

Abstract
BACKGROUND
Prodromal Alzheimer’s disease offers an opportunity to test the effect of drugs that modify the deposition of amyloid in the brain before the onset of dementia. Verubecestat is an orally administered β-site amyloid precursor protein-cleaving enzyme 1 (BACE-1) inhibitor that blocks production of amyloid-beta (Aβ). The drug did not prevent clinical progression in a trial involving patients with mild-to-moderate dementia due to Alzheimer’s disease.
METHODS
We conducted a randomized, double-blind, placebo-controlled, 104-week trial to evaluate verubecestat at doses of 12 mg and 40 mg per day, as compared with placebo, in patients who had memory impairment and elevated brain amyloid levels but whose condition did not meet the case definition of dementia. The primary outcome was the change from baseline to week 104 in the score on the Clinical Dementia Rating Scale-Sum of Boxes (CDR-SB; scores range from 0 to 18, with higher scores indicating worse cognition and daily function). Secondary outcomes included other assessments of cognition and daily function.
RESULTS
The trial was terminated for futility after 1454 patients had been enrolled; 485 had been assigned to receive verubecestat at a dose of 12 mg per day (the 12-mg group), 484 to receive verubecestat at a dose of 40 mg per day (the 40-mg group), and 485 to receive placebo. A total of 234 patients, 231 patients, and 239 patients per group, respectively, completed 104 weeks of the trial regimen. The estimated mean change from baseline to week 104 in the CDR-SB score was 1.65 in the 12-mg group, 2.02 in the 40-mg group, and 1.58 in the placebo group (P=0.67 for the comparison between the 12-mg group and the placebo group and P=0.01 for the comparison between the 40-mg group and the placebo group), suggesting a worse outcome in the higher-dose group than in the placebo group. The estimated rate of progression to dementia due to Alzheimer’s disease was 24.5, 25.5, and 19.3 events per 100 patient-years in the 12-mg group, the 40-mg group, and the placebo group, respectively (hazard ratio for 40 mg vs. placebo, 1.38; 97.51% confidence interval, 1.07 to 1.79, not adjusted for multiple comparisons), favoring placebo. Adverse events were more common in the verubecestat groups than in the placebo group.
CONCLUSIONS
Verubecestat did not improve clinical ratings of dementia among patients with prodromal Alzheimer’s disease, and some measures suggested that cognition and daily function were worse among patients who received verubecestat than among those who received placebo. (Funded by Merck Sharp & Dohme; ClinicalTrials.gov number, .)
THE AMYLOID HYPOTHESIS OF ALZHEImer’s disease proposes that accumulation of amyloid-beta (Aβ) in the brain triggers the spread of tau-related neurofibrillary tangles, neuroinflammation, and neuronal degeneration.1,2 Aβ is produced when amyloid precursor protein (APP) is cleaved sequentially by β-site APP-cleaving enzyme 1 (BACE-1; also referred to as β-secretase) and γ-secretase.3 Inhibition of BACE-1 potentially slows the progression of Alzheimer’s disease by reducing the production of Aβ and limiting the deposition of amyloid plaques.
Verubecestat is a BACE-1 inhibitor that reduces the level of Aβ by more than 60% in the cerebrospinal fluid of healthy persons and patients with Alzheimer’s disease.4–6 Verubecestat also inhibits BACE-2, an enzyme that has uncertain physiologic functions.7,8 Although BACE-2 is present at low levels in the brain of healthy persons, its expression is increased in persons with Alzheimer’s disease.9
A trial of verubecestat did not show slowing of disease progression in patients with mild-to-moderate Alzheimer’s disease despite inhibition of Aβ in the cerebrospinal fluid and some regression of amyloid plaques in the brain.6 However, initiation of treatment at the stage of mild-to-moderate dementia may be too late in the disease process to alter outcomes.10,11 We conducted a trial to determine whether verubecestat could slow disease progression in patients at a prodromal stage of Alzheimer’s disease, which is characterized by mild cognitive impairment with evidence of elevated brain amyloid levels.12 This stage precedes dementia due to Alzheimer’s disease but occurs years after deposition of amyloid begins.10,11
METHODS
PATIENTS
Patients were eligible for enrollment in the trial if they were between 50 and 85 years of age and if they did not meet criteria for dementia13,14 but had had a subjective decrease in memory for at least 1 year corroborated by an informant. Other eligibility criteria were a score on the Repeatable Battery for the Assessment of Neuropsychological Status Delayed Memory Index of at least 1 SD below the age- and education-appropriate population mean, corresponding to a score of 85 or less (scores range from 40 to 160, with lower scores indicating worse memory),15 and the presence of brain amyloid as gauged by a radiologist’s visual inspection of amyloid-ligand positronemission tomography (PET) imaging. All the patients underwent medical and neurologic evaluations, including magnetic resonance imaging (MRI) (or computed tomography if MRI was contraindicated). Other entry criteria included a score of 24 to 30 on the Mini–Mental State Examination (MMSE; scores range from 0 to 30, with lower scores indicating poorer cognitive performance).16 Patients could have been receiving an acetylcholinesterase inhibitor, memantine, or both, provided that they had received a stable dose for at least 3 months before screening. The diagnosis of prodromal Alzheimer’s disease was confirmed by independent review. The protocol is available with the full text of this article at NEJM.org.
OVERSIGHT
The trial was conducted in accordance with the principles of Good Clinical Practice guidelines and approved by the relevant institutional review boards. Written informed consent was provided by the patients or their legal representatives. The sponsor (Merck Sharp & Dohme [MSD]) designed the trial in consultation with the academic authors. Data were collected by the investigators and analyzed by the sponsor. The first draft of the manuscript was written by the first author and a professional medical writer who was employed by the sponsor. All the authors approved the manuscript, had full access to the trial data, and vouch for the accuracy and completeness of the data, for the fidelity of the trial to the protocol, and for the reporting of adverse events. Confidentiality agreements were in place between the sponsor and the authors. The trial was governed by three committees as described in the trial protocol.
TRIAL DESIGN
The trial was conducted at 238 centers in 22 countries from November 2013 through April 2018. A list of investigators is provided in the Supplementary Appendix, available at NEJM.org. The trial consisted of a randomized, double-blind, placebo-controlled, parallel group, 104-week trial period (part 1), followed by an optional extension period with a planned duration of up to 5 years (part 2). In part 1, patients were randomly assigned in a 1:1:1 ratio to receive, once daily, oral verubecestat at a dose of 12 mg, oral verubecestat at a dose of 40 mg, or oral placebo. The dose of verubecestat was based on data from phase 1 trials indicating that doses of 12 mg and 40 mg reduced levels of Aβ40 and Aβ42 in cerebrospinal fluid by 60% (12 mg) to 75% (40 mg).5 All the assigned trial regimens were administered as identical-appearing tablets. Patients who completed part 1 could enter the part 2 extension period in which patients in the placebo group were switched to the 40-mg dose of verubecestat and those who had been receiving the 12-mg or 40-mg dose continued to receive the same dose to which they had been assigned, with preserved masking of doses.
An interactive response system randomly assigned patients to trial groups according to a computer-generated schedule. Randomization was stratified according to geographic region, baseline MMSE score (24 to 26 or 27 to 30), and use of cholinesterase-inhibiting medications. We also performed biomarker substudies of brain-volume measures (with the use of MRI) and amyloid burden (with the use of PET and cerebrospinal fluid analysis) (see the Supplementary Appendix).
OUTCOMES
The primary efficacy outcome was the change from baseline to week 104 in the Clinical Dementia Rating Scale–Sum of Boxes score (CDR-SB; scores range from 0 to 18, with higher scores indicating worse cognition and daily function).17 There were seven secondary outcomes: the progression to the diagnosis of dementia due to Alzheimer’s disease; the change from week 13 to week 104 in the CDR-SB score; the change from baseline to week 104 in the three-domain composite cognition score (CCS-3D; derived from the z scores [mean of 0, standard deviation of 1, with higher scores indicating worse cognition] of tests of episodic memory, executive function, and attention and processing speed); the total hippocampal volume on MRI; the cortical amyloid load on PET; the score on the Alzheimer’s Disease Cooperative Study Activities of Daily Living for Mild Cognitive Impairment scale (ADCS-ADLMCI; scores range from 0 to 53, with lower scores indicating worse function)18; and the concentration of total tau in cerebrospinal fluid.
Exploratory outcomes included the change from baseline to week 104 in scores on the 13-item cognitive subscale version of the Alzheimer’s Disease Assessment Scale (ADAS-cog13; scores range from 0 to 85, with higher scores indicating worse cognition). The ADAS-cog13 adds delayed word recall and number cancellation tasks to the standard 11-item version (ADAS-cog11; scores range from 0 to 70, with higher scores indicating worse cognition).19 Other exploratory outcomes included the scores on the MMSE and the Neuropsychiatric Inventory (NPI; scores range from 1 to 144, with higher scores indicating more severe symptoms).20 Assessments were audiorecorded, and a subgroup of assessments underwent quality review by independent central raters, who provided feedback to the site raters (see the Supplementary Appendix).
Safety assessments included evaluation of adverse events, laboratory analyses, electrocardiography, and physical examinations performed as indicated in the protocol. MRI was initially performed to assess for possible amyloid-related imaging abnormalities but was subsequently discontinued on the basis of feedback from the independent monitoring committee and regulatory recommendations. Dermatologic examinations were performed at the clinic visits as described in the protocol. Suicidality was assessed at every clinic visit with the use of the Columbia Suicide Severity Rating Scale.21
Hippocampal volume on MRI was assessed with the use of an automated FreeSurfer-based segmentation method. The change in hippocampal volume was determined with a tensor-based morphometry algorithm developed by Bioclinica. Brain amyloid load was assessed by means of 18F-flutemetamol PET imaging. The composite cortical index of amyloid deposition was computed as the mean of the regional standardized uptake value ratio (SUVR) in the frontal, temporal, and parietal lobes; the anterior and posterior cingulate cortex; and the precuneus, with a subcortical white-matter region used as the reference.22 No partial-volume correction was applied. Cerebrospinal fluid concentrations of Aβ40, Aβ42, sAPPβ, total tau, and phosphorylated tau were measured in patients who underwent lumbar puncture.5,23
STATISTICAL ANALYSIS
The modified intention-to-treat approach was used for efficacy analyses involving patients who had received at least one dose of verubecestat or placebo and who had both a baseline outcome measurement and at least one postrandomization observation within a window of 6 weeks before to 6 weeks after a planned assessment visit. We used a longitudinal analysis of covariance (ANCOVA) model to evaluate all changes in scores, with time considered to be a categorical variable. The model was adjusted for geographic region, trial-group assignment, sex, APOE4 genotype (carrier or noncarrier), baseline use of medication for Alzheimer’s disease (use or no use), baseline use of vitamin E (0 to 400 IU per day or >400 IU per day), and the interaction between time and trial-group assignment, with the baseline values of MMSE score and age included as continuous covariates. The baseline value of the dependent variable and the interaction between the baseline value and time were also included. The mean differences between the trial groups (each verubecestat group vs. placebo) in the change from baseline to week 104, as well as the corresponding confidence intervals and two-sided P values, were estimated from this model. An unstructured covariance matrix was used to model the correlation among repeated measurements.
A Bonferroni approach (splitting the overall α between the two dose levels of verubecestat) in conjunction with a hierarchical sequential testing approach of outcomes was used to control for the type 1 error rate, with testing of outcomes as described in the order listed in the Outcomes section, beginning with the CDR-SB score, and in the statistical plan (available in the protocol at NEJM.org). Separately for each dose level, if significant superiority was not shown, all subsequent outcomes were assumed not to have differed significantly between the groups and are reported as point estimates with 97.51% confidence intervals or, for exploratory outcomes, 95% confidence intervals that were not adjusted for multiple comparisons. We also performed a sensitivity analysis excluding assessments conducted after notification of the termination of the trial.
For the analysis of the secondary outcome of progression to dementia due to Alzheimer’s disease, we used a Cox proportional-hazards model with adjustment for the same variables as the ANCOVA model to compare the hazard functions of each verubecestat group with placebo, as well as to compute the hazard ratios (verubecestat vs. placebo) and confidence intervals. Patients who reached the 104-week maximum duration of treatment were considered to have completed the trial, and data were censored at that time point. Data on patients who dropped out were censored at the time of last contact. Methods for imputation of missing data are described in the protocol. All patients who received at least one dose of verubecestat or placebo were included in the safety analyses. Prespecified adverse events of interest included amyloid-related imaging abnormalities, delirium, and clinically significant rash. All statistical analyses were performed with the use of SAS software, versions 9.3 and 9.4 (SAS Institute).
We calculated that 450 patients per trial group would be needed to provide the trial with 90% power to show a significant difference between at least one of the dose levels of verubecestat and placebo in the primary outcome. This calculation was based on an estimated cumulative dropout rate of 25% (i.e., 383 patients who completed the trial per group at week 104) and on an assumed drug effect of 35% for both dose levels of verubecestat (which would correspond to a 0.64-point difference in the CDR-SB score between patients receiving verubecestat and those receiving placebo). The rate of progression (projected worsening of 1.8 points) at 104 weeks in the placebo group was estimated with the use of data from the Alzheimer’s Disease Neuroimaging Initiative (ClinicalTrials.gov number, NCT01231971). Analyses at other time points and subgroup anaIyses were prespecified, but the triaI was not powered for such anaIyses. Interim safety anaIyses are described in the protocoI.
RESULTS
PATIENT CHARACTERISTICS
A total of 1454 patients were enrolled; 485 were randomly assigned to receive oral verubecestat at a dose of 12 mg per day (the 12-mg group), 484 to receive verubecestat at a dose of 40 mg per day (the 40-mg group), and 485 to receive matching placebo (Fig. 1). The number of patients in each group in the modified intention-to-treat population differed from the number of patients who were randomly assigned to a trial group (Table 1) because of differences in the numbers of missing data in each group. A total of 704 patients (47.7 to 49.3% of the patients in each group) completed part 1 of the trial (Fig. 1), and 593 of these patients entered part 2 (the extension phase) (Fig. S1 in the Supplementary Appendix).
Figure 1. Randomization, Trial-Group Assignment, and Follow-up in Part 1 of the Trial.
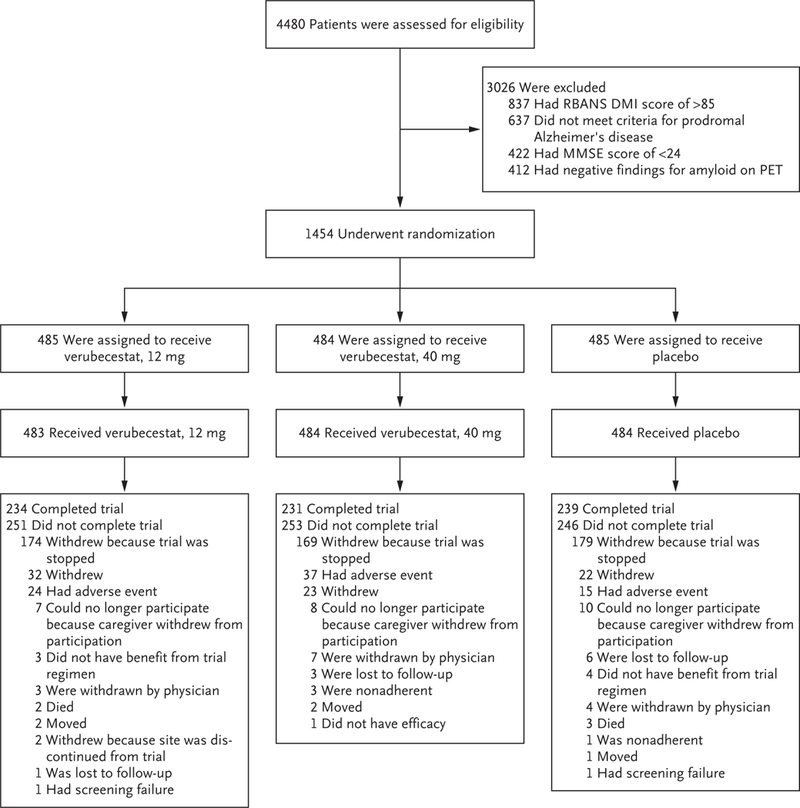
Only the most common reasons for exclusion from the trial are shown. Scores on the Repeatable Battery for the Assessment of Neuropsychological Status Delayed Memory Index (RBANS DMI) range from 40 to 160, with lower scores indicating worse memory. Scores on the Mini-Mental State Examination (MMSE) range from 0 to 30, with lower scores indicating poorer cognitive performance. PET denotes positron-emission tomography.
Table 1.
Demographic and Baseline Clinical Characteristics ofthe Patients.*
| Characteristic | Verubecestat 12-mg Group (N = 485) |
Verubecestat 40-mg Group (N = 484) |
Placebo Group (N = 485) |
|---|---|---|---|
| Demographic and clinical characteristics† | |||
| Age — yr | 71.7±7.1 | 71.0±7.4 | 71.6±7.1 |
| Female sex — no. (%) | 229 (47.4) | 244 (50.4) | 213 (44.0) |
| Race — no. (%)‡ | |||
| White | 396 (82.0) | 392 (81.0) | 390 (80.6) |
| Asian | 79 (16.4) | 85 (17.6) | 84 (17.4) |
| Other | 8 (1.7) | 7 (1.4) | 10 (2.1) |
| Geographic region — no. (%) | |||
| United States or Canada | 226 (46.8) | 224 (46.3) | 226 (46.7) |
| Europe, Australia, or New Zealand | 163 (33.7) | 163 (33.7) | 161 (33.3) |
| Japan | 57 (11.8) | 60 (12.4) | 59 (12.2) |
| Other | 37 (7.7) | 37 (7.6) | 38 (7.9) |
| APOE4 carrier — no. (%) | 328 (67.9) | 337 (69.6) | 335 (69.2) |
| MMSE score of ≥27 — no. (%)§ | 212 (43.9) | 211 (43.6) | 214 (44.2) |
| Treatment with acetylcholinesterase inhibitor or memantine for Alzheimer’s disease — no. (%) | 222 (46.0) | 225 (46.5) | 222 (45.9) |
| Education level below undergraduate degree — no. (%) | 243 (50.3) | 240 (49.6) | 250 (51.7) |
| Baseline scores on clinical outcome measures¶ | |||
| CDR-SB | |||
| No. of patients assessed | 465 | 458 | 469 |
| Score | 2.74±1.31 | 2.67±1.26 | 2.58±1.19 |
| CCS-3D | |||
| No. of patients assessed | 441 | 424 | 440 |
| Score | 0.03±1.04 | 0.02±0.99 | −0.06±0.98 |
| ADCS-ADLMCI | |||
| No. of patients assessed | 469 | 462 | 472 |
| Score | 42.2±5.9 | 43.1±5.4 | 42.8±5.9 |
| ADAS-cog13 | |||
| No. of patients assessed | 468 | 465 | 475 |
| Score | 22.1±6.6 | 22.6±6.5 | 21.9±6.4 |
| ADAS-cog11 | |||
| No. of patients assessed | 468 | 465 | 475 |
| Score | 13.4±4.9 | 13.8±4.8 | 13.4±4.7 |
| MMSE | |||
| No. of patients assessed | 466 | 463 | 475 |
| Score | 26.3±1.8 | 26.4±1.7 | 26.3±1.8 |
| NPI | |||
| No. of patients assessed | 463 | 459 | 472 |
| Score | 5.9±8.6 | 5.5±8.2 | 5.5±8.3 |
| Biomarker values¶ | |||
| Hippocampal volume on MRI — microliters | |||
| No. of patients assessed | 168 | 181 | 191 |
| Value | 6448±1107 | 6469±1106 | 6435±987 |
| Cortical amyloid load on PET — SUVR | |||
| No. of patients assessed | 63 | 59 | 65 |
| Value | 0.86±0.07 | 0.87±0.07 | 0.85±0.06 |
Plus-minus values are means ±SD. Percentages may not total 100 because of rounding. MRI denotes magnetic resonance imaging, PET positron-emission tomography, and SUVR standardized uptake value ratio.
Shown are data from patients who received at least one dose of the trial regimen (483 patients in the verubecestat 12-mg group, 484 patients in the verubecestat 40-mg group, and 485 patients in the placebo group).
Race was reported by the patients.
Scores on the Mini-Mental State Examination (MMSE) range from 0 to 30, with lower scores indicating poorer cognitive performance.
Shown are data from patients who were included in the modified intention-to-treat population. This population included patients who received at least one dose of the trial regimen and had a baseline measure and at least one valid follow-up outcome measure after randomization and positive findings for Alzheimer’s disease on PET (10 patients who had negative findings on PET but who had a positive tau:Aβ42 ratio in cerebrospinal fluid were also enrolled to validate a companion diagnostic test, but they are not included in the analyses). The number of patients evaluated for each outcome differed depending on which outcome measure was obtained. MRI and PET measures were obtained from a subgroup of patients. Scores on the Clinical Dementia Rating Scale-Sum of Boxes (CDR-SB) range from 0 to 18, with higher scores indicating worse cognition and daily function. Scores on the Composite Cognition Score-3 Domain (CCS-3D) are the mean z scores from multiple individual cognitive tests. Scores on the Alzheimer’s Disease Cooperative Study Activities of Daily Living for Mild Cognitive Impairment Inventory scale (ADCS-ADLMCI) range from 0 to 53, with lower scores indicating worse function. Scores on the 13-item cognitive subscale of the Alzheimer’s Disease Assessment Scale (ADAS-cog13) range from 0 to 85, with higher scores indicating worse cognition. Scores on the 11-item cognitive subscale of the Alzheimer’s Disease Assessment Scale (ADAS-cog11) range from 0 to 70, with higher scores indicating worse cognition. Neuropsychiatric Inventory (NPI) scores range from 1 to 144, with higher scores indicating more severe symptoms.
A decision to stop the trial was made in February 2018 at the recommendation of the data and safety monitoring committee on the basis of futility of finding superiority for either verubecestat dose over placebo (see the Supplementary Appendix for a description of the stopping rules for futility). At the time of trial termination, enrollment had been completed and approximately 12 months remained before the scheduled completion of part 1. Because of the early termination of the trial, the number of patients who completed the trial at week 104 in each group (231 in the 12-mg group, 234 in the 40-mg group, and 239 patients in the placebo group) (Fig. 1) was less than the 338 patients per group who had been expected to complete the trial. None of the patients completed the extension phase because of the early termination (Fig. S1 in the Supplementary Appendix).
Patient characteristics and baseline scores on efficacy measures were similar among the three trial groups (Table 1). A total of 69% of the patients were APOE4 carriers, and 46% were taking concurrent medication for Alzheimer’s disease.
CLINICAL OUTCOMES
The model-based mean change score from baseline to week 104 in the CDR-SB score (the primary outcome) was 1.65 in the 12-mg group, 2.02 in the 40-mg group, and 1.58 in the placebo group (P = 0.67 for the comparison between the 12-mg group and the placebo group and P = 0.01 for the comparison between the 40-mg group and the placebo group, favoring the placebo group) (Table 2). In an exploratory analysis according to time point, scores on the CDR-SB were also higher (signifying more impairment of cognition and daily functioning) in the 40-mg group than in the placebo group at 13, 26, and 52 weeks, with the lower limit of unadjusted confidence intervals greater than 0, suggesting but not confirming the possibility of worse performance at these earlier time points in the high-dose verubecestat group (Fig. 2A, and Table S1 in the Supplementary Appendix). Exploratory subgroup analyses did not suggest that the effect of verubecestat on the CDR-SB score was altered by baseline APOE4 gene-carrier status, age, sex, MMSE score, or PET SUVR (Table S2 in the Supplementary Appendix). CDR-SB results were similar in the sensitivity analysis, which excluded assessments performed after announcement of the trial termination (Table S3 in the Supplementary Appendix). The failure to support the primary hypothesis of superiority of verubecestat over placebo with respect to the CDR-SB score precluded formal statistical inferences for the remaining outcomes.
Table 2.
Change in Primary and Secondary Outcomes from Baseline to Week 104.*
| Outcome | Change from Baseline (95% Cl) | Difference in Mean Change | |||||
|---|---|---|---|---|---|---|---|
| Verubecestat 12 mg |
Verubecestat 40 mg |
Placebo | Verubecestat 12 mg vs. Placebo (97.51% Cl or 95%CI)† |
P Value ‡ | Verubecestat 40 mg vs. Placebo (97.51% Cl or 95%CI)† |
P Value ‡ | |
| Primary outcome | |||||||
| CDR-SB score | 1.65 (1.42 to 1.87) | 2.02 (1.75 to 2.28) | 1.58 (1.34 to 1.81) | 0.07 (−0.30 to 0.44) | 0.67 | 0.44 (0.04 to 0.84) | 0.01 |
| Secondary outcomes | |||||||
| Progression to dementia — no. of events/ 100 patient-yr§ |
24.5 | 25.5 | 19.3 | — | — | — | — |
| Change in CDR-SB score wk 13 to wk 104 |
— | — | — | 0.0 (−0.3 to 0.4) | 0.3 (−0.1 to 0.7) | ||
| CCS-3D score | 0.77 (0.66 to 0.87) | 0.78 (0.66 to 0.89) | 0.77 (0.65 to 0.89) | 0.00 (−0.18 to 0.17) | 0.01 (−0.18 to 0.20) | ||
| Hippocampal volume on MRI — % change¶ |
−6.5 (−6.9 to −6.2) | −6.7 (−7.1 to−6.3) | −6.1 (−6.5 to−5.7) | −0.4 (−1.0 to 0.2) | −0.6 (−1.2 to 0.0) | ||
| Cortical amyloid load on PET —SUVR¶ |
−0.03 (−0.04 to −0.03) | −0.04 (−0.05 to −0.04) | 0.02 (0.02 to 0.03) | −0.05 (−0.06 to −0.04) | −0.06 (−0.07 to −0.05) | ||
| ADCS-ADLMCI score | −5.2 (−6.1 to−4.3) | −5.8 (−6.8 to −4.8) | −4.1 (−5.0 to−3.3) | −1.0 (−2.4 to 0.4) | −1.7 (−3.2 to−0.2) | ||
| Total tau in CSF | NA | NA | NA | NA | NA | ||
| Exploratory outcomes | |||||||
| ADAS-cog13 score | 8.0 (7.2 to 8.9) | 8.2 (7.3 to 9.1) | 6.9 (6.1 to 7.7) | 1.1 (−0.1 to 2.3) | 1.3 (0.1 to 2.6) | ||
| ADAS-cog11 score | 6.3 (5.5 to 7.0) | 6.5 (5.7 to 7.3) | 5.2 (4.5 to 5.9) | 1.1 (0.1 to 2.1) | 1.3 (0.2 to 2.3) | ||
| MMSE score | −3.8 (−4.2 to−3.4) | −4.7 (−5.3 to−4.2) | −3.9 (−4.4 to −3.5) | 0.1 (−0.5 to 0.8) | −0.8 (−1.5 to−0.1) | ||
| NPI score | 2.7 (1.4 to 3.9) | 3.5 (2.1 to 4.8) | 1.5 (0.5 to 2.5) | 1.1 (−0.5 to 2.7) | 1.9 (0.2 to 3.6) | ||
Values shown are least-squares means with confidence intervals, with the exception of progression to dementia, for which the numbers of events per 100 patient-years are shown. All analyses except progression to dementia due to Alzheimer’s disease are based on longitudinal analysis of covariance with categorical factors of geographic region, time, trial-group assignment, sex, APOE4 genotype, baseline use of Alzheimer’s disease medication, baseline use of vitamin E, and the interaction of time according to trial-group assignment with baseline value, the interaction of baseline value and time, the baseline value ofthe MMSE score, and the baseline value of age included as continuous covariates. For CDR-SB, CCS-3D, ADAS-cog, and NPI, a higher positive mean change-from-baseline score corresponds to a greater decline relative to baseline, and a positive treatment difference indicates a greater decline with verubecestat than with placebo. For ADCS-ADLmci and MMSE, a higher negative mean change-from-baseline score corresponds to a greater decline relative to baseline, and a negative treatment difference indicates a greater decline with verubecestat than with placebo. For the change in CDR-SB score from week 13 to week 104, a positive treatment difference indicates a greater worsening at week 104 than at week 13 with verubecestat than with placebo. Progression to dementia due to Alzheimer’s disease is based on a Cox pro-portional-hazards model with adjustment for APOE4 genotype, sex, geographic region, baseline use of medication for Alzheimer’s disease, and baseline use of vitamin E as categorical terms, with baseline age and baseline MMSE score as continuous covariates. A higher change-from-baseline score corresponds to more events (greater decline) relative to baseline. CSF denotes cerebrospinal fluid, and NA not applicable.
The 97.51% confidence intervals are presented for primary and secondary outcomes, and the 95% confidence intervals are presented for exploratory outcomes (unadjusted for multiple comparisons).
P values for the primary outcome are two-sided. According to the strategy of adjustment for multiple testing, the primary hypothesis that verubecestat is superior to placebo with respect to the change from baseline in the CDR-SB score was not supported at either dose level; thus, further formal hypothesis testing was precluded.
In the category of progression to dementia due to Alzheimer’s disease, the hazard ratio for 12 mg vs. placebo was 1.30 (1.01 to 1.68), and the hazard ratio for 40 mg vs. placebo was 1.38 (1.07 to 1.79). A hazard ratio greater than 1 indicates more events (greater decline) with verubecestat than with placebo.
These analyses were based on smaller or substantially smaller sample sizes than the analyses for the clinical outcome scales. For biomarkers, a negative mean change corresponds to a reduction in the biomarker value relative to baseline and a positive mean change indicates an increase in the biomarker value relative to baseline.
Figure 2. Mean Change from Baseline in the CDR-SB, CCS-3D, and ADCS-ADLMCI Scores over 104 Weeks.
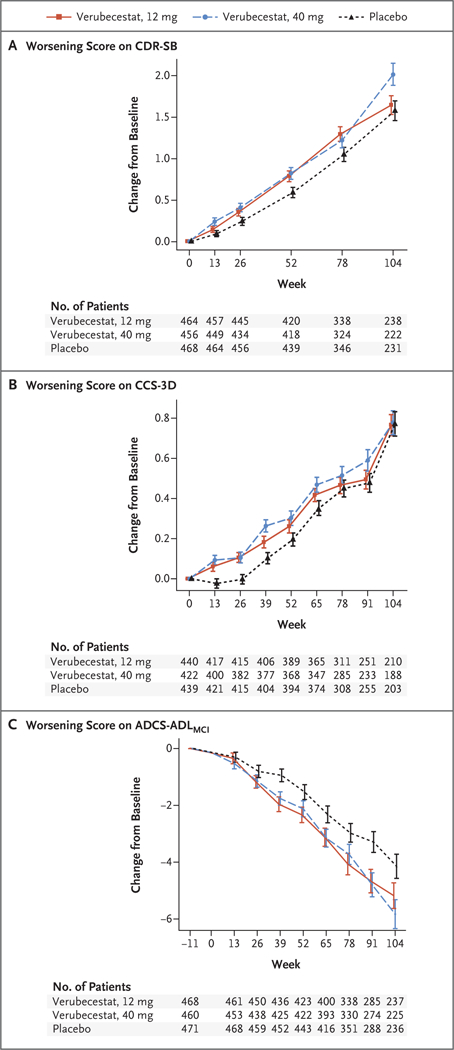
Panel A shows the mean change from baseline in the score on the Clinical Dementia Rating Scale-Sum of Boxes (CDR-SB); scores range from 0 to 18, with higher scores indicating worse cognition and daily function. Panel B shows the mean change from baseline in the score on the three-domain composite cognition score (CCS-3D; derived from the z scores [mean of 0, standard deviation of 1, with higher scores indicating worse cognition] of tests of episodic memory, executive function, and attention and processing speed). Panel C shows the mean change from baseline in the score on the Alzheimer’s Disease Cooperative Study Activities of Daily Living for Mild Cognitive Impairment Inventory scale (ADCS-ADLMCI); scores range from 0 to 53, with lower scores indicating worse function. Baseline is plotted at week −11, which is the mean assessment time of the baseline measurement as offset from the first dose of trial agent at week 0. As a result, there are no data plotted at week 0. The time course of the verubecestat groups between week −11 and week 0 was assumed to follow the same course as the placebo group. From this week 0 placebo coordinate, the time course for each respective verubecestat group was extended to the estimate at the first scheduled postdose time point. I bars indicate standard errors.
The event rates for dementia due to Alzheimer’s disease per 100 patient-years were 24.5 in the 12-mg group, 25.5 in the 40-mg group, and 19.3 in the placebo group (hazard ratio, 1.30; 97.51% confidence interval [CI], 1.01 to 1.68, unadjusted for multiple comparisons, for the comparison between the 12-mg group and the placebo group; and hazard ratio, 1.38; 97.51% CI, 1.07 to 1.79, unadjusted for multiple comparisons, for the comparison between the 40-mg group and the placebo group). Results for the other secondary and exploratory outcomes of cognition (the CCS-3D, ADAS-cog, and MMSE scores), function (the ADCS-ADLMCI score), and neuropsychiatric symptoms (the NPI score) also suggested that verubecestat may be inferior to placebo, since the unadjusted confidence intervals excluded 0 for three of the five remaining secondary outcomes (this excludes concentrations of tau in cerebrospinal fluid, which were not analyzed) and all four exploratory outcomes (Table 2 and Fig. 2, and Tables S1 and Figs. S2 through S6 in the Supplementary Appendix). These comparisons are exploratory, and the confidence intervals are unadjusted; thus, the strength of any resulting inferences is limited.
BIOMARKERS
The hippocampal volume, as assessed by MRI, was lower at week 104 than at baseline, by 6.1% in the placebo group and by 6.5 to 6.7% in the verubecestat groups (Table 2, and Fig. S7 in the Supplementary Appendix). An increase from baseline to week 104 in the brain amyloid load, as assessed by PET, was observed in the placebo group; in contrast, there was a reduction from baseline in the brain amyloid load in both verubecestat groups (Table 2, and Figs. S8 and S9 in the Supplementary Appendix). Since there were few patients in the substudy of biomarkers in cerebrospinal fluid (five or six patients per group), planned analyses were not performed; however, greater than 60% reductions from baseline in concentrations of Aβ42, Aβ40, and sAPPβ in cerebrospinal fluid were seen with verubecestat (Table S4 in the Supplementary Appendix).
SAFETY
In part 1 of the trial, adverse events were more common with verubecestat than with placebo (Table 3). Serious adverse events that were reported in more than 1% of the patients in at least one trial group in this part of the trial included osteoarthritis, basal-cell carcinoma, squamouscell carcinoma, syncope, and prostate cancer, with similar incidences among the groups. In part 1 of the trial, there were three deaths in the placebo group, three in the 12-mg group, and one in the 40-mg group (Table 3, and Table S5 in the Supplementary Appendix).
Table 3.
Adverse Events That Occurred within 14 Days after the Last Dose over 104 Weeks in Part 1 of the Trial.
| Event | Verubecestat 12-mg Group (N = 483) |
Verubecestat 40-mg Group (N =484) |
Placebo Group (N =484) |
Absolute Difference between 12-mg Group and Placebo Group |
Absolute Difference between 40-mg Group and Placebo Group |
|---|---|---|---|---|---|
| number of patients (percent) | percentage points (95% CI) | ||||
| Any adverse event | 441 (91.3) | 446 (92.1) | 421 (87.0) | 4.32 (0.40 to 8.31) | 5.17 (1.33 to 9.09) |
| Any serious adverse event | 124 (25.7) | 101 (20.9) | 96 (19.8) | 5.84 (0.55 to 11.12) | 1.03 (−4.05 to 6.12) |
| Adverse event that resulted in discontinuation | 32 (6.6) | 49 (10.1) | 22 (4.5) | 2.08 (−0.84 to 5.10) | 5.58 (2.35 to 8.99) |
| of assigned trial regimen | |||||
| Death | 3 (0.6) | 1 (0.2) | 3 (0.6) | 0.00 (−1.26 to 1.26) | −0.41 (−1.62 to 0.59) |
| Prespecified adverse events | |||||
| ARIA-H* | 5 (1.0) | 3 (0.6) | 3 (0.6) | 0.42 (−0.89 to 1.85) | 0.00 (−1.26 to 1.26) |
| ARIA-E* | 0 | 0 | 0 | 0.00 (−0.79 to 0.79) | 0.00 (−0.79 to 0.79) |
| Delirium | 5 (1.0) | 6 (1.2) | 9 (1.9) | -0.82 (−2.58 to 0.77) | −0.62 (−2.41 to 1.05) |
| Rashi† | 21 (4.3) | 28 (5.8) | 6 (1.2) | 3.11 (1.11 to 5.45) | 4.55 (2.36 to 7.12) |
| Specific adverse events with incidence of>5.0% in a verubecestat group and with greater incidence in the verubecestat group than in the placebo group‡ | |||||
| Rash, dermatitis, or urticaria§ | 96 (19.9) | 101 (20.9) | 62 (12.8) | 7.07 (2.42 to 11.75) | 8.06 (3.37 to 12.78) |
| Depression | 32 (6.6) | 50 (10.3) | 25 (5.2) | 1.46 (−1.55 to 4.54) | 5.17 (1.83 to 8.65) |
| Anxiety | 33 (6.8) | 44 (9.1) | 21 (4.3) | 2.49 (−0.41 to 5.52) | 4.75 (1.64 to 8.04) |
| Sleep disturbance§ | 38 (7.9) | 44 (9.1) | 22 (4.5) | 3.32 (0.29 to 6.50) | 4.55 (1.40 to 7.85) |
| Weight loss | 27 (5.6) | 32 (6.6) | 10 (2.1) | 3.52 (1.16 to 6.16) | 4.55 (2.07 to 7.33) |
| Cough | 28 (5.8) | 30 (6.2) | 15 (3.1) | 2.70 (0.10 to 5.47) | 3.10 (0.46 to 5.92) |
| Psychotic symptoms§ | 15 (3.1) | 27 (5.6) | 11 (2.3) | 0.83 (−1.29 to 3.05) | 3.31 (0.90 to 5.96) |
| Pruritus | 28 (5.8) | 22 (4.5) | 15 (3.1) | 2.70 (0.10 to 5.47) | 1.45 (−1.02 to 4.02) |
| Other adverse events of interest¶ | |||||
| Falls and injuries§ | 124 (25.7) | 123 (25.4) | 100 (20.7) | 5.01 (−0.31 to 10.33) | 4.75 (−0.56 to 10.05) |
| Suicidal ideation | 33 (6.8) | 45 (9.3) | 31 (6.4) | 0.43 (−2.77 to 3.64) | 2.89 (−0.51 to 6.38) |
| Hair-color change | 12 (2.5) | 24 (5.0) | 0 | 2.48 (1.43 to 4.29) | 4.96 (3.35 to 7.27) |
| Syncope-like events§ | 15 (3.1) | 18 (3.7) | 14 (2.9) | 0.21 (−2.05 to 2.50) | 0.83 (−1.50 to 3.23) |
In the subgroup of patients in whom magnetic resonance imaging was performed before 14 days after the last dose, 4 of 89 patients (4.5%) in the 12-mg group, 2 of 80 patients (2.5%) in the 40-mg group, and 3 of 93 patients (3.2%) in the placebo group had ARIA-H (amyloid-related imaging abnormalities of incident microhemorrhage, superficial siderosis, or macrohemorrhage); no patients in this subgroup had ARIA-E (amyloid-related imaging abnormalities of incident vasogenic edema).
These adverse events of rash were determined by the investigator to be clinically significant.
The greater incidence in a verubecestat group than in the placebo group was defined as an adverse event in which the lower limit of the 95% confidence interval for the difference between the verubecestat group and the placebo group was greater than 0.
Several specific adverse event terms that appeared to be related were combined in a post hoc and unblinded fashion in the composite items shown; psychotic symptoms included adverse event terms of paranoia, delusion, and hallucination.
The adverse events in this category were based on data from previous clinical trials.
Among the prespecified adverse events of clinical interest in part 1, verubecestat was associated with a greater incidence of rash than placebo but not with a greater incidence of delirium or amyloid-related imaging abnormalities. Common adverse events (reported in >5% of patients in any trial group) that were reported more frequently in both verubecestat groups than in the placebo group (lower limit of the 95% confidence interval for the difference of >0) included rash, dermatitis, or urticaria; sleep disturbance; weight loss; and cough (Table 3). A change in hair color was observed in both the 12-mg group (2.5%) and the 40-mg group (5.0%) but not in the placebo group (Table 3). The incidence of falls and injuries and suicidal ideation was higher in the verubecestat groups than in the placebo group, but the lower limits of the 95% confidence intervals of differences between groups included zero for both doses as compared with placebo (Table 3, and Table S6 in the Supplementary Appendix). There was a mean (±SD) weight change at week 104 of −1.3±4.2 kg in the 12-mg group and −1.3±5.3 kg in the 40-mg group, as compared with 0.4±4.0 kg in the placebo group. Adverse events and deaths in the extension phase are summarized in Tables S7 and S8 in the Supplementary Appendix. Results of the pharmacokinetic analysis are provided in the Supplementary Appendix and are generally similar to those in previous studies.5,6
DISCUSSION
In this trial of two dose levels of a BACE inhibitor in patients with prodromal Alzheimer’s disease in whom deposition of brain amyloid had been detected on PET, verubecestat showed no benefit with respect to the primary clinical outcome (a change in the CDR-SB score from baseline to week 104), as compared with placebo. The 40-mg group, but not the 12-mg group, had a worse outcome on this measure. The increase in clinical decline attributable to verubecestat as compared with placebo, as measured with the CDR-SB score, was smaller than the clinically relevant threshold used in our power calculations. A formal statistical analysis was not performed because of the hierarchical analysis plan and failure to show superiority of the drug in the analysis of the primary outcome; however, the confidence intervals around the hazard ratios of difference between the 12-mg and 40-mg groups and the placebo group in event rates of dementia due to Alzheimer’s disease did not include 1, suggesting that verubecestat may have accelerated the progression to diagnosis of dementia due to Alzheimer’s disease. In exploratory analyses, both dose levels of verubecestat were associated with poorer outcomes on the CCS-3D and the ADAS-cog measures of cognition that, relative to placebo, appeared worse at week 13 and did not appear to progress thereafter on the basis of unadjusted confidence intervals that excluded zero for between-group differences.
In the PET substudy, the cortical amyloid load increased over time in the placebo group and declined in the verubecestat groups but did not reach the 0.69 SUVR threshold for being amyloid-negative. Although the substudy of cerebrospinal fluid findings included only five or six patients per group, the results were consistent with those of previous studies showing greater than 60% reductions in concentrations of Aβ42 and related APP metabolites.5,6 Taken together, these findings indicate that verubecestat was acting at its intended target.
Adverse events were more common with verubecestat than with placebo and were similar to those seen in the trial of the drug in patients with mild-to-moderate Alzheimer’s disease.6 As in the previous trial, verubecestat was not associated with amyloid-related imaging abnormalities that have been reported with antiamyloid immunotherapies.24–26
The results of the current trial differed from those in the verubecestat trial involving patients with mild-to-moderate dementia due to Alzheimer’s disease, in whom no overall treatment effects were seen on cognition or function at week 78.6 However, in that previous trial involving patients who had established dementia due to Alzheimer’s disease, a prespecified unadjusted analysis of changes from baseline in ADAS-cog scores showed that these scores were worse at 13 weeks in both dose groups than in the placebo group. Given baseline measures and rates of progression, we estimate that the patients in the current trial were on average 3 to 4 years earlier in the disease course than patients in the previous trial. Patients at an earlier stage of the disease may be more sensitive to the effects of substantial BACE-1 inhibition, perhaps because of a role of BACE-1 in normal synaptic function.27–31 It is also possible that the effects of verubecestat are due to inhibition of BACE-2.28,29
In conclusion, in patients with prodromal Alzheimer’s disease, verubecestat did not have a beneficial effect on clinical outcomes. Some measures suggested possible worsening of cognition and daily function.
Supplementary Material
Acknowledgments
Supported by Merck Sharp & Dohme (MSD).
We thank the patients, caregivers, and families for their participation in the trial; the site investigators and their staff for performance of the trial; the members of the trial committees for trial oversight; Julie Stone and Marissa Dockendorf (both from MSD) and David Jaworowicz and Rebecca Humphrey (both from Cognigen) for the pharmacokinetic analysis; Nicole Dupre, Theresa Taylor, Ingrid Banks, Regina Gottwald, Erin Paradis, Swati Mercer, Saheeda Jackson, Carol Yacik, Carolyn DaSilva, Jill Anderson, Julie Stromswold, Diana Asbjorn, Karen E. Ramsey, Kate Civello, Sylwia Kucharska, Magdalena Bik, Grzegorz Toma-sik, Anna Blonska, Brandy Cahill, Deborah Matzura-Wolfe, and Madhu Katragadda (all from MSD) for their administrative support; Christopher Randolph of MedAvante for his assistance with the central rater review process; Christopher Lines of MSD for his assistance with the preparation of an earlier version of the manuscript; Sheila Erespe of MSD for her assistance with the submission of the manuscript; and Matthew Kennedy of MSD for helpful comments on an earlier draft of the article.
Footnotes
REFERENCES
- 1.Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL. Alzheimer’s disease. Nat Rev Dis Primers 2015;1:15056. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 2016;8:595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yan R, Vassar R. Targeting the β secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol 2014;13:319–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scott JD, Li SW, Brunskill AP, et al. Discovery of the 3-imino-1,2,4-thiadiazi-nane 1,1-dioxide derivative verubecestat (MK-8931) — a 0-site amyloid precursor protein cleaving enzyme 1 inhibitor for the treatment of Alzheimer’s disease. J Med Chem 2016;59:10435–50. [DOI] [PubMed] [Google Scholar]
- 5.Kennedy ME, Stamford AW, Chen X, et al. The BACE1 inhibitor verubecestat (MK-8931) reduces CNS β-amyloid in animal models and in Alzheimer’s disease patients. Sci Transl Med 2016;8: 363ra150. [DOI] [PubMed] [Google Scholar]
- 6.Egan MF, Kost J, Tariot PN, et al. Randomized trial of verubecestat for mild-to-moderate Alzheimer’s disease. N Engl J Med 2018;378:1691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan R. Physiological functions of the β-site amyloid precursor protein cleaving enzyme 1 and 2. Front Mol Neurosci 2017; 10:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu F, Zhang Y, Liang Z, et al. Cleavage of potassium channel Kv2.1 by BACE2 reduces neuronal apoptosis. Mol Psychiatry 2018;23:1542–54. [DOI] [PubMed] [Google Scholar]
- 9.Holler CJ, Webb RL, Laux AL, et al. BACE2 expression increases in human neurodegenerative disease. Am J Pathol 2012;180:337–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jack CR Jr, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 2013;12:207–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sperling R, Mormino E, Johnson K. The evolution of preclinical Alzheimer’s disease: implications for prevention trials. Neuron 2014;84:608–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol 2010;9: 1118–27. [DOI] [PubMed] [Google Scholar]
- 13.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984; 34:939–44. [DOI] [PubMed] [Google Scholar]
- 14.Diagnostic and statistical manual of mental disorders, 4 th ed., rev.: DSM-IV-TR Arlington, VA: American Psychiatric Association, 2000. [Google Scholar]
- 15.Randolph C. RBANS: repeatable battery for the assessment of neuropsychological status. San Antonio, TX: Psychological Corporation, 1998. [Google Scholar]
- 16.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state:” a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12: 189–98. [DOI] [PubMed] [Google Scholar]
- 17.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412–4. [DOI] [PubMed] [Google Scholar]
- 18.Galasko D, Bennett D, Sano M, et al. An inventory to assess activities of daily living for clinical trials in Alzheimer’s disease. Alzheimer Dis Assoc Disord 1997; 11:Suppl 2:S33–S39. [PubMed] [Google Scholar]
- 19.Mohs RC, Knopman D, Petersen RC, et al. Development of cognitive instruments for use in clinical trials of antidementia drugs: additions to the Alzheimer’s Disease Assessment Scale that broaden its scope. Alzheimer Dis Assoc Disord 1997;11:Suppl 2:S13–S21. [PubMed] [Google Scholar]
- 20.Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology 1994;44: 2308–14. [DOI] [PubMed] [Google Scholar]
- 21.Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry 2011;168:1266–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Landau SM, Fero A, Baker SL, et al. Measurement of longitudinal β-amyloid change with 18F-florbetapir PET and standardized uptake value ratios. J Nucl Med 2015;56:567–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mo Y, Stromswold J, Wilson K, et al. A multinational study distinguishing Alzheimer’s and healthy patients using cerebrospinal fluid tau/Aβ42 cutoff with concordance to amyloid positron emission tomography imaging. Alzheimers Dement (Amst) 2017;6:201–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salloway S, Sperling R, Fox NC, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med 2014;370:322–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doody RS, Thomas RG, Farlow M, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med 2014;370:311–21. [DOI] [PubMed] [Google Scholar]
- 26.Sevigny J, Chiao P, Bussière T, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016;537:50–6. [DOI] [PubMed] [Google Scholar]
- 27.Zhu K, Peters F, Filser S, Herms J. Consequences of pharmacological BACE inhibition on synaptic structure and function. Biol Psychiatry 2018;84:478–87. [DOI] [PubMed] [Google Scholar]
- 28.Barao S, Moechars D, Lichtenthaler SF, De Strooper B. BACE1 physiological functions may limit its use as therapeutic target for Alzheimer’s disease. Trends Neurosci 2016;39:158–69. [DOI] [PubMed] [Google Scholar]
- 29.Vassar R, Kuhn PH, Haass C, et al. Function, therapeutic potential and cell biology of BACE proteases: current status and future prospects. J Neurochem 2014; 130:4–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blume T, Filser S, Jaworska A, et al. BACE1 inhibitor MK-8931 alters formation but not stability of dendritic spines. Front Aging Neurosci 2018;10:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Filser S, Ovsepian SV, Masana M, et al. Pharmacological inhibition of BACE1 impairs synaptic plasticity and cognitive functions. Biol Psychiatry 2015;77:729–39. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.