

ABSTRACT
Smoking accounts for approximately 52% of bladder cancer incidence among postmenopausal women, but the underlying mechanism is poorly understood. Our study investigates whether changes in DNA methylation, as measured in blood, mediate the impact of smoking on bladder cancer risk among postmenopausal women. We conducted analyses among 206 cases and 251 controls that were current or never smokers at baseline from a previous case-control study of bladder cancer and genome-wide DNA methylation nested within the Women’s Health Initiative. Separate mediation analyses were conducted for three CpG sites demonstrating robust associations with smoking in prior methylome-wide association studies: cg05575921 (AhRR), cg03636183 (F2RL3), and cg19859270 (GPR15). We estimated causal effects using the regression-based, four-way decomposition approach, which addresses the interaction between smoking and each CpG site. The overall proportion of the excess relative risk mediated by cg05575921 was 92% (p-value = 0.004) and by cg19859270 was 79% (p-value = 0.02). The largest component of the excess relative risk of bladder cancer due to 30 pack-years of smoking history in current smokers was the mediated interaction for both cg05575921 (72%, p = 0.02) and cg19859270 (72%, p-value = 0.04), where the mediated interaction is the effect of smoking on bladder cancer that both acts through differential methylation and depends on smoking history. There was little evidence that smoking was mediated through cg03636183. Our results suggest that differential methylation of cg05575921 and cg19859270 mediate the effects of smoking on bladder cancer, potentially revealing downstream effects of smoking relevant for carcinogenesis.
KEYWORDS: Cigarette smoking, DNA methylation, bladder cancer, aryl-hydrocarbon receptor repressor gene (AhRR), G protein-coupled receptor 15 gene (GPR15), F2R like thrombin or trypsin receptor 3 gene (F2RL3)
Introduction
Cigarette smoking is an established cause of bladder cancer [1]. Transitional cell carcinoma (TCC) is by far the most common type of bladder cancer, accounting for 94% of cases [2]. Based on results from the NIH-AARP cohort, smoking accounts for an estimated 52% of TCC bladder cancer incidence in US women that are 50 to 71 years of age [3]. Approximately 25% of cases are diagnosed with muscle-invasive bladder cancer (MIBC) [4,5], and smokers are more likely to present with MIBC as well as with other aggressive tumour characteristics [6,7].
The link between smoking and bladder cancer is thought to involve exposure to more than 60 carcinogens that promote DNA adduct formation and subsequent accumulation of somatic mutations [1,8–10] and epigenetic reprogramming [11]. Specifically, the aromatic amines 2‐naphthylamine and 4‐aminobiphenyl are combustion products of cigarette smoke that can cause bladder cancer, as established in the context of the dye industry [1,12]. However, the specific mechanisms related to the carcinogenicity of cigarette smoking are not well described and fail to explain why bladder tissue is particularly susceptible to the tumorigenic effects of smoking [10,13].
Since DNA methylation is involved in transcriptional regulation, alternative splicing, and genome integrity, it is a promising marker of the carcinogenicity of smoking [11]. Blood-based measures of DNA methylation at specific loci have been reliably associated with past and current smoking [14–16]. Further elucidation of these smoking-associated methylation changes may improve our understanding of bladder carcinogenesis and reveal new avenues for bladder cancer prevention, screening, and treatment.
We hypothesize that smoking has indirect effects on bladder cancer through changes in DNA methylation at specific CpG sites, as was previously observed for lung cancer; Fasanelli et al. observed that hypomethylation of cg05575921 and of cg03636183 in blood explained approximately 37% of the increased risk of lung cancer induced by smoking [17]. Our study quantifies mediation of the association between smoking and bladder cancer through differential DNA methylation, as measured in blood, at each of three smoking-associated CpG sites.
Results
Table 1 shows the distribution of relevant variables by case-control status. A much higher proportion of cases were current smokers and methylation levels of the three CpG sites were lower among cases. In addition, compared to controls, a greater proportion of cases were White.
Table 1.
Distribution of relevant demographic and clinical characteristics among included bladder cancer cases and controls nested within the Women’s Health Initiative.
| Cases | Controls | |
|---|---|---|
| Smoking status (N, %) | ||
| Never smoker | 153 (74%) | 233 (93%) |
| Current smoker | 53 (26%) | 18 (7%) |
| Mediators, M-values (mean, SD) | ||
| cg03636183 | 1.38 (0.54) | 1.49 (0.36) |
| cg05575921 | 3.20 (1.36) | 3.83 (0.84) |
| cg19859270 | 3.62 (0.49) | 3.79 (0.28) |
| WHI arm (N, %) | ||
| OS | 108 (52%) | 131 (52%) |
| CT: HRT and DM | 14 (7%) | 16 (6%) |
| CT: HRT only | 26 (13%) | 29 (12%) |
| CT: DM only | 58 (28%) | 75 (30%) |
| Age, years (N, %) | ||
| < 50–59 | 58 (28%) | 57 (23%) |
| 60–69 | 85 (41%) | 116 (46%) |
| 70–79+ | 63 (31%) | 78 (31%) |
| Year of enrollment (N, %) | ||
| 1994–1995 | 50 (24%) | 61 (24%) |
| 1996 | 74 (36%) | 79 (32%) |
| 1997 | 50 (24%) | 65 (26%) |
| 1998 | 32 (16%) | 46 (18%) |
| Follow-up time, years (mean, SD) | 13.29 (3.89) | 14.05 (3.29) |
| DNA extraction method (N, %) | ||
| 5-prime | 197 (96%) | 240 (96%) |
| Phenol | 9 (4%) | 11 (4%) |
| Race (N, %) | ||
| Asian/Pacific Islander | 3 (2%) | 10 (4%) |
| Black/African American | 12 (6%) | 25 (10%) |
| Hispanic/Latino | 4 (2%) | 13 (5%) |
| Non-Hispanic White | 186 (90%) | 196 (78%) |
| Other | 1 (<1%) | 7 (3%) |
| Education (N, %) | ||
| < High school | 9 (4%) | 12 (5%) |
| High school | 39 (19%) | 60 (24%) |
| Post-high school training | 76 (37%) | 89 (35%) |
| College degree | 23 (11%) | 28 (11%) |
| Post-college training | 59 (29%) | 62 (25%) |
Abbreviations: OS = observational study; CT = clinical trials; HRT = hormone therapy clinical trial; DM = dietary modification clinical trial
We verified previously reported associations between smoking and methylation β-values at cg05575921 (regression coefficient = −0.23; q-value = 1.3e-114), cg03636183 (regression coefficient = −0.13; q-value = 9.2e-66), and cg19859270 (regression coefficient = −0.05; q-value = 1.1e-64). Adjustment for cell mixture using the reference-free method had a negligible impact on the association between smoking and β-values at cg05575921, cg03636183, and cg19859270.
The results of mediation analyses for each of cg05575921, cg03636183, and cg19859270 are presented in Table 2. The excess relative risk for the total effect (ERRTE) of 30 pack-years of smoking in current smokers as compared to never smoking on bladder cancer slightly varied across CpG sites and ranged from 2.63 to 3.19 (all p < 0.05).
Table 2.
Estimated mediating effects of select CpG sites in the association of 30 pack-years of smoking among current smokers with bladder cancer risk in the Women’s Health Initiative.
| Component of ERR estimatea |
Percent of ERR estimatea |
||||
|---|---|---|---|---|---|
| CpG site | Component of smoking ERR | Estimate | 95% CI | Estimate | P-value |
| cg05575921 | CDE | 0.18 | (−1.94, 2.29) | 5% | 0.86 |
| INTref | 0.09 | (0, 0.17) | 3% | 0.05 | |
| INTmed | 2.29 | (0.02, 4.55) | 72% | 0.02 | |
| PIE | 0.64 | (−0.38, 1.66) | 20% | 0.21 | |
| TE | 3.19 | (0.43, 5.95) | 100% | - | |
| cg03636183 | CDE | 2.06 | (−1.47, 5.58) | 77% | 0.09 |
| INTref | 0.03 | (−0.15, 0.21) | 1% | 0.74 | |
| INTmed | 0.90 | (−1.16, 2.95) | 34% | 0.45 | |
| PIE | −0.32 | (−0.60, −0.03) | −12% | 0.15 | |
| TE | 2.67 | (0.21, 5.13) | 100% | - | |
| cg19859270 | CDE | 0.46 | (−1.66, 2.58) | 17% | 0.64 |
| INTref | 0.10 | (−0.03, 0.23) | 4% | 0.13 | |
| INTmed | 1.89 | (0.04, 3.75) | 72% | 0.04 | |
| PIE | 0.18 | (−0.38, 0.74) | 7% | 0.52 | |
| TE | 2.63 | (0.24, 5.03) | 100% | - | |
aMediation models were adjusted for race/ethnicity, education, WHI arm, age at baseline, year of enrollment, follow-up time, and DNA extraction method.
Abbreviations: ERR = excess relative risk; CDE = controlled direct effect; INTref = reference interaction; INTmed = mediated interaction; PIE = pure indirect effect; TE = total effect
Much of the excess relative risk associated with 30 pack-years of smoking in current smokers as compared to never smoking was mediated through cg05575921 [92% of ERRTE] and cg19859270 [79% of ERRTE] (Table 2), where the excess relative risk for the total indirect effect (ERRTIE) is the sum of the excess relative risk for the mediated interaction (ERRINTmed) and the excess relative risk for the pure indirect effect (ERRPIE). The relationship between components of the four-way decomposition is illustrated in Figure 1, which also shows how these results correspond to the causal relationships relevant for our study.
Figure 1.
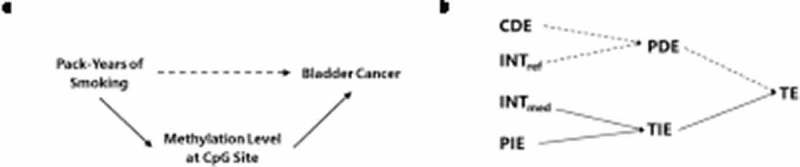
Graphical representation of the relationship between a causal directed acyclic graph (DAG) of the study and the results from the four-way decomposition method, where the sum of the mediated interaction (INTmed) and the pure indirect effect (PIE) corresponds to the indirect effect through the mediator (represented by solid arrows in A and B).
Abbreviations: CDE = controlled direct effect; INTref = reference interaction; INTmed = mediated interaction; PIE = pure indirect effect; PDE = pure direct effect; TIE = total indirect effect; TE = total effect
The mediated interaction was the largest component of the ERR of bladder cancer attributable to smoking for both cg05575921 (72%, p = 0.02) and cg19859270 (72%, p-value = 0.04). ERRINTmed captures the effect of smoking through differential methylation of the mediator that is both caused by smoking and has effects on bladder cancer that depend on the presence of smoking. There was little evidence that the effect of smoking on bladder cancer was mediated through cg03636183.
Discussion
Overall, our study suggests that the effects of smoking may be mediated through differential methylation of cg05575921 and cg19859270. Supporting the validity of our estimates, the total relative risks of bladder cancer associated with current smoking with a 30 pack-year smoking history as compared to never smoking ranged from 3.63 to 4.19, which is consistent with the adjusted hazard ratio of 4.65 for current smoking that was reported for women in the demographically similar NIH-AARP Diet and Health study [3]. The effect sizes for the association between baseline current smoking and methylation levels at our selected loci were also consistent with those reported by previous methylome-wide association studies [18–24].
Compared to non-smokers, smokers demonstrate decreased methylation at cg19859270, as observed in our study and as reported by previous methylome-wide studies of smoking [18–20,22–24]. This CpG site is located in the first exon of GPR15, a gene coding for a chemoattractant receptor [25]. First exon methylation is closely associated with transcriptional silencing and may be even more strongly associated with low gene expression than promoter methylation [26], so hypomethylation of GPR15 in leukocytes is expected to increase GPR15 expression in the blood of smokers. However, rather than an active causal change in methylation at cg19859270, there is evidence suggesting that the methylation change reflects a shift in the immune system. Specifically, smoking may trigger an inflammation response that substantially increases the proportion of T-cells expressing GPR15 (GPR15+ T-cells). In a study by Bauer et al., these GPR15+ T-cells were detected as an overall decrease in GPR15 methylation and increase in GPR15 expression in the blood of smokers [27]. In fact, Bauer et al. demonstrated that smoking was no longer associated with decreased methylation of cg19859270 after adjustment for GPR15+ T-cell subtype [27]. This finding of strongly differential expression of GPR15 in the blood of current versus never smokers was independently replicated (fold-change = 5.8, q-value = 0.004) [25]. GPR15 appears to regulate the homing of T-cells in epithelial tissue [28] and may be an indicator of chronic inflammation [25,28,29], and the inflammatory response has canonical relationships with the promotion of tumour initiation and progression [30,31]. Since the causal effect of smoking through cg19859270 was observed primarily as a mediated interaction, our results suggest that a pro-inflammatory change involving GPR15+ T-cells could be particularly carcinogenic in the context of other effects related to current smoking.
The cg05575921 CpG site is located in an enhancer-like regulatory element within AhRR, which is a putative tumour suppressor gene [32] whose expression down-regulates the aryl hydrocarbon receptor (AhR). This CpG site was found to be hypomethylated among smokers as compared to nonsmokers in our study and in prior methylome-wide studies [18–21]. Hypomethylation of cg05575921 in blood may reflect an inflammatory response mediated by white blood cells. There is convincing evidence that smoking is associated with hypomethylation of cg05575921, activates the AhRR enhancer, and up-regulates AhRR in a subset of monocytes [33], which may then promote inflammatory signalling in monocytes [34] and in monocyte-derived macrophages [33,34]. Alternatively, differential methylation of AhRR in blood may be associated with changes in the AhRR-ARNT complex in normal bladder tissue [11]. In fact, Wan et al. assert that this exposure-induced methylation change in AhRR occurs to some extent in all cell types, including those found in saliva (r 2 = 0.90 with monocytes) [33]. In data from The Cancer Genome Atlas (TCGA), current as compared to never smoking has a suggestive association with cg05575921 hypomethylation in normal bladder tissue when adjusting for age at diagnosis and gender (regression coefficient = −0.10; p-value = 0.07; N = 8) [35]. This change is part of a shift in the aryl hydrocarbon receptor signalling pathway, which may be particularly affected by smoking-induced methylation changes in blood [11]. This pathway is involved in the detoxification of chemicals in cigarette smoke, and aberrant AhRR methylation may enhance CYP1A1 expression, induce the formation of DNA adducts, and initiate smoking-related cancer [11]. Both proposed mechanisms are consistent with the strong mediated interaction of approximately 72% that we observed, since the effects of the methylation change at cg05575921 would depend on the continued presence of exposure to cigarette smoke.
Even though smoking was highly associated with hypomethylation of cg03636183, we found no evidence that the association between smoking and bladder cancer risk is mediated through cg03636183. We note that this locus is in the north shore of an intragenic CpG island in F2RL3, which encodes the thrombin protease-activated receptor-4 protein (PAR-4) [15].
While there have been prior methylome-wide association studies of smoking and bladder cancer [36–39], to our knowledge, there is no prior mediation analysis that assess smoking-associated methylation changes as potential mediators of the association between smoking and bladder cancer. We were uniquely positioned to conduct mediation analyses, since our study is nested in a prospective cohort that includes pre-diagnostic blood samples and comprehensive baseline information on potential confounders, which allowed us to address the temporality and no-confounding assumptions required for causal inference. Despite being nested in a large cohort study, our sample size did not allow us to explore whether these causal effects vary across subtypes of bladder cancer.
Since we only have single measures of smoking and DNA methylation at baseline, they may imprecisely capture smoking and methylation levels for the time periods etiologically relevant to bladder cancer carcinogenesis. These errors in our measures of smoking and methylation are likely to be non-differential and are expected to attenuate the reported direct and indirect effect estimates. Additional error was likely introduced when we calculated pack-years from self-reported categorical data. As such, it is also possible that the methylation changes at the smoking-associated mediators capture some of the direct effects of smoking, thereby leading to overestimation of indirect effects.
With the mechanisms related to the immune response and inflammation that we propose to underlie our observations, we would expect associations for current smoking with GPR15 and AhRR methylation to be largely attenuated by reference-free adjustment for cell-type composition. However, this was not the case. While this may suggest that smoking-related changes in methylation may be markers of mechanisms unrelated to immune response, it should be noted that the cell-type adjustment method is not based on actual reference data for white blood cell types and does not explicitly account for white blood cell subtypes, including GPR15+ T-cells or AhRR+ monocytes.
The methylation changes at each of cg05575921, cg03636183, and cg19859270 are closely associated with smoking and, as a result, are highly correlated with each other. As a result, the mediated causal effects estimated from the individual models for these CpG sites likely capture the same causal pathways. However, we were unable to model these sites together because methodology for extending the four-way decomposition approach to allow for multiple mediators is not yet available. When interpreting our results, it is also important to keep in mind that the ERRCDE captures some of the effect of the ERRINTref, due to limitations of this approach.
A substantial proportion of the effect of current smoking on bladder cancer may be mediated through methylation differences at cg05575921 and cg19859270 and may be particularly harmful in the context of continued smoking. These results may indicate the promotion of chronic inflammation through a higher proportion of GPR15+ T-cells in blood or through increased expression of AhRR in specific white blood cell subtypes or in bladder tissue. Further investigation of these possible mechanisms has the potential to expand our understanding of the relationship between bladder cancer and smoking, which is its strongest known risk factor.
Patients and methods
Study participants
Data for the current analyses were drawn from our case-control study of pre-diagnostic DNA methylation and bladder cancer [36], which was approved by the Fred Hutchinson Cancer Research Center Institutional Review Board. This case-control study was nested in the Women’s Health Initiative (WHI), which includes 161,808 postmenopausal women recruited from 1993 to 1998 across the US [40]. There are two arms of the WHI: the clinical trials (CT) and the observational study (OS). The CT involved concurrent-randomized controlled trials of hormone therapy, dietary modification, and subsequently calcium/vitamin D. Those not eligible or not willing to participate in the CT were asked to participate in the OS. A total of 440 cases diagnosed with urothelial carcinoma (i.e. transitional cell carcinoma) of the bladder during the WHI follow-up period were included in the case-control study, as were 440 cancer-free controls matched on year of enrollment, age at enrollment (± 2 years), follow-up time greater than or equal to their matched case, trial component, and DNA extraction method [36].
Since smoking-associated methylation changes are reversible [17,41], we expected the associations under investigation would be attenuated in former smokers. Despite the reduction in sample size, to most effectively examine the relevant causal relationships, we restricted our analyses to the 210 cases and 256 controls who were never smokers or current smokers with available pack-years data. To allow for covariate adjustment of our models, we also excluded the few participants whose DNA was extracted using BioServe or salt methods (N = 4) and who were missing data on race/ethnicity or education level (N = 5), leaving 206 cases (53 current smokers, 153 never smokers) and 251 controls (18 current smokers, 233 never smokers) for analyses, where 48 (23%) of the cases were diagnosed with muscle-invasive bladder cancer.
Data and biospecimen collection
Basic demographic information, including age and race/ethnicity, was reported during the WHI screening process [40]. On baseline questionnaires, participants reported if they had ever smoked at least 100 cigarettes (yes, no), currently smoked cigarettes (yes, no), or had ever smoked to lose weight (yes, no); this information was used to determine smoking status. Participants also disclosed the average number of cigarettes currently or previously smoked per day (<1, 1–4, 5–14, 15–24, 25–34, 35–44, 45+) and the number of years as a regular smoker (<5, 5–9, 10–19, 20–29, 30–39, 40–49, 50+). Pack-years were calculated by dividing the approximate midpoint of the cigarettes per day category by 20 and then multiplying by the approximate midpoint of the years of regular smoking category. For current smokers who did not provide information on years as a regular smoker or indicated over 50 years as a regular smoker, years of regular smoking were estimated based on the categorical age at which they started smoking regularly, if available. The time from enrollment to end of follow-up was based on the number of days between enrollment and death or last contact. In addition, blood samples were collected at baseline after at least 12 hours of fasting and stored at −70°C as buffy coats.
DNA methylation array
As described previously, we used the Illumina 450K Infinium HumanMethylation Bead Array (Illumina, San Diego, CA, USA) to interrogate methylation status at approximately 485,577 CpG sites among bladder cancer cases and controls [36]. We used the M-value to measure methylation at each CpG site to improve the heteroscedasticity of methylation levels [42]. To get the M-value, we calculated the base-2 logit of the β-value, where the β-value is the ratio of the methylated signal over the total signal and can be interpreted as the proportion of methylation at a specific site [43]. After reading in the raw image files, we checked for failed samples and then performed background correction and functional normalization of the methylation data [36]. We excluded any CpG sites that were undetected in at least 10% of samples, had a beadcount less than 3 in at least 10% of samples, were in any SNP or within 10 base pairs of SNPs with minor allele frequencies greater than 1%, were classified as cross-reactive probes, or were located on the sex chromosomes [36].
Identifying and validating smoking-associated loci
A systematic review of 14 methylome-wide studies in blood reported a total of 1,460 CpG sites associated with smoking [15]. Three CpG loci were most consistently associated with smoking: cg05575921 (located in the aryl-hydrocarbon receptor repressor gene; AhRR), cg03636183 (located in the F2R like thrombin receptor 3 gene; F2RL3), and cg19859270 (located in the G-protein receptor 15 gene; GPR15). These associations were subsequently verified in two large-scale studies [18,41].
Statistical analysis
To validate the previously reported associations between the selected CpG sites and current smoking, we used adjusted linear regression models to test associations between current smoking and methylation levels across the genome, focusing on three CpG sites of interest in our study population. The β-value, rather than the M-value, was used to allow direct comparisons with results from previous studies. We used the empirical Bayes approach to test the significance of these associations and adjusted for multiple testing using the FDR method [44]. To address the role of cell-type composition, we additionally adjusted these associations for latent variables related to cell mixture using the reference-free method [45], where the latent variable dimension was estimated using the random matrix theory method, standard errors were estimated based on 100 bootstrap samples, and p-values were adjusted for multiple testing using the FDR approach [45].
Separate mediation analyses were conducted with baseline methylation level at each of the three CpG sites (cg05575921, cg03636183, cg19859270) as a mediator, with baseline pack-years of smoking as the exposure among current or never smokers, and incident bladder cancer as the outcome. We estimated the causal effects using a regression-based approach for dichotomous outcomes [46,47], which is based on a counterfactual framework for causal inference. This method uses a logistic regression model to estimate the outcome and a linear regression model to estimate the mediator. The models were adjusted for a set of covariates that included our matching covariates (WHI arm, age at baseline, year of enrollment, follow-up time, DNA extraction method) and the following potential confounders: race/ethnicity (Asian/Pacific Islander, Black/African American, Hispanic/Latino, non-Hispanic White, other) and education (less than high school, high school, post-high school training, college degree, post-college training).
To account for an additive interaction between the exposure and the mediator, we used the four-way decomposition method, which addresses interaction and mediation simultaneously [47]. We estimated the relevant causal effects as components of the excess relative risk (ERR or RR-1) of bladder cancer associated with smoking using the coefficients from the regression models (Appendix 1, Supplemental Online Material), where these causal effects capture all possible combinations of mediation (absent, present) and interaction (absent, present). The calculations were based on setting smoking to a level of 30 pack-years as compared to 0 pack-years, where the median smoking history among current smokers was 37.5 pack-years. The meditator was considered absent at its average M-value in non-smoking controls (m*). Each continuous covariate and level of categorical covariates was held constant at the level observed in controls.
The analyses were conducted using SAS® software (version 9.3, SAS Institute Inc., Cary, NC, USA) and the four-way decomposition code published by VanderWeele [47].
Funding Statement
This work was supported by the American Cancer Society under Grant 125299-RSG-13-100-01-CCE; and Kristina M. Jordahl was supported by the National Cancer Institute (NCI) at the National Institutes of Health (NIH) under Training Grants R25 CA094880 and T32 CA094880. The contents of this work are solely the responsibility of the authors and do not necessarily represent the official views of the NCI, NIH. The WHI program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U. S. Department of Health and Human Services through contracts HHSN268201600018C, HHSN268201600001C, HHSN268201600002C, HHSN268201600003C, and HHSN268201600004C.
Acknowledgments
We would like to acknowledge Xiaoling Song for performing the bisulfite conversion and plating of DNA samples and Cassandra Sather for running the DNA methylation assays.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Centers for Disease Control and Prevention (US) How tobacco smoke causes disease: the biology and behavioral basis for smoking-attributable disease: a report of the surgeon general [Internet] Atlanta (GA): Centers for Disease Control and Prevention (US); 2010. Available from: http://www.ncbi.nlm.nih.gov/books/NBK53017/ [PubMed] [Google Scholar]
- [2].Zhang Y, Zhu C, Curado MP, et al. Changing patterns of bladder cancer in the USA: evidence of heterogeneous disease. BJU Int. 2012;109:52–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Freedman ND, Silverman DT, Hollenbeck AR, et al. Association between smoking and risk of bladder cancer among men and women. JAMA. 2011;306:737–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sanli O, Dobruch J, Knowles MA, et al. Bladder cancer. Nat Rev Dis Primer. 2017;3:17022. [DOI] [PubMed] [Google Scholar]
- [5].Barbosa ALA, Vermeulen SHHM, Aben KK, et al. Smoking intensity and bladder cancer aggressiveness at diagnosis. PLoS One. 2018;13:e0194039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pietzak EJ, Mucksavage P, Guzzo TJ, et al. Heavy cigarette smoking and aggressive bladder cancer at initial presentation. Urology. 2015;86:968–972. [DOI] [PubMed] [Google Scholar]
- [7].Soria F, Marra G, Capoun O, et al. Prevention of bladder cancer incidence and recurrence: tobacco use. Curr Opin Urol. 2018;28:80. [DOI] [PubMed] [Google Scholar]
- [8].Florescu A, Ferrence R, Einarson T, et al. Methods for quantification of exposure to cigarette smoking and environmental tobacco smoke: focus on developmental toxicology. Ther Drug Monit. 2009;31:14–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Besaratinia A, Tommasi S.. Genotoxicity of tobacco smoke-derived aromatic amines and bladder cancer: current state of knowledge and future research directions. Faseb J. 2013;27:2090–2100. [DOI] [PubMed] [Google Scholar]
- [10].Besaratinia A, Cockburn M, Tommasi S. Alterations of DNA methylome in human bladder cancer. Epigenetics. 2013;8:1013–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ma Y, Li MD. Establishment of a strong link between smoking and cancer pathogenesis through DNA methylation analysis. Sci Rep. [Internet] 2017. [cited 2018 September5];7 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5431893/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Luch A. Nature and nurture – lessons from chemical carcinogenesis. Nat Rev Cancer. 2005;5:113. [DOI] [PubMed] [Google Scholar]
- [13].Zeegers MPA, Tan FES, Dorant E, et al. The impact of characteristics of cigarette smoking on urinary tract cancer risk. Cancer. 2000;89:630–639. [DOI] [PubMed] [Google Scholar]
- [14].Shenker NS, Ueland PM, Polidoro S, et al. DNA methylation as a long-term biomarker of exposure to tobacco smoke. Epidemiol Camb Mass. 2013;24:712–716. [DOI] [PubMed] [Google Scholar]
- [15].Gao X, Jia M, Zhang Y, et al. DNA methylation changes of whole blood cells in response to active smoking exposure in adults: a systematic review of DNA methylation studies. Clin Clin Epigenet. 2015;7:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Philibert RA, Beach SRH, Lei M-K, et al. Changes in DNA methylation at the aryl hydrocarbon receptor repressor may be a new biomarker for smoking. Clin Clin Epigenet. 2013;5:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fasanelli F, Baglietto L, Ponzi E, et al. Hypomethylation of smoking-related genes is associated with future lung cancer in four prospective cohorts. Nat Commun. [Internet] 2015;6 Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4682166/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Joehanes R, Just AC, Marioni RE, et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet. 2016;9:436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zeilinger S, Kühnel B, Klopp N, et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PloS One. 2013;8:e63812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tsaprouni LG, Yang T-P, Bell J, et al. Cigarette smoking reduces DNA methylation levels at multiple genomic loci but the effect is partially reversible upon cessation. Epigenetics. 2014;9:1382–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Shenker NS, Polidoro S, van Veldhoven K, et al. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum Mol Genet. 2013;22:843–851. [DOI] [PubMed] [Google Scholar]
- [22].Sun YV, Smith AK, Conneely KN, et al. Epigenomic association analysis identifies smoking-related DNA methylation sites in African Americans. Hum Genet. 2013;132:1027–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wan ES, Qiu W, Baccarelli A, et al. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum Mol Genet. 2012;21:3073–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Harlid S, Xu Z, Panduri V, et al. CpG sites associated with cigarette smoking: analysis of epigenome-wide data from the sister study. Environ Health Perspect. 2014;122:673–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kõks G, Uudelepp M-L, Limbach M, et al. Smoking-induced expression of the GPR15 gene indicates its potential role in chronic inflammatory pathologies. Am J Pathol. 2015;185:2898–2906. [DOI] [PubMed] [Google Scholar]
- [26].Brenet F, Moh M, Funk P, et al. DNA methylation of the first exon is tightly linked to transcriptional silencing. Plos One. 2011;6:e14524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bauer M, Linsel G, Fink B, et al. A varying T cell subtype explains apparent tobacco smoking induced single CpG hypomethylation in whole blood. Clin Clin Epigenet. 2015;7:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kõks S, Kõks G. Activation of GPR15 and its involvement in the biological effects of smoking. Exp Biol Med. 2017;242:1207–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nguyen LP, Pan J, Dinh TT, et al. Role and species–specific expression of colon T cell homing receptor GPR15 in colitis. Nat Immunol. 2015;16:207–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. [DOI] [PubMed] [Google Scholar]
- [31].Qian B-Z, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zudaire E, Cuesta N, Murty V, et al. The aryl hydrocarbon receptor repressor is a putative tumor suppressor gene in multiple human cancers. J Clin Invest. 2008;118:640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wan M, Bennett BD, Pittman GS, et al. Identification of smoking-associated differentially methylated regions using reduced representation bisulfite sequencing and cell type-specific enhancer activation and gene expression. Environ Health Perspect. 2018;126:047015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang -D-D, Wang W-T, Xiong J, et al. Long noncoding RNA LINC00305 promotes inflammation by activating the AHRR-NF-κB pathway in human monocytes. Sci Rep. 2017;7:46204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Goldman M, Craft B, Brooks AN, et al. The UCSC xena platform for cancer genomics data visualization and interpretation. bioRxiv. 2018;326470 Available from: https://www.biorxiv.org/content/10.1101/326470v1.full [Google Scholar]
- [36].Jordahl KM, Randolph TW, Song X, et al. Genome-wide DNA methylation in pre-diagnostic blood and bladder cancer risk in the women’s health initiative. Cancer Epidemiol Prev Biomark. 2018;27(6):689–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Marsit CJ, Koestler DC, Christensen BC, et al. DNA methylation array analysis identifies profiles of blood-derived DNA methylation associated with bladder cancer. J Clin Oncol. 2011;29:1133–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Olkhov-Mitsel E, Savio AJ, Kron KJ, et al. Epigenome-wide DNA methylation profiling identifies differential methylation biomarkers in high-grade bladder cancer. Transl Oncol. 2017;10:168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yang X, Gao L, Zhang S. Comparative pan-cancer DNA methylation analysis reveals cancer common and specific patterns. Brief Bioinform. 2017;18:761–773. [DOI] [PubMed] [Google Scholar]
- [40].Hays J, Hunt JR, Hubbell FA, et al. The women’s health initiative recruitment methods and results. Ann Epidemiol. 2003;13:S18–77. [DOI] [PubMed] [Google Scholar]
- [41].Ambatipudi S, Cuenin C, Hernandez-Vargas H, et al. Tobacco smoking-associated genome-wide DNA methylation changes in the EPIC study. Epigenomics. 2016;8:599–618. [DOI] [PubMed] [Google Scholar]
- [42].Du P, Zhang X, Huang -C-C, et al. Comparison of beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Dedeurwaerder S, Defrance M, Bizet M, et al. A comprehensive overview of infinium human Methylation450 data processing. Brief Bioinform. 2014;15:929–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Li D, Xie Z, Le Pape M, et al. An evaluation of statistical methods for DNA methylation microarray data analysis. BMC Bioinformatics. 2015;16:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Houseman EA, Molitor J, Marsit CJ. Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics. 2014;30:1431–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].VanderWeele TJ, Vansteelandt S. Odds ratios for mediation analysis for a dichotomous outcome. Am J Epidemiol. 2010;172:1339–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].VanderWeele T. Explanation in causal inference: methods for mediation and interaction. Oxford (NY): Oxford University Press; 2015. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.