoxic effects of sodium arsenite and sodium arsenate on mouse embryos in culture. Teratology. 1990;41:105–112. doi: 10.1002/tera.1420410111. [DOI] [PubMed] [Google Scholar]
- 11.Cherrington JM, Mocarski ES. Human cytomegalovirus iel transactivates the a promoter-enhancer via an 18-base-pair repeat element. J Virol. 1989;63:1435–1440. doi: 10.1128/jvi.63.3.1435-1440.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clarkson TW. Metal toxicity in the central nervous system. Environ Health Perspect. 1987;75:59–64. doi: 10.1289/ehp.877559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clarkson TW, Nordberg GF, Sager PR. Reproductive and developmental toxicity of metals. Scand J Work Environ Health. 1985;11:145–154. doi: 10.5271/sjweh.2239. [DOI] [PubMed] [Google Scholar]
- 14.Clifton AD, Young PR, Cohen P. A comparison of the substrate specificity of MAPKAP kinase-2 and MAPKAP kinase-3 and their activation by cytokines and cellular stress. FEBS Lett. 1996;392:209–214. doi: 10.1016/0014-5793(96)00816-2. [DOI] [PubMed] [Google Scholar]
- 15.Coso OA, Chiariello M, Yu JC, Teramoto H, Crespo P, Xu N, Miki T, Gutkind JS. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell. 1995;81:1137–1146. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- 16.Cowan WM, Fawcett JW, O'Leary DDM, Stanfield BB. Regressive events in neurogenesis. Science. 1984;225:1258–1265. doi: 10.1126/science.6474175. [DOI] [PubMed] [Google Scholar]
- 17.Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, Young PR, Lee JC. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995;364:229–233. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- 18.Cuenda A, Cohen P, Buee-Scherrer V, Goedert M. Activation of stress-activated protein kinase-3 (SAPK3) by cytokines and cellular stresses is mediated via SAPKK3 (MKK6); Comparison of the specificities of SAPK3 and SAPK2 (RK/p38). EMBO J. 1997;16:295–305. doi: 10.1093/emboj/16.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davies AM. The Bcl-2 family of proteins, and the regulation of neuronal survival. Trends Neurosci. 1995;18:355–358. doi: 10.1016/0166-2236(95)93928-q. [DOI] [PubMed] [Google Scholar]
- 20.del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 21.Dérijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, Davis RJ. JNK1: a protein kinase stimulated by uv light and HA-ras binds to and activates the c-jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 22.Dickens M, Rogers J, Cavanagh J, Raitano A, Xia Z, Halpern JR, Greenberg ME, Sawyers C, Davis RJ. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science. 1997;277:693–696. doi: 10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- 23.D'Mello SR, Anelli R, Calissano P. Lithium induces apoptosis in immature cerebellar granule cells but promotes survival of mature neurons. Exp Cell Res. 1994;211:332–338. doi: 10.1006/excr.1994.1095. [DOI] [PubMed] [Google Scholar]
- 24.Domingo JL. Metal-induced developmental toxicity in mammals: a review. J Toxicol Environ Health. 1994;42:123–141. doi: 10.1080/15287399409531868. [DOI] [PubMed] [Google Scholar]
- 25.Eilers A, Whitfield J, Babij C, Rubin LL, Ham J. Role of the Jun kinase pathway in the regulation of c-Jun expression and apoptosis in sympathetic neurons. J Neurosci. 1998;18:1713–1724. doi: 10.1523/JNEUROSCI.18-05-01713.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Estus S, Zaks WJ, Freeman RS, Gruda M, Bravo R, Johnson EM. Altered gene expression in neurons during programmed cell death: identification of c-jun as necessary for neuronal apoptosis. J Cell Biol. 1994;127:1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Faris M, Kokot N, Latinis K, Kasibhatla S, Green DR, Koretzky GA, Nel A. The c-Jun N-terminal kinase cascade plays a role in stress-induced apoptosis in Jurkat cells by up-regulating Fas ligand expression. J Immunol. 1998;160:134–144. [PubMed] [Google Scholar]
- 28.Ferm VH, Saxon A, Smith BM. The teratogenic profile of sodium arsenate in the golden hamster. Arch Environ Health. 1971;22:557–560. doi: 10.1080/00039896.1971.10665901. [DOI] [PubMed] [Google Scholar]
- 29.Glicksman MA, Chiu AY, Dionne CA, Harty M, Kaneko M, Murakata C, Oppenheim RW, Prevette D, Sengelaub DR, Vaught JL, Neff NT. CEP-1347/KT7515 prevents motor neuronal programmed cell death and injury-induced dedifferentiation in vivo. J Neurobiol. 1998;35:361–370. [PubMed] [Google Scholar]
- 30.Gunn-Moore FJ, Tavare JM. Apoptosis of cerebellar granule cells induced by serum withdrawal, glutamate or beta-amyloid, is independent of Jun kinase or p38 mitogen activated protein kinase activation. Neurosci Lett. 1998;250:53–56. doi: 10.1016/s0304-3940(98)00438-8. [DOI] [PubMed] [Google Scholar]
- 31.Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B, Davis RJ. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996;15:2760–2770. [PMC free article] [PubMed] [Google Scholar]
- 32.Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- 33.Hansen MB, Nielsen SE, Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods. 1989;119:203–210. doi: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- 34.Hartwig A. Current aspects in metal genotoxicity. Biometals. 1995;8:3–11. doi: 10.1007/BF00156151. [DOI] [PubMed] [Google Scholar]
- 35.Hetman M, Kanning K, Smith-Cavanaugh JE, Xia Z. Neuroprotection by brain-derived neurotrophic factor is mediated by extracellular-signal-regulated kinase and phosphatidylinositol-3 kinase. J Biol Chem. 1999;274:22569–22580. doi: 10.1074/jbc.274.32.22569. [DOI] [PubMed] [Google Scholar]
- 36.Hetman M, Cavanaugh JE, Kimelman D, Xia Z. Role of glycogen synthase kinase-3b in neuronal apoptosis induced by trophic withdrawal. J Neurosci. 2000;20:2567–2574. doi: 10.1523/JNEUROSCI.20-07-02567.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hockenbery D, Nuñez G, Millman C, Schreiber RD, Korsmeyer SJ. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990;348:334–336. doi: 10.1038/348334a0. [DOI] [PubMed] [Google Scholar]
- 38.Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 39.Hood RD, Bishop SL. Teratogenic effects of sodium arsenate in mice. Arch Environ Health. 1972;24:62–65. doi: 10.1080/00039896.1972.10666051. [DOI] [PubMed] [Google Scholar]
- 40.Ichijo H, Nishida E, Irie K, tenDijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 41.Ip YT, Davis RJ. Signal transduction by the c-Jun N-terminal kinase (JNK)—from inflammation to development. Curr Opin Cell Biol. 1998;10:205–219. doi: 10.1016/s0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- 42.Johnson MD, Xiang H, London S, Kinoshita Y, Knudson M, Mayberg M, Korsmeyer SJ, Morrison RS. Evidence for involvement of Bax and p53, but not caspases, in radiation-induced cell death of cultured postnatal hippocampal neurons. J Neurosci Res. 1998;54:721–733. doi: 10.1002/(SICI)1097-4547(19981215)54:6<721::AID-JNR1>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 43.Johnson MD, Kinoshita Y, Xiang H, Ghatan S, Morrison RS. Contribution of p53-dependent caspase activation to neuronal cell death declines with neuronal maturation. J Neurosci. 1999;19:2996–3006. doi: 10.1523/JNEUROSCI.19-08-02996.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kanamoto T, Mota M, Takeda K, Rubin LL, Miyazono K, Ichijo H, Bazenet CE. Role of apoptosis signal-regulating kinase in regulation of the c-Jun N-terminal kinase pathway and apoptosis in sympathetic neurons. Mol Cell Biol. 2000;20:196–204. doi: 10.1128/mcb.20.1.196-204.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kawasaki H, Morooka T, Shimohama S, Kimura J, Hirano T, Gotoh Y, Nishida E. Activation and involvement of p38 mitogen-activated protein kinase in glutamate-induced apoptosis in rat cerebellar granule cells. J Biol Chem. 1997;272:18518–18521. doi: 10.1074/jbc.272.30.18518. [DOI] [PubMed] [Google Scholar]
- 46.Kuan CY, Yang DD, Roy DRS, Davis RJ, Rakic P, Flavell RA. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron. 1999;22:667–676. doi: 10.1016/s0896-6273(00)80727-8. [DOI] [PubMed] [Google Scholar]
- 47.Kunimoto M. Methylmercury induces apoptosis of rat cerebellar neurons in primary culture. Biochem Biophys Res Commun. 1994;204:310–317. doi: 10.1006/bbrc.1994.2461. [DOI] [PubMed] [Google Scholar]
- 48.Le Niculescu H, Bonfoco E, Kasuya Y, Claret FX, Green DR, Karin M. Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol Cell Biol. 1999;19:751–763. doi: 10.1128/mcb.19.1.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luo YQ, Umegaki H, Wang XT, Abe R, Roth GS. Dopamine induces apoptosis through an oxidation-involved SAPK/JNK activation pathway. J Biol Chem. 1998;273:3756–3764. doi: 10.1074/jbc.273.6.3756. [DOI] [PubMed] [Google Scholar]
- 50.Machado AF, Hovland DN, Jr, Pilafas S, Collins MD. Teratogenic response to arsenite during neurulation: relative sensitivities of C57BL/6J and SWV/Fnn mice and impact of the splotch allele. Toxicol Sci. 1999;51:98–107. doi: 10.1093/toxsci/51.1.98. [DOI] [PubMed] [Google Scholar]
- 51.Mao Z, Bonni A, Xia F, Nadal-Vicens M, Greenberg ME. Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science. 1999;286:785–790. doi: 10.1126/science.286.5440.785. [DOI] [PubMed] [Google Scholar]
- 52.Maroney AC, Glicksman MA, Basma AN, Walton KM, Knight E, Murphy CA, Bartlett BA, Finn JP, Angeles T, Matsuda Y, Neff NT, Dionne CA. Motoneuron apoptosis is blocked by CEP-1347 (KT 7515), a novel inhibitor of the JNK signaling pathway. J Neurosci. 1998;18:104–111. doi: 10.1523/JNEUROSCI.18-01-00104.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maroney AC, Finn JP, Bozyczko-Coyne D, O'Kane TM, Neff NT, Tolkovsky AM, Park DS, Yan CY, Troy CM, Greene LA. CEP-1347 (KT7515), an inhibitor of JNK activation, rescues sympathetic neurons and neuronally differentiated PC12 cells from death evoked by three distinct insults. J Neurochem. 1999;73:1901–1912. [PubMed] [Google Scholar]
- 54.Martin JH, Mohit AA, Miller CA. Developmental expression in the mouse nervous system of the p49(3F12) SAP kinase. Mol Brain Res. 1996;35:1–2. doi: 10.1016/0169-328x(95)00181-q. [DOI] [PubMed] [Google Scholar]
- 55.Mayford M, Wang J, Kandel ER, O'Dell TJ. CaMKII regulates the frequency-response function of hippocampal synases for the production of both LTD and LTP. Cell. 1995;81:891–904. doi: 10.1016/0092-8674(95)90009-8. [DOI] [PubMed] [Google Scholar]
- 56.Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- 57.Mesner PW, Epting CL, Hegarty JL, Green SH. A time table of events during programmed cell death induced by trophic factor withdrawal from neuronal PC12 cells. J Neurosci. 1995;15:7357–7366. doi: 10.1523/JNEUROSCI.15-11-07357.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Migheli A, Piva R, Atzori C, Troost D, Schiffer D. c-Jun, JNK/SAPK kinases and transcription factor NF-kappa B are selectively activated in astrocytes, but not motor neurons, in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 1997;56:1314–1322. doi: 10.1097/00005072-199712000-00006. [DOI] [PubMed] [Google Scholar]
- 59.Minden A, Lin A, McMahon M, Lange Carter C, D'Erijard B, Davis RJ, Johnson GL, Karin M. Differential activation of ERK and JNK mitogen-activated protein kinases by Raf-1 and MEKK. Science. 1994;266:1719–1723. doi: 10.1126/science.7992057. [DOI] [PubMed] [Google Scholar]
- 60.Minden A, Lin A, Cjaret FX, Abo A, Karin M. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell. 1995;81:1147–1157. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- 61.Morrissey RE, Mottet NK. Arsenic-induced exencephaly in the mouse and associated lesions occurring during neurulation. Teratology. 1983;28:399–411. doi: 10.1002/tera.1420280311. [DOI] [PubMed] [Google Scholar]
- 62.Oberto A, Marks N, Evans HL, Guidotti A. Lead (Pb+2) promotes apoptosis in newborn rat cerebellar neurons: pathological implications. J Pharmacol Exp Ther. 1996;279:435–442. doi: 10.1163/2211730x96x00234. [DOI] [PubMed] [Google Scholar]
- 63.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 64.Park DS, Stefanis L, Yan CYI, Farinelli SE, Greene LA. Ordering the cell death pathway—differential effects of BCL2, an interleukin-1-converting enzyme family protease inhibitor, and other survival agents on JNK activation in serum nerve growth factor-deprived PC12 cells. J Biol Chem. 1996;271:21898–21905. doi: 10.1074/jbc.271.36.21898. [DOI] [PubMed] [Google Scholar]
- 65.Pirvola U, Xing-Qun L, Virkkala J, Saarma M, Murakata C, Camoratto AM, Walton KM, Ylikoski J. Rescue of hearing, auditory hair cells, and neurons by CEP-1347/KT7515, an inhibitor of c-Jun N-terminal kinase activation. J Neurosci. 2000;20:43–50. doi: 10.1523/JNEUROSCI.20-01-00043.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Raff MC, Barres BA, Burne J, Coles HS, Ishizaki Y, Jacobson MD. Programmed cell death and the control of cell survival: lessons from the nervous system. Science. 1993;262:695–700. doi: 10.1126/science.8235590. [DOI] [PubMed] [Google Scholar]
- 67.Rapp UR, Troppmair J, Beck T, Birrer MJ. Transformation by raf and other oncogenes renders cells differentially sensitive to growth inhibition by a dominant negative c-jun mutant. Oncogene. 1994;9:3493–3498. [PubMed] [Google Scholar]
- 68.Reed JC. Double identity for proteins of the Bcl-2 family. Nature. 1997;387:773–776. doi: 10.1038/42867. [DOI] [PubMed] [Google Scholar]
- 69.Roy TS, Andrews JE, Seidler FJ, Slotkin TA. Chlorpyrifos elicits mitotic abnormalities and apoptosis in neuroepithelium of cultured rat embryos. Teratology. 1998;58:62–68. doi: 10.1002/(SICI)1096-9926(199808)58:2<62::AID-TERA7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 70.Sarafian TA, Vartavarian L, Kane DJ, Bredenden DE, Verity MA. bcl-2 expression decreases methyl mercury-induced free-radical generation and cell killing in a neural cell line. Toxicol Lett. 1994;74:149–155. doi: 10.1016/0378-4274(94)90093-0. [DOI] [PubMed] [Google Scholar]
- 71.Shalat SL, Walker DB, Finnell RH. Role of arsenic as a reproductive toxin with particular attention to neural tube defects. J Toxicol Environ Health. 1996;48:253–272. doi: 10.1080/009841096161320. [DOI] [PubMed] [Google Scholar]
- 72.Snow ET. Metal carcinogenesis: mechanistic implications. Pharmacol Ther. 1992;53:31–65. doi: 10.1016/0163-7258(92)90043-y. [DOI] [PubMed] [Google Scholar]
- 73.Stefanis L, Burke RE, Greene LA. Apoptosis in neurodegenerative disorders. Curr Opin Neurol. 1997;10:299–305. doi: 10.1097/00019052-199708000-00004. [DOI] [PubMed] [Google Scholar]
- 74.Stefanis L, Park DS, Friedman WJ, Greene LA. Caspase-dependent and -independent death of camptothecin-treated embryonic cortical neurons. J Neurosci. 1999;19:6235–6247. doi: 10.1523/JNEUROSCI.19-15-06235.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tabocova S, Hunter ES, III, Gladen BC. Developmental toxicity of inorganic arsenic in whole embryo: culture oxidation state, dose, time, and gestational age dependence. Toxicol Appl Pharmacol. 1996;138:298–307. doi: 10.1006/taap.1996.0128. [DOI] [PubMed] [Google Scholar]
- 76.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 77.Wang JH, Redmond HP, Watson RWG, Bouchier Hayes D. Induction of human endothelial cell apoptosis requires both heat shock and oxidative stress responses. Am J Physiol. 1997;41:C1543–C1551. doi: 10.1152/ajpcell.1997.272.5.C1543. [DOI] [PubMed] [Google Scholar]
- 78.Wang TH, Popp DM, Wang HS, Saitoh M, Mural JG, Henley DC, Ichijo H, Wimalasena J. Microtubule dysfunction induced by paclitaxel initiates apoptosis through both c-Jun N-terminal kinase (JNK)-dependent and -independent pathways in ovarian cancer cells. J Biol Chem. 1999;274:8208–8216. doi: 10.1074/jbc.274.12.8208. [DOI] [PubMed] [Google Scholar]
- 79.Watson A, Eilers A, Lallemand D, Kyriakis J, Rubin LL, Ham J. Phosphorylation of c-Jun is necessary for apoptosis induced by survival signal withdrawal in cerebellar granule neurons. J Neurosci. 1998;18:751–762. doi: 10.1523/JNEUROSCI.18-02-00751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Watson RW, Redmond HP, Wang JH, Bouchier Hayes D. Mechanisms involved in sodium arsenite-induced apoptosis of human neutrophils. J Leukoc Biol. 1996;60:625–632. doi: 10.1002/jlb.60.5.625. [DOI] [PubMed] [Google Scholar]
- 81.Whitmarsh AJ, Shore P, Sharrocks AD, Davis RJ. Integration of MAP kinase signal transduction pathways at the serum response element. Science. 1995;269:403–407. doi: 10.1126/science.7618106. [DOI] [PubMed] [Google Scholar]
- 82.Whitmarsh AJ, Cavanagh J, Tournier C, Yasuda J, Davis RJ. A mammalian scaffold complex that selectively mediates MAP kinase activation. Science. 1998;281:1671–1674. doi: 10.1126/science.281.5383.1671. [DOI] [PubMed] [Google Scholar]
- 83.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 84.Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xu X, Raber J, Yang DS, Su B, Mucke L. Dynamic regulation of c-Jun N-terminal kinase activity in mouse brain by environmental stimuli. Proc Natl Acad Sci USA. 1997;94:12655–12660. doi: 10.1073/pnas.94.23.12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yamamoto K, Ichijo H, Korsmeyer SJ. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol Cell Biol. 1999;19:8469–8478. doi: 10.1128/mcb.19.12.8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]