

Abstract
Children who survive premature birth often exhibit reductions in hippocampal volumes and deficits in working memory. However, it is unclear whether synaptic plasticity and cellular mechanisms of learning and memory can be elicited or disrupted in the preterm fetal hippocampus. CA1 hippocampal neurons were exposed to two common insults to preterm brain: transient hypoxia-ischemia (HI) and hypoxia (Hx). We used a preterm fetal sheep model using both sexes in twin 0.65 gestation fetuses that reproduces the spectrum of injury and abnormal growth in preterm infants. Using Cavalieri measurements, hippocampal volumes were reduced in both Hx and HI fetuses compared with controls. This volume loss was not the result of neuronal cell death. Instead, morphometrics revealed alterations in both basal and apical dendritic arborization that were significantly associated with the level of systemic hypoxemia and metabolic stress regardless of etiology. Anatomical alterations of CA1 neurons were accompanied by reductions in probability of presynaptic glutamate release, long-term synaptic plasticity and intrinsic excitability. The reduction in intrinsic excitability was in part due to increased activity of the channels underlying the fast and slow component of the afterhyperpolarization in Hx and HI. Our studies suggest that even a single brief episode of hypoxemia can markedly disrupt hippocampal maturation. Hypoxemia may contribute to long-term working memory disturbances in preterm survivors by disrupting neuronal maturation with resultant functional disturbances in hippocampal action potential throughput. Strategies directed at limiting the duration or severity of hypoxemia during brain development may mitigate disturbances in hippocampal maturation.
SIGNIFICANCE STATEMENT Premature infants commonly sustain hypoxia-ischemia, which results in reduced hippocampal growth and life-long disturbances in learning and memory. We demonstrate that the circuitry related to synaptic plasticity and cellular mechanisms of learning and memory (LTP) are already functional in the fetal hippocampus. Unlike adults, the fetal hippocampus is surprisingly resistant to cell death from hypoxia-ischemia. However, the hippocampus sustains robust structural and functional disturbances in the dendritic maturation of CA1 neurons that are significantly associated with the magnitude of a brief hypoxic stress. Since transient hypoxic episodes occur commonly in preterm survivors, our findings suggest that the learning problems that ensue may be related to the unique susceptibility of the hippocampus to brief episodes of hypoxemia.
Keywords: dendritic morphology, developmental neuroanatomy, electrophysiology, hippocampus, LTP, synaptic plasticity
Introduction
Survivors of preterm birth commonly sustain persistent unexplained disturbances in learning and memory (Vohr, 2014). Vulnerability of the developing hippocampus is supported by several human volumetric MRI studies, which identified that persistent reductions in hippocampal volumes were associated with verbal and working memory disturbances in former premature neonates (Peterson et al., 2000; Beauchamp et al., 2008; Thompson et al., 2008). A role for hypoxia-ischemia (HI) in disrupted hippocampal development is consistent with observations in neonatal (McDonald and Johnston, 1990) and adult (Choi and Rothman, 1990) rodents that the CA1 subfields are particularly susceptible to HI via glutamate-mediated excitotoxicity. Paradoxically, in utero models of HI in fetal sheep similarly found that the near-term hippocampus is particularly susceptible to ischemic neuronal degeneration (Mallard et al., 1992), whereas the preterm hippocampus displays more variable and unexplained responses to HI that range from extensive (Reddy et al., 1998; George et al., 2004) to minimal injury (Riddle et al., 2006).
To address these disparate observations, we tested the hypothesis that the preterm fetal hippocampus is susceptible to HI-mediated disturbances in neuronal maturation that disrupt the structural and functional organization of hippocampal learning and memory circuits. We focused on the developing CA1 subfields given their aforementioned susceptibility to HI and the role of CA1 circuitry in cellular mechanisms of learning and memory, including LTP. Induction of LTP involves CA1 stimulation protocols that coordinate presynaptic and postsynaptic mechanisms that induce activity-dependent increases in synaptic activity (Kennedy, 2013). During development, LTP mediates activity-dependent refinement of neural circuit formation (Yasuda et al., 2003). The developmental onset of LTP in rodents varies with the induction protocol used and occurs between approximately the first and second postnatal weeks (Harris and Teyler, 1984; Palmer et al., 2004; Cao and Harris, 2012). The onset of LTP is coincident with changes in AMPA receptor composition, the appearance of dendritic spines, and the release of glial-derived factors that support the maturational onset of LTP (Henley and Wilkinson, 2016; Nicoll, 2017). Although considerable rodent data suggest that the onset of LTP is a postnatal phenomenon, there are limited data regarding the developmental maturation of LTP in higher mammals. Although LTP can be induced in human hippocampal slices (Bliss and Cooke, 2011) by repetitive sensory stimulation (Clapp et al., 2012), the timing of onset during fetal primate development is unclear.
Here we studied preterm (0.65 gestation) fetal sheep models of HI and hypoxemia (Hx), which reproduce major features of cerebral injury in preterm human neonates (Back et al., 2012). We demonstrate that neither fetal HI nor Hx caused significant degeneration of hippocampal neurons. However, fetal Hx alone was sufficient to disrupt the complexity of the CA1 dendritic arbor in a dose-dependent fashion. Using a tetanic stimulation protocol, LTP was robustly induced in the preterm fetal hippocampus. Hx caused highly significant reductions in LTP and intrinsic excitability (IE) that manifested as reduced action potential (AP) firing. Consistent with a more refractory synaptic firing rate, Hx induced an elevation in the current underlying the neuronal slow after-hyperpolarization (sAHP) channel mediated by potassium currents. Hence, Hx disrupts maturation of preterm CA1 neurons, leading to persistent structural and electrophysiological disturbances in learning and memory circuits.
Materials and Methods
Ethics statement.
All experiments performed on animals adhere to strict protocols approved by the Oregon Health and Science University Institutional Animal Care and Use Committee.
Instrumented fetal sheep surgery.
Sterile surgeries on time-bred pregnant ewes were performed between 88 and 92 d gestational age (term 145 d) at Oregon Health and Science University as previously described (Hagen et al., 2014; McClendon et al., 2017). Briefly, a midline laparotomy and hysterotomy were used to access each of the twin fetuses. A vinyl catheter was placed nonocclusively in a carotid, brachial, or femoral artery to measure arterial blood gases (ABGs) and monitor fetal heart rate, and an inflatable silastic occluder was placed around the brachiocephalic artery to control blood flow to the brain through the common carotid and vertebral arteries. In the sheep, all collateral circulation to the brain, except the spinal arteries, is provided through branches of the brachiocephalic artery. An additional catheter was sewn to the fetal skin to allow monitoring of the fetal amniotic fluid pressure. Following surgical instrumentation, the fetus was returned to the uterus, the maternal incision was closed, and the ewe was allowed to recover from surgery for 3 d after which HI studies were conducted (see Fig. 4A). Fetuses of both sexes were included in the study.
Figure 4.

Fetal ABG values for treatment groups during experimental paradigm. A, Flow chart of the experimental protocol described in Materials and Methods. B, Fetal oxygen content (vol %) initially drops in response to maternal hypoxia. With the onset of fetal ischemia, fetal oxygen content in both HI groups (red and solid black lines) attempts to rebound back to baseline levels, while remaining relatively constant in the fetuses exposed only to maternal hypoxia (Hx group blue line). Within 10 min of the cessation of maternal hypoxia and fetal ischemia, fetal oxygen content returns to baseline levels. The differences in oxygen content between the HI and HI-HC groups reflect the sampling site from which the blood gas was collected; in the fetus, oxygen content is higher in carotid arteries than in femoral arteries. Black dotted lines indicate normal control fetuses (TC). C, Fetal lactate concentration increases markedly in HI groups (red and solid black lines) by the end of the ischemic period and continues to rise 10 min after the cessation of fetal ischemia and maternal hypoxia compared with fetuses exposed only to maternal hypoxia (Hx group, blue lines). D, Fetal glucose concentration rises markedly in HI groups (red and solid black lines) compared with Hx group (blue line). E, The basal dendritic complexity of HI-treated CA1 hippocampal neurons is plotted versus the fetal systemic oxygen content 5 min into maternal hypoxia. F, Basal dendritic complexity of HI-treated CA1 hippocampal neurons is positively associated with the magnitude of the initial fetal response to maternal hypoxia defined as the difference between the pre-ABG (baseline) and Hx-ABG oxygen content values. The greater the drop in the oxygen content value from baseline for the HI-HC fetus, basal dendritic complexity is observed to increase in CA1 neurons. G, Basal dendritic complexity of HI-treated CA1 hippocampal neurons is positively associated with the magnitude of change in lactate recovery defined as the difference between post-ABG (recovery) lactate value and the pre-ABG (baseline) lactate value. H, Basal dendritic complexity of HI-treated CA1 hippocampal neurons versus the magnitude of change in fetal systemic glucose, defined as the difference between the HI-ABG glucose value and the Hx-ABG glucose value.
Cerebral HI studies and physiological monitoring.
Maternal hypoxia (Hx) was induced by supplying the pregnant ewe with a mixture of 50% room air and 50% nitrogen gas for an inhaled oxygen fraction of 10.5%. Following 5 min of maternal hypoxia, cerebral ischemia was initiated in one of the twin fetuses (designated HI fetus), and maintained by inflating the brachiocephalic occluder for 25 min. Four ABG samples were collected from each fetus during the study (see Fig. 4A): prehypoxia (baseline or pre-ABG), at 5 min after initiation of maternal hypoxia (Hx-ABG), 25 min after initiation of ischemia-induction (HI-ABG), and 10 min after cessation of Hx and HI (recovery or post-ABG). Blood samples were analyzed using an ABL800 flex blood gas analyzer (Radiometer Medical, A/S RRID:SCR_014772). If the fetal arterial oxygen concentration (ctO2) fell to <5.0 vol% after a 5 min exposure to maternal Hx, the maternal inhaled oxygen fraction was enriched to 14% for the duration of the experiment. Fetuses exposed exclusively to 30 min of maternal Hx were designated hypoxic controls (Hx), whereas fetuses exposed to maternal hypoxia in combination with cerebral hypoperfusion were designated hypoxic-ischemic (HI) animals (see Fig. 4A). Each experimental twin pregnancy was comprised of an Hx and HI pair. Both instrumented (surgically operated, n = 12) and noninstrumented (no surgery, n = 15) fetuses, which were not subjected to either maternal Hx or HI were designated true controls (TC), since a two-way ANOVA revealed no effect of sex or surgical status for LTP, paired-pulse ratio (PPR), fEPSP input-output slope (I/O slope), IE slope, or IE integral. For the basal dendritic studies using the Golgi stain, only noninstrumented fetuses (n = 6) were used as TC.
Tissue collection and processing.
To ensure the viability of the fetal brains for concurrent electrophysiological studies, the ewe was placed under general anesthesia and ventilated. After fetal harvest, ewes were killed with a 12 ml intravenous bolus of a pentobarbital sodium and phenytoin sodium solution (Euthasol NAC, #10230760, Virbac). Fetal brains were harvested 4 weeks after HI exposure and other experimental studies, with the exception of brains collected at 24 h, 4 d, and 7 d for cell death studies. Fetal brains were removed from the cranial vault, and the cerebellum and brainstem were removed by cutting the cerebral peduncles at the pontopeduncular junction. The cerebrum and diencephalon were divided into anteroposterior pieces with a single coronal cut at the rostral end of the ectomarginal sulcus, caudal to the ansate (cruciate) sulcus. Next, the anterior and posterior blocks were hemisected by a single sagittal cut through the midline. The posterior block (posterior to section 0640 in the Michigan State University online sheep brain atlas: https://msu.edu/∼brains/brains/sheep/index.html) of one hemisphere containing the entire hippocampus (Simpson and King, 1911; Vanderwolf and Cooley, 1990) was immersed in cold, aerated, artificial cerebrospinal fluid (aCSF) and used for the CA1 electrophysiological and neurobiotin morphometric studies, whereas the opposite hemisphere was immersed in fresh Golgi-Cox stain and used for Golgi-filled CA1 morphometric studies. Tissues destined for cell death or other immunohistochemical analyses were initially immersed in 4% PFA in 0.1 m phosphate buffer (pH 7.4, 4°C) for 3 d, then stored in PBS with 0.05% sodium azide at 4°C until use.
Cavalieri volumetric analysis.
To prepare the hippocampal and dentate gyri for volumetric analysis, the hippocampal formation was initially dissected from the surrounding cortex and diencephalon of PFA-fixed hemispheres. By accessing the hippocampus medially through the lateral ventricle of the hemisphere, the structure could be rolled toward the midline and dissected free from the cortex by a cut through the parahippocampal gyrus. The entire hippocampal formation was then imbedded in 5% agarose in ddH2O. Next, the hippocampus was transected into dorsal and ventral halves and cut into 200 μm slices tangential to the longitudinal axis of the dentate and hippocampal gyri, which produced the “jelly-roll” configuration necessary for the identification of hippocampal neuronal subfields (see Fig. 1A). All slices were incubated for 2 weeks in mouse anti-NeuN antibody (1:500, Millipore, catalog #MAB377) to ensure antibody penetration of the thick slices, and were visualized using biotinylated goat anti-mouse IgG (1:200, Jackson ImmunoResearch Laboratories, catalog # 115-065-071, RRID:AB_2338564) followed by dichlorotriazinylamino fluorescein conjugated streptavidin (1:200, Jackson ImmunoResearch Laboratories, catalog #016-010-084, RRID:AB_2337236). All slices were mounted on subbed slides and imaged in anatomical order using a Carl Zeiss AxioImager M2 upright microscope controlled by the StereoInvestigator software (RRID:SCR_002526, MBF Bioscience) with a 5×/NA = 0.16 Plan NeoFluar objective. Half of the sections sampled at 400 μm intervals were chosen for Cavalieri probe analysis. If a chosen slice could not be analyzed, the adjacent slice was used. For all image slices analyzed, a contour was precisely drawn enclosing both the hippocampal and dentate gyri, but excluding the subiculum at a line drawn tangential to the end of the CA1 neuronal subfield between the hippocampal fissure and the ependymal surface of the hippocampal gyrus, and excluding the alveus where it coalesces to form the fimbria fibers (see Fig. 1A). Using a 100 μm point grid, the number of points falling within the boundaries of the drawn contour was used to calculate the image slice area, which was then used with the slice thickness and section interval to calculate the hippocampal volume (http://www.stereology.info/cavalieri-estimator/). All estimated volume calculations were corrected for overprojection due to the thickness of the slices, and this corrected value was used in the statistical analyses of the results. For all evaluated structures, the Gundersen coefficient of error (shape factor m = 0) was <0.018 (n = 18, range: 0.010–0.018, mean ± SD: 0.0139 ± 0.00245), and the Gundersen coefficient of error (shape factor m = 1) was <0.004 (n = 18, range: 0.002–0.004, mean ± SD: 0.00311 ± 0.00058).
Figure 1.

Volumetric changes were observed in the hippocampal and dentate gyri in response to HI calculated by the Cavalieri estimation. A, NeuN staining of a transverse slice through the hippocampal formation reveals the neuronal subfields of the fetal sheep hippocampal and dentate gyri in the stereotypical “jelly-roll” configuration. Areal calculations of the CA1-CA4 subfields and the dentate gyrus of each slice excluded the subiculum and the entorhinal cortex at a border drawn tangential to the end of the CA1 neuronal subfield (red solid line) between the ependymal surface of the hippocampal gyrus and the hippocampal fissure (yellow dotted line), and the alveus was also excluded where it coalesces to form the fimbria fibers (orange dotted line). B, After 4 weeks, the hippocampal volume of fetuses exposed to Hx or HI was reduced versus controls (TC). No significant differences were observed between Hx and HI treatments. *p < 0.05 (Tukey's post hoc multiple-comparison test).
Cell death immunohistochemistry.
All sections were immersion fixed for 3 d in 4% PFA and cut into 50-μm-thick slices using a vibrating microtome (VT1000S, Leica Microsystems). No antigen retrieval was performed to protect the fragile 0.65 gestation tissue. The mouse monoclonal O4 antibody used to visualize preoligodendrocytes was a gift from Dr. Steven E. Pfeiffer (University of Connecticut Medical School, Farmington, CT), and its purification and visualization have been described previously (Bansal et al., 1989; Back et al., 2001). Sections were counterstained with the nuclear marker Hoechst 33342 (1:1000, Invitrogen, catalog #H3570) and mounted on slides with VectaShield Antifade (Vector Laboratories, catalog #H-1000, RRID:AB_2336789).
Brains harvested both at 4 and 7 d after Hx and HI treatment protocol were examined using the TACS-XL DAB apoptosis detection kit or TUNEL assay (Trevigen, catalog #4828–30-DK) according to the manufacturer's packaged instructions to look for any evidence of delayed cell death. This methodology included tissue treated with TACS-nuclease as a positive control for DNA fragmentation.
Golgi-Cox staining.
We initially used the FD Rapid Golgi Stain Kit (FD Neurotechnologies) as previously described (Dean et al., 2013; McClendon et al., 2014), but the amount of background precipitation and capillary filling hampered detection of neurons, which met our tracing criteria. We then switched to a modified Golgi-Cox stain (https://cornell.flintbox.com/public/project/22082) produced by Drs. Francis Lee and Deqiang Jing of Cornell University (New York) that yielded a cleaner tissue preparation in a shorter time period. No differences were observed in any morphometric features of normal CA1 neurons stained with the two methods (e.g., basal complexity mean ± SE; 34,739 ± 6468, n = 71 neurons FD method vs 33,307 ± 4753, n = 100 neurons Cornell method; p = 0.4587, Mann–Whitney test). Consequently, we were able to pool our two populations.
All Golgi-stained brain blocks containing the hippocampal formation were cut into 200 μm coronal slices using a vibrating microtome. All tissue slices containing the hippocampus were mounted on subbed slides, processed for Golgi visualization, dehydrated in a graded alcohol series, and coverslipped.
Neurobiotin staining.
To visualize the morphology of a subset of CA1 neurons filled with 0.125% neurobiotin during the electrophysiological recording sessions, 300-μm-thick hippocampal slices were transferred into fixative with 2% PFA and 15% picric acid (Sigma-Aldrich, catalog #239801) in a 0.1 m phosphate buffer, pH 7.4, overnight. Sections were washed in PBS for ∼1 h until the solution was cleared of yellow picric acid fixative. Sections were incubated overnight in 0.4% PBS-Tris with FITC-conjugated streptavidin (1:400, Jackson ImmunoResearch Laboratories, catalog #016-010-084, RRID:AB_2337236), washed with PBS 3 times for 10 min, counterstained with Hoechst 33342, and mounted on slides with VectaShield.
Hippocampal CA1 neuronal reconstruction and analyses.
The soma and complete basal dendritic arbor of Golgi-stained CA1 hippocampal neurons were reconstructed with Neurolucida software (MBF Bioscience, RRID:SCR_001775) connected to a DMREII inverted microscope (Leica Microsystems). Hippocampal neurons were traced under a 63× air objective (NA = 0.70, Leica Microsystems) if they met the following criteria: (1) location within the CA1 hippocampal layer, (2) adequate filling of the cell body and basal dendritic processes with Golgi stain, (3) presence of the entire basal dendritic arbor without significant process truncation, and (4) no obstruction of the neuron by other Golgi-filled structures. The outline of the neuronal soma was traced at its widest point in the 2D plane to provide an estimate of cross-sectional area, and the dendritic arbor was traced in the x-, y-, and z-coordinate planes. The basal dendritic arbor was not followed into adjacent slices, and the dendritic diameter was not studied. The Golgi stain tended to produce sparse and incomplete filling of the apical dendrites; consequently, they were not analyzed. Using these selection criteria, a total of 602 Golgi-filled CA1 neurons (n = 171 TC neurons, 6 fetuses, 5 female, 1 male; n = 211 Hx neurons, 10 fetuses, 5 female, 5 male; n = 220 HI neurons, 9 fetuses, 3 female, 6 male) were examined.
Using similar criteria as for the Golgi-filled population, the complete dendritic arbor (both basal and apical dendrites) and soma of the neurobiotin-filled CA1 neurons were traced. To minimize quenching of the fluorescent FITC label, the neurons were imaged on an AxioImager M2 upright microscope (Carl Zeiss) controlled by the Neurolucida software (MBF Bioscience). Initially, an overview image of the entire hippocampal slice containing the labeled cells was acquired with a 2.5×/NA = 0.085 Plan apochromatic objective. Using a 20×/NA = 0.8 Plan apochromatic objective, a perimeter contour was draw around the labeled neuron to limit the x-y plane of the image while ensuring that the entire neuron was included in the acquisition. Finally, the image stacks were acquired at 1 μm z steps using a 40×/NA = 0.95 Plan apochromatic objective. Image stacks were saved as TIFF files (MBF Bioscience) and later traced using either the Neurolucida or NL360 software (MBF Bioscience) by an observer blinded to animal and treatment group. A total of 81 neurobiotin-filled CA1 neurons (n = 25 TC neurons, 10 fetuses, 3, female, 7 male; n = 34 Hx neurons, 14 fetuses, 7 female, 7 male; n = 22 HI neurons, 12 fetuses, 8 female, 4 male) were examined.
As previously described (Dean et al., 2013), the Neuroexplorer software (MBF Bioscience) was used to generate a morphometric analysis of total dendritic length (summed lengths of all basal or apical dendritic branches per cell) and dendritic complexity. Included in these analyses were the total number of arbor branches, branch points (nodes), and terminal tips (endings). Branch order analysis for all these parameters was also performed according to a centrifugal nomenclature (Uylings et al., 1975, 1986), where dendritic branches arising from the soma are considered first-order (primary) segments until they branch into second-order segments, which branch into third-order segments, etc. To further assess dendritic complexity, we performed Sholl analysis on all reconstructed neurons to calculate the number of intersections of dendrites per each Sholl ring (5 μm interval concentric spheres centered on the soma). To estimate arbor complexity, a value was computed by Neurolucida software by the following equation: [Sum of the terminal orders + Number of terminals] × [Total dendritic length/Number of primary dendrites] (Pillai et al., 2012). Basal and apical dendritic analyses were evaluated separately.
Dendritic spine analysis.
Using the Neurolucida and Neuroexplorer software, the density of the basal dendritic spines was quantified on the same population of Golgi-filled CA1 hippocampal neurons sampled for dendritic morphology. With a 63× Plan Apochromat oil objective (NA = 1.4; Carl Zeiss), spine density was determined by counting all visible spines on all terminal third-order branches of basal dendrites that were at least 25 μm long. Approximately 6% TC, 10% Hx, and 14% HI CA1 neurons did not have dendrites that met these criteria because they were either more complex than or not as complex as the rest of the population. Other neuronal exclusion criteria included basal dendrites that were incompletely filled or that were obscured by other processes. No distinction was made between subtypes of spines. Spine densities were assessed on a total of 354 basal dendrites on 162 CA1 neurons in the control group, 343 basal dendrites on 190 CA1 neurons in the Hx group, and 341 basal dendrites on 189 CA1 neurons in the HI group. Spine data were calculated on a per-dendrite basis by averaging the spine densities from all assessed dendrites. Data are presented as the number of spines per micrometer.
Fetal ovine hippocampal slice preparation.
After rapid removal of the brain, one hemisphere of the posterior cerebral block was rapidly isolated and immersed in ice-cold (0°C–2°C), low-sodium, aCSF composed of the following (in mm): 234 sucrose, 3.2 KCl, 17 NaH2PO4, 6 MgSO4, 10 d-glucose, 27.7 NaHCO3, and 0.216 CaCl2, and transported to the electrophysiology laboratory (total transport time 5 min). The hippocampus was microdissected from the surrounding cortex and mounted in a slicing chamber filled with ice-cold (0°C-2°C), low sodium aCSF; 300 μm slices from the middle of the hippocampus were cut using a vibrating microtome (VT1200S; Leica Microsystems). Slices were held in carbogen-saturated aCSF of the following composition (in mm): 119 NaCl, 26 NaHCO3, 2.5 KCl, 1 NaH2PO4, 1 MgCl2, 2 CaCl2, and 25 dextrose (saturated with a carbogen mixture of 95% O2/5% CO2) at 35°C for 30 min, and then at room temperature (22°C-24°C) for at least 1 h before recording.
Electrophysiology.
Experiments were performed at 32°C. Hippocampal slices and individual CA1 pyramidal neurons were visualized using a fixed-stage, upright microscope (Axioskop; Carl Zeiss) equipped with infrared differential interference contrast optics. The recording chamber was continuously superfused with carbogen-saturated aCSF flowing at a rate of 2–4 ml/min. Recording electrodes were pulled from filamented borosilicate pipettes (BF150-86-10, Sutter Instruments) and had tip resistances of 2–3 mΩ when filled with the following internal solution for whole-cell patch-clamp recordings (mm): 140 KMeSO4, 10 KCl, 10 HEPES, 2 MgATP, 0.4 Na2GTP, and 10 Tris-phosphocreatine, pH adjusted to 7.25 with KOH, with a final osmolarity of 290 mOsm. The internal solution also contained 0.125% neurobiotin in 50% of the recordings for histology. Results were not corrected for a liquid junction potential of ∼15 mV between the internal pipette and bath aCSF solutions.
Whole-cell patch-clamp recordings were obtained using a Multiclamp 700B amplifier (Molecular Devices). Signals were filtered at 5 kHz, digitized at 20 kHz, and transferred to a computer using an ITC-18 (Instrutech) and Patchmaster (Heka Instruments). For current-clamp recordings of IE, membrane potentials were maintained at −65 mV with bias current injection. Series resistance (10.1 ± 0.2 mΩ, n = 95) was electronically compensated in bridge mode. Passive membrane properties, apparent resting membrane potential (RMPapp), input resistance (Rinput), and capacitance (Cap) were monitored with a hyperpolarizing somatic injection of 40 pA. Apparent RMP was determined from reversal of the test current membrane voltage relative to the bias holding current and resting potential. Cap was determined from the time constant (tau = Cap × Rinput) of hyperpolarization determined by fitting a single exponential to the step. For voltage-clamp experiments, the membrane holding potential was −50 mV. Series resistance and whole-cell capacitance were electronically compensated. Rinput was continuously measured with a 5 mV hyperpolarizing test pulse.
All experiments were performed in the presence of SR95531 (5 μm) and CGP55845 (2.5 μm) to block inhibitory synaptic transmission.
fEPSP recordings were obtained using a GeneClamp amplifier (Molecular Devices). Signals were filtered at 5 kHz, digitized at 20 kHz, and transferred to a computer using an ITC-18 (Instrutech) and Patchmaster (Heka Instruments). Hippocampal slices were visualized using a fixed-stage, upright microscope (Axioskop, Carl Zeiss) with a 5× objective. The recording chamber was continuously perfused at 32°C with oxygenated aCSF flowing at a rate of 1–2 ml/min. Recording electrodes were pulled from borosilicate pipettes (Sutter Instruments) and had tip resistances of 1–2 mΩ when filled with aCSF for extracellular field recordings. Glass stimulating electrodes of approximate resistance of 1 mΩ, were filled with aCSF, connected to a Digitimer constant current stimulus isolation unit (AutoMate Scientific). The stimulating and recording electrodes were placed in the CA1 stratum radiatum approximately equal distance 100–200 μm from stratum pyramidal separated by 100–300 μm. Stimulus duration was 0.1 ms allowing for clear separation of fiber volley (FV) from the preceding stimulus artifact. Field responses were elicited at 0.1 Hz. All experiments were performed in the presence of SR95531 (5 μm) and CGP55845 (2.5 μm) to block inhibitory synaptic transmission. The CA3 axons were severed to eliminate recurrent excitation within the CA3 subfield.
LTP at CA3-CA1 excitatory synapses was induced by stimulating CA3 axons with 3 sets of 100 stimuli at 50 Hz delivered at 0.08 Hz. The fEPSP slope was analyzed for individual slices and normalized to baseline before LTP induction. Diary plots of LTP were obtained by binning all slices for each treatment at 1 min intervals. Data are displayed as mean ± SEM.
Statistical analyses.
All morphometric data analyses were performed with either SAS/STAT (SAS Institute, RRID:SCR_008567) or Prism 6 statistical software (GraphPad Software, RRID:SCR_002798). Data are expressed as mean ± SEM. ANOVA followed by Tukey's post hoc correction for multiple pairs was used for volumetric comparisons. Comparisons of group means for dendritic morphology among age-matched TC, hypoxic control (Hx), and HI animals were performed with the Kruskal–Wallis (one-way ANOVA) test followed by the Dunn post hoc test for multiple pairs. Sholl (radius × treatment group) and branch order (branch order × treatment group) analyses of dendritic morphology parameters between age-matched TC, Hx, and HI animals were performed by two-way ANOVA, followed by Bonferroni post hoc tests. ABG parameters among age-matched TC, Hx, and HI animals were performed with the SAS/STAT GLIMMIX mixed-effects model, which tested and controlled for any effects of ewe, fetus, or gender, followed by the Bonferroni post hoc test for multiple treatment pairs. No effects of ewe, fetus, or gender were observed unless otherwise noted.
Whole-cell and fEPSP traces were analyzed using custom macros written in Igor Pro (WaveMetrics). A two-way ANOVA GLM using SPSS version 25 (IBM) was initially used to determine the effects of sex versus treatment. When no effects of sex were present, sexes were pooled to increase statistical power, and a nonparametric ANOVA (Kruskal–Wallis) was used to examine the effects of multiple treatments followed by the Dunn post hoc correction for multiple pairs. A value of p < 0.05 was considered statistically significant.
Results
Hippocampal volume is reduced in response to fetal Hx and HI
Consistent with prior human volumetric MRI studies, exposure of the developing brain to HI at ∼0.65 gestation produced a significant reduction in the volume of the hippocampal and dentate gyri when measured 4 weeks later at ∼0.85 gestation as estimated by the Cavalieri method (Fig. 1B; TC 2.492 × 1011 ± 3.633 × 1010 μm3 vs Hx 1.865 × 1011 ± 3.951 × 1010, mean ± SD, Tukey's multiple-comparison p < 0.05; TC 2.492 × 1011 ± 3.633 × 1010 vs HI 1.911 × 1011 ± 2.710 × 1010, Tukey p < 0.05, n = 7 TC, n = 5 Hx, n = 6 HI fetuses). Surprisingly, no difference in volume loss was observed between the fetal groups that had been exposed to Hx versus HI. To determine whether the volume loss was related to early cell death, we first screened for degenerating Hoechst 33342-labeled nuclei at 24 h after HI or Hx. Although no degenerating nuclei were detected in the CA1 subfield in response to HI or Hx, scattered degenerating nuclei were detected in the adjacent white matter of the HI group. These nuclei coincided with O4 antibody-labeled cells derived from the oligodendrocytes lineage (Fig. 2A–F). Staining for NeuN supported a lack of degeneration of CA1 neurons in response to HI (Fig. 2G,H) or Hx (data not shown). TUNEL staining at 4 and 7 d after HI also showed no evidence of neuronal degeneration in the hippocampus or adjacent regions (data not shown).
Figure 2.
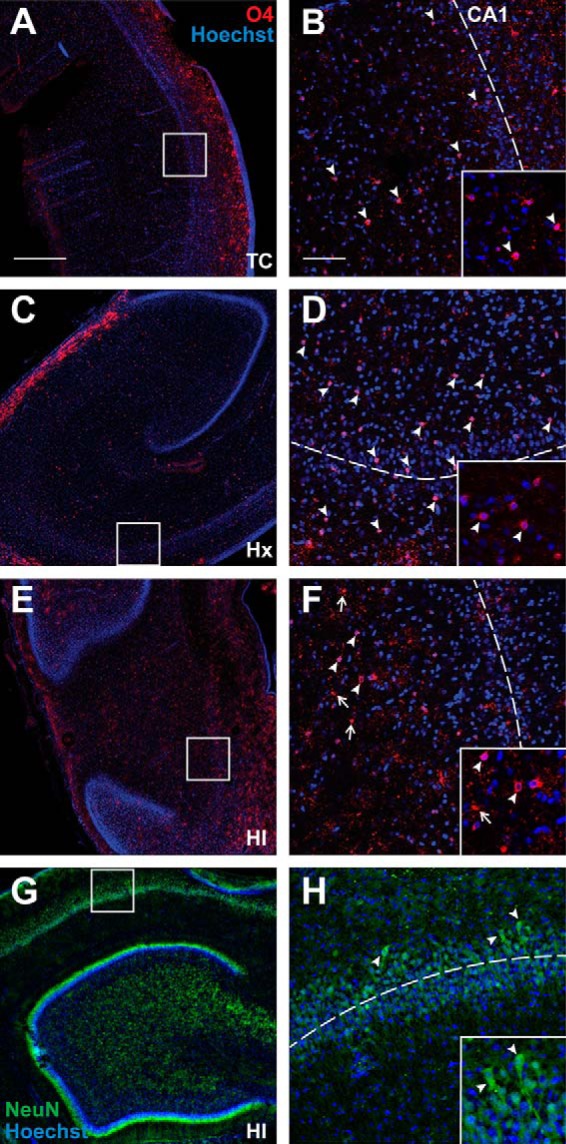
Hippocampal neurons and oligodendrocyte lineage cells are largely intact 24 h after exposure to HI or Hx at 94 d (∼0.65) gestation. A–F, Low (A,C,E) and high (B,D,F) power images of the CA1 subfield (visualized with Hoechst 33342; blue) and oligodendrocyte lineage cells stained with O4 antibody (red) in the adjacent white matter. A, C, E, Boxes represent the regions of higher-power detail in B, D, F where the dotted lines indicate the approximate location through the middle of CA1. White arrowheads indicate that O4+ cells were typically intact in the control (TC) and Hx groups (see insets), but scattered degenerating O4+ cells were visualized in response to HI (F, white arrows; inset). G, H, Low (G) and high (H; see box in G) power images of NeuN staining in the CA1 (green) with Hoechst counterstain (blue). H, Inset, The typical morphology of intact-appearing CA1 pyramidal neurons. Scale bars: A, C, E, 500 μm; B, D, F, 100 μm.
CA1 basal dendritic maturation is augmented in response to fetal Hx
Although CA1 pyramidal neurons appeared to be resistant to degeneration in response to Hx or HI, we next determined whether CA1 neurons displayed disturbances in maturation of the dendritic arbor as visualized by Golgi stain. The basal dendrites of a total of 602 CA1 neurons (n = 171 TC, 211 Hx, and 220 HI cells; n = 6 TC, 10 Hx, and 9 HI fetal brains) were traced and quantified by Neurolucida after a 4 week survival period following experimental treatment (∼0.85 gestation). For multiple measures of basal dendritic complexity, CA1 neurons from fetuses exposed only to maternal Hx generally displayed greater complexity than either the TC or HI groups (Fig. 3). This trend was evident across most global measures of complexity (Fig. 3B,E,G,H,J,K), as well as individual dendritic branches when complexity metrics were explored by branch order (Fig. 3C,F,I,L). As a population, the mean area of the cell soma of Hx CA1 neurons was greater relative to normal controls (Fig. 3A: TC 595.1 ± 13.91 vs Hx 650.7 ± 13.04, mean ± SEM, Dunn's multiple-comparison p < 0.01; TC 595.1 ± 13.91 vs HI 623.2 ± 10.54, not significant; Hx 650.7 ± 13.04 vs HI 623.2 ± 10.54, not significant). No differences among the experimental groups were observed in the number of primary (first-order) dendrites emerging from the cell soma (Fig. 3D: TC 4.947 ± 0.1177 vs Hx 4.735 ± 0.1123 vs HI 4.855 ± 0.1139, Kruskal–Wallis test p = 0.3829). However, for every other global measure of neuronal complexity, the total population of Hx CA1 neurons displayed increased dendritic complexity relative to both the TC and HI neurons: total number of dendritic branches (Fig. 3B: TC 38.41 ± 1.143 vs Hx 45.67 ± 1.277, Dunn p < 0.001; Hx 45.67 ± 1.277 vs HI 40.41 ± 1.268, Dunn p < 0.01), total dendritic length (Fig. 3E: TC 1549 ± 61.93 vs Hx 2116 ± 73.35, Dunn p < 0.0001; Hx 2116 ± 73.35 vs HI 1757 ± 62.68, Dunn p < 0.001), total number of dendritic ends (Fig. 3G: TC 22.43 ± 0.6036 vs Hx 25.86 ± 0.6835, Dunn p < 0.01; Hx 25.86 ± 0.6835 vs HI 23.29 ± 0.6762, Dunn p < 0.05), mean dendritic length (Fig. 3H: TC 340.6 ± 16.81 vs Hx 485.9 ± 18.37, Dunn p < 0.0001; Hx 485.9 ± 18.37 vs HI 397.3 ± 16.42, Dunn p < 0.001), dendritic complexity (Fig. 3J: TC 33,901 ± 3853 vs Hx 57,824 ± 4177, Dunn p < 0.0001; Hx 57,824 ± 4177 vs HI 41,875 ± 3455, Dunn p < 0.001), and total number of dendritic nodes (Fig. 3K: TC 15.98 ± 0.5476 vs Hx 19.81 ± 0.5996, Dunn p < 0.0001; Hx 19.81 ± 0.5996 vs HI 17.13 ± 0.5977, Dunn p < 0.001). No differences in global complexity metrics were observed between the total TC and HI populations, except for mean dendritic length (Fig. 3H: TC 340.6 ± 16.81 vs HI 397.3 ± 16.42, Dunn p < 0.05) where complexity was again greater in the HI group versus controls.
Figure 3.

Increased basal dendritic complexity found in the total population of CA1 neurons after exposure to Hx. Paired comparisons are made to normal controls (TC) unless designated by brackets. A, Mean cell body area, (B) total number of basal dendritic branches, (C) number/quantity of basal dendritic branches by branch order, (D) number of primary (first-order) basal dendrites, (E) total basal dendritic length, (F) total basal dendritic length by branch order, (G) basal dendritic ends (terminals), (H) mean basal dendritic length, (I) mean basal dendritic length by branch order, (J) computed complexity index for basal dendrites, (K) total number of basal dendritic nodes (branch points), (L) basal dendritic nodes (branch points) by branch order, (M) Golgi-stained control (TC) CA1 neuron, (N) Golgi-stained CA1 neuron exposed to maternal hypoxia only (Hx), and (O) Sholl analysis (number of basal dendritic intersections of Sholl sphere). Black bars/symbols represent controls (TC). Slate blue bars/symbols represent Hx. Mauve/magenta bars/symbols represent HI. Error bars indicate SE. p values were calculated using Dunn's post hoc test for multiple comparisons after Kruskal–Wallis one-way ANOVA for global metrics. Branch order and Sholl analyses: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; Bonferroni's post hoc test for multiple comparisons. Scale bars: M, N, 50 μm.
Branch order analyses revealed that the increased complexity of the Hx-treated CA1 neurons was prevalent primarily in branch orders 3–6 (Fig. 3C,F,I,L). Consistent with the global metric findings, no differences in branch orders were generally observed between the TC and HI groups, except within the fourth and seventh branch orders of the mean dendritic length metric (Fig. 3I).
Although the global complexity metrics and branch order analyses revealed little effect of HI, the Sholl analysis indicated that the dendritic response to HI was similar to Hx. Both Hx- and HI-treated CA1 neurons display increased Sholl intersections throughout the middle range of radii relative to TC (Fig. 3O: TC vs Hx radii 70–190, Bonferroni p < 0.0001; TC vs HI radii 85–140, p < 0.001). Consistent with global complexity metrics and branch order analyses, Hx CA1 neurons display increased Sholl intersections throughout the middle range of radii relative to HI (Fig. 3O: Hx vs HI radii 85–180, p < 0.0001).
CA1 neuronal complexity is directly related to fetal systemic oxygenation
Since we often paradoxically observed that the HI group was less significantly affected than the Hx group, we determined whether systemic oxygen status affected hippocampal dendricity. We instrumented 25 sets of twins to collect ABG values on both Hx (8 females/17 males) and HI (11 females/14 males) fetuses. Blood samples were collected (Fig. 4A) at the start of the experiment (pre-ABG), 5 min after the onset of maternal hypoxia (Hx-ABG), 25 min later at the end of Hx or HI (HI-ABG), and 10 min after the end of Hx or HI (post-ABG). The mean oxygen content, lactate, and glucose values of the 25 twin pairs (Fig. 4B—D: blue represents Hx line; red represents HI lines) were compared with the mean values of 8 of the 10 HI fetuses used in this study to analyze CA1 hippocampal anatomy (Fig. 4B–D; solid black represents HI-HC lines; data on two fetuses were not available due to failure of the sampling catheter). Figure 4B shows that all fetal groups had the same mean baseline (pre-ABG) values for oxygen content. Five minutes into maternal hypoxia (Hx-ABG), all fetal groups responded with a similar drop in oxygen content (vol %, Table 1: Hx-ABG Hx vs HI, Bonferroni's multiple comparisons, not significant; Hx vs HI-HC, not significant; HI vs HI-HC, not significant). Twenty-five minutes later (HI-ABG), the two fetal HI groups mounted a compensatory response that elevated their oxygen content back to baseline levels, whereas the Hx group continued to display reduced oxygen content until maternal Hx was terminated.
Table 1.
The ABG values of 25 twin pairs (one Hx and one HI fetus/pair) and 7 normal control fetuses (TC) used to study the hippocampal electrophysiology are compared with the ABG values of the 8 HI fetuses used to study hippocampal anatomy in this paper (HI-HC)
| Treatment |
p (Bonferroni's multiple-comparisons test) |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| ABG parameter | ABG time | TC (n = 7) | Hx (n = 24) | HI (n = 21) | HI-HC (n = 8) | TC versus Hx | Hx versus HI | Hx versus HI-HC | HI versus HI-HC |
| O2 content (vol %) | Pre-ABG | 7.1 ± 1.2 | 7.0 ± 1.1 | 7.0 ± 1.0 | 8.6 ± 1.2 | NS | NS | <0.05 | <0.05 |
| Hx-ABG | 7.3 ± 1.2 | 4.8 ± 1.7 | 4.4 ± 1.4 | 5.6 ± 1.8 | <0.001 | NS | NS | NS | |
| HI-ABG | 7.4 ± 1.2 | 4.0 ± 1.7 | 5.4 ± 2.0 | 7.1 ± 1.9 | <0.0001 | <0.01 | <0.0001 | <0.05 | |
| Post-ABG | 7.2 ± 1.8 | 7.1 ± 1.0 | 7.5 ± 0.9 | 8.8 ± 1.5 | NS | NS | <0.05 | NS | |
| Lactate (mm) | Pre-ABG | 1.4 ± 0.2 | 1.5 ± 0.3 | 1.5 ± 0.2 | 1.3 ± 0.3 | NS | NS | NS | NS |
| Hx-ABG | 1.4 ± 0.2 | 1.5 ± 0.4 | 1.6 ± 0.2 | 1.4 ± 0.2 | NS | NS | NS | NS | |
| HI-ABG | 1.3 ± 0.2 | 2.3 ± 1.2 | 3.2 ± 0.9 | 3.3 ± 0.8 | <0.05 | <0.001 | <0.01 | NS | |
| Post-ABG | 1.4 ± 0.2 | 2.3 ± 1.1 | 3.8 ± 1.1 | 4.1 ± 0.9 | <0.05 | <0.0001 | <0.0001 | NS | |
| Glucose (mm) | Pre-ABG | 1.4 ± 0.1 | 1.3 ± 0.1 | 1.2 ± 0.1 | 1.4 ± 0.1 | NS | NS | NS | NS |
| Hx-ABG | 1.4 ± 0.1 | 1.3 ± 0.1 | 1.3 ± 0.1 | 1.5 ± 0.2 | NS | NS | NS | NS | |
| HI-ABG | 1.4 ± 0.2 | 1.3 ± 0.1 | 1.6 ± 0.1 | 1.8 ± 0.2 | NS | <0.05 | <0.001 | NS | |
| Post-ABG | 1.5 ± 0.2 | 1.5 ± 0.1 | 1.5 ± 0.1 | 1.8 ± 0.2 | NS | NS | NS | NS | |
| Hct (%) | Pre-ABG | 29 ± 2.2 | 27 ± 2.7 | 27 ± 2.3 | 28 ± 3.6 | NS | NS | NS | NS |
| Hx-ABG | 29 ± 2.2 | 27 ± 2.9 | 27 ± 2.5 | 28 ± 3.9 | NS | NS | NS | NS | |
| HI-ABG | 28 ± 3.0 | 28 ± 3.1 | 29 ± 2.9 | 31 ± 3.9 | NS | NS | NS | NS | |
| Post-ABG | 29 ± 2.0 | 27 ± 2.8 | 27 ± 3.3 | 29 ± 4.0 | NS | NS | NS | NS | |
| Hb (g/dl) | Pre-ABG | 9.4 ± 0.7 | 8.7 ± 0.9 | 8.8 ± 0.8 | 9.2 ± 1.2 | NS | NS | NS | NS |
| Hx-ABG | 9.4 ± 0.7 | 8.7 ± 1.0 | 8.8 ± 0.8 | 9.2 ± 1.3 | NS | NS | NS | NS | |
| HI-ABG | 9.1 ± 1.0 | 8.9 ± 1.0 | 9.4 ± 0.9 | 9.9 ± 1.3 | NS | NS | <0.05 | NS | |
| Post-ABG | 9.3 ± 0.7 | 8.8 ± 0.9 | 9.0 ± 0.8 | 9.4 ± 1.3 | NS | NS | NS | NS | |
| sO2 (%) | Pre-ABG | 56 ± 8.1 | 59 ± 7.6 | 59 ± 7.4 | 70 ± 4.9 | NS | NS | NS | NS |
| Hx-ABG | 57 ± 8.5 | 40 ± 13 | 37 ± 12 | 46 ± 14 | <0.01 | NS | NS | NS | |
| HI-ABG | 61 ± 9.1 | 33 ± 14 | 42 ± 16 | 53 ± 14 | <0.0001 | <0.05 | <0.0001 | NS | |
| Post-ABG | 57 ± 12 | 60 ± 7.4 | 62 ± 8.2 | 70 ± 8.7 | NS | NS | NS | NS | |
| paO2 (mmHg) | Pre-ABG | 23 ± 2.3 | 23 ± 2.6 | 23 ± 3.0 | 25 ± 1.7 | NS | NS | NS | NS |
| Hx-ABG | 23 ± 2.5 | 18 ± 3.2 | 17 ± 2.6 | 17 ± 3.6 | <0.001 | NS | NS | NS | |
| HI-ABG | 24 ± 2.4 | 16 ± 4.4 | 19 ± 3.4 | 20 ± 4.4 | <0.0001 | NS | <0.05 | NS | |
| Post-ABG | 23 ± 3.2 | 23 ± 3.0 | 25 ± 3.2 | 26 ± 3.1 | NS | NS | NS | NS | |
| paCO2 (mmHg) | Pre-ABG | 47 ± 2.2 | 48 ± 2.3 | 48 ± 2.8 | 45 ± 2.9 | NS | NS | <0.05 | <0.05 |
| Hx-ABG | 47 ± 2.5 | 45 ± 2.4 | 46 ± 2.9 | 41 ± 2.9 | NS | NS | <0.01 | <0.01 | |
| HI-ABG | 45 ± 6.3 | 46 ± 3.1 | 44 ± 3.0 | 41 ± 5.3 | NS | NS | <0.001 | NS | |
| Post-ABG | 48 ± 2.6 | 47 ± 2.0 | 48 ± 2.8 | 44 ± 3.3 | NS | NS | NS | <0.05 | |
| pH | Pre-ABG | 7.4 ± 0.04 | 7.4 ± 0.02 | 7.4 ± 0.02 | 7.4 ± 0.02 | NS | NS | NS | NS |
| Hx-ABG | 7.4 ± 0.04 | 7.4 ± 0.02 | 7.4 ± 0.01 | 7.4 ± 0.02 | <0.01 | NS | NS | NS | |
| HI-ABG | 7.4 ± 0.05 | 7.4 ± 0.02 | 7.4 ± 0.03 | 7.4 ± 0.03 | NS | NS | <0.05 | <0.05 | |
| Post-ABG | 7.4 ± 0.05 | 7.4 ± 0.02 | 7.4 ± 0.03 | 7.3 ± 0.03 | NS | <0.01 | <0.001 | NS | |
To better understand the source of the rise in mean oxygen content, we measured hemoglobin (Hb), hematocrit (Hct), oxygen saturation (sO2 %), and partial pressure (PaO2 mmHg) values. While there were no differences in Hb or Hct levels across the groups, sO2 and PaO2 values had a corresponding rise in the HI group relative to Hx (Table 1). Hence, this response of the HI group to the severity of the insult resulted in normalization of fetal systemic Hx and appeared to be related to the generally more normal maturation of the basal dendritic arbor in the HI group relative to the Hx. To test this hypothesis, we analyzed the complexity of the basal dendritic arbors of CA1 neurons for the 8 HI animals for which blood gas values were available relative to several indices of fetal oxygenation and metabolism. We first asked whether dendritic complexity was associated with the fetal capacity to raise oxygen content during the HI insult. To test this, we calculated an oxygen content rebound ratio, defined as the ratio of the HI-ABG/Hx-ABG oxygen content values. No correlation was observed between dendritic complexity and the systemic oxygen rebound ratio (r2 = 0.1781, p = 0.2976; data not shown). Dendricity was also not associated with the absolute value of the systemic fetal oxygen content measured at 5 min into maternal hypoxia (Fig. 4E: r2 = 0.3541, p = 0.1197). However, basal dendritic complexity was positively associated with the magnitude of the change in fetal systemic oxygen content expressed as the difference between the baseline (pre-ABG) and the Hx-ABG O2 content values (Fig. 4F: r2 = 0.8038, p = 0.0026). Hence, with a greater initial fall in fetal oxygenation after the onset of Hx, dendritic complexity increased. In addition, dendritic complexity was positively associated with changes in the fetal systemic lactate, defined as the difference in post-ABG minus pre-ABG lactate values (Fig. 4G: r2 = 0.5814, p = 0.0278). Hence, greater fetal metabolic stress, as indicated by a higher fetal blood lactate level, was significantly associated with increased dendritic complexity. Similar to the lactate change, we analyzed for an association between dendritic complexity and a change in systemic glucose, defined as the difference between the HI-ABG and Hx-ABG glucose values, which trended toward significance (Fig. 4H: r2 = 0.4837, p = 0.0555).
To further analyze the influence of fetal systemic oxygen content on basal dendritic complexity, we analyzed only those CA1 neurons from both Hx and HI animals where ABG oxygen content values were available for the HI fetus. We then parsed this dataset into two groups based on the initial fetal response to maternal hypoxia, and normalized the change in oxygen content. The Hx and HI fetuses that were able to maintain at least 78% of their baseline systemic oxygen content (mean 81.5%, range 78%–87%) were designated as “high O2.” All others were designated “low O2” (mean 50.5%, range 41%–59%). Consequently, we were left with 65 Hx (low O2), 71 HI (low O2), 78 Hx (high O2), and 84 HI (high O2) CA1 neurons to compare to 171 TC.
In all measures of complexity, CA1 hippocampal neurons from fetuses in the high oxygen content group showed no anatomical differences relative to controls (Fig. 5). In contrast, CA1 hippocampal neurons from fetuses who fell below this threshold of 78%, displayed increased basal dendritic complexity values in all global and branch order metrics, except the number of primary dendrites (Fig. 5D). No significant differences were observed between Hx and HI treatments in either of the high or low O2 groups.
Figure 5.

Basal dendritic complexity in CA1 neurons after exposure to maternal hypoxia (Hx) and/or ischemia is related to the fetal systemic oxygen content. Paired comparisons are made to normal controls (TC) unless designated by brackets. A, Mean cell body area, (B) total number of basal dendritic branches, (C) number/quantity of basal dendritic branches by branch order, (D) number of primary (first order) basal dendrites, (E) total basal dendritic length, (F) total basal dendritic length by branch order, (G) basal dendritic ends (terminals), (H) mean basal dendritic length, (I) mean basal dendritic length by branch order, (J) computed complexity index for basal dendrites, (K) total number of basal dendritic nodes (branch points), (L) basal dendritic nodes (branch points) by branch order, (M) Golgi-stained control (TC) CA1 neuron, (N) Golgi-stained CA1 neuron exposed to maternal hypoxia only (Hx), and (O) Sholl analysis (number of basal dendritic intersections of Sholl sphere). Black bars/symbols represent true normal controls (TC). Slate blue and mauve/magenta bars/symbols represent fetuses that received maternal hypoxia only (hypoxic control, Hx) and hypoxia-ischemia (HI), respectively, but were able to maintain oxygen content values within 78% of baseline values (high O2). Bright blue and red bars/symbols represent Hx and HI fetuses, respectively, that fell below the 78% threshold (low O2). Error bars indicate SE. p values were calculated using Dunn's post hoc test for multiple comparisons after Kruskal–Wallis one-way ANOVA for global metrics. **p < 0.01; ***p < 0.001; ****p < 0.0001; Branch order and Sholl analysis: Bonferroni's post hoc test for multiple comparisons. Scale bars: M, N, 50 μm.
Increased CA1 basal dendritic complexity is accompanied by no change in basal dendritic spine density
We next analyzed whether the increase in basal dendritic complexity of CA1 neurons was accompanied by disturbances in spine density. Because CA1 basal dendritic arbors were generally complex, we chose to sample spine density on terminal tertiary branches (Fig. 6A–D). Neurons exposed to both Hx and HI revealed no changes in spines/μm relative to controls (Fig. 6D: mean ± SEM, TC 0.2601 ± 0.006 vs Hx 0.2581 ± 0.006, not significant; Dunn's multiple-comparison, TC 0.2601 ± 0.006 vs HI 0.2896 ± 0.008, not significant, Hx 0.2581 ± 0.006 vs HI 0.2896 ± 0.008, p < 0.05).
Figure 6.

Augmented basal dendritic arbor maturation is accompanied by no changes in basal dendritic spine density. For the same population of CA1 neurons that were sampled for dendritic morphology, spine density was quantified on third-order terminal dendritic branches of all experimental groups of neurons. A, B, Example of third-order terminal branches (yellow arrows) that were present on the Golgi-impregnated neuron (A) and the corresponding Neurolucida tracing (B, purple/hot pink lines). B, Branch order rank is indicated by color: bright yellow represents first order; white represents second order; purple/hot pink represents third order; bright green represents fourth order; cyan blue represents fifth order; orange represents sixth order; slate gray represents seventh order. C, Dendritic spines visualized on a Golgi-impregnated CA1 hippocampal pyramidal neuron. D, Spine density in control, hypoxia-only (Hx), and HI groups at 4 weeks after treatment. Preterm neurons in both the Hx and HI groups (blue and red bars) revealed no significant change in the number of spines versus controls (black bars). However, Hx- versus HI-treated groups were significant at p < 0.05 (Dunn's correction for multiple pairs, black horizontal bar with asterisk). n = 354 TC, 343 Hx, and 341 HI tertiary basal dendrites. *p < 0.05.
CA1 apical dendritic maturation is reduced in response to fetal HI
We next analyzed the maturation of the CA1 apical dendrites from which electrophysiological recordings were obtained. Since Golgi stain often resulted in truncation and incomplete filling of apical dendrites, we analyzed a total of 80 CA1 pyramidal neurons (n = 24 TC, n = 34 Hx, n = 22 HI) that were filled with neurobiotin (Fig. 7). Unlike the basal dendrites, the apical dendritic arbors were reduced in response to HI (Fig. 8). This response was found in all global measures of complexity: nodes (Fig. 8A: mean ± SEM, TC 37.58 ± 1.89 vs HI 30.64 ± 2.41, Dunn p < 0.05), ends (Fig. 8B: mean ± SEM, TC 39.54 ± 1.92 vs HI 32.64 ± 2.53, Dunn p < 0.05), total length (Fig. 8C: mean ± SEM, TC 8193 ± 452.2 vs HI 6310 ± 536.9, Dunn p < 0.01), total number of dendritic branches (Fig. 8D: mean ± SEM, TC 77.13 ± 3.80 vs HI 63.27 ± 4.93, Dunn p < 0.05), and complexity (Fig. 8E: mean ± SEM, TC 2.73 × 106 ± 322,594 vs HI 1.70 × 106 ± 299,545, Dunn p < 0.05), as well as the Sholl analysis (TC vs HI Bonferroni p < 0.001 from intersections 685–775). While the Hx-treated neurons showed a similar trend toward a reduction in complexity with respect to TC neurons, this trend only reached statistical significance for total length (Fig. 8C: mean ± SEM, TC 8193 ± 452.2 vs Hx 6515 ± 338.5, Dunn p < 0.05) and at Sholl intersection 275 (Bonferroni p < 0.05).
Figure 7.

A–C, Top row, Maximum projection images of z stacks of representative neurobiotin-filled CA1 pyramidal neurons from TC (A), Hx (B), and HI (C) brains. Second row, The 3D reconstructions of the soma and full basal and apical dendritic arbors of the representative neurons in the top row were obtained by tracing in Neurolucida z stack images collected at 1 μm steps. Branch order rank is indicated by color: bright yellow represents first order; white represents second order; purple/hot pink represents third order; bright green represents fourth order; cyan blue represents fifth order; orange represents sixth order; slate gray represents seventh order; salmon pink represents eighth order; forest green represents ninth order; bright blue represents 10th order; olive green represents 11th order; purple represents 12th order; red represents 13th order; plum represents 14th order. Third row, Neurolucida tracings of only the apical dendritic arbors and soma of the representative TC, Hx, and HI neurons. Fourth row, Neurolucida tracings of only the basal dendritic arbors and soma of the representative TC, Hx, and HI neurons.
Figure 8.

A trend toward decreased apical dendritic complexity was seen in neurobiotin-filled CA1 pyramidal neurons after exposure to maternal Hx but only reached significance in global measures within the HI-treated group. Dendritic differences were apparent when analyzed by Sholl intersections. A, Total number of apical dendritic nodes (branch points). B, Total number of apical dendritic endings (terminals). C, Total length of the apical dendritic arbor. D, The total number of apical branches. E, Computed complexity index for apical dendrites. F, Sholl analysis, number of apical dendritic intersections of Sholl sphere. Black bars/symbols represent true normal controls (TC). Blue bars/symbols represent maternal Hx only (hypoxic control, Hx). Red bars/symbols represent HI. Error bars indicate SEM. p values were calculated using Dunn's post hoc test for multiple comparisons after one-way ANOVA for global metrics. F, For the Sholl analyses, p values were calculated using Bonferroni's post hoc test for multiple comparisons following two-way ANOVA. *p < 0.05. **p < 0.01.
Synaptic transmission following Hx and HI
Neuronal communication in hippocampal CA1 neurons depends on the efficiency of synaptic transmission at Schaffer collateral CA3-CA1 synapses to initiate output APs at CA1 somata. Synaptic strength can be enhanced by stimulation protocols that induce LTP. To determine whether the reduction in apical dendritic maturation affected communication in the hippocampus, we first evaluated synaptic strength using extracellular field recordings on slices obtained from TC, Hx, and HI. To quantify the strength of CA3-CA1 synaptic transmission, extracellular fEPSPs were evoked at CA3-CA1 synapses by synaptic stimuli of varying intensities. Increasing stimulus intensities (20–100 μA) resulted in an increase in FV and fEPSP amplitudes (Fig. 9A). The FV reflects activation of the CA3 axons (presynaptic effect), and the initial slope of the fEPSPs (shaded area) reflects predominately activation of AMPA receptors (postsynaptic effect). The fEPSP recordings of FV allowed for an examination of the CA3 axon recruitment and excitability as described by plotting the peak of the FV against the stimulus intensity (Stim, Fig. 9B). The FV-Stim intensity relationship was described by a linear relation yielding a slope of 3.2 μV/μA for this representative slice, which reflects the FV IE. A scatterplot of FV IE was generated for TC, Hx, and HI (Fig. 9D). Sex was denoted by open (female) and closed (male) symbols. There was no difference between sex (p = 0.44) and treatment (p = 0.17). Grouping sex for each treatment revealed an average FV IE of 3.56 ± 0.24 μV/μA (n = 51 slices from 14 TC brains), 3.63 ± 0.19 μV/μA (70 slices from 23 Hx brains), and 3.11 ± 0.19 μV/μA (70 slices from 20 HI brains). These results suggest that IE and recruitment of the CA3 axon collaterals was not affected by Hx or HI treatment.
Figure 9.

Field recording of synaptic strength in CA1 region of hippocampus. A, Representative fEPSPs evoked by increasing stimulation intensities (20–100 μA at 20 μA interval). Each trace is the average of 5 consecutively recorded voltage traces at each stimulation intensity. Gray bars represent the regions where fEPSP initial slopes were measured. FV indicates FV peak. The stimulus artifact preceding the FV was blanked out. B, Plot of FV versus stimulus intensity (Stim) from A. The FV-Stim relationship was fit with a linear function without constraints, yielding a slope of 3.2 μV/μA. The slope of the FV-Stim relationship is a reflection of the IE of Schaffer collateral neurons and the density of neurons being excited (FV IE). C, Plot of fEPSP slope versus FV relation from A. The fEPSP initial slope-FV relationship was fit with linear functions without constraints, yielding a slope of 0.92 ms−1. The slope derived from the fits reflects the input–output relation of synaptic transmission (fEPSP I/O) at CA3-CA1 synapses. D, E, Scatterplot of FV IE and fEPSP I/O determined in B and C for individual slices for TC (n = 52 slices from 15 TC brains), Hx (n = 70 slices from 23 Hx brains), and HI (n = 72 from 20 HI brains). Open and closed symbols represent female and male data, respectively. Horizontal error bar in each scatterplot indicates the mean. F, Average FV versus Stim for all slices in D. Data are mean ± SEM. G, Average of fEPSP slope plotted versus average FV from all slices in E. Average fEPSP slope was determined by dividing the FV from each experiment into 50 μV bins, and averaging these across all experiments for both FV and fEPSP slope. Data are mean ± SEM.
The fEPSP input–output relationships (fEPSP I/O) were determined from plots of fEPSP slope versus FV (Fig. 9C). The maximal initial slope of the fEPSP was measured to monitor the strength of synaptic transmission mediated primarily by AMPA, minimizing contamination by voltage-dependent events. The maximal initial slope was determined as the maximum rate of rise of the fEPSP that usually occurs between 25% and 50% of the rise time (∼3 ms into the fEPSP; Fig. 9A, shaded area). The fEPSP slope, determined from an average of 10 fEPSPs, was plotted against the FV amplitudes to yield a fEPSP I/O relation (Fig. 9C). The fEPSP I/O relations constructed from individual experiments were fit with linear functions, the slope of which reflects efficiency of presynaptic AP generation and the postsynaptic membrane response due to presynaptic AP-induced glutamate release (Fig. 9C). The slope of the fEPSP I/O relationship was used as an indicator of synaptic strength, which was 0.92 (ms−1) for this representative example. A scatterplot of fEPSP I/O was generated for TC, Hx, and HI, with open and closed symbols denoting female and male, respectively (Fig. 9E). There was no difference between sex (p = 0.77) and treatment (p = 0.51). Grouping sex for each treatment yielded an fEPSP I/O of 0.977 ± 0.048 (n = 51 slices from 14 TC brains), 1.055 ± 0.053 (70 slices from 23 Hx brains), and 1.056 ± 0.052 (ms−1) (70 slices from 20 HI brains). These results demonstrate that Hx and HI do not have long term effects on synaptic strength at hippocampal CA3-CA1 synapse. This is further demonstrated in Figure 9F, G, which shows the average FV IE for each stimulus intensity and average fEPSP slope versus average FV across all slices from Figure 9D and Figure 9E, respectively.
Probability of glutamate release is decreased following Hx and HI treatment.
To determine whether Hx or HI affects presynaptic glutamate release or short-term synaptic plasticity, extracellular paired-pulse ratio (PPR) measurements were made by giving two stimuli in close succession (50 ms) (Fig. 10A). PPR was determined as the ratio of the initial slope of the average of 10 fEPSPs for the second and the first stimuli (shaded area in representative traces for TC, Hx, and HI, Fig. 10B–D, respectively). A scatter plot of PPR was generated for TC, Hx, and HI (Fig. 10E). There was no difference between sex (p = 0.78), but there was a significant effect of treatment on PPR (p < 0.0001). Grouping sex for each treatment yielded an average PPR of 1.68 ± 0.04 (43 slices from 12 TC brains), 2.07 ± 0.04 (65 slices from 23 Hx brains), and 1.98 ± 0.04 (61 slices from 20 HI brains). Probing for multiple pairs using the Dunn correction revealed that the PPR was increased in Hx (p < 0.001) and HI (p < 0.001). There was no difference between Hx and HI. Even though the fEPSP I/O was not affected by Hx or HI treatment, the increase in PPR suggests that Hx and HI reduce the probability of glutamate release at CA3-CA1 synapses. This may indicate that postsynaptic mechanisms are altered by Hx or HI to compensate for the reduced probability of release.
Figure 10.

In utero Hx and HI reduce PPR. A, Representative fEPSPs evoked by paired pulse stimulation separated by 50 ms. Top, Stimulus timing. B–D, Overlay of first and second pulse for representative trace from TC (B), Hx (C), and HI (D). Each trace is the average of 10 consecutively recorded voltage traces. Overlay of first (black) and second (red) fEPSP. Gray bars represent the regions where fEPSP initial slopes were measured. E, Scatter plot of PPR determined by measuring the slope of the average of 10 individual trials. Open and closed symbols represent female and male data, respectively. Horizontal error bar in each scatterplot indicates the mean. ***p < 0.001.
Long-term synaptic potentiation is impaired following short-term Hx in fetal hippocampus
The hippocampus is required for memory formation, and LTP of the CA3-CA1 excitatory synapses is thought to be the cellular correlate that mediates hippocampus-dependent memory formation. LTP at these synapses involves a rapid increase in synaptic strength that is largely attributed to increased postsynaptic AMPA receptor insertion. The degree of LTP induced at these synapses was compared between the TC-, Hx-, and HI-treated brains. Extracellular fEPSPs were measured using a stimulus intensity set to give 25%–50% of the maximal field response determined in Figure 9 for each slice. After achieving 5 min of stable baseline responses, a high-frequency train of stimuli (3 sets at 0.1 Hz of 100 pulses at 50 Hz; Fig. 11A) was administered to induce LTP. The degree of LTP was measured from the average of 10 traces from before and 14–15 min after delivery of the high-frequency train (Fig. 11B). Figure 11D shows a scatterplot of LTP versus treatment, with open and closed symbols indicating female and male, respectively. There was a trend for a difference between sex (p = 0.076), and there was a significant effect of treatment (p = 0.0002). Given the lack of a significant sex difference, the data for sexes were combined for each treatment, which yielded an average LTP of 177.4 ± 1.6% (48 slices from 14 TC brains), 142.7 ± 4.8% (65 slices from 23 Hx brains), and 163.3 ± 6.9% (62 slices from 20 HI brains). Probing for multiple pairs using the Dunn correction revealed that LTP was decreased in Hx (p < 0.001) and HI (p < 0.05) compared with TC. LTP in HI was greater than that in Hx (p < 0.05). These results suggest inappropriate information storage/coding following in utero hypoxia and hypoxia/ischemia.
Figure 11.

In utero hypoxia reduces LTP. A, Top, Induction protocol for LTP. A 50 Hz stimulation train and typical response elicited by LTP stimulation protocol (3 × 100 pulses at 50 Hz). Bottom, fEPSP response to first LTP protocol. The stimulus artifact preceding each FV was blanked out. B, Representative average of 10 fEPSPs from a single slice before (baseline, black) and after LTP induction (LTP, red). Gray bars represent the regions where fEPSP initial slopes were measured. C, Time course (mean ± SEM) of fEPSP slope response normalized to baseline before LTP induction for TC, Hx, and HI. D, Scatterplot represents the reduced levels of LTP observed in Hx. Open and closed symbols represent female and male data, respectively, for TC (n = 48 slices from 15 TC brains), Hx (n = 65 slices from Hx 23 brains), and HI (n = 62 slices from 20 HI brains). Horizontal error bar in each scatterplot indicates the mean. *p < 0.05. ***p < 0.001.
Short-term Hx and HI decrease IE of CA1 neurons
The likelihood that an EPSP will produce an AP is dependent on the IE of CA1 neurons, as defined by propensity to generate APs in response to somatic current injection. Whole-cell current-clamp recordings were performed on CA1 pyramidal neurons from TC, Hx, and HI fetal sheep hippocampi.
Passive membrane properties (RMPapp, Rinput, and Cap) were determined from a −40 pA somatic current injection. No differences were observed between sex (p = 0.93) or treatment (p = 0.36) for RMPapp. The RMPapp was −48.6 ± 1.3 mV (n = 16 TC neurons), −50.2 ± 0.9 mV (n = 40 Hx), and −48.5 ± 0.9 (n = 31 HI). Correcting for liquid junction potential between pipette and bath solution (15 mV) yields an average RMPapp of −64.3 ± 0.6 mV (n = 87 CA1 neurons from 20 fetal hippocampi). Analysis of Rinput revealed a treatment difference (p = 0.025) but no sex difference (p = 0.6). Recombining by treatment yielded a Rinput of 156.3 ± 9.4 (n = 24 TC neurons), 129.0 ± 6.1 (n = 40 Hx), and 150.5 ± 7.9 (n = 31 HI). Rinput was reduced in Hx-treated neurons compared with TC (p < 0.05). Analysis of Cap revealed no differences observed between sex (p = 0.93) or treatment (p = 0.36). The Cap was 245.0 ± 8.5 pF (n = 24 TC), 249.8 ± 7.6 mV (n = 40 Hx), and 255.6 ± 7.6 (n = 31 HI).
IE was assessed with a series of 1 s, incrementing depolarizing current steps from 50 to 350 pA at 50 pA increments (Fig. 12A–C). Hx and HI showed reduced excitability and more irregular firing (Fig. 12B,C). The number of APs evoked versus somatic injection current (ΔI) relationship was reduced in Hx and HI compared with TC for these representative neurons (Fig. 12D). Two measurements were made to quantify IE. The number of APs versus ΔI for each neuron was fit to a linear function between 50 and 150 pA (Fig. 12D). The initial slope determined from the fit reflects the initial rate of AP recruitment versus current injection. Figure 12E shows a scatterplot of the initial slope for TC, Hx, and HI. No difference was observed between sexes (p = 0.31). The data were recombined by treatment, and a nonparametric ANOVA (Kruskal–Wallis) revealed a significant treatment difference (p < 0.0001). Probing for multiple pairs revealed that the recruitment of APs was significantly reduced in Hx (p < 0.001) and HI (p < 0.001) compared with TC. No difference was observed between Hx versus HI. The mean initial slope was 0.0894 ± 0.0045 for TC, 0.0589 ± 0.0038 for Hx, and 0.0591 ± 0.0040 #AP/pA for HI. IE measured as the initial slope of the number of APs versus ΔI relation was reduced by 33% in Hx and HI.
Figure 12.

Hx and HI decrease IE of hippocampal CA1 pyramidal neurons. A–C, Representative voltage traces from a TC, Hx, and HI neuron, respectively, in response to incrementing depolarizing current injections in whole-cell current-clamp mode. Top, Pulse timing. D, The number of evoked APs (number of APs) for the recordings in A–C plotted against current injection (ΔI, pA). The I/O relationships were fit with a linear function from 50 to 150 pA, yielding a slope in number of APs/pA of 0.11 (TC), 0.05 (Hx), and 0.06 (HI). The slope is a measure of IE reflecting the initial recruitment of AP firing in CA1 neurons. E, F, Summary scatterplot of IE measured as the initial slope of the number of APs versus ΔI injection (C) and the integral of number of APs versus ΔI injection relationship (D). Open and closed symbols represent female and male data, respectively. Data from the TC brains (n = 24 slices from 4 TC brains), Hx brains (n = 44 slices from 11 Hx brains), and HI brains (n = 36 slices from 8 HI brains). Horizontal error bar in each scatterplot indicates the mean. **p < 0.01. ***p < 0.001. G, Summary plot of number of APs evoked at each current injection from the slices in C, D. Data are mean ± SEM. Data were fit with a linear function from 50 to 150 pA, yielding an average slope in number of APs/pA of 0.084 (TC), 0.050 (Hx), and 0.055 (HI). The integral under the I/O relationship from 50 to 350 pA was 3119 (TC), 1936 (Hx), and 2232 (HI).
The integral of the AP versus ΔI relationship from 50 to 350 pA (Fig. 12D) is another measure of IE. This relationship is illustrated in the scatterplot in Figure 12F. Two-way ANOVA revealed no difference in sex (p = 0.36). The data were recombined by treatment, and a nonparametric ANOVA (Kruskal–Wallis) revealed a significant treatment difference (p < 0.0001). Probing for multiple pairs using the Dunn correction revealed that the integral was significantly reduced in Hx (p < 0.001) and HI (p < 0.01) compared with TC. No difference was observed between Hx versus HI. The mean integral is as follows: TC, 3242 ± 171; Hx, 1872 ± 150; HI, 2254 ± 163 number of AP × pA. IE measured as the integral of the number of APs versus ΔI relation was reduced by 37% and 29% for Hx and HI, respectively. For comparison, the average of the number of APs for all neurons was plotted versus ΔI for TC, Hx, and HI (Fig. 12G). Figure 12G demonstrates that Hx and HI reduce the IE of CA1 pyramidal neurons in fetal sheep, which could contribute to reduced output from the hippocampus.
Increase in currents underlying the AHP in Hx and HI
The pause in AP firing during the current injections in Hx (Fig. 12B) and HI (Fig. 12C) is consistent with an increase in spike-frequency adaption, which is strongly regulated by the AHP following each AP. In many central neurons, repetitive AP firing activates several K+ conductances that give rise to an AHP that lasts beyond cessation of firing. These K+ conductances exert a negative feedback onto IE, limiting both firing frequency and duration. In hippocampal CA1 pyramidal neurons, the AHP includes three kinetically distinct components: the fast AHP, lasting 1–5 ms that is a continuation of spike repolarization due to BK channel activity (Lancaster and Nicoll, 1987; Gu et al., 2007); the medium AHP, lasting 50–150 ms, due to M-channel activity (Williamson and Alger, 1990; Gu et al., 2005) and the Ca2+-activated K+ channel SK2 (Bond et al., 2004); and the sAHP, which presents markedly slow activation (tact ∼600 ms at room temperature) (Gerlach et al., 2004) and decay kinetics on the order of seconds (Madison and Nicoll, 1984; Storm, 1990). Due to its long-lasting nature, the sAHP exerts the predominant influence on limiting repetitive firing in response to a sustained depolarization (Madison and Nicoll, 1984; Lancaster and Adams, 1986; Hille, 1992; Sah, 1996). The current underlying the AHP (IAHP) was measured in voltage clamp (Fig. 13A). Depolarization to 20 mV for 100 ms from a holding potential of −50 mV elicits a slowly decaying outward tail current on repolarization to −50 mV. To quantify the fast component of the AHP (IfAHP), the peak outward tail current following repolarization to −50 mV was taken as a measure of the IfAHP (Fig. 13B). To quantify the medium and slow component of the AHP, the current at 100 ms and 1 s following repolarization to −50 mV was taken as the ImAHP (Fig. 13B, vertical dashed line) and IsAHP (Fig. 13C, vertical dashed line), respectively. Figure 13D–F shows a scatter plot of the results for TC, Hx, and HI, with open and closed symbols indicating female and male, respectively. There were not enough females for TC and males for HI to analyze sex differences. There was a treatment difference in the IfAHP (p = 0.0167, two-way ANOVA). The IfAHP was 1153.0 ± 110.9 pA (n = 24 TC neurons), 1516.8 ± 107.9 pA (n = 33 Hx), and 1559.1 ± 109.5 pA (n = 32 HI). The IfAHP was increased 32% in Hx (p < 0.05) and 35% in HI (p < 0.05) treated neurons compared with TC (Fig. 13D). There was not a treatment (p = 0.0583) difference in the ImAHP (Fig. 13E). The average ImAHP was 524.4 ± 42.2 pA (n = 24 TC neurons), 629.6 ± 49.5 pA (n = 33 Hx), and 612.4 ± 42.0 pA (n = 32 HI). There was a treatment difference in the IsAHP (p = 0.0004), and the average IsAHP was 85.3 ± 6.4 pA (n = 24 TC neurons), 141.8 ± 14.3 pA (n = 33 Hx), and 121.1 ± 11.5 pA (n = 32 HI). The IsAHP was increased by 66% in Hx (p < 0.001) and 42% in HI (p < 0.05) neurons compared with TC (Fig. 13F). Together, these results show that IE is reduced in Hx and HI neurons, and the reduction in IE is in part due to increased activity of the channels underlying the fAHP and sAHP in Hx and HI.
Figure 13.

Voltage-clamp analysis of the currents underlying the AHP. A, Voltage-clamp recordings of CA1 neurons after a 100 ms depolarizing pulse to 20 mV from a holding potential of −50 mV for a representative TC neuron. Top, Pulse protocol. B, C, Average of all tail currents following repolarization to −50 mV from TC (24 slices from 4 hippocampi), Hx (33 slices from 8 hippocampi), and HI (32 slices from 7 hippocampi) brains. Shaded area represents SEM for each average. IfAHP is measured as the peak outward tail current (B). Dashed vertical line indicates 100 ms time point (B) and 1 s time point (C) after repolarization for measure of the current underlying the mAHP (ImAHP) and the sAHP (IsAHP), respectively. D–F, Scatterplot of the IfAHP, ImAHP, and IsAHP for TC (24 slices from 4 hippocampi), Hx (33 slices from 8 hippocampi), and HI (32 slices from 7 hippocampi) brains. Open and closed symbols represent female and male data. *p < 0.05. ***p < 0.001. Horizontal error bar in each scatterplot indicates the mean.
Discussion
Exposure to Hx during preterm brain development is a significant risk factor for persistent neurodevelopmental disabilities (Poets et al., 2015; Bonkowsky and Son, 2018). Preterm neonates may sustain up to 600 transient hypoxic events per week (Di Fiore et al., 2010). A third or more of preterm survivors develop significant neurodevelopmental challenges with learning and memory, attention, and socialization, which coincide with markedly higher rates of autism and major mental illnesses, including anxiety disorders, depression, and schizophrenia (Nosarti et al., 2012; Vohr, 2014). We identified a critical window at 0.65 gestation when transient disturbances in fetal ovine systemic oxygenation persistently disrupted the structural and functional maturation of the hippocampus. We determined that synaptic plasticity, integral to cellular mechanisms of learning and memory, is established in the preterm fetal brain during a window in development when the risk for neuronal dysmaturation is high (Back and Miller, 2014). At this time, the fetal hippocampus was highly sensitive to reduced cerebral oxygenation and sustained persistent disturbances in LTP and IE, which suggests a novel mechanism for the life-long memory deficits in human preterm survivors.
The 0.65 gestation fetus, like the term fetus (Acheson et al., 1957), was poorly adapted to transient Hx and sustained a persistent depression in systemic oxygenation during the insult. By contrast, HI-exposed fetuses compensated by gradually elevating their systemic oxygenation during the course of HI. One explanation for this unexpected fetal response to Hx is the observation that preterm fetal sheep respond to acute Hx with an initial increase in fetal cerebral blood flow, which is not sufficient to maintain cerebral oxygen transport as cerebral oxygen content falls below a critical threshold (Gleason et al., 1990). Moreover, before 0.7 gestation, the preterm cerebral vasculature may be unable to fully increase cerebral perfusion in response to global Hx (Brew et al., 2014), such that brainstem sparing occurs at the expense of cerebral perfusion (Cavazzuti and Duffy, 1982).
In 4 week chronic survivors, a transient reduction in fetal oxygenation at 0.65 gestation resulted in persistent disturbances in the maturation of apical and basal dendritic arbors, which were in opposing directions. In response to Hx or HI, basal dendrites were more complex, whereas apical dendrites displayed more simplified distal arbors. Moreover, more severe compromise in fetal oxygenation resulted in greater dose-dependent disruption in basal arbor complexity for both the Hx and HI groups. Notably, an extensive arborization analysis of multiple datasets has suggested that the dendritic morphology of hippocampal neurons may be under intrinsic homeostatic control such that fluctuations in dendritic arbor size in one portion of the neuron are systematically counterbalanced by the remaining dendrites in the same cell (Samsonovich and Ascoli, 2006). While our study focused solely on the CA1 pyramidal cell, other hippocampal neuronal populations may have been similarly affected, which may provide an explanation for the impaired hippocampal growth observed here and in human preterm survivors (Peterson et al., 2000; Beauchamp et al., 2008; Thompson et al., 2008). These persistent morphometric disturbances were accompanied by functional disturbances in LTP and short-term facilitation, which were also more disrupted in Hx animals. These persistent dysmaturational responses to a transient compromise in systemic oxygenation are similar to the response of subplate neurons (McClendon et al., 2017). Together, these findings support that a brief episode of fetal Hx alone is sufficient to chronically disrupt preterm dendritic maturation of CA1 neurons.
Although the developmental maturation of LTP appears to be a critical mechanism to establish prenatal connectivity, there is little known about the timing of appearance and features of synaptic plasticity in the fetal brain (Lohmann and Kessels, 2014). Our results support that cellular mechanisms of synaptic plasticity are fully expressed in the fetal ovine hippocampus at 0.85 gestation. Notably, fetal LTP was inducible within 2 weeks after Hx at 0.75 gestation (data not shown). Moreover, these mechanisms were vulnerable to disruption from Hx or HI at 0.65 gestation. We observed multiple features of fetal synaptic plasticity that were consistent with classical LTP. Although we did not confirm NMDA receptor dependence of LTP, prior studies confirmed in the P11 rat that the late phase of LTP was blocked with an NMDA receptor antagonist (Cao and Harris, 2012).
The efficiency of synaptic transmission assessed by the fEPSP I/O relationship was not compromised by Hx or HI, even though the PPR was increased in both Hx and HI. An increase in PPR is consistent with a decrease in probability of release of glutamate at the CA3-CA1 synapses, which should be reflected as a decrease in synaptic transmission, fEPSP I/O. The lack of a decrease in synaptic transmission thus suggests that postsynaptic AMPA and/or NMDA receptors could be upregulated as a result of the Hx or HI insult, similar to anoxia or ischemic-induced potentiation of synaptic transmission due to increased glutamate release (Hammond et al., 1994; Lyubkin et al., 1997). The ensuing decrease in probability of glutamate release therefore most likely reflects a form of homeostatic plasticity to maintain functional stability of synaptic transmission (Delvendahl and Müller, 2019). Similarly, the decrease in IE in Hx and HI reflects a form of homeostatic plasticity to promote stability of firing (Desai et al., 1999). The recovery following Hx and HI reflects a coordination of changes in synaptic transmission and excitability to stabilize circuit function (Tien and Kerschensteiner, 2018).
Human preterm survivors sustain chronic disturbances in episodic and working memory that persist into adulthood (Nosarti and Froudist-Walsh, 2016). These memory deficits are related to MRI-defined structural and functional connectivity disturbances in large-scale networks that involve the hippocampus. Prior experimental studies supported that hypoxia disrupts cerebral connectivity through disturbances in axon pathfinding that disrupt the formation of commissural fiber tracts. During specific developmental windows in Caenorhabditis elegans (Pocock and Hobert, 2008) and zebrafish embryogenesis, Hx disrupted axon pathfinding via ephrin-dependent mechanisms that were mediated independently of apoptosis (Pocock and Hobert, 2008; Stevenson et al., 2012; Xing et al., 2015). It remains to be determined whether these findings contribute to neurodevelopmental defects in higher mammals. In contrast to the response of axons to Hx, the disturbances in CA1 dendritic maturation were diffusely induced by Hx in a gyrencephalic fetal brain that follows a maturational trajectory similar to human (Back et al., 2012). Our findings to date support that the preterm brain sustains persistent neuronal dysmaturation that not only targets the hippocampus, but diffusely involves the cerebral cortex (Dean et al., 2013), caudate nucleus (McClendon et al., 2014), and subplate (McClendon et al., 2017). Hence, further study is needed to confirm that other cortical and subcortical brain regions integral to episodic and working memory circuits are similarly sensitive to Hx-mediated neuronal dysmaturation.
In response to HI or Hx, hippocampal neurons in our model of relatively selective white matter injury appear to display resistance to ischemic neuronal degeneration in the first week after HI in contrast to preoligodendrocytes, which diffusely degenerate in response to similar insults (Riddle et al., 2006; McClendon et al., 2017). In contrast, the susceptibility of hippocampal neurons to cell death was more pronounced in models where white matter injury was more severe and accompanied by neuronal degeneration in other gray matter regions, such as the cerebral cortex and basal ganglia (Reddy et al., 1998; Matsuda et al., 1999; Petersson et al., 2002).
The response of the fetal and neonatal mammalian brain to transient versus chronic Hx appears to differ markedly in terms of the magnitude of cell death. Whereas we found that transient exposure to Hx caused minimal CA1 neuronal degeneration, exposure of preterm fetal sheep to 12 h of chronic Hx at mid-gestation resulted in significant hippocampal injury that reduced the density of CA1 neurons and induced astrogliosis (Rees et al., 1999). Notably, dendritic arborization of cerebellar Purkinje cells was reduced, but CA1 dendrites were unaffected, which suggests that the duration and/or timing of Hx may be critical factors related to the magnitude of the disturbance in CA1 dendritic arborization. Chronic sublethal Hx also resulted in loss of GABAergic interneurons and simplification of cerebellar Purkinje cell dendrites in neonatal mice (Zonouzi et al., 2015). Chronic hypobaric Hx in adult rats is a model of high altitude Hx exposure that similarly caused progressive neuronal degeneration and reversible simplification of dendritic arbors with reduction in spine numbers of CA1 and CA3 pyramidal neurons (Maiti et al., 2008). Hence, chronic Hx in postnatal neonates or adults can drive susceptible populations of neurons toward cell death rather than dendritic dysmaturation.
We found that HI and Hx induced chronic disturbances in preterm CA1 dendrites that were distinct from those observed in the adult CNS, where diffuse swelling and beading of dendrites and loss of spines are early degenerative responses that precede HI-mediated cell death (Hori et al., 1998; Brown et al., 2008; Li and Murphy, 2008). Several factors may contribute to the resistance of preterm CA1 neurons to ischemic degeneration. In contrast to near term fetal and adult sheep, cerebral oxygenation consumption and transport are significantly lower in the preterm fetus (Gleason et al., 1990). Notably, human preterm infants were similarly shown to have minimal or no brain injury despite cerebral oxygen consumption below the level of brain viability in adults (Altman et al., 1993). This suggests that fetal CA1 neurons may display greater tolerance for Hx or HI due to lower energy requirements, which may be met by nonoxidative metabolism. The resistance of CA1 neurons to cell death appears unrelated to greater exposure to ischemia, since the fetal ovine hippocampus, parietal cortex, and white matter displayed similar blood flow responses under basal and ischemic conditions (Riddle et al., 2006). High expression of the calcium-impermeable AMPA glutamate receptor 2 subunit could also reduce the susceptibility of fetal CA1 neurons to excitotoxic degeneration. Although AMPA and NMDA receptor expression has not been studied in fetal hippocampus, at 0.65 gestation, glutamate receptor 2 subunit and NR1 expression is low in cerebral white matter but displays peak expression in cerebral cortex (Dean et al., 2005). No patterns of gene expression have been identified in mammals that may promote resistance to Hx-induced dendritic degeneration. In Drosophila, prolonged exposure to severe Hx does not disrupt dendritic integrity due to partial neuroprotection conferred by Nmnat (Coleman and Freeman, 2010; Wen et al., 2013).
In conclusion, we have defined a novel mechanism of fetal neuronal dysmaturation that involves persistent morphometric and electrophysiological disturbances in CA1 neuron maturation. The particular sensitivity of fetal CA1 neurons to transient disturbances in oxygen delivery in utero is clinically relevant in several settings. During human pregnancy, transient or chronic intrauterine Hx may arise from maternal, placental, or fetal causes. Transient or chronic episodes of Hx are also commonly encountered in some preterm neonates in association with critical illness, cardiopulmonary disease, or sepsis. Our studies thus suggest that transient hippocampal Hx may contribute to adverse neurodevelopmental outcomes by disrupting neuronal maturation through persistent functional disturbances in hippocampal circuitry linked to the widespread cognitive and learning disabilities in preterm survivors. Our findings suggest that persistent hippocampal neuronal dysmaturation may contribute to long-term working memory deficits in preterm survivors.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke and Office of Research on Women's Health Grants NS045737 and NS054044.
The authors declare no competing financial interests.
References
- Acheson GH, Dawes GS, Mott JC (1957) Oxygen consumption and the arterial oxygen saturation in foetal and new-born lambs. J Physiol 135:623–643. 10.1113/jphysiol.1957.sp005735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman DI, Perlman JM, Volpe JJ, Powers WJ (1993) Cerebral oxygen metabolism in newborns. Pediatrics 92:99–104. [PubMed] [Google Scholar]
- Back SA, Miller SP (2014) Brain injury in premature neonates: a primary cerebral dysmaturation disorder? Ann Neurol 75:469–486. 10.1002/ana.24132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Luo NL, Borenstein NS, Levine JM, Volpe JJ, Kinney HC (2001) Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci 21:1302–1312. 10.1523/JNEUROSCI.21-04-01302.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Riddle A, Dean J, Hohimer AR (2012) The instrumented fetal sheep as a model of cerebral white matter injury in the premature infant. Neurotherapeutics 9:359–370. 10.1007/s13311-012-0108-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal R, Warrington AE, Gard AL, Ranscht B, Pfeiffer SE (1989) Multiple and novel specificities of monoclonal antibodies O1, O4, and R-mAb used in the analysis of oligodendrocyte development. J Neurosci Res 24:548–557. 10.1002/jnr.490240413 [DOI] [PubMed] [Google Scholar]
- Beauchamp MH, Thompson DK, Howard K, Doyle LW, Egan GF, Inder TE, Anderson PJ (2008) Preterm infant hippocampal volumes correlate with later working memory deficits. Brain 131:2986–2994. 10.1093/brain/awn227 [DOI] [PubMed] [Google Scholar]
- Bliss TV, Cooke SF (2011) Long-term potentiation and long-term depression: a clinical perspective. Clinics (Sao Paulo) 66 [Suppl 1]:3–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond CT, Herson PS, Strassmaier T, Hammond R, Stackman R, Maylie J, Adelman JP (2004) Small conductance Ca2+-activated K+ channel knock-out mice reveal the identity of calcium-dependent afterhyperpolarization currents. J Neurosci 24:5301–5306. 10.1523/JNEUROSCI.0182-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonkowsky JL, Son JH (2018) Hypoxia and connectivity in the developing vertebrate nervous system. Dis Model Mech 11:dmm037127. 10.1242/dmm.037127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brew N, Walker D, Wong FY (2014) Cerebral vascular regulation and brain injury in preterm infants. Am J Physiol Regul Integr Comp Physiol 306:R773–R786. 10.1152/ajpregu.00487.2013 [DOI] [PubMed] [Google Scholar]
- Brown CE, Wong C, Murphy TH (2008) Rapid morphologic plasticity of peri-infarct dendritic spines after focal ischemic stroke. Stroke 39:1286–1291. 10.1161/STROKEAHA.107.498238 [DOI] [PubMed] [Google Scholar]
- Cao G, Harris KM (2012) Developmental regulation of the late phase of long-term potentiation (L-LTP) and metaplasticity in hippocampal area CA1 of the rat. J Neurophysiol 107:902–912. 10.1152/jn.00780.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavazzuti M, Duffy TE (1982) Regulation of local cerebral blood flow in normal and hypoxic newborn dogs. Ann Neurol 11:247–257. 10.1002/ana.410110304 [DOI] [PubMed] [Google Scholar]
- Choi DW, Rothman SM (1990) The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu Rev Neurosci 13:171–182. 10.1146/annurev.ne.13.030190.001131 [DOI] [PubMed] [Google Scholar]
- Clapp WC, Hamm JP, Kirk IJ, Teyler TJ (2012) Translating long-term potentiation from animals to humans: a novel method for noninvasive assessment of cortical plasticity. Biol Psychiatry 71:496–502. 10.1016/j.biopsych.2011.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman MP, Freeman MR (2010) Wallerian degeneration, wld(s), and nmnat. Annu Rev Neurosci 33:245–267. 10.1146/annurev-neuro-060909-153248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean JM, Fraser M, Shelling AN, Bennet L, George S, Shaikh S, Scheepens A, Gunn AJ (2005) Ontogeny of AMPA and NMDA receptor gene expression in the developing sheep white matter and cerebral cortex. Brain Res Mol Brain Res 139:242–250. 10.1016/j.molbrainres.2005.05.026 [DOI] [PubMed] [Google Scholar]
- Dean JM, McClendon E, Hansen K, Azimi-Zonooz A, Chen K, Riddle A, Gong X, Sharifnia E, Hagen M, Ahmad T, Leigland LA, Hohimer AR, Kroenke CD, Back SA (2013) Prenatal cerebral ischemia disrupts MRI-defined cortical microstructure through disturbances in neuronal arborization. Sci Transl Med 5:168ra167. 10.1126/scitranslmed.3004669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delvendahl I, Müller M (2019) Homeostatic plasticity: a presynaptic perspective. Curr Opin Neurobiol 54:155–162. 10.1016/j.conb.2018.10.003 [DOI] [PubMed] [Google Scholar]
- Desai NS, Rutherford LC, Turrigiano GG (1999) Plasticity in the intrinsic excitability of cortical pyramidal neurons. Nat Neurosci 2:515–520. 10.1038/9165 [DOI] [PubMed] [Google Scholar]
- Di Fiore JM, Bloom JN, Orge F, Schutt A, Schluchter M, Cheruvu VK, Walsh M, Finer N, Martin RJ (2010) A higher incidence of intermittent hypoxemic episodes is associated with severe retinopathy of prematurity. J Pediatr 157:69–73. 10.1016/j.jpeds.2010.01.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George S, Gunn AJ, Westgate JA, Brabyn C, Guan J, Bennet L (2004) Fetal heart rate variability and brain stem injury after asphyxia in preterm fetal sheep. Am J Physiol Regul Integr Comp Physiol 287:R925–R933. 10.1152/ajpregu.00263.2004 [DOI] [PubMed] [Google Scholar]
- Gerlach AC, Maylie J, Adelman JP (2004) Activation kinetics of the slow afterhyperpolarization in hippocampal CA1 neurons. Pflugers Arch 448:187–196. 10.1007/s00424-003-1237-2 [DOI] [PubMed] [Google Scholar]
- Gleason CA, Hamm C, Jones MD Jr (1990) Effect of acute hypoxemia on brain blood flow and oxygen metabolism in immature fetal sheep. Am J Physiol 258:H1064–H1069. 10.1152/1jpheart.1990.258.4.H1064 [DOI] [PubMed] [Google Scholar]
- Gu N, Vervaeke K, Hu H, Storm JF (2005) Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J Physiol 566:689–715. 10.1113/jphysiol.2005.086835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu N, Vervaeke K, Storm JF (2007) BK potassium channels facilitate high-frequency firing and cause early spike frequency adaptation in rat CA1 hippocampal pyramidal cells. J Physiol 580:859–882. 10.1113/jphysiol.2006.126367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen MW, Riddle A, McClendon E, Gong X, Shaver D, Srivastava T, Dean JM, Bai JZ, Fowke TM, Gunn AJ, Jones DF, Sherman LS, Grafe MR, Hohimer AR, Back SA (2014) Role of recurrent hypoxia-ischemia in preterm white matter injury severity. PLoS One 9:e112800. 10.1371/journal.pone.0112800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond C, Crépel V, Gozlan H, Ben-Ari Y (1994) Anoxic LTP sheds light on the multiple facets of NMDA receptors. Trends Neurosci 17:497–503. 10.1016/0166-2236(94)90140-6 [DOI] [PubMed] [Google Scholar]
- Harris KM, Teyler TJ (1984) Developmental onset of long-term potentiation in area CA1 of the rat hippocampus. J Physiol 346:27–48. 10.1113/jphysiol.1984.sp015005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley JM, Wilkinson KA (2016) Synaptic AMPA receptor composition in development, plasticity and disease. Nat Rev Neurosci 17:337–350. 10.1038/nrn.2016.37 [DOI] [PubMed] [Google Scholar]
- Hille B. (1992) Ionic channels of excitable membranes, Ed 2 Oxford: Sinauer. [Google Scholar]
- Hori N, Wiest R, Groszmann RJ (1998) Enhanced release of nitric oxide in response to changes in flow and shear stress in the superior mesenteric arteries of portal hypertensive rats. Hepatology 28:1467–1473. 10.1002/hep.510280604 [DOI] [PubMed] [Google Scholar]
- Kennedy MB. (2013) Synaptic signaling in learning and memory. Cold Spring Harb Perspect Biol 8:a016824. 10.1101/cshperspect.a016824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster B, Adams PR (1986) Calcium-dependent current generating the afterhyperpolarization of hippocampal neurons. J Neurophysiol 55:1268–1282. 10.1152/jn.1986.55.6.1268 [DOI] [PubMed] [Google Scholar]
- Lancaster B, Nicoll RA (1987) Properties of two calcium-activated hyperpolarizations in rat hippocampal neurones. J Physiol 389:187–203. 10.1113/jphysiol.1987.sp016653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Murphy TH (2008) Two-photon imaging during prolonged middle cerebral artery occlusion in mice reveals recovery of dendritic structure after reperfusion. J Neurosci 28:11970–11979. 10.1523/JNEUROSCI.3724-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmann C, Kessels HW (2014) The developmental stages of synaptic plasticity. J Physiol 592:13–31. 10.1113/jphysiol.2012.235119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyubkin M, Durand DM, Haxhiu MA (1997) Interaction between tetanus long-term potentiation and hypoxia-induced potentiation in the rat hippocampus. J Neurophysiol 78:2475–2482. 10.1152/jn.1997.78.5.2475 [DOI] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA (1984) Control of the repetitive discharge of rat CA 1 pyramidal neurones in vitro. J Physiol 354:319–331. 10.1113/jphysiol.1984.sp015378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti P, Muthuraju S, Ilavazhagan G, Singh SB (2008) Hypobaric hypoxia induces dendritic plasticity in cortical and hippocampal pyramidal neurons in rat brain. Behav Brain Res 189:233–243. 10.1016/j.bbr.2008.01.007 [DOI] [PubMed] [Google Scholar]
- Mallard EC, Gunn AJ, Williams CE, Johnston BM, Gluckman PD (1992) Transient umbilical cord occlusion causes hippocampal damage in the fetal sheep. Am J Obstet Gynecol 167:1423–1430. 10.1016/S0002-9378(11)91728-1 [DOI] [PubMed] [Google Scholar]
- Matsuda T, Okuyama K, Cho K, Hoshi N, Matsumoto Y, Kobayashi Y, Fujimoto S (1999) Induction of antenatal periventricular leukomalacia by hemorrhagic hypotension in the chronically instrumented fetal sheep. Am J Obstet Gynecol 181:725–730. 10.1016/S0002-9378(99)70519-3 [DOI] [PubMed] [Google Scholar]
- McClendon E, Chen K, Gong X, Sharifnia E, Hagen M, Cai V, Shaver DC, Riddle A, Dean JM, Gunn AJ, Mohr C, Kaplan JS, Rossi DJ, Kroenke CD, Hohimer AR, Back SA (2014) Prenatal cerebral ischemia triggers dysmaturation of caudate projection neurons. Ann Neurol 75:508–524. 10.1002/ana.24100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClendon E, Shaver DC, Degener-O'Brien K, Gong X, Nguyen T, Hoerder-Suabedissen A, Molnár Z, Mohr C, Richardson BD, Rossi DJ, Back SA (2017) Transient hypoxemia chronically disrupts maturation of preterm fetal ovine subplate neuron arborization and activity. J Neurosci 37:11912–11929. 10.1523/JNEUROSCI.2396-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JW, Johnston MV (1990) Physiological and pathophysiological roles of excitatory amino acids during central nervous system development. Brain Res Brain Res Rev 15:41–70. 10.1016/0165-0173(90)90011-C [DOI] [PubMed] [Google Scholar]
- Nicoll RA. (2017) A brief history of long-term potentiation. Neuron 93:281–290. 10.1016/j.neuron.2016.12.015 [DOI] [PubMed] [Google Scholar]
- Nosarti C, Froudist-Walsh S (2016) Alterations in development of hippocampal and cortical memory mechanisms following very preterm birth. Dev Med Child Neurol 58 [Suppl 4]:35–45. 10.1111/dmcn.13042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosarti C, Reichenberg A, Murray RM, Cnattingius S, Lambe MP, Yin L, MacCabe J, Rifkin L, Hultman CM (2012) Preterm birth and psychiatric disorders in young adult life. Arch Gen Psychiatry 69:E1–E8. 10.1001/archgenpsychiatry.2011.1374 [DOI] [PubMed] [Google Scholar]
- Palmer MJ, Isaac JT, Collingridge GL (2004) Multiple, developmentally regulated expression mechanisms of long-term potentiation at CA1 synapses. J Neurosci 24:4903–4911. 10.1523/JNEUROSCI.0170-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson BS, Vohr B, Staib LH, Cannistraci CJ, Dolberg A, Schneider KC, Katz KH, Westerveld M, Sparrow S, Anderson AW, Duncan CC, Makuch RW, Gore JC, Ment LR (2000) Regional brain volume abnormalities and long-term cognitive outcome in preterm infants. JAMA 284:1939–1947. 10.1001/jama.284.15.1939 [DOI] [PubMed] [Google Scholar]
- Petersson KH, Pinar H, Stopa EG, Faris RA, Sadowska GB, Hanumara RC, Stonestreet BS (2002) White matter injury after cerebral ischemia in ovine fetuses. Pediatr Res 51:768–776. 10.1203/00006450-200206000-00019 [DOI] [PubMed] [Google Scholar]
- Pillai AG, de Jong D, Kanatsou S, Krugers H, Knapman A, Heinzmann JM, Holsboer F, Landgraf R, Joëls M, Touma C (2012) Dendritic morphology of hippocampal and amygdalar neurons in adolescent mice is resilient to genetic differences in stress reactivity. PLoS One 7:e38971. 10.1371/journal.pone.0038971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocock R, Hobert O (2008) Oxygen levels affect axon guidance and neuronal migration in Caenorhabditis elegans. Nat Neurosci 11:894–900. 10.1038/nn.2152 [DOI] [PubMed] [Google Scholar]
- Poets CF, Roberts RS, Schmidt B, Whyte RK, Asztalos EV, Bader D, Bairam A, Moddemann D, Peliowski A, Rabi Y, Solimano A, Nelson H (2015) Association between intermittent hypoxemia or bradycardia and late death or disability in extremely preterm infants. JAMA 314:595–603. 10.1001/jama.2015.8841 [DOI] [PubMed] [Google Scholar]
- Reddy K, Mallard C, Guan J, Marks K, Bennet L, Gunning M, Gunn A, Gluckman P, Williams C (1998) Maturational change in the cortical response to hypoperfusion injury in the fetal sheep. Pediatr Res 43:674–682. 10.1203/00006450-199805000-00017 [DOI] [PubMed] [Google Scholar]
- Rees S, Breen S, Loeliger M, McCrabb G, Harding R (1999) Hypoxemia near mid-gestation has long-term effects on fetal brain development. J Neuropathol Exp Neurol 58:932–945. 10.1097/00005072-199909000-00004 [DOI] [PubMed] [Google Scholar]
- Riddle A, Luo NL, Manese M, Beardsley DJ, Green L, Rorvik DA, Kelly KA, Barlow CH, Kelly JJ, Hohimer AR, Back SA (2006) Spatial heterogeneity in oligodendrocyte lineage maturation and not cerebral blood flow predicts fetal ovine periventricular white matter injury. J Neurosci 26:3045–3055. 10.1523/JNEUROSCI.5200-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P. (1996) Ca(2+)-activated K+ currents in neurones: types, physiological roles and modulation. Trends Neurosci 19:150–154. 10.1016/S0166-2236(96)80026-9 [DOI] [PubMed] [Google Scholar]
- Samsonovich AV, Ascoli GA (2006) Morphological homeostasis in cortical dendrites. Proc Natl Acad Sci U S A 103:1569–1574. 10.1073/pnas.0510057103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson S, King JL (1911) Localisation of the motor area in the sheep. Q J Exp Physiol 4:53–65. 10.1113/expphysiol.1911.sp000083 [DOI] [Google Scholar]
- Stevenson TJ, Trinh T, Kogelschatz C, Fujimoto E, Lush ME, Piotrowski T, Brimley CJ, Bonkowsky JL (2012) Hypoxia disruption of vertebrate CNS pathfinding through ephrinB2 Is rescued by magnesium. PLoS Genet 8:e1002638. 10.1371/journal.pgen.1002638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm JF. (1990) Potassium currents in hippocampal pyramidal cells. Prog Brain Res 83:161–187. 10.1016/S0079-6123(08)61248-0 [DOI] [PubMed] [Google Scholar]
- Thompson DK, Wood SJ, Doyle LW, Warfield SK, Lodygensky GA, Anderson PJ, Egan GF, Inder TE (2008) Neonate hippocampal volumes: prematurity, perinatal predictors, and 2-year outcome. Ann Neurol 63:642–651. 10.1002/ana.21367 [DOI] [PubMed] [Google Scholar]
- Tien NW, Kerschensteiner D (2018) Homeostatic plasticity in neural development. Neural Dev 13:9. 10.1186/s13064-018-0105-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uylings HB, Smit GJ, Veltman WA (1975) Ordering methods in quantitative analysis of branching structures of dendritic trees. Adv Neurol 12:347–354. [PubMed] [Google Scholar]
- Uylings HB, Ruiz-Marcos A, van Pelt J (1986) The metric analysis of three-dimensional dendritic tree patterns: a methodological review. J Neurosci Methods 18:127–151. 10.1016/0165-0270(86)90116-0 [DOI] [PubMed] [Google Scholar]
- Vanderwolf C, Cooley R (1990) The sheep brain: a photographic series, Ed 2 London, Ontario, Canada: Kirby. [Google Scholar]
- Vohr BR. (2014) Neurodevelopmental outcomes of extremely preterm infants. Clin Perinatol 41:241–255. 10.1016/j.clp.2013.09.003 [DOI] [PubMed] [Google Scholar]
- Wen Y, Zhai RG, Kim MD (2013) The role of autophagy in Nmnat-mediated protection against hypoxia-induced dendrite degeneration. Mol Cell Neurosci 52:140–151. 10.1016/j.mcn.2012.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson A, Alger BE (1990) Characterization of an early afterhyperpolarization after a brief train of action potentials in rat hippocampal neurons in vitro. J Neurophysiol 63:72–81. 10.1152/jn.1990.63.1.72 [DOI] [PubMed] [Google Scholar]
- Xing L, Son JH, Stevenson TJ, Lillesaar C, Bally-Cuif L, Dahl T, Bonkowsky JL (2015) A serotonin circuit acts as an environmental sensor to mediate midline axon crossing through EphrinB2. J Neurosci 35:14794–14808. 10.1523/JNEUROSCI.1295-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda H, Barth AL, Stellwagen D, Malenka RC (2003) A developmental switch in the signaling cascades for LTP induction. Nat Neurosci 6:15–16. 10.1038/nn985 [DOI] [PubMed] [Google Scholar]
- Zonouzi M, Scafidi J, Li P, McEllin B, Edwards J, Dupree JL, Harvey L, Sun D, Hübner CA, Cull-Candy SG, Farrant M, Gallo V (2015) GABAergic regulation of cerebellar NG2 cell development is altered in perinatal white matter injury. Nat Neurosci 18:674–682. 10.1038/nn.3990 [DOI] [PMC free article] [PubMed] [Google Scholar]