

Abstract
Mitochondrial metabolism plays a central role in insulin secretion in pancreatic beta-cells. Generation of protonmotive force and ATP synthesis from glucose-originated pyruvate are critical steps in the canonical pathway of glucose-stimulated insulin secretion. Mitochondrial metabolism is intertwined with pathways that are thought to amplify insulin secretion with mechanisms distinct from the canonical pathway, and the relative importance of these two pathways is controversial. Here I show that glucose-induced mitochondrial membrane potential (MMP) hyperpolarization is necessary for, and predicts, the rate of insulin secretion in primary cultured human beta-cells. When glucose concentration is elevated, increased metabolism results in a substantial MMP hyperpolarization, as well as in increased rates of ATP synthesis and turnover marked by faster cell respiration. Using modular kinetic analysis I explored what properties of cellular energy metabolism enable a large glucose-induced change in MMP in human beta-cells. I found that an ATP-dependent pathway activates glucose or substrate oxidation, acting as a positive feedback in energy metabolism. This activation mechanism is essential for concomitant fast respiration and high MMP, and for a high magnitude glucose-induced MMP hyperpolarization and therefore for insulin secretion.
Keywords: oxidative phosphorylation, mitochondrial membrane potential, cell respiration, metabolic control analysis, glucose-stimulated insulin secretion, diabetes
1. Introduction
Mitochondrial energization, more precisely the protonmotive force across the mitochondrial inner membrane, or its readily measurable major component, the mitochondrial membrane potential (ΔψM), and the downstream ATP/ADP, play a central signaling role in glucose-stimulated insulin secretion (GSIS) in pancreatic β-cells. When glucose concentration is increased, its metabolism results in substantial hyperpolarization of ΔψM [1–3]. A concomitant increase in ATP/ADP, or effectively a drop in [ADP] [4,5], closes ATP-sensitive K-channels on the plasma membrane and depolarizes plasma membrane potential (ΔψP). The resulting Ca2+ influx through voltage-dependent Ca2+ channels triggers exocytosis. This is the triggering or canonical pathway of GSIS [6]. Mitochondrial metabolism has also been linked to insulin secretion by a set of intermediates, metabolic coupling factors, that are thought to mediate amplification of secretion, possibly in a KATP-independent manner [7,8]. However, the roles and contributions of these pathways are controversial [5,9,10].
The ongoing importance of this topic is marked by the pandemic prevalence of type 2 diabetes (T2D) [11]. The first-phase of insulin secretion is impaired in T2D and the pre-diabetic state, indicating β-cell dysfunction [12–18]. Bioenergetic compromise has been demonstrated in type 2 diabetic human pancreatic islets [19–21], in primary cultured human β-cells [2] and also in rodent models of diabetes [22–25].
Glucose-induced ΔψM hyperpolarization [9,26,27], and increase in ATP/ADP [28,29] or in ATP production [20,30] have been highlighted as good predictors of insulin secretion rates in rodent pancreatic β-cells and insulinoma cell lines. In contrast, others reported saturation of the ΔψM response at a lower glucose concentration than for insulin secretion [31,32]. Amplification of insulin secretion by metabolic coupling factors is thought to explain energization-independent actions of glucose [7,8]. Some of these metabolic coupling factors have been indicated to amplify insulin secretion at the level of exocytosis, such as glutamate ([33] but see [34]), monoacylglycerol [35], GSH and NADPH ([36] but see [30]). Others have no known target in the exocytotic machinery but may modulate metabolism, such as malonyl-CoA ([37] but see [38]), and mitochondrial GTP [39,40]. Inhibition of metabolic pathways that produce these factors lowered GSIS, while no effects were detected on glucose-induced ΔψM hyperpolarization and on ATP/ADP [41–43], or ATP levels did not correlate with secretion [39]. The limitations of assay technologies used to reach these conclusions are considered in the Discussion. Altogether, there is a generic disagreement not just between the relative importance of the canonical and metabolic coupling pathways, but also whether particular metabolites act as coupling factors [5,9,10]. Here I reevaluate and demonstrate the predictive role of ΔψM in GSIS using current state-of-the-art bioenergetic assay technologies in human primary β-cells.
A major (~30 mV) hyperpolarization of ΔψM [2] and about a doubling of the cytosolic ATP/ADP [28,29] concomitant with increased ATP usage, marked by almost doubled cell respiration [20,21,44–47] during increased availability of a single metabolic substrate has no trivial explanation. This is currently an unexplained paradox of β-cell bioenergetics [5,48]. In contrast to β-cells, many other tissues maintain stable ATP/ADP as workload or nutrient conditions change [49]. This β-cell-specific regulation of metabolism is possibly allowed by β-cell-specific gene expression [50].
Glucose stimulation of the β-cell changes virtually all variables of cellular energy metabolism [48,51,52], making it challenging to define which processes drive observed changes. Metabolic control analysis with appropriate modularization is a powerful method for understanding this through simplification [53–55]. Using control analysis of cellular energy metabolism we have previously described a positive feedback amplification of glucose metabolism in rodent insulinoma cells [56]. Here I show that this feedback mechanism has a particularly strong effect on glucose-stimulated ΔψM hyperpolarization in non-diabetic human β-cells, and works in an ATP-dependent manner. I propose that the hyperpolarization of ΔψM during GSIS is a result of activation of glucose oxidation and the observed positive feedback is responsible for this.
2. Materials and Methods
2.1. Materials
The ΔψP indicator (PMPI; #R8126 FLIPR Membrane Potential Assay Explorer Kit; red version) was from Molecular Devices (Sunnyvale, CA). AccuMAX was from Innovative Cell Technologies, Inc. (San Diego, CA). A custom RPMI 1640 medium formulated as powder without NaCl, NaHCO3, Ca(NO3)2, glucose, glutamine, biotin, riboflavin, folic acid and phenol red and with Na2HPO4 decreased to 0.91 mM; Geltrex (A1413302), tetramethylrhodamine methyl ester (TMRM) and other fluorescence probes were from Life Technologies (Carlsbad, CA); other fine chemicals were from Cayman Chemical Company (Ann Arbor, MI), Sigma-Aldrich (St. Louis, MO) or Santa Cruz Biotechnology (Dallas, TX), unless otherwise noted. The growth medium for dispersed islet cultures comprised PIM(S) medium (Prodo Laboratories Inc., Irvine, CA) supplemented with human serum (5%, Corning CV35–060CI), glutamine plus glutathione (PIM(G) from Prodo) according to the supplier’s recommendations, plus 100 units/ml penicillin and 100 μg/ml streptomycin. The potentiometric medium (PM) was made from a 2× stock (2×PM) of the modified RPMI 1640 containing (as final concentrations) 7.5 nM TMRM, 1:100 PMPI, 1 μM tetraphenylborate, 2.5 CaCl2, 3 glucose, 2 glutamine, 5 NaHCO3, 20 TES, which was then diluted to final volume using 240 mM NaCl (resulting in PM), 240 mM KCl (resulting in PMK) or 8 % paraformaldehyde (PFA) solution (resulting in PM-PFA, which was further supplemented with 120 mM KCl). All variants of the PM were kept in glass vials to prevent depletion of the probes. The respirometry medium (RM) closely matched the composition of the PM, contained the modified RPMI 1640, and in mM, 120 NaCl, 2.5 CaCl2, 3 glucose, 2 glutamine, 5 NaHCO3, 20 TES. Both PM and RM were free of serum or BSA unless otherwise noted, were set to pH 7.4 at 37 °C, and were supplemented with 1 μM zosuquidar [56] during assays to mitigate possible pumping of fluorescence probes by multidrug resistance proteins. PM and RM were used at 37°C in air.
2.2. Islet dispersion and cell culture
Human islets (3000–4000 islet equivalents; Prodo) were dispersed in a single batch upon receipt after overnight cold shipping, for each donor. Islets were incubated with AccuMAX (1.5 ml, for 10 min at 37°C), then triturated 30× with a 1 ml pipette tip in the presence of serum, and then with a 200 μl tip in growth medium. Dispersed islet cells for microscopy were plated at 5–6×103 cells per well in ice-cold growth medium in the presence of 5 v/v % Geltrex into coverglass-bottomed, half-area 96-well microplates (Corning 4580) previously coated overnight with polyethyleneimine (1:15000 w/v %). Plates were spun for 5 min at 500 g to attach cells before warming. Typically about total 100 wells were plated distributed between 5 plates. Culturing was performed at 37°C and 5% CO2. Cultures were fed with half replacement of the growth medium every other day.
2.3. Cell sorting
About 1–1.6×106 dispersed islet cells from above were re-suspended in RM containing 0.5 mM CaCl2 and 5.6 mM glucose (sorting medium), and stained with 3 μM FluoZin3-AM [57] for 40 min at 37°C on a rocker. Then the cell suspension was incubated in sorting medium with 5% human serum without the dye for 30 min at 37°C. Cells were pelleted and rigorously re-suspended in Ca2+-free sorting medium containing 5% human serum. Beta-cells were enriched using a BD FACSAria II cell sorter (BD Biosciences, San Jose CA) with a 130 μm nozzle aperture. Single cells were identified by forward and side scatter, and zinc-dependent fluorescence of FluoZin3 was measured by excitation at 488 nm and using a 530/30 nm band pass filter. Beta-cells were identified as a shoulder population comprising the highest 45% of cells in this channel. Sorted cells were washed 3 times with growth medium and plated into Seahorse XFe96 plates that were coated overnight with polyethyleneimine (1:15,000 w/v %) at 1.7×104 cells per well in the presence of 5 v/v % Geltrex in ice-cold growth medium. Plates were spun for 5 min at 500 g to attach cells before warming. Typical yields allowed recording from up to 12 wells distributed between two plates.
2.4. Mitochondria:cell volume fraction
Mitochondria:cell volume fractions (VF) were determined as described [2,58,59], with modifications. Briefly, dispersed islet cells in coverglass-bottomed 96-well microplates were loaded with MitoTracker Red CMXRos (60 nM) and calcein-AM (0.25 μM) in RM containing 5.6 mM glucose and imaged on a Zeiss LSM780 laser scanning confocal microscope using a Plan-Apochromat 63×/1.4 oil lens, 488 nm and 561 nm lasers and simultaneous detection at 493–556 nm and 566–690 nm. These, together with insulin immunofluorescence (see below) of the same view fields, were analyzed using the “Mitochondria:cell volume fractionator (basic with post-hoc staining)” image analysis pipeline in Image Analyst MKII (Image Analyst Software, Novato, CA) to yield mitochondria:cell volume fractions.
2.5. Determination of ΔψP and ΔψM
ΔψP and ΔψM were determined in absolute millivolts as previously described [2,58,59], with modifications. Time-lapse recordings were performed on a Nikon Ti-Eclipse Perfect Focus System fully motorized wide-field fluorescence microscope with Elements 4.2 software (Nikon) using an S-Fluor 10× air lens. Experiments were performed at 37 °C in air. Potentiometric experiments were started after 4 h preincubation of dispersed islet cell cultures in microplates in PM with 3 mM glucose. The long preincubation and subsequent washes aimed to equilibrate cells but also to saturate the microplate walls with the probes. 18 wells per plate, with two view fields per well, were recorded with 100 s cycle time. All treatments were performed by part-replacement of PM in the wells with supplemented PM previously loaded in a deep well 96-well reservoir lined with 0.7 ml glass inserts and kept at 37 °C within the environment chamber of the microscope. Using glass reservoirs was a critical innovation to avoid loss of potentiometric probes into the reservoir walls and also to enable the use of low nanomolar concentrations of rotenone and FCCP. Successive additions were pre-mixed with previous additions in a way that kept constant concentrations of already-added compounds. The PM included 1:5300 DMSO and when DMSO was used as vehicle, 1:4000 final dilution was used for treatments. The calibration of ΔψP and ΔψM followed the “Complete with known kP” and “Complete” methods, respectively [59], or in conditions in which ΔψM was largely depolarized before calibration, the “Complete (known k)” method was used to calibrate ΔψM. The corresponding kT value (see [58]) was 0.014±0.001 s−1 (n=4 individuals) on average. First the mitochondrial depolarization cocktail (MDC) containing in 1 μM valinomycin, 100 nM FCCP, 2 μg/ml oligomycin, 2 μM antimycin A and 2 μM myxothiazol and the anti-swelling cocktail [58], containing 100 μM IAA-94, 10 μM DIOA, and 80 μM bumetanide plus 1 μM tetrodotoxin were applied and the decay of TMRM fluorescence intensity was recorded for 30 min. Then PM was part replaced with PMK supplemented with the above cocktails in 3 steps with 3 cycles of recordings for each (required for calibration of ΔψP), and finally the assay medium was half replaced with PM-PFA supplemented with 5 μg/ml gramicidin and the recording was carried on for another 30 min to record fluorescence when both potentials are zero. At the end of the potentiometric recording, media were replaced with PBS containing 100 μg/ml dithizone and 5 μg/ml Hoechst 33342, and bright field images at 510 nm and the fluorescence of the nuclear marker were recorded in the view fields matching those of the potentiometric recordings. Finally the entire surface area of each well was recorded with tiled imaging for cell counting (see below). Automated image analysis and conversion of fluorescence intensities to millivolt potentials was carried out in Image Analyst MKII with the “Mitochondrial membrane potential measurement (TMRM/PMPI) with post-hoc absorbance classifier” standard pipeline, using dithizone optical density (OD) to distinguish β-cells from other islet cells. Cells in the upper 45 percentile of dithizone ODs were taken as β-cells in each view field. The apparent activity coefficient ratio (aR’) is a parameter required for the calculation of ΔψM in millivolts [58]. A value of 0.359, previously measured in human β-cells was used, because it did not show donor to donor variations [2]. The mitochondria:cell volume fraction was determined for each donor as described above (Table 1) and the matrix:mitochondrion volume fraction (VFM) of 0.63 was from [58].
Table 1.
Donor Table.
| Donor codea | Age, gender, racea | BMIa | HbA1ca (%) | Cause / mechanism / circumstance of deatha | Ischemic downtimea | Hours between operation and islet isolationa | Days between islet isolation and dispersion | Stimulation index in isletsa,b | Stimulation index in dispersed culturesc | Mitochondria:cell volume fraction (%) | basal ΔψM (mV) | basal ΔψP (mV) | basal respiration (pmol/min/105 cells) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HP17250 | 32yr, M, C | 26.3 | 4.9 | head trauma / GSW / suicide | - | <19 | 5.0 | 1.8 | 6.9±0.9(5) | 8.35±0.76 (2) | −157.6±3.3 (5) | −57.7±1.9 (5) | 19.2d |
| HP17264 | 57yr, F, C | 27.5 | 5.9 | anoxia (pulmonary embolism) / CV | 5 min | 10 | 5.0 | 2.3 | 5.9±0.7(4) | 8.20±0.91 (2) | −146.9±1.7 (4) | −63.1±1.6 (4) | 16.4e |
| HP17281 | 55yr, M, C | 30.0 | 5.6 | head trauma / GSW / suicide | - | <20 | 3.0 | 2.5 | 4.2±0.4 (3) | 8.22±0.23 (2) | −149.8±2.7 (4) | −61.8±1.5 (4) | 18.6±2.8e (2) |
| HP17342 | 38yr, M, C | 30.0 | 5.8 | head trauma / blunt injury / MVA | 2 hr, cold | 10 | 3.6 | 5.9 | 3.4±0.3 (4) | 7.82±0.20 (2) | −139.6±1.4 (4) | −56.9±1.2 (4) | 10.6±0.9e (2) |
, Data provided by Prodo Laboratories;
, Stimulation index in islets reflects insulin secretion in 28 mM glucose (1 hr) compared to 3 mM glucose, performed by Prodo Laboratories;
, Stimulation index in dispersed islet cell cultures reflects insulin secretion in 16 mM glucose (30 min) compared to 3 mM glucose. These and basal bioenergetic parameters were determined in 3 mM glucose, 2 mM glutamine in modified RPMI 1640 medium and data are mean±SE of (n) experiments performed between days 3 and 9 after dispersion;
, Mitochondrial respiration in non-sorted islet cell cultures;
, Mitochondrial respiration in β-cell enriched islet cell cultures. Respiration was normalized to total cell count after recording. C, Caucasian; CV, cardiovascular; GSW, gunshot wound; MVA, motor vehicle accident.
2.6. Insulin secretion
Insulin secretion was measured in samples resulting from part-replacement of PM during potentiometric recordings. Samples were mixed 1:1 with 0.2% casein, 120 mM NaCl, 30 mM TRIS, pH 7.4 buffer and spun 10 min at 3220 g in V-bottom microplates. Insulin content was determined by AlphaLISA using the kit AL350C (PerkinElmer Inc., Waltham, MA) in an Enspire Alpha Plate Reader (PerkinElmer) with 12 μl assay volume in 384-well ProxiPlates. A BSA-free insulin standard was used, because an interaction between BSA and PMPI interfered with the assay. The modified RPMI 1640 used for making the PM did not contain biotin, to allow the use of the AlphaLISA technology. Rates of insulin secretion were normalized to the total cell counts described below.
2.7. Respirometry
Cell respirometry was performed after 5 days of β-cell-enriched dispersed islet cell culturing using a Seahorse XFe96 Analyzer (Agilent, Santa Clara, CA). Cultures were preincubated for 4 h in 3 mM glucose, 2 mM glutamine containing RM. The medium was also supplemented with 1 μM zosuquidar and 1 μM tetraphenylborate to match composition of the potentiometric recordings, but fluorophores were omitted. Experiments were performed with 160 μl starting volume and 20 μl port injection volumes, and followed modified (1 min mix, 5 min measure) acquisition cycles to improve sensitivity. Additions were pre-mixed with previous additions as they were during potentiometric recordings to ensure constant concentrations of already-added compounds. All recordings were started at the common 3 mM glucose baseline, and for modular kinetic analysis mitochondrial respiration was expressed as a % of this baseline. To show absolute respiration (Table 1), respiration per well was normalized to total cell counts (see below).
2.8. Immunocytochemistry
After volume fraction and cell respirometry measurements, cultures were fixed in 4% PFA and stained for insulin using guinea pig polyclonal anti-insulin primary (AR029–5R 1:5; Biogenex, San Ramon, CA) and anti-guinea pig IgG – Alexa 488 (1:500; Life Technologies) secondary antibodies, and with Hoechst 33342 (2 μg/ml) for nuclei.
2.9. Cell counting
Total cell numbers in microplate wells for insulin secretion and cell respirometry were obtained by tiled imaging of Hoechst 33342 fluorescence in the entire surface area of the well using the microscope setup described for the potentiometric assay. For respirometry, insulin immunofluorescence was also captured to enable calculation of the purity of cultures. Recorded images were analyzed in an automated fashion with custom pipelines in Image Analyst MKII based on the standard pipeline “Seahorse well cell count with nuclear stain with ICC”.
2.10. Statistics
Wherever possible mean±SE for n=human individuals are shown, and experimental replicates from repeated microplates were considered as technical replicates. All assays were performed in partial microplate formats and associations between conditions and wells were randomly scrambled for each experiment. Data analysis was performed in Microsoft Excel and Graphpad Prism.
3. Results
3.1. Properties of human dispersed islet cell cultures were similar between donors and in time
To study the bioenergetic control of GSIS, I used human (Table 1) dispersed islet cell cultures, where detection was gated to β-cells for membrane potential measurements using post-hoc staining (Fig. 1A-B). For cell respirometry, cultures were enriched in β-cells using cell sorting (see below; Fig. 4). These dispersed islet cell cultures were not strictly single-cell cultures as β and other islet cells spontaneously aggregated into “mini islets” during culturing (Fig. 1B, Fig. 4B), thus may retain some aspects of cell-to-cell cooperativity observed in intact islets.
Fig. 1.
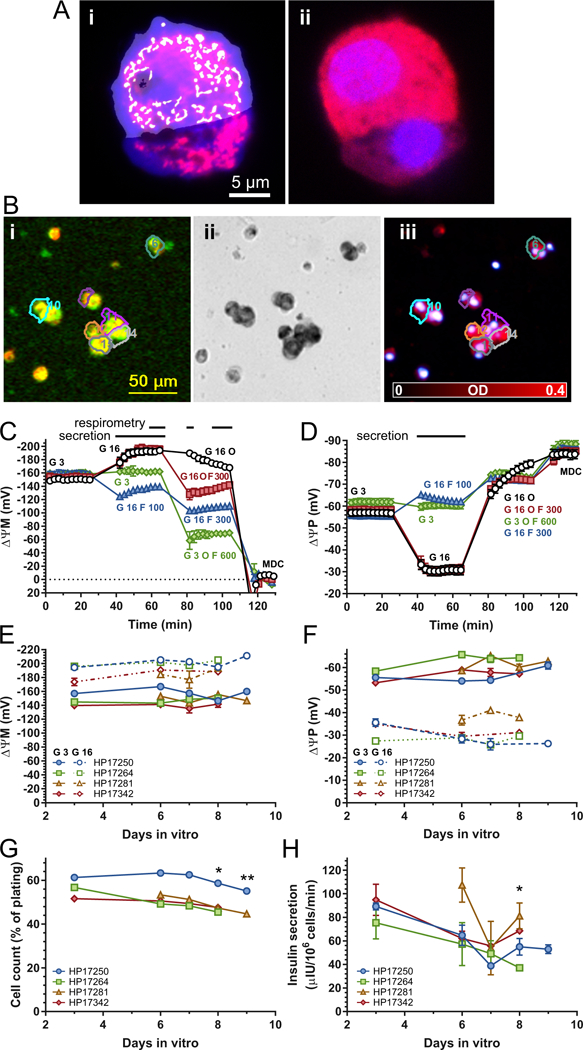
Dispersed islet cell cultures from non-diabetic human organ donors. (A) Mitochondria:cell volume fraction measurement with confocal microscopy. Representative micrographs of MitoTracker Red (red) and calcein-AM (dark blue) stained dispersed islet cells (Ai). Mitochondrial (white) and cellular profiles (light blue) were computationally determined in images of live cells (Ai) and were manually gated to insulin immunopositivity (Aii red) recorded after fixation and staining of the same cells. B) ΔψP and ΔψM measurement with fluorescence time-lapse microscopy. Bi) Cropped view field (~5% of total area) of TMRM (red) and PMPI (green) fluorescence of dispersed islet cell cultures at 16 mM glucose. At the end of the time-lapse recording, cultures were stained with dithizone (Bii) and captured dithizone images were converted to optical density (OD) images (Biii). Regions of interests mark β-cells detected by the software that also passed QC of the potentiometric calibration. (C-D) Time courses of ΔψM and ΔψP. Data were recorded as TMRM and PMPI fluorescence intensities and converted to millivolt values based on the calibration applied at the end of each time course (partly shown; MDC, mitochondrial depolarization cocktail; see Methods). Data points are mean±SE of n=4 individuals (except for the “G 3 O F 600” trace where n=2). Horizontal bars on the top indicate the time intervals in which potentials were averaged to match data with respirometry and secretion measurements. The total of additions is marked by labels matching trace colors for each segment of the time course. G, glucose in mM; O, oligomycin (2 μg/ml); F, FCCP in nM. (E-F) Dependence of ΔψM and ΔψP on the duration of dispersed islet cell culturing. Closed and open symbols mark potentials measured at 3 and 16 mM glucose for each donor. Data are mean±SE of technical replicates (wells) for each day. (G) Survival of dispersed islet cells during culturing. Cell counts in microplate wells used for potentiometric and insulin secretion assays were expressed as a percent of plating density. *, p<0.05; **, p<0.01 significance by ANOVA comparing cell counts at indicated time points to cell counts at 3 days with Tukey’s multiple comparison on data additionally normalized to day 6. (H) Dependence of 16 mM-glucose stimulated insulin secretion on the duration of dispersed islet cell culturing. *, p<0.05 significance by ANOVA comparing to secretion at 3 days with Tukey’s multiple comparison on data additionally normalized to day 6.
Determinations of ΔψP and ΔψM were performed using the absolute potentiometric fluorescence microscopic technology we have previously developed [58] and optimized for pancreatic β-cells [59]. For each donor mitochondria:cell volume fractions were determined (Fig. 1A, Table 1), and used to calculate ΔψM. Millivolt potentials were obtained from calibration of time courses of TMRM and PMPI fluorescence intensities (represented by Fig. 1C-D), and indicate average potentials in the specified time intervals marked over the time courses.
Previously we found that the magnitude of ΔψM drifted during whole islet culturing [59], therefore here I dispersed all islet cells immediately after cold, overnight transport, without recovery at 37°C. This resulted in stable potentials within the observation window of 3–9 days after dispersion. The baseline ΔψM and ΔψP after 4 h incubation in 3 mM glucose, 2 mM glutamine containing modified RPMI 1640 medium (Fig. 1C and D black circles “G 3” and Fig. 1E and F, solid lines) did not change significantly during culturing. This was also the case for their magnitudes following a 30 min elevation of glucose concentration to 16 mM in the same medium (Fig. 1C and D black circles “G 16” and Fig. 1E and F, dashed lines). Cell counts slightly (Fig. 1G), and insulin secretion more substantially, decayed during this period, however insulin secretion was not significantly different during the 6–8 days of cell culturing in which most of the assays were performed (Fig. 1H).
3.2. Insulin secretion correlates with ΔψM hyperpolarization
Dispersed islet cell cultures were exposed to a step increase in glucose concentration, from 3 mM to up to 16 mM, and the mean ΔψP, ΔψM and insulin secretion for the following 30 min were determined. The latter was detected by sampling the assay medium in the same experiments (Fig. 2A-C). Half-maximal activations were at 6.7±0.5 mM glucose for secretion, 7.06±0.5mM for ΔψP and 6.8±0.2 mM for ΔψM (n=4; no significant differences by ANOVA). ΔψP depolarization correlated with ΔψM hyperpolarization (Fig. 2D; black circles) as we have previously reported using single cell analysis [2]. This correlation is the hallmark of KATP operation as glibenclamide (1 μM; a KATP inhibitor) or diazoxide (50 μM; a KATP activator) in the presence of 3 or 16 mM glucose, respectively, broke the correlation (Fig. 2D; red triangles). This submaximal dose of diazoxide was not expected to have direct mitochondrial effects [60]. Importantly, Fig. 2E and F indicate that GSIS correlated both with ΔψM and ΔψP. Glibenclamide and diazoxide broke this observed relationship between insulin secretion and ΔψM (Fig. 2E), but not between insulin secretion and ΔψP (Fig. 2F). Thus, insulin secretion correlates with ΔψM, as long as the downstream canonical pathway is intact.
Fig. 2.
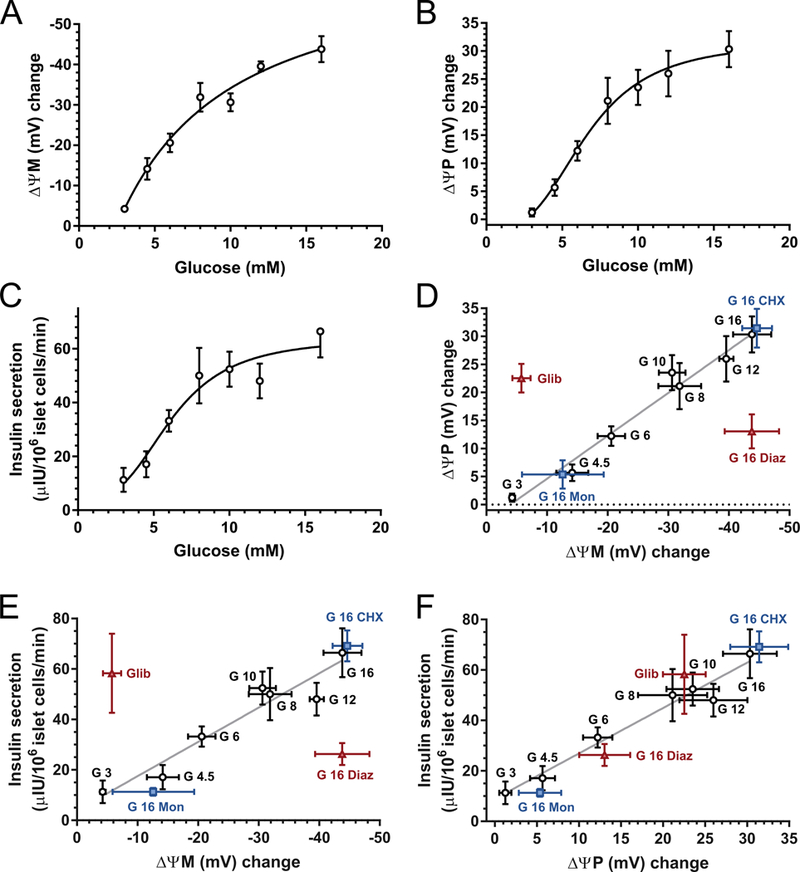
Glucose concentration dependence of ΔψM, ΔψP and insulin secretion. (A-C) Average ΔψM, ΔψP and insulin secretion were measured during a 30-min period after elevation of glucose from the basal 3 mM to the indicated level, corresponding to the horizontal bars shown at the top of Fig. 1C-D. Potentials are given as the change compared to the average of the 30-min baseline at 3 mM glucose. Corresponding absolute values are shown in Table 1. Insulin release into the assay medium was measured in the same time period and normalized to the total islet cell count at the end of the time course. (D-F) Pairwise correlation analysis of ΔψM, ΔψP and insulin secretion. Black circles correspond to A-C. Glib, glibenclamide (1 μM) was given in the presence of 3 mM glucose. Diaz, diazoxide (50 μM); Mon, monensin (1 μM); and CHX, cycloheximide (35 μM) were given with 16 mM glucose. These recordings followed identical timing to the glucose titration. Data are mean±SE of n=4 individuals.
3.3. ΔψM hyperpolarization is required for insulin secretion
First, consider the possibility that insulin secretion is predicted by ΔψM (or downstream ATP/ADP), and effects of other glycolytic or mitochondrial metabolic coupling factors are small. To test this, ΔψM was manipulated below, using complex I inhibition by rotenone, which inhibits both metabolic fluxes (indirectly by increasing NADH/NAD+ in the mitochondrial matrix) and ΔψM, or uncoupling by FCCP, which speeds up metabolic fluxes while decreasing ΔψM (see cell respiration data in Fig. 4D below). In the above scenario partially diminishing ΔψM by respiratory inhibition or by uncoupling at constantly high glucose concentration is expected to have similar effects to decreasing glucose concentration. Conversely, if mitochondrial or glycolytic metabolic coupling factors play a significant role in the current paradigm, then partial uncoupling is expected to have a smaller effect on GSIS than partial respiratory inhibition at the same ΔψM.
Dispersed islet cell cultures were treated with rotenone (1–5 nM) for 30 min before glucose stimulation to allow time for the onset of inhibition at the low nanomolar concentrations used (Fig. 3A). FCCP titration (10–100 nM; Fig. 3B) followed the same paradigm as glucose titration in Fig. 2. Both rotenone and FCCP dose-dependently decreased the response of ΔψM, ΔψP and the insulin secretion to 16 mM glucose (Fig. 3C-F). In the presence of 100 nM FCCP or 5 nM rotenone, raising glucose from 3 to 16 mM did not evoke insulin secretion and ΔψM and ΔψP were more de- and hyperpolarized, respectively, than at the 3 mM glucose baseline (Fig. 3C-F open symbols). To address whether changing ΔψM by glucose or inhibitor concentrations results in the same or different insulin secretion, secretion was compared at identical ΔψM by inter- or extrapolation. When ΔψM was “depolarized” to the same level as observed at 3 mM glucose (“G 3 DMSO”), but by using rotenone or FCCP in the presence of 16 mM glucose, estimated insulin secretion (Fig. 3C-F red diamonds) was not different from that observed at 3mM glucose. Therefore a ΔψM hyperpolarization is required for insulin secretion, and the magnitude of ΔψM predicts insulin secretion. Notably, because 100 nM FCCP or 5 nM rotenone caused substantial mitochondrial depolarization below the 3 mM glucose baseline level, these were excluded from the estimation because these non-permissive ranges of depolarized potentials are not expected to maintain a linear relationship to secretion.
Fig. 3.
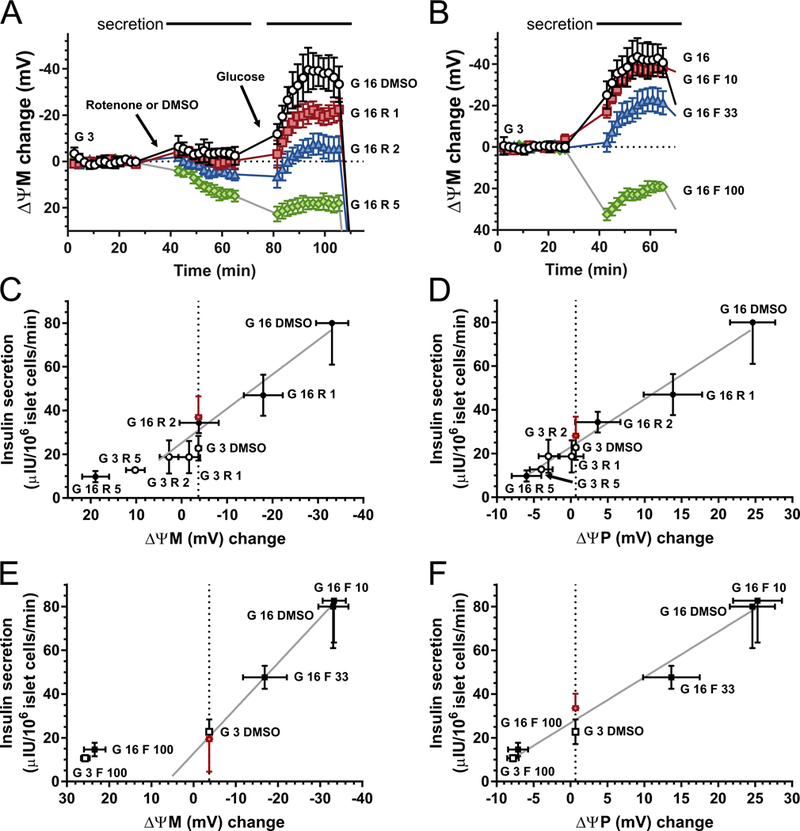
ΔψM predicts glucose-stimulated insulin secretion. (A-B) Time courses of ΔψM in β-cells stimulated with glucose (G in mM) in the presence of rotenone (R in nM) or FCCP (F in nM). Horizontal bars on the top indicate the time intervals where potentials were averaged to match data with secretion measurements. See Table 1 for the absolute millivolt values corresponding to the baseline. (C-D) Effects of increasing concentrations of rotenone on the relationship of ΔψM or ΔψP to insulin secretion, in the presence of 3 mM (open circles) or 16 mM (closed circles) glucose. The red diamond indicates estimated insulin secretion at 16 mM glucose plus rotenone at a potential (vertical dotted line) matching that at 3 mM glucose without inhibitor. The estimated secretion was not different from the vehicle-treated control (“G 3 DMSO”) by paired t-test. E-F) Effects of increasing concentrations of FCCP on the relationship of ΔψM or ΔψP to insulin secretion, in the presence of 3 mM (open squares) or 16 mM (closed squares) glucose. The red diamond indicates estimated insulin secretion at 16 mM glucose plus FCCP at a potential (vertical dotted line) matching that at 3 mM glucose without inhibitor. For ΔψM the highest inhibitor concentrations, which result in ΔψM depolarization, were excluded from the estimation. The estimated secretion was not different from the vehicle-treated control (“G 3 DMSO”) by paired t-test. Estimations were performed for each individual and all shown data are mean±SE of n=4 individuals. Gray trend lines indicate the relationship of secretion and ΔψM or ΔψP at 16-mM glucose and changing inhibitor concentration.
3.4. Micro-scale respirometry of human dispersed β-cells
I showed above that the glucose-induced ΔψM hyperpolarization is required and predicts insulin secretion in human β-cells. Here I assess how β-cells achieve ΔψM hyperpolarization in response to glucose while both supply and demand on ΔψM increases. The protonmotive force, and therefore the ΔψM, is generated by proton pumping by three of the respiratory complexes. Proton pumping is propelled by electrons originating from the metabolism of glucose and other substrates, traveling through the respiratory complexes and finally reacting with O2. The flow of electrons to react with O2, and therefore the flux of substrate oxidation, is conveniently measured as mitochondrial respiration. When β-cells are not momentarily perturbed, ΔψM and mitochondrial respiration are constant (neglecting oscillations, which were not common). This is a steady state, where electron flow supplying ΔψM is equivalent to the demand. When glucose concentration is increased from 3 to 16 mM a new steady state develops. The mechanism that establishes that the new steady state develops at a more negative ΔψM is expected to be critical to GSIS because ΔψM in turn predicts secretion. To tease apart effects of changing supply and demand I use metabolic control analysis below. In order to do this, cell respirometry was performed in conditions matching those of ΔψM measurements.
For respirometry, dispersed islet cell cultures were enriched in β-cells using fluorescence-activated cell sorting. This was required because in contrast to the ΔψM assay where analysis was gated to β-cells, cell respiration is detectable only in cell populations (Fig. 4A-B). Cell sorting was based on the Zn2+ content of insulin granules marked by the Zn2+-sensitive fluorescence probe FluoZin-3, and increased the β-cell content of cultures (Fig. 4C). Positive selected cultures responded to elevation of glucose from 3 mM to 16 mM by a twofold increase in mitochondrial respiration, slightly greater than previous reports in whole human islets [20,21,46]. In contrast, the complement cell population did not respond significantly to glucose (Fig. 4D-E). Fig. 4D shows a set of respirometry time courses with timing closely matching that of ΔψM determinations (Fig. 1C). The few differences between the two recording modalities were due to technical limitations imposed by differing temporal resolutions, liquid handling and specimen mass requirement. Recordings were started in RM containing 3 mM glucose, 2 mM glutamine. Then, glucose was elevated to 16 mM with or without modulators (Fig. 4D; Fig. 5A). The next two additions were oligomycin (2 μg/ml) to inhibit ATP synthesis and low concentrations of FCCP to partially speed up respiration and substrate metabolism. The orders of these additions in different paradigms were designed to allow recording of data points for Fig. 5 below in minimal numbers of microplate wells. Experiments were concluded by addition of antimycin A (2 μM) plus myxothiazol (2 μM), and the remaining non-mitochondrial respiration was subtracted from all data.
Fig. 4.
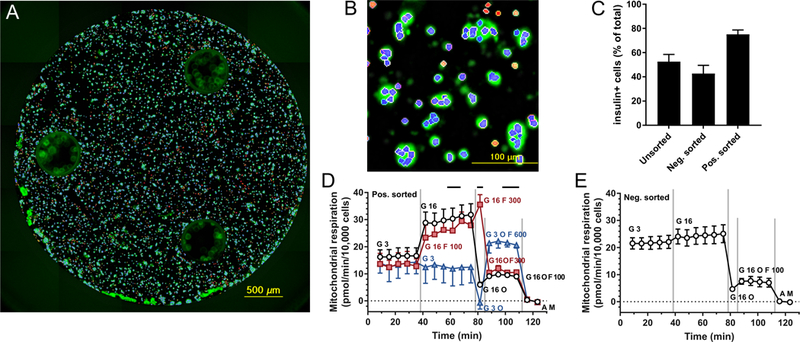
Cell respirometry in β-cell-enriched cultures. (A-B) Low (A) and high (B) magnification processed image of insulin immunofluorescence (green) in β-cell-enriched islet cell culture in a Seahorse XFe96 well after respirometry. Counted insulin-positive cell nuclei are marked by blue and insulin-negative by red color based on segmentation and classification of nuclear (Hoechst 33342) staining. (C) Fraction of insulin-positive cells based on insulin immunostaining and cell counting after respirometry. Data are mean±SE, n= 3 individuals for positively sorted cultures and n=2 for negatively or unsorted cultures. (D-E) Time courses of cell respiration in β-cell enriched islet cultures. Non-mitochondrial respiration was measured after antimycin plus myxothiazol addition (“A M”, 2 μM each) and subtracted from all data points. Four additions were performed during the time course marked by the vertical gray lines. Different symbols indicate different paradigms, and the total of additions is marked by matching color labels for each period between the additions. G, glucose in mM; O, oligomycin (2 μg/ml); F, FCCP in nM. Data are mean±SE, n=3 individuals, except for blue triangles and (E) where n=2.
Fig. 5.
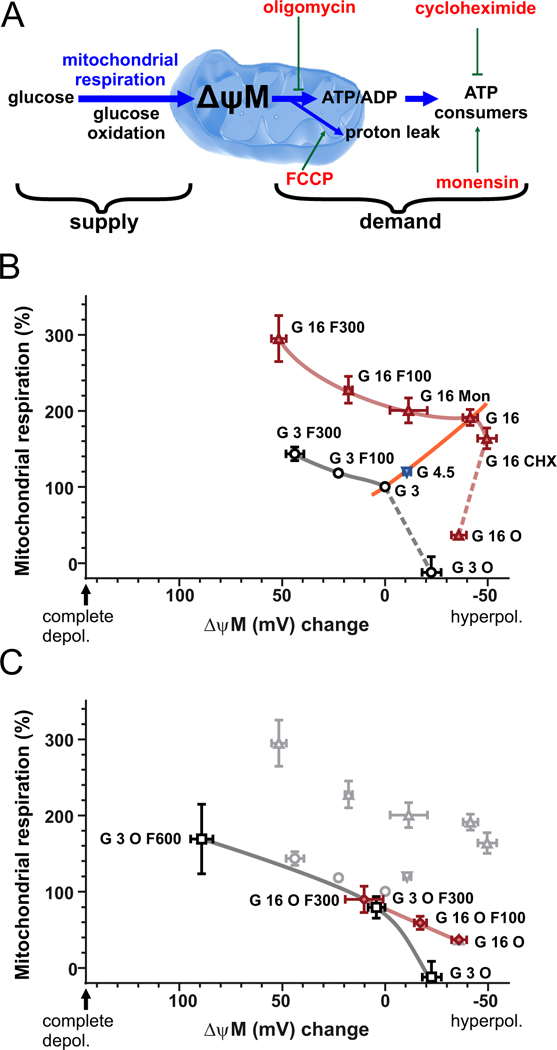
Modular kinetic analysis of β-cell energy metabolism. (A) Modularization of energy metabolism. The supply module comprises all processes from glucose uptake to proton pumping in the respiratory chain. The demand module encompasses all processes that drain ΔψM: ATP synthesis and utilization, and proton leak. The supply and demand modules are assumed to communicate directly or indirectly by changes of the level of their common intermediate, ΔψM. Modulators of the demand module used below are indicated by red labels. (B) Modular kinetic analysis of β-cell energy metabolism. The kinetic response of substrate oxidation to ΔψM is shown at 3 mM glucose (black circles) and 16 mM glucose (red triangles), and illustrated by gray and red curves, respectively. Dashed curves indicate uncertainty. The response of demand is shown by connecting the 3, 4.5 and 16 mM glucose points with no modulators of demand present (orange curve). Data points show mitochondrial respiration from experiments represented by Fig. 4D as a function of ΔψM from experiments represented by Fig. 1C in matching conditions, indicated by the labels. (C) Modular kinetic analysis of β-cell substrate oxidation with ATP synthesis inhibited by oligomycin, at 3 mM glucose (black squares) and 16 mM glucose (red diamonds). Data from (B) is shown in grey in the background. G, glucose in mM; O, oligomycin (2 μg/ml); F, FCCP in nM; Mon, monensin (1 μM); CHX, cycloheximide (35 μM). Data are mean±SE, n=3 individuals, except for the “G 3 O” and “G 3 O F 600” points where n=2.
3.5. Substantial reserve capacity and small proton leak in human dispersed β-cells.
Uncoupling by 300 nM FCCP increased mitochondrial respiration in the presence of 16 mM glucose indicating that glucose oxidation was not running at its maximal capacity, because of submaximal demand (Fig. 4D red squares; see Fig. 5B for comparison of relative changes; p<0.05 by paired t-test; n=3). This is consistent with data reported in human islets [20]. Notably, I did not attempt to determine maximal respiratory capacity here, which may be larger than the observed highest value. The leak respiration, measured in the absence of the uncoupler but in the presence of oligomycin, was immeasurably low at 3mM glucose and was only 19.5±1.3% (n=3) of mitochondrial respiration at 16 mM glucose. This indicates that dispersed human β-cells are very well coupled, in contrast to the well-established observations of high proton leak (~50%) in rodent insulinoma cells [45,56,61,62] and in rodent islets (~40%) [21,47].
3.6. Modular kinetic analysis of cellular energy metabolism
To understand how β-cells hyperpolarize ΔψM when glucose concentration is elevated and both supply and demand activities increase, I studied substrate oxidation in isolation in intact cells using modular kinetic analysis. To this end cellular energy metabolism was divided into supply and demand modules connected by ΔψM (Fig. 5A): substrate oxidation (including glycolysis, glutamine and β-oxidation, the TCA cycle and the respiratory chain) and phosphorylation (ATP synthesis and cellular ATP consumption) plus proton leak. The kinetic response of cellular substrate oxidation to ΔψM was determined by observing how the flux changed as a function of the common intermediate ΔψM (Fig. 5B-C), when the activity of the demand module was modulated (Fig. 5A red labels) without interfering with the supply module. Notably, steady state, thus constant ΔψM and respiration developed in minutes after glucose or modulator challenge (including for oligomycin; data not shown) in dispersed β-cell cultures (Fig. 1C and Fig. 4D), in contrast to studies on intact islets [5,21,47]. To work with steady-state values, ΔψM and respiration were evaluated at 15–30 min after glucose stimulation below, as indicated by horizontal bars in Fig. 1C and Fig. 4D.
3.7. Kinetic properties of substrate oxidation in 3mM glucose
Fig. 5B shows kinetic curves of substrate oxidation (gray and red curves) compiled from ΔψM and mitochondrial respiration determinations represented by Fig. 1C and Fig. 4D. Data shown are from 3 human individuals (mean±SE for the last 3 donors from Table 1), and to decrease variation all values are expressed relative to an identical baseline condition, 3 mM glucose (see corresponding absolute values in Table 1). In low glucose (Fig. 5B, black circles) starting from the baseline condition (“G 3”) ΔψM hyperpolarized and respiration decreased as the demand module was largely obliterated by inhibition of ATP synthesis (“G 3 O”). From the “G 3” point respiration increased and ΔψM depolarized as the demand was increased by increasing concentrations of FCCP (100 nM and 300 nM, marked by “G 3 F 100” and “G 3 F 300”, respectively). Altogether, these four data points in the force-flux space outline the kinetic response of substrate oxidation to ΔψM at 3 mM glucose (gray curve). Notably, whether the “G 3 O” point is part of the curve, or not, is considered below.
3.8. Kinetic properties of substrate oxidation in 16mM glucose
At 16 mM glucose (Fig. 5B red triangles) I utilized two further modulators to outline the substrate oxidation curve (red). Monensin (1 μM; Mon), a Na+/H+ exchanger, induces ATP demand by Na+/K+-ATPase [63] as transported Na+ is pumped back to the medium. Cycloheximide (35 μM; CHX) inhibits protein synthesis, a major contributor to the cellular ATP budget [64], and therefore decreases demand. Notably, monensin almost completely inhibited ΔψP and insulin secretion responses to glucose, while CHX had no significant effect on them (Fig. 2F). These, together with the oligomycin-inhibited (“G 16 O”) and FCCP-induced (“G 16 F100 and 200”) demand, outline the kinetic response of substrate oxidation to ΔψM at 16 mM glucose (red trace).
Comparison of the 16 mM and 3 mM glucose curves indicates a higher flux in high glucose at each ΔψM, thus glucose oxidation worked against the identical force faster in high glucose. This marks the activation of the supply pathway by glucose. This activation may be a direct kinetic effect of increased glucose availability, or due to other, indirect effects, and this is considered below. The glucose oxidation curve in high glucose had a marked “elbow” around the “G 16” point, which functionally maximizes the ΔψM that β-cells can achieve when the flux is high. In contrast, currently there are no data on the shape of the low-glucose curve between the “G 3” and “G 3 O” points (dashed gray line in Fig. 5B), preventing precise comparison of the shapes of the two curves.
3.9. Substrate oxidation is deactivated when ATP synthesis is inhibited by oligomycin
In classical isolated non-β-cell mitochondrial systems, the kinetic response of substrate oxidation to ΔψM is obtained by uncoupler titration of the non-phosphorylating state (state IV) in the presence of oligomycin, and typically this curve also encompasses the state III, thus uninhibited, phosphorylating state data point [65]. In contrast, substrate oxidation behaved distinctly in β-cells, being suppressed in the non-phosphorylating state. In Fig. 5C the demand was increased starting from the “G 3 O” and “G 16 O” points by uncoupling in the presence of ATP synthase inhibition. The kinetic curve of substrate oxidation, independently of glucose concentration, closely resembled the curve obtained in the presence of 3 mM glucose without ATP synthase inhibition, and the respiration was lower than measured at 16 mM glucose without ATP synthase inhibition at any ΔψM.
The activity of substrate oxidation is expressed and compared by looking up its rate that works against a specific force. To this end mitochondrial respiration was calculated for each substrate oxidation curve (Fig. 5B and C) at the ΔψM corresponding to the 3 mM glucose baseline (0 mV; Fig. 6A). The calculated activities indicate the absence of activation of substrate oxidation when ATP synthesis is inhibited, even though kinetic effects of increased substrate availability will still be present. Notably, because of the observed effects of ATP synthesis inhibition, the “G 3 O” and “G 16 O” points likely underestimate the uninhibited substrate oxidation activity in Fig. 5B. The dashing in the last segments of these curves also expresses this uncertainty.
Fig. 6.
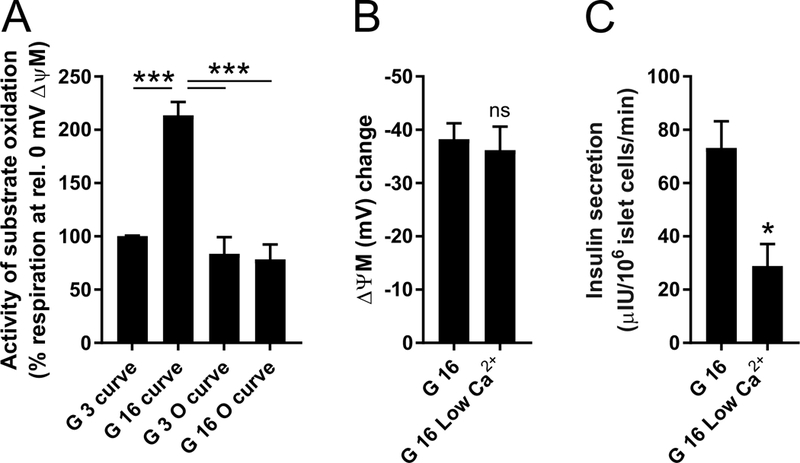
Glucose-activation of β-cell energy metabolism is dependent on ATP synthesis, but not on Ca2+. (A) Activity of substrate oxidation was calculated from Fig. 5B and C, by calculating mitochondrial respiration at 0 mV ΔψM change using linear interpolation of nearby data points on each substrate oxidation kinetic curve. ***, p<0.001 significance by ANOVA followed by Tukey’s post hoc test (n=3 individuals). (B) ΔψM hyperpolarization by 16 mM glucose compared to the 3 mM glucose baseline at normal 2.5 mM extracellular Ca2+ and when Ca2+ was lowered to 0.25 mM. ns, not significant by paired t-test (n=3 individuals). (C) Insulin secretion in 16 mM glucose (30 min) at normal 2.5 mM extracellular Ca2+ and when Ca2+ was lowered to 0.25 mM. *, p<0.05 singificance by paired t-test (n=3 individuals).
3.10. Activation of substrate oxidation is an ATP-dependent, positive feedback mechanism
In conclusion, data in Fig. 5 and Fig. 6A show that upon elevation of glucose concentration from 3 to 16 mM, substrate oxidation was activated, but not when ATP synthesis was inhibited. Thus, substrate oxidation is activated by, or its activation requires, its downstream product, forming a positive feedback loop. The kinetic curves in Fig. 5B indicate that the observed ~40 mV of ΔψM hyperpolarization is not possible without this activation, even if demand is inhibited to the extent of complete shutdown of ATP turnover (e.g. “3 G O”). Thus inhibition of any mechanism contributing to the activation should decrease glucose-evoked ΔψM hyperpolarization. Partial opening of KATP by diazoxide did not decrease 16 mM glucose-induced ΔψM hyperpolarization (Fig. 2E; not significant by paired t-test, n=4). In addition, lowering extracellular [Ca2+] did not decrease 16 mM glucose-induced ΔψM hyperpolarization (Fig. 6B), whereas it decreased insulin secretion (Fig. 6C). These data indicate that the feedback is not mediated by effects of ATP downstream of KATP or by Ca2+.
4. Discussion
84 million Americans were estimated in 2015 to have prediabetes, and annually 5–10% of them develop type 2 diabetes (T2D) [11,66]. An important factor in the progression from prediabetes to diabetes is the deterioration of insulin secretion in pancreatic β-cells, which is at least partly caused by a functional decline in the β-cells [18,67]. Because β-cell bioenergetics is demonstrably involved in β-cell dysfunction in human T2D [2,19–21], it is prompting to understand the role of bioenergetics in human β-cell function and dysfunction. This work establishes basic relationships between glucose stimulation, mitochondrial energization and insulin secretion in healthy human β-cells.
By assessing bioenergetic prerequisites of GSIS in non-diabetic human β-cells, I found that glucose-evoked ΔψM hyperpolarization is required for GSIS, and the rate of GSIS is predicted by the magnitude of ΔψM hyperpolarization. Importantly, the increasing glucose concentration is able to greatly hyperpolarize ΔψM because of an ATP-dependent activation of glucose oxidation. This activation changes glucose oxidation so mitochondria can respire and turn ATP over faster at higher ΔψM (Fig. 5B). This can be also conceptualized as an amplification of metabolism by a positive feedback loop in β-cell energy metabolism.
To my knowledge this is the first report using human primary β-cells to indicate a predictive relationship between glucose-induced ΔψM hyperpolarization and insulin secretion. ΔψM or ATP/ADP have been previously shown to correlate with secretion while spanning physiological glucose concentrations in rodents [26–29]. Bioenergetic inhibitor and uncoupler titrations in the presence of high glucose have also been used to confirm these relationships in rodents [68,69]. However, a reevaluation of GSIS using human β-cells is mandated by findings of differing mitochondrial coupling pathways between rodents and humans [70,71].
To obtain data relevant to human health and disease I studied primary dispersed β-cells from non-diabetic human organ donors. Dispersed cell cultures, in contrast to intact islets, offered: 1) reduction of selection bias of individual islets as cells from each batch of ~4000 islets were evenly mixed for all assay points [2]; 2) elimination of cells damaged during isolation and transport by culturing and single cell quality control during data analysis [59]; 3) no diffusion limitation for oxygen, nutrients or acute pharmacologic challenges reaching the β-cells. The latter may explain quicker steady states in our specimens than observed in whole islets [5,21,47]. Our experimental paradigms avoided deenergization of β-cells in pre-stimulation conditions by using growth media-like assay media with amino acids, including glutamine, present. Secretion was observed for 30 min encompassing both first and second phases of insulin secretion.
I did not observe a ΔψM-independent component in GSIS that could be attributed to the effect of metabolic coupling factors. In contrast, about 50% of insulin secretion has been estimated to be due to KATP- or Ca2+-independent metabolic amplification in both phases of secretion in rodents [72]. Importantly, this does not contradict ΔψM predicting GSIS, because ATP/ADP may control amplification of secretion as well [6]. The large contribution of metabolic amplification to GSIS was determined based on saturation of cytosolic [Ca2+] responses at lower glucose concentration than that of secretion [72], but [Ca2+] responses have been also shown to saturate at lower glucose concentration than that of the ΔψM response [27]. Nevertheless, in our study the changing insulin secretion with days spent in cell culture and the small number of studied individuals may limit the sensitivity to detect subtle ΔψM-independent contributors to GSIS. During the rotenone and FCCP titrations (Fig. 3C-F) insulin secretion was generally higher than during glucose titrations shown in Fig. 2, but I attribute this to a cell culture age or a vehicle effect.
An argument against mitochondrial energization being a major regulator of GSIS may originate from observations that mitochondrial energization saturates at lower glucose concentration than does secretion [31,32] and from data indicating no effect of interfering with metabolic coupling pathways on glucose-induced ΔψM hyperpolarization [41,42,73]. However, these observations were made with the quench-mode ΔψM probe rhodamine-123, which loses sensitivity at more negative ΔψM. The current study utilized a non-quench mode, absolute calibrated ΔψM assay that does not lose sensitivity with hyperpolarization [58,59]. A similar argument may be made based on ATP or ATP/ADP determinations, as ATP levels were found not to correlate to GSIS [68,74,75]. Importantly, ATP levels change very little at high ATP/ADP ratios [5,9], therefore total ATP levels are expected to be very insensitive to resolve the range of cellular energization states relevant to GSIS. Furthermore, determinations of β-cell ATP/ADP are blunted by binding to proteins and by the nucleotide pool of insulin granules [49]. Conversely, degranulation of islets resulted in a good correlation between ATP/ADP and secretion [29]. In contrast, measuring ΔψM is devoid of this compartmentalization problem, and therefore ΔψM is a more practical indicator of cellular energization than ATP/ADP. The estimated ΔψM is tightly linked to ATP synthesis and therefore to ATP/ADP, because in β-cells glycolytic ATP production cannot exist independently of oxidative phosphorylation due to the lack of ability to form lactate [76].
Mitochondrial ATP synthesis relies on the protonmotive force across the mitochondrial inner membrane. I used ΔψM as a surrogate for protonmotive force, assuming that ΔψM is its larger component, and ΔψM and ΔpH change proportionally under the conditions studied. ΔpH has been shown to increase during GSIS with different kinetics compared to ΔψM in insulinoma cells [77], however the latter was detected with the ΔψM probe JC-1, which has a quantitatively unpredictable response to potential [65]. If ΔpH plays a disproportionally stronger role in GSIS than ΔψM [77,78], then the data shown here underestimates mitochondrial energization. However, this would result in the ΔψM response saturating at lower glucose concentrations than the ΔψP and secretion responses, because the latter two are linked to ATP/ADP and therefore to protonmotive force, but no difference in saturation was observed (Fig. 2).
High glucose increases both cell respiration and the magnitude of ΔψM. Why doesn’t the high ΔψM (or ATP/ADP) inhibit respiration? The control analysis presented here explains this with an activation of glucose oxidation. When glucose concentration increases from 3 to 16 mM, a new steady state forms between the activated supply and the demand. Importantly, the demand is also activated as a result of ΔψP depolarization and downstream ion cycles. In this top-down aspect, there is a single demand kinetic curve spanning different glucose concentrations (Fig. 5B orange line). This is valid if all effects of glucose on ATP turnover are mediated by ATP/ADP, therefore indirectly by ΔψM, and I have shown here that this is true for GSIS. Therefore it is safe to assume that this is also true for the GSIS-related ATP turnover. An increase in ATP turnover encompasses the downstream GSIS pathway, an ATP/ADP-dependent activation of secretion-related consumers, such as ion pumping, as well as kinetic effects of higher ATP/ADP on consumers. Critical to GSIS is where the new steady state sets in the force-flux space when glucose concentration is elevated. The observed “elbow” shape in the activated supply kinetics (around the “G 16” point in Fig. 5) results in a new steady state set at a very high ΔψM. In contrast, with ATP-synthase inhibited in the presence of high glucose, no such “elbow” was observed, and the activity of glucose oxidation (the flux compared at the same force) was less than half of the activity observed without oligomycin (Fig. 5C), suggesting that the activation is not simply a kinetic effect of increased glucose availability. Thus, an activation mechanism dependent on ATP or a related downstream product activates and alters the kinetics of glucose oxidation in high glucose, and this results in a large hyperpolarization of ΔψM. The causation is implicit to the control analysis. I propose that this observation explains the paradox of β-cell energy metabolism [5,48], thus an increased ATP/ADP and ATP turnover can coexist during GSIS due to activation of supply.
An important question is whether the observed ATP-dependent activation marks physiological signaling taking place during GSIS or alternatively, is a permissive effect of ATP, which is negated by ATP-synthase inhibition. The physiological role may be supported by the differing shapes of uninhibited and oligomycin-inhibited glucose oxidation kinetic curves in 16 mM glucose (Fig. 5B vs C red curves). The “elbow” around the “G 16” point in Fig. 5B is a unique feature of the activated kinetics, and in contrast the oligomycin-inhibited kinetic curve is straight (Fig. 5C red trace). The “elbow” in contrast to the straight kinetics may indicate that glucose oxidation is activated more at more negative ΔψM than at depolarized potentials, thus the activation is ΔψM-dependent (or ATP-dependent as ATP synthase is operational). This argues for a graded, physiological effect. Alternative possibilities are that different supply pathways with differing kinetic response to ΔψM are operating in low glucose (or high glucose + oligomycin) and high glucose conditions, or the rate control moves to different components of the same pathway. These could indicate either physiological or permissive effects. Notably, the force-flux space in the right side of “G 3” was not explorable with the currently used modulators, because CHX was without effects in 3 mM glucose and oligomycin was not titratable (data not shown).
Conceptually, the “glucose sensor” of β-cells converts the rate of metabolism to the level of one or more intermediates that act as coupling factors controlling secretion. The rate of ATP synthesis, typically measured as oligomycin-sensitive respiration, has been often correlated to the rate of insulin secretion [30,47,79,80]. Because of this conversion process, rates of metabolism and levels of coupling factors are naturally linked, but only the latter controls secretion. Importantly, if ΔψM and ATP/ADP are major predictors of GSIS, then not the rate of ATP synthesis, but the levels that are determined both by synthesis and utilization are critical to secretion. Conversely, if insulin secretion is governed mostly by metabolic coupling factors, then intermediate levels linked to glucose utilization, rather than oxidation rates, may be of importance as oxidation and utilization fluxes are different [30]. That fluxes and levels are interchangeably used to describe GSIS may originate from a seemingly tight relationship between them as indicated in Fig. 5B (phosphorylation + leak; orange curve) that has been also predicted on a theoretical basis [81]. Even though this relationship exists during normal GSIS, it is not necessarily the case when supply or demand are experimentally disturbed. Therefore it is important to distinguish bioenergetic forces and fluxes when the mechanism of insulin secretion is sought.
Rotenone half-inhibited GSIS between 1 and 2 nM, consistently with observations in rat islets [82], while the Ki of rotenone for complex I NADH:ubiquinone oxidoreductase activity is 4 nM in bovine heart submitochondrial particles [83]. I found that ΔψM was less sensitive to rotenone in 3 mM glucose, and markedly sensitive in 16 mM glucose (Fig. 3A). While glucokinase is thought to be the major rate controlling step in β-cell glucose oxidation [84] (although the control analysis that was performed did not account for oxidative phosphorylation), the high rotenone-sensitivity suggests that in 16 mM glucose complex I gains substantial control over glucose oxidation. This supports findings in INS-1 cells with varying glucokinase activity [85], showing that in high glucose glucokinase loses control over glucose oxidation. A possible explanation for the high rotenone-sensitivity of glucose-induced ΔψM hyperpolarization and insulin secretion is that complex I inhibition may interfere with the positive feedback amplification leading to a supralinear response.
Reactive oxygen species (ROS), particularly superoxide and H2O2, are controversial coupling factors ([86] but see [87,88]), and their chronic elevation may mediate glucotoxic effects [89]. The magnitude of 16 mM glucose-stimulated ΔψM was −180 to −200 mV, notably larger than I previously reported in human β-cells (−160 to −180 mV in [2]). The difference may be a result of improved assay accuracy, as previously-experienced probe depletion during the experiment was prevented here using glass containers (see Methods), but culturing conditions were also slightly different. Nevertheless, the high magnitude of ΔψM raises the possibility of increased formation of superoxide and H2O2 in mitochondria during GSIS. Importantly, formation of mitochondrial superoxide and H2O2 depends on the reduction state of specific mitochondrial redox centers, which can sometimes be differentially influenced by ΔψM and ΔpH [90]. While more-reduced redox centers drive hyperpolarization of ΔψM in general, fluxes through mitochondrial metabolism and the respiratory chain are also greatly increased during GSIS, making predictions of the redox states of actual ROS-forming centers difficult. ROS levels in turn are also defined by their removal, which is known to increase when glucose is present [91].
In conclusion, data presented here explains how a central component of the canonical pathway of GSIS operates in human β-cells. ΔψM hyperpolarization is not just a simple kinetic response to increased substrate availability, but is strongly amplified by a more complicated feedback process. The activation of glucose oxidation is dependent on ATP or a related intermediate, and is responsible for the high magnitude hyperpolarization of ΔψM in response to elevation of glucose concentration. ΔψM, in turn, predicts GSIS at physiologically relevant glucose concentrations. Future studies will need to identify the molecular players of this activation mechanism and their subcellular localization. Data from human T2D β-cells [2] are consistent with the possibility that this activation mechanism is impaired in T2D [56].
Acknowledgements
The author thanks Dr Martin Brand for careful reading of the manuscript and providing facilities for this work, Herbert Kasler for cell sorting, and Chad Lerner for immunocytochemistry. This work was supported by a Sponsored Research Agreement from Calico Life Sciences LLC and National Institutes of Health grant 1R41DA043369. The author declares a financial interest in Image Analyst Software.
References
- [1].Duchen MR, Smith PA, Ashcroft FM, Substrate-dependent changes in mitochondrial function, intracellular free calcium concentration and membrane channels in pancreatic beta-cells., Biochem. J. 294 (Pt 1) (1993) 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gerencser AA, Bioenergetic Analysis of Single Pancreatic β-Cells Indicates an Impaired Metabolic Signature in Type 2 Diabetic Subjects., Endocrinology. 156 (2015) 3496–503. doi: 10.1210/en.2015-1552. [DOI] [PubMed] [Google Scholar]
- [3].Kindmark H, Köhler M, Brown G, Bränström R, Larsson O, Berggren PO, Glucose-induced oscillations in cytoplasmic free Ca2+ concentration precede oscillations in mitochondrial membrane potential in the pancreatic beta-cell., J. Biol. Chem. 276 (2001) 34530–6. doi: 10.1074/jbc.M102492200. [DOI] [PubMed] [Google Scholar]
- [4].Fridlyand LE, Ma L, Philipson LH, Adenine nucleotide regulation in pancreatic beta-cells: modeling of ATP/ADP-Ca2+ interactions., Am J Physiol Endocrinol Metab. 289 (2005) E839–48. doi: 10.1152/ajpendo.00595.2004. [DOI] [PubMed] [Google Scholar]
- [5].Nicholls DG, The Pancreatic β-Cell: A Bioenergetic Perspective., Physiol. Rev. 96 (2016) 1385–447. doi: 10.1152/physrev.00009.2016. [DOI] [PubMed] [Google Scholar]
- [6].Henquin JC, Triggering and amplifying pathways of regulation of insulin secretion by glucose, Diabetes. 49 (2000) 1751–1760. [DOI] [PubMed] [Google Scholar]
- [7].Gembal M, Gilon P, Henquin JC, Evidence that glucose can control insulin release independently from its action on ATP-sensitive K+ channels in mouse B cells, J.Clin.Invest. 89 (1992) 1288–1295. doi: 10.1172/JCI115714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Prentki M, Matschinsky FM, Madiraju SRM, Metabolic signaling in fuel-induced insulin secretion., Cell Metab. 18 (2013) 162–85. doi: 10.1016/j.cmet.2013.05.018. [DOI] [PubMed] [Google Scholar]
- [9].Lamontagne J, Al-Mass A, Nolan CJ, Corkey BE, Murthy Madiraju SR, Joly E, Prentki M, Identification of the signals for glucose-induced insulin secretion in INS1 (832/13) β-cells using metformin-induced metabolic deceleration as a model, J. Biol. Chem. 292 (2017) 19458–19468. doi: 10.1074/jbc.M117.808105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mulder H, Transcribing β-cell mitochondria in health and disease., Mol. Metab. 6 (2017) 1040–1051. doi: 10.1016/j.molmet.2017.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tabák AG, Herder C, Rathmann W, Brunner EJ, Kivimäki M, Prediabetes: a high-risk state for diabetes development., Lancet (London, England). 379 (2012) 2279–90. doi: 10.1016/S0140-6736(12)60283-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yalow RS, Berson SA, Plasma insulin concentrations in nondiabetic and early diabetic subjects. Determinations by a new sensitive immuno-assay technic., Diabetes. 9 (1960) 254–60. [DOI] [PubMed] [Google Scholar]
- [13].Del Prato S, Marchetti P, Bonadonna RC, Phasic insulin release and metabolic regulation in type 2 diabetes., Diabetes. 51 Suppl 1 (2002) S109–16. [DOI] [PubMed] [Google Scholar]
- [14].Varsano-Aharon N, Echemendia E, Yalow RS, Berson SA, Early insulin responses to glucose and to tolbutamide in maturity-onset diabetes., Metabolism. 19 (1970) 409–17. [DOI] [PubMed] [Google Scholar]
- [15].Kahn SE, The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes., Diabetologia. 46 (2003) 3–19. doi: 10.1007/s00125-002-1009-0. [DOI] [PubMed] [Google Scholar]
- [16].Brunzell JD, Robertson RP, Lerner RL, Hazzard WR, Ensinck JW, Bierman EL, Porte D, Relationships between fasting plasma glucose levels and insulin secretion during intravenous glucose tolerance tests., J. Clin. Endocrinol. Metab. 42 (1976) 222–9. doi: 10.1210/jcem-42-2-222. [DOI] [PubMed] [Google Scholar]
- [17].Haffner SM, Miettinen H, Gaskill SP, Stern MP, Decreased insulin action and insulin secretion predict the development of impaired glucose tolerance., Diabetologia. 39 (1996) 1201–7. [DOI] [PubMed] [Google Scholar]
- [18].Bergman M, Pathophysiology of prediabetes and treatment implications for the prevention of type 2 diabetes mellitus., Endocrine. 43 (2013) 504–13. doi: 10.1007/s12020-012-9830-9. [DOI] [PubMed] [Google Scholar]
- [19].Anello M, Lupi R, Spampinato D, Piro S, Masini M, Boggi U, Del Prato S, Rabuazzo AM, Purrello F, Marchetti P, Del PS, Rabuazzo AM, Purrello F, Marchetti P, Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients., Diabetologia. 48 (2005) 282–9. doi: 10.1007/s00125-004-1627-9. [DOI] [PubMed] [Google Scholar]
- [20].Doliba NM, Qin W, Najafi H, Liu C, Buettger CW, Sotiris J, Collins HW, Li C, Stanley CA, Wilson DF, Grimsby J, Sarabu R, Naji A, Matschinsky FM, Glucokinase activation repairs defective bioenergetics of islets of Langerhans isolated from type 2 diabetics, AJP Endocrinol. Metab. 302 (2012) E87–E102. doi: 10.1152/ajpendo.00218.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wikstrom JD, Sereda SB, Stiles L, Elorza A, Allister EM, Neilson A, Ferrick DA, Wheeler MB, Shirihai OS, A novel high-throughput assay for islet respiration reveals uncoupling of rodent and human islets, PLoS One. 7 (2012) e33023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li F, Porterfield DM, Zheng X-Y, Wang W-J, Xu Y, Zhang Z-M, Abnormal mitochondrial function impairs calcium influx in diabetic mouse pancreatic beta cells., Chin. Med. J. (Engl). 125 (2012) 502–10. doi: 10.3760/cma.j.issn.0366-6999.2012.03.019. [DOI] [PubMed] [Google Scholar]
- [23].Mizukami H, Wada R, Koyama M, Takeo T, Suga S, Wakui M, Yagihashi S, Augmented beta cell loss and mitochondrial abnormalities in sucrose-fed GK rats., Virchows Arch. 452 (2008) 383–92. doi: 10.1007/s00428-007-0508-2. [DOI] [PubMed] [Google Scholar]
- [24].Pepin É, Al-Mass A, Attané C, Zhang K, Lamontagne J, Lussier R, Madiraju SRM, Joly E, Ruderman NB, Sladek R, Prentki M, Peyot M-L, Pancreatic β-Cell Dysfunction in Diet-Induced Obese Mice: Roles of AMP-Kinase, Protein Kinase Cε, Mitochondrial and Cholesterol Metabolism, and Alterations in Gene Expression., PLoS One. 11 (2016) e0153017. doi: 10.1371/journal.pone.0153017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lu H, Koshkin V, Allister EM, Gyulkhandanyan AV, Wheeler MB, Molecular and metabolic evidence for mitochondrial defects associated with beta-cell dysfunction in a mouse model of type 2 diabetes., Diabetes. 59 (2010) 448–59. doi: 10.2337/db09-0129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Antinozzi PA, Ishihara H, Newgard CB, Wollheim CB, Mitochondrial metabolism sets the maximal limit of fuel-stimulated insulin secretion in a model pancreatic beta cell: a survey of four fuel secretagogues, J Biol. Chem. 277 (2002) 11746–11755. doi: 10.1074/jbc.M108462200. [DOI] [PubMed] [Google Scholar]
- [27].Heart E, Corkey RF, Wikstrom JD, Shirihai OS, Corkey BE, Glucose-dependent increase in mitochondrial membrane potential, but not cytoplasmic calcium, correlates with insulin secretion in single islet cells., Am J Physiol Endocrinol Metab. 290 (2006) E143–E148. doi: 10.1152/ajpendo.00216.2005. [DOI] [PubMed] [Google Scholar]
- [28].Ohta M, Nelson D, Nelson J, Meglasson MD, Erecińska M, Oxygen and temperature dependence of stimulated insulin secretion in isolated rat islets of Langerhans., J. Biol. Chem. 265 (1990) 17525–32. [PubMed] [Google Scholar]
- [29].Detimary P, Jonas JC, Henquin JC, Possible links between glucose-induced changes in the energy state of pancreatic B cells and insulin release. Unmasking by decreasing a stable pool of adenine nucleotides in mouse islets, J. Clin. Invest. 96 (1995) 1738–45. doi: 10.1172/JCI118219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mugabo Y, Zhao S, Lamontagne J, Al-Mass A, Peyot ML, Corkey BE, Joly E, Madiraju SRM, Prentki M, Metabolic fate of glucose and candidate signaling and excess-fuel detoxification pathways in pancreatic β-cells, J. Biol. Chem. 292 (2017) 7407–7422. doi: 10.1074/jbc.M116.763060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Merglen A, Theander S, Rubi B, Chaffard G, Wollheim CB, Maechler P, Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells., Endocrinology. 145 (2004) 667–78. doi: 10.1210/en.2003-1099. [DOI] [PubMed] [Google Scholar]
- [32].Sun MY, Yoo E, Green BJ, Altamentova SM, Kilkenny DM, V Rocheleau J, Autofluorescence imaging of living pancreatic islets reveals fibroblast growth factor-21 (FGF21)-induced metabolism., Biophys. J. 103 (2012) 2379–88. doi: 10.1016/j.bpj.2012.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Maechler P, Wollheim CB, Mitochondrial glutamate acts as a messenger in glucose-induced insulin exocytosis, Nature. 402 (1999) 685–689. [DOI] [PubMed] [Google Scholar]
- [34].MacDonald MJ, Fahien LA, Glutamate is not a messenger in insulin secretion., J. Biol. Chem. 275 (2000) 34025–7. doi: 10.1074/jbc.C000411200. [DOI] [PubMed] [Google Scholar]
- [35].Zhao S, Mugabo Y, Iglesias J, Xie L, Delghingaro-Augusto V, Lussier R, Peyot M-L, Joly E, Taïb B, Davis MA, Brown JM, Abousalham A, Gaisano H, Madiraju SRM, Prentki M, α/β-Hydrolase domain-6-accessible monoacylglycerol controls glucose-stimulated insulin secretion., Cell Metab. 19 (2014) 993–1007. doi: 10.1016/j.cmet.2014.04.003. [DOI] [PubMed] [Google Scholar]
- [36].Ivarsson R, Quintens R, Dejonghe S, Tsukamoto K, in ‘t Veld P, Renström E, Schuit FC, Redox control of exocytosis: regulatory role of NADPH, thioredoxin, and glutaredoxin., Diabetes. 54 (2005) 2132–42. [DOI] [PubMed] [Google Scholar]
- [37].Prentki M, Vischer S, Glennon MC, Regazzi R, Deeney JT, Corkey BE, Malonyl-CoA and long chain acyl-CoA esters as metabolic coupling factors in nutrient-induced insulin secretion., J. Biol. Chem. 267 (1992) 5802–10. [PubMed] [Google Scholar]
- [38].Mulder H, Lu D, Finley J, An J, Cohen J, Antinozzi PA, McGarry JD, Newgard CB, Overexpression of a modified human malonyl-CoA decarboxylase blocks the glucose-induced increase in malonyl-CoA level but has no impact on insulin secretion in INS-1-derived (832/13) beta-cells, J Biol. Chem. 276 (2001) 6479–6484. doi: 10.1074/jbc.M010364200. [DOI] [PubMed] [Google Scholar]
- [39].Kibbey RG, Pongratz RL, Romanelli AJ, Wollheim CB, Cline GW, Shulman GI, Mitochondrial GTP regulates glucose-stimulated insulin secretion., Cell Metab. 5 (2007) 253–64. doi: 10.1016/j.cmet.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Stark R, Pasquel F, Turcu A, Pongratz RL, Roden M, Cline GW, Shulman GI, Kibbey RG, Phosphoenolpyruvate cycling via mitochondrial phosphoenolpyruvate carboxykinase links anaplerosis and mitochondrial GTP with insulin secretion., J. Biol. Chem. 284 (2009) 26578–90. doi: 10.1074/jbc.M109.011775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Casimir M, Rubi B, Frigerio F, Chaffard G, Maechler P, Silencing of the mitochondrial NADH shuttle component aspartate-glutamate carrier AGC1/Aralar1 in INS-1E cells and rat islets, Biochem J. 424 (2009) 459–466. [DOI] [PubMed] [Google Scholar]
- [42].Joseph JW, V Jensen M, Ilkayeva O, Palmieri F, Alárcon C, Rhodes CJ, Newgard CB, Alarcon C, Rhodes CJ, Newgard CB, The mitochondrial citrate/isocitrate carrier plays a regulatory role in glucose-stimulated insulin secretion., J. Biol. Chem. 281 (2006) 35624–32. doi: 10.1074/jbc.M602606200. [DOI] [PubMed] [Google Scholar]
- [43].Fransson U, Rosengren AH, Schuit FC, Renstrom E, Mulder H, Anaplerosis via pyruvate carboxylase is required for the fuel-induced rise in the ATP:ADP ratio in rat pancreatic islets, Diabetologia. 49 (2006) 1578–1586. [DOI] [PubMed] [Google Scholar]
- [44].Erion KA, Berdan CA, Burritt NE, Corkey BE, Deeney JT, Chronic Exposure to Excess Nutrients Left-shifts the Concentration Dependence of Glucose-stimulated Insulin Secretion in Pancreatic β-Cells., J. Biol. Chem. 290 (2015) 16191–201. doi: 10.1074/jbc.M114.620351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Goehring I, Sharoyko VV, Malmgren S, Andersson LE, Spegel P, Nicholls DG, Mulder H, Chronic high Glucose and Pyruvate Levels differentially affect Mitochondrial Bioenergetics and Fuel-stimulated insulin secretion from Clonal INS-1 832/13 Cells, J. Biol. Chem. 289 (2014) 3786–3798. doi: 10.1074/jbc.M113.507335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sweet IR, Gilbert M, Scott S, Todorov I, Jensen R, Nair I, Al-Abdullah I, Rawson J, Kandeel F, Ferreri K, Glucose-stimulated increment in oxygen consumption rate as a standardized test of human islet quality., Am J Transplant. 8 (2008) 183–92. doi: 10.1111/j.1600-6143.2007.02041.x. [DOI] [PubMed] [Google Scholar]
- [47].Schulz N, Kluth O, Jastroch M, Schürmann A, Minor Role of Mitochondrial Respiration for Fatty-Acid Induced Insulin Secretion, Int. J. Mol. Sci. 14 (2013) 18989–18998. doi: 10.3390/ijms140918989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Spacek T, Santorová J, Zacharovová K, Berková Z, Hlavatá L, Saudek F, Jezek P, Santorova J, Zacharovova K, Berkova Z, Hlavata L, Saudek F, Jezek P, Glucose-stimulated insulin secretion of insulinoma INS-1E cells is associated with elevation of both respiration and mitochondrial membrane potential., Int. J. Biochem. Cell Biol. 40 (2008) 1522–35. doi: 10.1016/j.biocel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- [49].Affourtit C, Alberts B, Barlow J, Carré JE, Wynne AG, Control of pancreatic β-cell bioenergetics, Biochem. Soc. Trans. (2018) BST20170505. doi: 10.1042/BST20170505. [DOI] [PubMed] [Google Scholar]
- [50].Pullen TJ, Rutter GA, When less is more: the forbidden fruits of gene repression in the adult β-cell., Diabetes. Obes. Metab. 15 (2013) 503–12. doi: 10.1111/dom.12029. [DOI] [PubMed] [Google Scholar]
- [51].Spégel P, Andersson LE, Storm P, Sharoyko V, Göhring I, Rosengren AH, Mulder H, Unique and Shared Metabolic Regulation in Clonal β-Cells and Primary Islets Derived From Rat Revealed by Metabolomics Analysis., Endocrinology. 156 (2015) 1995–2005. doi: 10.1210/en.2014-1391. [DOI] [PubMed] [Google Scholar]
- [52].Spégel P, V Sharoyko V, Goehring I, Danielsson APH, Malmgren S, Nagorny CLF, Andersson LE, Koeck T, Sharp GWG, Straub SG, Wollheim CB, Mulder H, Time-resolved metabolomics analysis of β-cells implicates the pentose phosphate pathway in the control of insulin release., Biochem. J. 450 (2013) 595–605. doi: 10.1042/BJ20121349. [DOI] [PubMed] [Google Scholar]
- [53].Hafner RP, Brown GC, Brand MD, Analysis of the control of respiration rate, phosphorylation rate, proton leak rate and protonmotive force in isolated mitochondria using the “top-down” approach of metabolic control theory., Eur. J. Biochem. 188 (1990) 313–9. [DOI] [PubMed] [Google Scholar]
- [54].Ainscow EK, Brand MD, Internal regulation of ATP turnover, glycolysis and oxidative phosphorylation in rat hepatocytes., Eur. J. Biochem. 266 (1999) 737–49. [DOI] [PubMed] [Google Scholar]
- [55].Brand MD, Regulation analysis of energy metabolism., J. Exp. Biol. 200 (1997) 193–202. [DOI] [PubMed] [Google Scholar]
- [56].Gerencser AA, Mookerjee SA, Jastroch M, Brand MD, Positive Feedback Amplifies the Response of Mitochondrial Membrane Potential to Glucose Concentration in Clonal Pancreatic Beta Cells, Biochim. Biophys. Acta - Mol. Basis Dis. 1863 (2017) 1054–1065. doi: 10.1016/j.bbadis.2016.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Jayaraman S, A novel method for the detection of viable human pancreatic beta cells by flow cytometry using fluorophores that selectively detect labile zinc, mitochondrial membrane potential and protein thiols., Cytometry. A. 73 (2008) 615–25. doi: 10.1002/cyto.a.20560. [DOI] [PubMed] [Google Scholar]
- [58].Gerencser AA, Chinopoulos C, Birket MJ, Jastroch M, Vitelli C, Nicholls DG, Brand MD, Quantitative measurement of mitochondrial membrane potential in cultured cells: calcium-induced de- and hyperpolarization of neuronal mitochondria., J. Physiol. 590 (2012) 2845–71. doi: 10.1113/jphysiol.2012.228387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Gerencser AA, Mookerjee SA, Jastroch M, Brand MD, Measurement of the Absolute Magnitude and Time Courses of Mitochondrial Membrane Potential in Primary and Clonal Pancreatic Beta-Cells., PLoS One. 11 (2016) e0159199. doi: 10.1371/journal.pone.0159199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Grimmsmann T, Rustenbeck I, Direct effects of diazoxide on mitochondria in pancreatic B-cells and on isolated liver mitochondria., Br. J. Pharmacol. 123 (1998) 781–8. doi: 10.1038/sj.bjp.0701663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Affourtit C, Brand MD, Stronger control of ATP/ADP by proton leak in pancreatic beta-cells than skeletal muscle mitochondria., Biochem. J. 393 (2006) 151–9. doi: 10.1042/BJ20051280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Barlow J, Affourtit C, Novel insights in pancreatic beta cell glucolipotoxicity from real-time functional analysis of mitochondrial energy metabolism in INS-1E insulinoma cells, Biochem. J. 456 (2013) 417–426. [DOI] [PubMed] [Google Scholar]
- [63].Mookerjee SA, Nicholls DG, Brand MD, Determining Maximum Glycolytic Capacity Using Extracellular Flux Measurements, PLoS One. 11 (2016) e0152016. doi: 10.1371/journal.pone.0152016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Birket MJ, Orr AL, Gerencser AA, Madden DT, Vitelli C, Swistowski A, Brand MD, Zeng X, A reduction in ATP demand and mitochondrial activity with neural differentiation of human embryonic stem cells., J. Cell Sci. 124 (2011) 348–58. doi: 10.1242/jcs.072272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Brand MD, Nicholls DG, Assessing mitochondrial dysfunction in cells., Biochem. J. 435 (2011) 297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Estimates of Diabetes and Its Burden in the United States, Natl. Diabetes Stat. Rep. (2017). https://www.cdc.gov/diabetes/pdfs/data/statistics/national-diabetes-statistics-report.pdf.
- [67].Weir GC, Bonner-Weir S, Five stages of evolving beta-cell dysfunction during progression to diabetes., Diabetes. 53 Suppl 3 (2004) S16–21. doi: 10.2337/diabetes.53.suppl_3.S16. [DOI] [PubMed] [Google Scholar]
- [68].Ashcroft SJ, Weerasinghe LC, Randle PJ, Interrelationship of islet metabolism, adenosine triphosphate content and insulin release., Biochem. J. 132 (1973) 223–31. doi: 10.1042/bj1320223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Detimary P, Gilon P, Nenquin M, Henquin JC, Two sites of glucose control of insulin release with distinct dependence on the energy state in pancreatic B-cells, Biochem. J. 297 ( Pt 3 (1994) 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].MacDonald MJ, Longacre MJ, Stoker SW, Kendrick M, Thonpho A, Brown LJ, Hasan NM, Jitrapakdee S, Fukao T, Hanson MS, a Fernandez L, Odorico J, Differences between human and rodent pancreatic islets: low pyruvate carboxylase, atp citrate lyase, and pyruvate carboxylation and high glucose-stimulated acetoacetate in human pancreatic islets., J. Biol. Chem. 286 (2011) 18383–96. doi: 10.1074/jbc.M111.241182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Skelin Klemen M, Dolenšek J, Slak Rupnik M, Stožer A, The triggering pathway to insulin secretion: Functional similarities and differences between the human and the mouse β cells and their translational relevance., Islets. 9 (2017) 109–139. doi: 10.1080/19382014.2017.1342022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Henquin JC, Regulation of insulin secretion: a matter of phase control and amplitude modulation, Diabetologia. 52 (2009) 739–751. doi: 10.1007/s00125-009-1314-y. [DOI] [PubMed] [Google Scholar]
- [73].Mármol P, Pardo B, Wiederkehr A, del Arco A, Wollheim CB, Satrústegui J, Marmol P, Pardo B, Wiederkehr A, Del AA, Wollheim CB, Satrustegui J, Requirement for aralar and its Ca2+-binding sites in Ca2+ signal transduction in mitochondria from INS-1 clonal beta-cells., J. Biol. Chem. 284 (2009) 515–24. doi: 10.1074/jbc.M806729200. [DOI] [PubMed] [Google Scholar]
- [74].Maechler P, Wang H, Wollheim CB, Continuous monitoring of ATP levels in living insulin secreting cells expressing cytosolic firefly luciferase, FEBS Lett. 422 (1998) 328–332. [DOI] [PubMed] [Google Scholar]
- [75].Heart E, Cline GW, Collis LP, Pongratz RL, Gray JP, Smith PJS, Role for malic enzyme, pyruvate carboxylation, and mitochondrial malate import in glucose-stimulated insulin secretion., Am J Physiol Endocrinol Metab. 296 (2009) E1354–62. doi: 10.1152/ajpendo.90836.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Sekine N, Cirulli V, Regazzi R, Brown LJ, Gine E, Tamarit-Rodriguez J, Girotti M, Marie S, MacDonald MJ, Wollheim CB, Rutter GA, Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic beta-cells. Potential role in nutrient sensing., J. Biol. Chem. 269 (1994) 4895–902. [PubMed] [Google Scholar]
- [77].Wiederkehr A, Park K-SS, Dupont O, Demaurex N, Pozzan T, Cline GW, Wollheim CB, Matrix alkalinization: a novel mitochondrial signal for sustained pancreatic beta-cell activation., EMBO J. 28 (2009) 417–28. doi: 10.1038/emboj.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Akhmedov D, Braun M, Mataki C, Park KS, Pozzan T, Schoonjans K, Rorsman P, Wollheim CB, Wiederkehr A, Mitochondrial matrix pH controls oxidative phosphorylation and metabolism-secretion coupling in INS-1E clonal {beta} cells, FASEB J. 24 (2010). [DOI] [PubMed] [Google Scholar]
- [79].De Marchi U, Hermant A, Thevenet J, Ratinaud Y, Santo-Domingo J, Barron D, Wiederkehr A, A novel ATP-synthase-independent mechanism coupling mitochondrial activation to exocytosis in insulin-secreting cells, J. Cell Sci. 130 (2017) 1929–1939. doi: 10.1242/jcs.200741. [DOI] [PubMed] [Google Scholar]
- [80].Sweet IR, Gilbert M, Contribution of calcium influx in mediating glucose-stimulated oxygen consumption in pancreatic islets., Diabetes. 55 (2006) 3509–19. doi: 10.2337/db06-0400. [DOI] [PubMed] [Google Scholar]
- [81].Wilson DF, Cember ATJ, Matschinsky FM, The thermodynamic basis of glucose-stimulated insulin release: a model of the core mechanism, Physiol. Rep. 5 (2017) 1–13. doi: 10.14814/phy2.13327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].MacDonald MJ, Differences between mouse and rat pancreatic islets: succinate responsiveness, malic enzyme, and anaplerosis., Am J Physiol Endocrinol Metab. 283 (2002) E302–10. doi: 10.1152/ajpendo.00041.2002. [DOI] [PubMed] [Google Scholar]
- [83].Degli Esposti M, Ghelli A, Ratta M, Cortes D, Estornell E, Natural substances (acetogenins) from the family Annonaceae are powerful inhibitors of mitochondrial NADH dehydrogenase (Complex I), Biochem. J. 301 (1994) 161–167. doi: 10.1042/bj3010161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Sweet IR, Matschinsky FM, Mathematical model of beta-cell glucose metabolism and insulin release. I. Glucokinase as glucosensor hypothesis., Am. J. Physiol. 268 (1995) E775–88. [DOI] [PubMed] [Google Scholar]
- [85].Wang H, Iynedjian PB, Modulation of glucose responsiveness of insulinoma beta-cells by graded overexpression of glucokinase., Proc. Natl. Acad. Sci. U. S. A. 94 (1997) 4372–4377. doi: 10.1073/pnas.94.9.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Pi J, Bai Y, Zhang Q, Wong V, Floering LM, Daniel K, Reece JM, Deeney JT, Andersen ME, Corkey BE, Collins S, Reactive oxygen species as a signal in glucose-stimulated insulin secretion., Diabetes. 56 (2007) 1783–91. doi: 10.2337/db06-1601. [DOI] [PubMed] [Google Scholar]
- [87].Lortz S, Gurgul-Convey E, Naujok O, Lenzen S, Overexpression of the antioxidant enzyme catalase does not interfere with the glucose responsiveness of insulin-secreting INS-1E cells and rat islets., Diabetologia. 56 (2013) 774–82. doi: 10.1007/s00125-012-2823-7. [DOI] [PubMed] [Google Scholar]
- [88].Benhamou PY, Moriscot C, Richard MJ, Beatrix O, Badet L, Pattou F, Kerr-Conte J, Chroboczek J, Lemarchand P, Halimi S, Adenovirus-mediated catalase gene transfer reduces oxidant stress in human, porcine and rat pancreatic islets., Diabetologia. 41 (1998) 1093–100. doi: 10.1007/s001250051035. [DOI] [PubMed] [Google Scholar]
- [89].Bindokas VP, Kuznetsov A, Sreenan S, Polonsky KS, Roe MW, Philipson LH, Visualizing superoxide production in normal and diabetic rat islets of Langerhans, J. Biol. Chem. 278 (2003) 9796–9801. doi: 10.1074/jbc.M206913200. [DOI] [PubMed] [Google Scholar]
- [90].Wong H-S, Dighe PA, Mezera V, Monternier P-A, Brand MD, Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions., J. Biol. Chem. 292 (2017) 16804–16809. doi: 10.1074/jbc.R117.789271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Liu X, Han S, Yang Y, Kang J, Wu J, Glucose-induced glutathione reduction in mitochondria is involved in the first phase of pancreatic β-cell insulin secretion., Biochem. Biophys. Res. Commun. 464 (2015) 730–6. doi: 10.1016/j.bbrc.2015.07.016. [DOI] [PubMed] [Google Scholar]