

Abstract
Protein binding specificities can be manipulated by redesigning contacts that already exist at an interface or by expanding the interface to allow interactions with residues adjacent to the original binding site. Previously, we developed a strategy, called AnchorDesign, for expanding interfaces around linear binding epitopes. The epitope is embedded in a loop of a scaffold protein, in our case a monobody, and then surrounding residues on the monobody are optimized for binding using directed evolution or computational design. Using this strategy, we have increased binding affinities by over 100-fold, but we have not tested whether it can be used to control protein binding specificities. Here, we test whether AnchorDesign can be used to engineer a monobody that binds specifically to the Mitogen-activated protein kinase (MAPK) p38α, but not to the related MAPKs ERK2 and JNK. To anchor the binding interaction, we used a small (D) docking motif from the Mitogen-activated protein kinase kinase (MAP2K) MKK6 that interacts with similar affinity to p38α and ERK2. Our hypothesis was that by embedding the motif in a larger protein that we could expand the interface and create contacts with residues that are not conserved between p38α and ERK2. Molecular modeling was used to inform insertion of the D motif into the monobody and a combination of phage and yeast display were used to optimize the interface. Binding experiments demonstrate that the engineered monobody binds to the target surface on p38α and does not exhibit detectable binding to ERK2 or JNK.
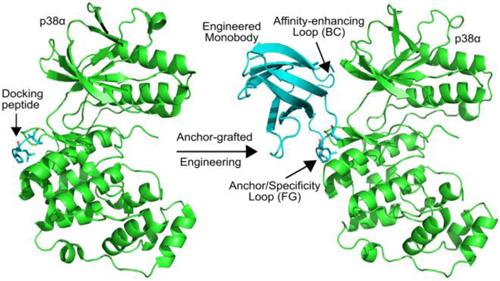
Graphical Abstract
Introduction
Protein-protein interactions are essential to almost all biological processes. Engineered proteins with novel binding properties are used as therapeutics and are important tools for cellular and biomolecular research1,2. The improvement of binding affinity and/or binding specificity is a frequent goal for projects in structure-based interface design. One approach to manipulate affinity and specificity is to mutate amino acids already at the interface3, but in some cases these residues may already be highly optimized for the target interaction. Another strategy is to redesign the structure of one or both partners to expand the interface and allow the formation of new contacts4,5. New contacts can help tighten the interaction and they can provide specificity if the newly buried surface area is unique to the binding partners.
Recently, we developed a design strategy aimed at expanding the set of contacts at a protein-protein or peptide-protein interface6. The approach begins with the identification of a set of amino acids linear in primary sequence that contribute significantly to an interaction. This string of “anchor” residues is then embedded in a loop of a monobody. Monobodies are antibody-like domains derived from fibronectin type III domains2. They contain several loops that are amenable to substitution and can be redesigned to form interactions with a diversity of proteins. After the anchor loop is inserted in the monobody, directed evolution and/or computational design is used to redesign neighboring loops in the monobody to create additional contacts at the interface. Previously, we demonstrated that we could use this approach to create a competitive inhibitor against the KEAP1-NRF2 interaction4. By inserting a peptide from NRF2 into the FG-loop of the monobody and optimizing the neighboring BC-loop with phage display we were able to lower the dissociation constant from 141 nM to 300 pM. Here, we explore whether a similar strategy can be used to manipulate binding specificity. As a model system, we study a peptide that binds to the Common Docking (CD) groove of MAP family kinases (MAPKs)7,8.
MAPKs regulate a variety of cellular processes including cell proliferation, differentiation, and apoptosis9,10,11,12. MAPKs are activated by mitogen-activated protein kinase kinases (MAP2Ks). The specificity of MAP2K/MAPK interactions are determined in part by linear motifs on the MAP2Ks (docking (D) motifs) which bind in the CD groove of MAPKs7,8. Binding studies have been used to characterize the specificity of different D motifs for MAPKs, and while several D motif sequences bind exclusively to the MAPK JNK1, many of the D motif sequences that bind to the MAPK p38α also bind to the MAPK ERK213. This multi-specificity reflects the close structural similarity between the CD groove in p38α and ERK2. However, a superposition of the p38α and ERK2 crystal structures shows that adjacent to the CD groove there is a patch of residues that differ between the two proteins13 (Figure 1). We hypothesized that interface expansion with our monobody-based strategy could be used to expand the D motif/MAPK interaction to encompass this dissimilarity patch and create binders specific for p38α. Here, we describe the engineering and characterization of a monobody specific for p38α.
Figure 1.

A comparison of D motif binding sites on p38α and ERK2. (A) A crystal structure (PDB ID: 2y8o) of a docking peptide derived from MKK6 (purple) bound to p38α (gray). p38α residues adjacent to the binding site that differ from ERK2 are shown in green. (B) A superposition of the MKK6 D-motif (purple) on a crystal structure of ERK2 (gray, PDB ID: 2y9q). Residues in green differ from those on p38α. *Residue numbers from ERK2 are shifted up by one to match structurally equivalent positions on p38α.
Materials and Methods
Monobody modeling
All modeling work was performed using the Rosetta application AnchoredDesign and its components6. The goal of the modeling was to determine which loop in a monobody was compatible with insertion of the MKK6 D motif and would place a neighboring loop of the monobody adjacent to the target surface on p38α. First, AnchoredPDBCreator was used to create starting models for the simulation. Next, flexible design loop simulations were performed with AnchoredDesign to search for monobody loop sequences that are compatible with the grafted anchor conformations and p38α binding. AnchoredPDBCreator enabled the creation of intermediate models using structural data of monobody (PDB ID: 1fna) and the p38α/pep-MKK6 complex (PDB ID: 2y8o)13.
AnchoredPDBCreator was used to combine the aforementioned structures in a manner that loop residues of monobody were replaced with the identities and conformations of peptides derived from the pep-MKK6 chain. This anchor-grafted monobody was then docked against p38α by superimposing the anchor residues on the monobody with the D motif from the p38α/pep-MKK6 crystal structure. Non-anchor portions of the monobody were then checked for any major clashes between the monobody and p38α. Since we did not have any data a priori as to which anchor sequence, loop length or anchor placement within a loop was optimal, we performed multiple pep-MKK6-derived anchor sequences in several loop placements and lengths [Supplementary Table 1].
The second step of the computational work involved design simulations using AnchoredDesign. In these design simulations, anchor-residues were held fixed internally as well as relative to the monobody. The p38α/anchor residues of the interface was held fixed while non-anchor residues in the anchor-containing loop and non-anchor loops were remodeled with Rosetta’s implementation of the cyclic coordinate descent and kinematic loop closure14. All other residues near the interface were given the freedom to relax their side-chains but no sequence alterations were allowed. Residues away from the interface were not modified. All simulations were performed on the UNC research computing cluster using command line options as described in Guntas et al.4. Examination of the best models as adjudged by the Rosetta energy and visual inspection [Supplementary Figure 1] suggested that anchors placed in the FG loop (anchor-loop) with the PGLKIP sequence grafted at either position 79 or 80 (numbering as in PDB 1fna) and complete randomization of the BC loop (non-anchor loop) had a better likelihood of generating monobody binders.
Synthesis of peptide anchor in phage display
The Pep-MKK6 sequence NPGLKIP was cloned as a N-terminal fusion to the phage pIII-coat protein into the pFNOM6-tat-pIII plasmid as described before4. Briefly, 100 pmols of the oligonucleotide (Eurofins Genomics) encoding the peptide sequence [5’-CTCGCGGCC GCAGGATCCAACCCGGGCCTGAAAATTCCGAAACTCGAGTCTAGAGGGCCC-3’] flanking 18 bases matching with the pFNOM6-tat-pIII on each side was first phosphorylated with 20 units of T4 polynucleotide kinase (NEB) for 1 hr at 37° C, followed by heat inactivation at 65° C for 10 min. In parallel, single-stranded template uracil-DNA (pFNOM6-tat-pIII) was amplified and purified as described15. Next, 0.4 pmol of template DNA was combined with 5-fold excess phosphorylated peptide oligo for template-oligo annealing followed by polymerase extension and ligation of covalently closed circular dsDNA (CCC-dsDNA) as described previously15. 2 μL Purified CCC-dsDNA was mixed with 30 μL electrocompetent E. coli SS320 cells into a pre-chilled 0.2 cm gap cuvette [USA Scientific] followed by electroporation by Biorad Genepulser II set at E. coli transformation setting. Transformed SS320 cells were first recovered after electroporation by growing them at 37° C for 1 hr at 200 rpm and subsequent plating in LB+ampicillin agar plates. Peptide sequence was verified by isolating phagemids for DNA sequencing (Genewiz) using the reverse sequencing primer 5’-CACCCTCAGAGCCGCCACCAGAAC’−3.
Library construction and affinity-selection by phage display
The phage display monobody library, Lib1 containing the PGLKIP anchor in the FG loop and the randomized BC loop was constructed using Kunkel mutagenesis as N-terminal fusions to the pIII coat-protein of the filamentous bacteriophage M13. 300 pmol of each oligonucleotide i.e. the oligonucleotide encoding 7 NNK degenerate codons to create the randomized-BC library and a second oligonucleotide encoding the PGLKIP anchor flanked by one NNK codon on each side of the anchor within the FG loop were 5′-phosphorylated in a single reaction with 20 units of T4 polynucleotide kinase in the presence of 1 mM ATP, 5 mM DTT, 10 mM MgCl2 and 50 mM Tris–HCl pH 7.5. Phosphorylated oligos were annealed to 20 μg of uracil-incorporated single-stranded phagemid with 5:1 oligo:template ratio in the presence of 10 mM MgCl2 and 50 mM Tris–HCl pH 7.5 by cooling from 90°C to 30°C over 50 min followed by incubation for 5 min at 4°C. Annealed oligonucleotides were extended with 30 units of T7 DNA polymerase and the CCC-dsDNA was ligated with 2 kU of T4 DNA ligase by incubation at 20°C for 3 h in the presence of 0.85 mM each dNTP, 0.33 mM ATP, 5 mM DTT, 10 mM MgCl2 and 50 mM Tris–HCl pH 7.5. Desalted reaction products were transformed into 350 μL electrocompetent SS320 cells as described in the previous section. Pooled transformed bacteria were grown for 1 hr in 12 mL SOC media and finally expanded into 500 ml 2YT medium supplemented with 100 μg/mL ampicillin (for phagemid selection), 1010 pfu/mL M13K07 helper phage and 25 μg/mL kanamycin (helper phage selection marker). Prior to superinfection with the helper phage, serial dilutions were plated on LB+Ampicillin plates to estimate library size. Phage library, Lib1 was produced by shaking (225 rpm) overnight at 37° C. Next day, phage from Lib1 was precipitated, quantified and stored at −80° C as described15. Lib1 diversity was estimated to be > 3×108 from cfu count on serially diluted plates. For phage panning, Lib1 was first incubated for 2 hrs at room temperature with a Maxisorp plate coated overnight with 5% BSA (in PBS) as a negative selection step. Supernatant from BSA incubation was then incubated for 2 hrs at room temperature with Maxisorp plate pre-coated with recombinantly expressed and purified p38α (as in previous section) and pre-blocked with BSA. Phage supernatant was removed, followed by washing the well 10x with the wash buffer, WB (1% BSA and 0.05% Tween 20 in PBS). Subsequently, bound phage was eluted with 100 μL 0.2 M glycine/HCl/0.1% BSA, pH 2.2 buffer followed by immediate neutralization with 15 μL 1M Tris –HCl, pH 9.1 buffer. Actively growing, log-phase SS320 cells were infected with the eluted phage followed by superinfection with the helper phage to amplify for the next round. Serially diluted infected SS320 cells were also plated on LB+Ampicillin plates before superinfection with helper phage. Four rounds of phage panning were carried out in this fashion followed by phage ELISA. To perform phage ELISA, each well of a 96-well Maxisorp plate (ThermoFisher) was coated overnight at 4° C with 0.5 μg/100 μL recombinantly purified p38α (purification protocol is given below) or BSA control in 50 mM sodium bicarbonate buffer, pH 9.6. Next day, the 96-well plates were blocked for approximately 2 hours at room temperature with blocking buffer (0.5% BSA in PBS). In parallel, colonies harboring phagemid encoding the peptide anchor and 93 colonies from the 4th round of panning were inoculated in 450 μL aliquots of 2YT medium supplemented with 50 μg/mL Ampicillin and 1010 pfu/mL M13K07 helper phage in a 96-well microtubes-rack (National Scientific) for overnight culture in a 37° C incubator with shaking at 200 rpm. Supernatant from the overnight cultures were 3-fold diluted in PBT buffer (PBS+0.05% Tween 20+0.5% BSA) and 100 μL of this dilute supernatant was added to each well of pre-blocked, p38α-coated 96-well plate. After 1 hr of incubation at room-temperature, supernatant from each well was removed, followed by 8X wash with PT buffer (PBS+0.05% Tween 20) and then incubation with 1:2500 diluted (in PBT) HRP-conjugated anti-M13 antibody (GE Healthcare Life Sciences) for 30 min. Next, the 96-well plates were washed 6X with PT buffer followed by the addition of freshly prepared TMB substrate (KPL Inc.) for incubation with 5–10 min until color development. The reaction was quenched with 100 μL 1M H3PO4 and read in a microplate reader at 450 nm (SpectraMax, Molecular Devices). Signal from the control wells were subtracted from p38α-coated wells for plotting the histogram in Supplementary Figure 3. In parallel, SS320 cells were infected with phage supernatant used in the phage ELISA followed by overnight culture in 2YT medium supplemented with 5 μg/mL tetracycline, miniprepping (Qiagen) and subsequent DNA sequencing with the reverse sequencing primer as described in the previous section.
Library construction in yeast display
To generate a yeast-display library (Lib2), error-prone PCR was performed separately on G4mb and C6mb monobody clones as template DNA. For each template DNA, varying concentrations of nucleotide analogs and number of PCR cycles were employed as described16. Ten and 20 cycles of PCRs with both nucleotide analogs (8-oxo-dGTP and dPTP, Trilink Biotechnologies) at 10 μM each as well as 20 and 30 cycles of PCRs with both nucleotide analogs at 2 μM each was performed. In addition to the nucleotide analogs, each 50 μL PCR reaction was composed of 1X Thermopol buffer (NEB), 200 μM dNTP mix, 20 ng template DNA, 0.1 μM each of yeast display (YSD) forward and reverse primers (Supplementary Table 2, Eurofins Genomics) and 2.5 U Taq DNA polymerase (NEB). The reaction mixture was denatured at 95° C for 30 sec followed by 10/20/30 cycles of denaturation at 95° C for 30 sec, annealing at 62° C for 45 sec, extension at 68° C for 45 sec and a final extension at 68° C for 5 min in an Eppendorf Mastercycler Pro machine. To get a high yield, 5×50 μL PCR for each of the four combination PCRs were carried out. PCR-products were gel-extracted (Qiagen Gel extraction kit) and 200 ng DNA from each combination of PCR were mixed to give a template DNA mixture at 25 ng/μL. A final DNA amplification reaction (50 μL PCR) was composed of 1X Q5 reaction buffer (NEB), 200 μM dNTP mix, 25 ng template DNA, 0.1 μM each of the yeast display forward and reverse primers (Eurofins Genomics) and 1 U high- fidelity Q5 DNA polymerase (NEB). Product from this final round of PCR was also gel-extracted and pellet-painted (EMD Millipore) to 1.5 μg/μL and 1.8 μg/μL final insert DNA originating from C6mb and G4mb respectively. Although both of these clones were isolated from a single phage display library, different set of primers were used for these two genes because the template for G4mb was inadvertently ordered as a gblock dsDNA (IDT DNA) and hence codons did not match with C6mb. Lib2 was generated by co-transformation of insert DNA and linearized plasmid in yeast as described previously with slight modifications17. The yeast display vector pETCON and the yeast strain S. cerevisia EBY100 were gifts from the Baker Laboratory (University of Washington, USA). The pETCON vector was digested with NheI, BamHI and SalI restriction endonuleases (Fermentas). Each of the twenty 0.2 cm gap cuvette for electroporation contained freshly prepared 50 μL EBY100 electrocompetent cells containing 1 μg vector and 2×2.8 μg of insert DNA (2.8 μg from each of the G4mb and C6mb error-prone PCR product). Electroporation was carried out in an electroporator (Biorad Gene Pulser II) set at 0.54 kV, 25 μF and 1000 Ω with a pulse controller. All transformations were pooled to a total of 40 mL YPD medium (10 g/L yeast extract, 20 g/L peptone, 20 g/L dextrose), grown for an hour at 30° C incubator with shaking at 250 rpm. Cells were then pelleted and resuspended in 10 mL SDCAA (20 g/L dextrose, 5 g/L Casamino acids, 6.7 g/L yeast nitrogen base, 5.40 g/L Na2HPO4, 7.45 g/L NaH2PO4) medium for serial dilution in SDCAA plates followed by >48 hours of growth in 250 mL SDCAA medium supplemented with Penicillin+Streptomycin (ThermoFisher Scientific) in a shaker at 30° C and 250 rpm. Library diversity was estimated to be ~1.65×107 based on the number of yeast transformants on SDCAA plates.
Yeast display library screening by FACS
Expression of the yeast library was induced by switching yeast cell culture medium from SDCAA to SGCAA (20 g/L galactose, 2 g/L dextrose, 5 g/L Casamino acids, 6.7 g/L yeast nitrogen base, 5.40 g/L Na2HPO4, 7.45 g/L NaH2PO4) for 20–24 hours at 20° C and 250 rpm. A negative selection step was performed using paramagnetic streptavidin beads (ThermoFisher Scientific) as described previously18. Briefly, yeast cells (30x the library diversity) from SGCAA culture was pelleted and washed with 0.1% BSA in PBS (PBSA) followed by a 30 min incubation with 100 μL pre-washed streptavidin beads with gentle rotation at 4° C. Supernatant from this incubation was subjected to six rounds of FACS. For each round of sorting, induced yeast cells from SGCAA culture was first washed with PBSA and labeled with varying concentrations of biotinylated p38α (starting at 1μM in the first sort down to 5 nM in the last sort) and 4 μg/mL Chicken anti-c-Myc IgY (ThermoFisher scientific) for an hour of incubation at 4° C with gentle rotation. After washing off unbound reagents, secondary labeling was performed with 4 μg/mL goat anti-chicken Alexa-633 conjugate (ThermoFisher scientific) and Streptavidin-Phycoerythrin (SA-PE) to achieve immunofluorescent detection. All sorts were performed in a Beckman Coulter MoFlo XDP flow cytometer located at the UNC Flow Cytometry core facility. Cells washed after secondary labeling were pelleted and kept on ice until they were analyzed in the flow cytometer. Expression of c-Myc fusions were analyzed by similar labeling with Chicken anti-c-Myc IgY only followed by goat anti-chicken Alexa-633 conjugate. All appropriate controls were performed as described previously17. The population isolated after the last sort was plated on an SDCAA plate followed by culture into SDCAA media. Plasmid DNA was isolated from ten clones (Zymoprep kit) and sequenced (MWG Operon) using the forward primer: 5’-GTTCCAGACTACGCTCTGCAGG-3’. The single monobody sequence that emerged from Lib2 in this fashion is hereafter termed ‘αp38mb’.
Cloning, Expression, Purification & Biotinylation of proteins
The WTmb construct (UniProtKB-P02751) from our previous work was expressed and purified as before4. All monobody mutants were cloned into pQE80L vector between the BamHI and HindIII restriction sites with an N-terminal 6XHis tag, except pepMKK6mb which was cloned by Gibson assembly at the C-terminus of a fusion protein containing SUMO and a 10X His tag. The p38α construct (UniProtKB-Q16539) for E. coli expression of un-phosphorylated p38α (a bicistronic construct with a lambda phosphatase) with a N-terminal 6XHis tag was a generous gift from the Reményi Laboratory (Eötvös Loránd University, Hungary). ERK2 (UniProtKB-P28482) and JNK1 (UniProtKB-P45983) genes were cloned into NpT7–5 and pRSETB vector with an N-terminal 6XHIS tag and were gifts from Klaus Hahn’s Laboratory (UNC, USA). Expression of all proteins were carried out in BL21(DE3)pLysS cells in LB media with IPTG induction. All MAPK’s were expressed for 4 hrs at 25° C except ERK2 which was expressed at 30° C. All other proteins were expressed at 25° C for 18–20 hrs. Supernatant from the sonicated (Fisher) cell pellets was purified by a 5 mL Ni-NTA affinity column in an FPLC system (BioLogic LP, Biorad). Upon purification by Ni-NTA resins, SUMO fusion of pepMKKmb was cleaved with SUMO protease UlpI (1 mg UlpI/20 mg fusion) in PBS/1 mM BME dialysis buffer overnight at 4 °C followed by collection of the cleaved monobody as a flow through from Ni-NTA resins. The 6X His tag from the Ni-NTA purified p38α was cleaved with TEV protease (mixed at 1:100 ratio of TEV protease:p38α). Cleaved p38α and all other Ni-NTA purified proteins were further purified by an S75 gel filtration column (GE) in a GE AKTA FPLC system followed by dialysis in PBS/5 mM β-mercaptoethanol (BME), pH 7.4. Biotinylation of p38α was carried out in PBS/5 mM BME, pH 7.4 using 20-fold molar excess of EZ-link-sulfo-NHS-LC-Biotin using manufacturer’s protocol (ThermoFisher Scientific) followed by extensive dialysis in PBS/5 mM BME, pH 7.4 to remove any trace of unreacted biotin.
Isothermal Titration Calorimetry (ITC)
For any given ITC experiment, each pair of proteins were dialyzed in the same container against the dialysis buffer PBS/1–5 mM BME, pH 7.4. All titrations and measurements were performed in either MicroCal Auto-iTC system (GE Healthcare) or Malvern automatic PEAQ-ITC system. Protein with the lower concentration (12–35 μM) was stored in the ITC-cell followed by 20 injections (2 μL/injection) of the higher concentration protein (225–540 μM). Data acquired from the titrations were fit to a one-site binding curve in the Microcal Origin 5.0 software.
Circular Dichroism (CD) Spectroscopy
Identification of the secondary structure of proteins was performed by CD in a JASCO J-815 CD spectrometer. Data were acquired using protein samples (30–100 μM) stored in a 1 mm cuvette at 20° C with a scanning speed of 50 nm/min between 190 to 260 nm wavelength using 4 sec D.I.T., 2 nm bandwidth and 1 nm pitch. Ellipticity data were normalized to mean residue ellipticity, θ [deg.cm2.dmol−1] using the equation: where c, N, and l refer to concentration (mM), number of amino acid residues and optical path length (cm).
Fluorescence Polarization (FP)
All FP experiments were conducted using a Jobin Yvon Horiba FluoroMax3 fluorescence spectrometer. N-terminus TAMRA-labeled peptides (Synthesized by the UNC Peptide Core Facility) were excited with polarized light at 555 nm, and the polarization of emitted light was measured at 584 nm. For direct binding experiments between p38α and TAMRA-labeled peptide, starting peptide concentration was 50 nM (in PBS/5 mM BME, pH 7.4). Data for direct FP binding was fitted to one-site binding model using SigmaPlot software. For direct binding experiments between ERK2 and JNK1 with N-terminus TAMRA lebeled pep-MKK6 and pep-MKK4, starting peptide concentrations were 1 μM (in PBS/1 mM BME, pH 7.4). For competitive FP assays, peptide and p38α were first brought to a concentration of 10 μM each. Titration of monobody was then performed at the concentrations indicated in Figure 4. Data were numerically fit to a competitive binding model as described previously19.
Figure 4.

Competitive Fluorescence Polarization (FP) binding assays to determine epitope specificity and binding affinity. (a) Schematic representation of the FP assay. Pep-MKK6 labeled with TAMRA dye was first equilibrated with p38α in solution. Subsequently, αp38mb was titrated into the solution of pep-MKK6/p38α complex, thereby displacing pep-MKK6 from its binding site on p38α. (b) FP values were recorded (circular data points) and fit with a competitive binding model (solid line). Loss of polarization with increasing amounts of αp38mb indicates that αp38mb competes for binding to p38α with pep-MKK6 at the same binding site, confirming the epitope-specificity of αp38mb. KD values from three independent experiments were 600±298 nM.
Plasmids for cell-culture work
Monobodies were cloned into the pLEX-MCS vector (Thermo Scientific) between BamHI and XhoI restriction sites. pDONR223-MAPK14 (p38α) was a gift from William Hahn & David Root (Addgene plasmid # 23865). pLX302 was a gift from David Root (Addgene plasmid # 25896)20. MAPK14/p38α was recombined into pLX302 using Gateway LR Clonase II (Invitrogen) according to the manufacturer’s protocol.
Immunoprecipitation (IP) and Immunoblotting (IB)
HEK293T cells were grown in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin and cultured at 37°C in 5% CO2. Cell line was tested for mycoplasma. HEK293T cells cultured in 60mm dishes were transfected with 9 ug of the pLEX monobody and 1 ug pLX302-MAPK14/p38 (V5 epitope tag) using jetPRIME transfection reagent (Polyplus) according to the manufacturer’s protocol. Forty-eight hours, post-transfection, cells were washed in cold phosphate buffered saline (PBS), pH 7.4, and scraped in cold NETN lysis buffer (20mM Tris-HCl, pH8.0; 100mM NaCl; 1mM EDTA, pH8.0, and 0.5% (v/v) NP-40) plus inhibitors (1%). Following 20 min incubation on ice, lysates were clarified by centrifugation and protein concentration was determined by Bradford assay. After normalization, 10% input protein was boiled in SDS-PAGE sample buffer. One mg of protein lysate per IP was pre-cleared with rotation for 60 min at 4°C with 30 ul of a 50% slurry of SureBeads Protein A magnetic beads (Bio-Rad) that had been equilibrated in NETN lysis buffer plus inhibitors. IPs were performed overnight at 4°C using approximately 1–2 ug GFP antibody (Cell Signaling, #2555) as a control or V5-Tag antibody (Cell Signaling, #13202). Thirty microliters of equilibrated Protein A SureBeads were added to each IP and incubated with rotation for 60 min at 4°C. IPs were washed in 5 × 5 min with 1ml NETN lysis buffer.
Lysates and IPs were separated by 8% or 15% SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked in 5% (w/v) milk in Tris-buffered saline (TBS: 50 mM Tris-HCl, pH7.6; 150 mM NaCl) for 1 h at room temperature and incubated in primary antibodies overnight at 4°C. Primary antibodies used were from Cell Signaling: V5-Tag (#13202), and HA-Tag (#3724). After washing in TBS + 0.1% (v/v) Tween-20 (TBS-T), membranes were incubated with peroxidase-conjugated donkey anti-rabbit IgG (Jackson Immunoresearch) for 45 min at room temperature, washed with TBS-T, and detected using SuperSignal West Pico Chemiluminescent Substrate (Thermo). Images were collected on a Bio-Rad ChemiDoc MP Imaging System.
Results
Anchor selection and monobody modeling with Rosetta
Canonical D motifs bind similarly to the MAPKs p38α and ERK2. For instance, a D motif peptide (SKGKKRNPGLKIPK) from the MAP2K MKK6 binds to p38α with an affinity of 7.5 μM and ERK2 with an affinity of 9.7 μM13. A crystal structure has been solved of the MKK6 D motif bound to p38α, and the residues from the D motif that are resolved in the structure (NPGLKIPK) adopt a conformation similar to that observed for other D motifs bound to p38α and ERK213. Superimposing the structures of p38α and ERK2 reveals that residues in the CD groove are highly conserved, but that there is a cluster of three residues adjacent to the MKK6 binding site that are dissimilar (Figure 1). In p38α the residues are Gly110, Glu160 and Asp161. In ERK2 theses residues are Glu, Thr, and Thr respectively. Given these differences, we embedded the MKK6 D motif in a monobody and redesigned surrounding residues in the monobody in an effort to gain specific affinity for p38α over ERK2.
There are two loops in monobodies, the FG- and BC-loops, that are frequently mutated by protein engineers to gain affinity for other proteins21. In order to pick the loop to insert the D motif, we used the molecular modeling program Rosetta to build models of a monobody binding p38α with either the FG- or BC-loop used as the anchor loop. In these simulations, the anchor loop in the monobody was constrained to adopt the conformation and sequence of the core portion of the MKK6 D motif (PGLKIP, hereafter referred to as pep-MKK6), and then the monobody was docked onto p38α so that the anchor loop mimics D motif binding to p38α. This starting model was then refined in Rosetta by allowing the monobody to pivot around the anchor loop and adjusting the conformation of the neighboring loop (BC or FG) to see if additional contacts could be made with p38α. In these simulations, we also tested alternative cut points for inserting the anchor residues in the monobody loops. Final models were visually inspected to identify which loop and insertion point most effectively positioned the monobody over the residues that differ between p38α and ERK2. Figure 2 illustrates a model of the anchor loop we decided to move forward with (also see Supp. Fig. 1). In this construct, Gly79 through Ser84 (GDSPAS) from the FG-loop is replaced with the D motif sequence PGLKIP. Anchoring the monobody in this fashion places the BC loop adjacent to Gly 110, Glu 160 and Asp 161 from p38α.
Figure 2.

Overview of anchor-guided engineering (A) Structure of a monobody (1fna) with the FG loop removed to allow insertion of the anchor peptide and the BC loop colored in cyan. (B) Crystal structure of p38α (gray) bound to the MKK6 D motif (PDB ID: 2y8o). (C) Model generated with the AnchorDesign protocol in Rosetta showing that insertion of the D motif in the FG loop of the monobody is predicted to place the BC loop adjacent to a patch of residues (green) that differ between p38α and ERK2.
Anchor insertion alone does not provide tight binding to the MAPKs
Based on the modeling we chose to replace residues 79–84 from the wildtype monobody with the pep-MKK6 residues PGLKIP (we named this construct pepMKK6mb). Binding experiments by isothermal titration calorimetry (ITC) showed no detectable binding with either p38α or ERK2 and weak binding to JNK1 (>100 μM) [Supplementary Figure 2]. The weak binding to JNK1 suggested that the anchor residues can provide some affinity to the CD groove, but that further optimization of the binding loop and neighboring residues would be needed to achieve tighter binding and specificity for p38α.
Library design and screening by phage display
To optimize the sequence of the BC loop for binding to p38α, we generated a combinatorial library (Lib1) of monobody variants in phage display. In Lib1, residues 79–84 from the FG loop were constrained to the pep-MKK6 sequence, while the residues flanking the anchor, 78 and 85, were randomized using a NNK degenerate codon. The BC loop was extended by one residue and the seven residues between D23 and R30 (numbering as in pdb:1fna) were randomized using NNK degenerate codons [Table 1] giving a theoretical diversity of ~1011. The DNA for Lib1 was synthesized by Kunkel mutagenesis in the pFN-OM6 vector to enable expression of mutants as fusions to the phage pIII-coat protein. Lib1 was subsequently screened against solubly expressed and purified unphosphorylated p38α. In each round of selection, 0.5 μg p38α was immobilized in a 96-well plate followed by elution of p38α-bound phage particles. Clones isolated after four rounds of selection were assessed for binding to p38α by phage ELISA [Supplementary Figure 3]. Sequencing of clones with the highest binding signal revealed two unique monobody sequences, G4mb and C6mb [Table 1]. An anchor-only control peptide was also tested by phage ELISA side-by-side [A1(p3-pep) in Supplementary Figure 3]. Notably, the binding signals from G4mb and C6mb were significantly higher than that from the anchor-only peptide suggesting higher binding affinity of the monobody clones than the anchor alone. G4mb and C6mb were then recombinantly expressed in E. coli. Circular dichroism spectra of both variants were consistent with that expected for a β-sheet protein [Supplementary Figure 4]. The variants were then tested for binding to p38α with ITC, both G4mb and C6mb showed low micromolar affinity [Supplementary Figure 5]. Additionally, we tested the binding of G4mb for ERK2 and JNK1 by ITC [Supplementary Figure 7]. While G4mb had no detectable binding to JNK1, weak binding to ERK2 (58 μM) was observed. This indicated that our strategy of interface expansion was beginning to generate specificity for p38α over ERK2. To further improve affinity and specificity we decided to optimize additional residues in the monobody with yeast display. As a control experiment, we also confirmed that the wild type monobody had no detectable binding to p38α [Supplementary Figure 5c].
Table 1.
Sequence alignment of the BC and FG loops of monobody. ‘X’ represents positions completely randomized in library design. Mutations differing between G4mb and αp38mb are highlighted green. Position 29a refers to the extension of the BC loop for library design and subsequent mutants isolated from library screening.
| Monobody | BC loop | FG loop |
|---|---|---|
| 22 . . .26 . . .29a30 | 75 . . . .80 . . . .85 . .88 | |
| Wild type monobody | W D A P A V T V - R | V T G R G D S P A S S K P I |
| 674mb | W V P P W R P V - R | V N G I G P Q L T I P D G I |
| Monobody Library | W D X X X X X X X R | V T G X P G L K I P X K P I |
| C6mb | W D S P R W W W V R | V T G M P G L K I P L K P I |
| G4mb | W D P P R C R R I R | V T G M P G L K I P R K P I |
| αp38mb | W D P P R C R R A R | V T S M P R L G I P R K S I |
Affinity maturation by yeast display
To further optimize the binding of G4mb and C6mb for p38α, we performed affinity maturation using error prone PCR combined with yeast display. Error prone PCR was conducted with the entire G4mb and C6mb genes, and the resulting DNA were pooled to create a random mutagenesis library termed Lib2. In Lib2, monobody mutants were expressed as a C-terminal fusion to the yeast cell wall protein Aga2 and were flanked by an N-terminal HA epitope tag and a C-terminal c-myc epitope tag. Immunofluorescent labeling of the c-myc tag of Lib2 followed by flow cytometric analysis showed that approximately 40% of the mutants in Lib2 expressed as full-length monobody variants [Figure 3]. This result suggested that a very large fraction of monobody mutants were folded correctly since yeast endoplasmic reticulum tightly regulates the export of only correctly folded proteins to the cell surface, with the exception of some small, thermally stable proteins22,23. To isolate high affinity binders from Lib2, we screened the library against biotinylated p38α using fluorescent assisted cell sorting (FACS). Six rounds of selection were performed with decreasing concentrations of p38α used at each round, starting with 1 μM and finishing with 5 nM [Figure 3]. Following the sixth round of selection, ten clones from the population were randomly selected for sequencing and biophysical analysis. All ten clones corresponded to the same sequence, which was named αp38mb [Table 1].
Figure 3.

Expression and screening analysis of monobody yeast library (Lib2). (a) Yeast cells displaying monobody mutants were labeled with an anti-cmyc antibody followed by a secondary antibody conjugated to Alexa Fluor-633 (red histogram) or the secondary antibody only (blue histogram). Approximately 40% of the cells expressed the cmyc tag and therefore full-length monobody mutants as cell surface fusions. (b), (c) and (d) show flow cytometry plots from the screening of monobody yeast library by FACS. Yeast cells displaying monobody mutants were simultaneously labeled with a chicken anti-cmyc antibody and 0 μM (b, no p38α control), 1 μM (c, sort 1) or 5 nM (d, sort 6) biotinylated p38α followed by secondary labeling with a goat anti-chicken antibody conjugated to Alexa Fluor 633 (to detect expression) and streptavidin conjugated to PE (to detect binding) and analyzed by flow cytometry. Cells in the polygon in (c) were sorted from round 1, grown and further sorted by labeling at successively lower concentrations of p38α until sort 6 (d). Yeast cells from round 6 were plated to isolate individual clones for further analysis.
The sequence of αp38mb indicates that it is derived from G4mb. The BC loop in αp38mb only has one mutation compared to G4mb and is highly basic with the sequence PPRCRRA, which is presumably favorable for binding the negative patch adjacent to the CD grove in p38α. Two mutations were observed in the anchor region. The first glycine from the anchor, PGLKIP, was mutated to an arginine and the lysine was mutated to a glycine. Both of these mutations are compatible with canonical D motif binding as these residues point away from the interface, and are not conserved in an alignment of D motif sequences. Three additional mutations outside the targeted FG and BC loops are also present in αp38mb. Two of these mutations are on β strands A (T14A) and B (S21A) and a third mutation (S43G) occurred in the CD loop.
Binding Studies
αp38mb was recombinantly expressed in E. coli with an N-terminal 6xHis tag. Its circular dichroism spectra closely matched that of the starting monobody and is consistent with a folded protein [Supplementary Figure 4]. To measure the binding affinity of αp38mb for p38α and determine if it binds to the CD groove, we performed competitive fluorescence polarization (FP) experiments. A MKK6-derived 12-residue peptide GKKRNPGLKIPK was chemically synthesized with a TAMRA fluorophore at its N-terminus. First, the affinity of the MKK6 peptide for p38α was determined by measuring fluorescence polarization as a function of the concentration of p38α. The data fit well to a single-site binding model with a dissociation constant of 3.9 μM ± 0.4 μM [Supplementary Figure 8]. Next, αp38mb was titrated into a solution of the TAMRA-labeled peptide and p38α. Consistent with binding to the CD groove, the monobody displaced the peptide and the resulting data were fit to a competitive binding model to determine the dissociation constant between the monobody and p38α, which was 600 ± 298 nM [Figure 4]. Additionally, we showed that αp38mb and its precursor G4mb have no binding to BSA and streptavidin, which were used throughout the library screening process [Supplementary Figure 6].
To determine if αp38mb binds preferentially to p38α, we used isothermal titration calorimetry (ITC) to measure the binding affinity of αp38mb for p38α, ERK2, and JNK1 [Figure 5]. The KD measured by ITC for p38α, 649 ± 100 nM, was very similar to what we measured with the competitive FP experiment. In the experiments with ERK2 and JNK1, there was no detectable binding by ITC, indicating that if there is binding that the KD is greater than ~100 μM. These results indicate that although we did not explicitly disfavor binding to ERK2 during the design process, that the process of embedding the anchor peptide in the monobody and then optimizing neighboring residues led to a binder that is specific for p38α. Notably, we verified the quality of all three MAPK samples that were used in our experiments by testing their binding interactions with peptides that are known to bind to the MAPKs13 [Supplementary Figures 8 and 9].
Figure 5.

Affinity and specificity analysis of αp38mb by ITC. ITC experiments are reported for the titration of αp38mb into solutions of 12.5 μM p38α (a), 13 μM ERK2 (b), and 15 μM JNK1 (c). The binding affinity (KD) of αp38mb for p38α was measured to be 649±100 nM, whereas no specific binding was detected of αp38mb to either ERK2 or JNK1.
We also tested if αp38mb binds to p38α in cells. We transiently transfected HEK293 cells with αp38mb (or the wild type monobody) and p38α as fusions with the HA and V5 epitope tags, respectively. First, we confirmed co-expression of both monobodies and p38α by immunoblotting (IB) [Figure 6a], and then used immunoprecipation (IP) to test for binding. When p38α was precipitated from cell lysate, it co-precipitated αp38mb, but not the wild type monobody [Figure 6b].
Figure 6:

The binding interaction between the engineered monobody and p38α is retained in cell culture. (a) Monobodies were co-expressed with p38α in cells. Wildtype monobody (WTmb) or engineered monobody (αp38mb) containing the HA-epitope tag was transiently co-transfected with V5-tagged p38α in HEK293 cells. Immunoblotting shows co-expression of p38α with either WTmb or αp38mb. (b) Constructs from (a) were used in a pull-down assay where p38α was immunoprecipitated with anti-V5 antibody followed by Immunoblotting analysis of V5 and HA-epitope tags. Co-immunoprecipitation of αp38mb with p38α was observed, but WTmb was not co-immunoprecipitated.
Discussion
Our approach for designing a monobody that binds specifically to p38α involved three primary steps: (1) computer-guided insertion of a MAPK binding motif (D motif) into the FG loop of the monobody, (2) optimization of the adjacent BC loop with a combinatorial library and phage display, and (3) final optimization of the monobody sequence with PCR mutagenesis and yeast display. Step 1, insertion of the D motif into the FG loop, was only able to provide weak binding to the MAPKs, but was an important step in the protocol because it predisposed the engineered monobodies to bind the CD groove on the MAPKs. The modeling also predicted that insertion into the FG loop would place the BC loop adjacent to residues on p38α that differ from the other MAPKs. Ideally, we would have also used computational protein design to optimize the sequence of the BC to provide tight binding against p38α. Unfortunately, the computational design of “antibody-like” loops that bind to target protein surfaces remains a very challenging problem for protein design algorithms24,25. For this reason, for stage 2 of the protocol, we choose to use phage display to identify binders from a combinatorial library. Phage display was used for this step in the protocol because it can be used to probe larger libraries than can generally be handled with yeast display, and other labs have found that it is a powerful technique when starting with a naïve library or with weak binders26. For stage 3, we switched to yeast display because it can be used to simultaneously select for high levels of protein expression as well as binding27.
One interesting result from the design process was that the binding affinities of the engineered monobody for the MAPKs after anchor insertion (stage 1) were weaker than what the isolated D motif exhibits for MAPKs. This suggests that placing the D motif in the monobody restricted its ability to adopt a conformation suitable for binding the kinases. This was one of the reasons that we allowed residues flanking the D motif to vary in the phage display. Notably, these flanking residues changed during phase display, and yeast display identified further mutations near the binding residues that presumably allow the loop to adopt a conformation more favorable for binding. We noticed a similar result in a previous study in which we embedded a binding motif in a monobody for the purpose of binding the protein Keap14. Simply placing the motif a loop of the monobody reduced binding affinity relative to the free peptide, but subsequent optimization of the monobody resulted in tighter binding than exhibited by the free peptide.
Other strategies have also been used to engineer monobodies that bind to specific MAPKs. Mann et al. used a naïve monobody library with variations in the BC, DE and FG loop, and then screened for library members that bound to wild type ERK2 but did not bind to an ERK2 variant with mutations in the CD groove28. With this approach, they identified nanomolar binders that bound to ERK2 in the vicinity of the CD groove, and did not interact with p38α or JNK1. Our approach differs from that used by Mann and co-workers in that we did not need to use negative selection to direct binding towards a specific surface patch, rather made use of a pre-established peptide anchor. In sum, these studies demonstrate the utility of larger protein surfaces to discriminate between highly similar proteins in situations where peptide binders do not show specific binding.
Supplementary Material
Acknowledgments
We thank Joseph Harrison for help in performing selected isothermal titration calorimetry experiments. We thank the UNC Flow Cytometry Core Facility which is supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center and in part by the North Carolina Biotech Center Institutional Support Grant 2005-IDG-1016.
Funding Information
This work was supported by the grant R01GM073960 from the National Institutes of Health.
Footnotes
Supporting information
Supporting information for this manuscript includes molecular models of monobodies docked against p38α, binding studies with pepMKK6mb, phage ELISA results with clones isolated from phage panning, circular dichroism spectra of the monobodies, binding studies with monobodies discovered after phage display, binding studies with reagents used during library screening, and binding studies to establish that ERK2 and JNK1 were properly folded.
References
- (1).Plückthun A (2015) Designed ankyrin repeat proteins (DARPins): binding proteins for research, diagnostics, and therapy. Annu. Rev. Pharmacol. Toxicol 55, 489–511. [DOI] [PubMed] [Google Scholar]
- (2).Sha F, Salzman G, Gupta A, and Koide S (2017) Monobodies and other synthetic binding proteins for expanding protein science. Protein Science 26, 910–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Arkadash V, Yosef G, Shirian J, Cohen I, Horev Y, Grossman M, Sagi I, Radisky ES, Shifman JM, and Papo N (2017) Development of High Affinity and High Specificity Inhibitors of Matrix Metalloproteinase 14 through Computational Design and Directed Evolution. J Biol Chem 292, 3481–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Guntas G, Lewis SM, Mulvaney KM, Cloer EW, Tripathy A, Lane TR, Major MB, and Kuhlman B (2016) Engineering a genetically encoded competitive inhibitor of the KEAP1-NRF2 interaction via structure-based design and phage display. Protein Eng Des Sel 29, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Berger S, Procko E, Margineantu D, Lee EF, Shen BW, Zelter A, Silva D-A, Chawla K, Herold MJ, Garnier J-M, Johnson R, MacCoss MJ, Lessene G, Davis TN, Stayton PS, Stoddard BL, Fairlie WD, Hockenbery DM, and Baker D (2016) Computationally designed high specificity inhibitors delineate the roles of BCL2 family proteins in cancer. eLife 5, 1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lewis SM, and Kuhlman BA (2011) Anchored design of protein-protein interfaces. PLoS ONE 6, e20872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Tanoue T, and Nishida E (2002) Docking interactions in the mitogen-activated protein kinase cascades. Pharmacol. Ther 93, 193–202. [DOI] [PubMed] [Google Scholar]
- (8).Reményi A, Good MC, and Lim WA (2006) Docking interactions in protein kinase and phosphatase networks. Curr. Opin. Struct. Biol 16, 676–685. [DOI] [PubMed] [Google Scholar]
- (9).Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, and Cobb MH (2001) Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr. Rev 22, 153–183. [DOI] [PubMed] [Google Scholar]
- (10).Arthur JSC, and Ley SC (2013) Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol 13, 679–692. [DOI] [PubMed] [Google Scholar]
- (11).Cargnello M, and Roux PP (2011) Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev 75, 50–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Johnson GL, and Lapadat R (2002) Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298, 1911–1912. [DOI] [PubMed] [Google Scholar]
- (13).Garai A, Garai A, Zeke A, Zeke A, Gógl G, Gogl G, Toro I, Töro I, Fordos F, Fördos F, Blankenburg H, Blankenburg H, Bárkai T, Barkai T, Varga J, Varga J, Alexa A, Alexa A, Emig D, Emig D, Albrecht M, Albrecht M, Remenyi A, and Reményi A (2012) Specificity of Linear Motifs That Bind to a Common Mitogen-Activated Protein Kinase Docking Groove. Science Signaling 5, ra74–ra74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Mandell DJ, Coutsias EA, and Kortemme T (2009) Sub-angstrom accuracy in protein loop reconstruction by robotics-inspired conformational sampling. Nat Methods 6, 551–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tonikian R, Zhang Y, Boone C, and Sidhu SS (2007) Identifying specificity profiles for peptide recognition modules from phage-displayed peptide libraries. Nat Protoc 2, 1368–1386. [DOI] [PubMed] [Google Scholar]
- (16).Gera N, Hill AB, White DP, Carbonell RG, and Rao BM (2012) Design of pH sensitive binding proteins from the hyperthermophilic Sso7d scaffold PLoS ONE (Karnik S, Ed.) 7, e48928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Gera N, Hussain M, and Rao BM (2013) Protein selection using yeast surface display. Methods 60, 15–26. [DOI] [PubMed] [Google Scholar]
- (18).Hussain M, Lockney D, Wang R, Gera N, and Rao BM (2013) Avidity-mediated virus separation using a hyperthermophilic affinity ligand. Biotechnol Prog 29, 237–246. [DOI] [PubMed] [Google Scholar]
- (19).Purbeck C, Purbeck C, Eletr ZM, Eletr ZM, and Kuhlman B (2010) Kinetics of the Transfer of Ubiquitin from UbcH7 to E6AP. Biochemistry 49, 1361–1363. [DOI] [PubMed] [Google Scholar]
- (20).Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, Lubonja R, Thomas SR, Alkan O, Bhimdi T, Green TM, Johannessen CM, Silver SJ, Nguyen C, Murray RR, Hieronymus H, Balcha D, Fan C, Lin C, Ghamsari L, Vidal M, Hahn WC, Hill DE, and Root DE (2011) A public genome-scale lentiviral expression library of human ORFs. Nat Methods 8, 659–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Batori V, Koide A, and Koide S (2002) Exploring the potential of the monobody scaffold: effects of loop elongation on the stability of a fibronectin type III domain. Protein Engineering, Design and Selection 15, 1015–1020. [DOI] [PubMed] [Google Scholar]
- (22).Shusta EV, Kieke MC, Parke E, Kranz DM, and Wittrup KD (1999) Yeast polypeptide fusion surface display levels predict thermal stability and soluble secretion efficiency. J Mol Biol 292, 949–956. [DOI] [PubMed] [Google Scholar]
- (23).Park S, Xu Y, Stowell XF, Gai F, Saven JG, and Boder ET (2006) Limitations of yeast surface display in engineering proteins of high thermostability. Protein Engineering Design and Selection 19, 211–217. [DOI] [PubMed] [Google Scholar]
- (24).Baran D, Pszolla MG, Lapidoth GD, Norn C, Dym O, Unger T, Albeck S, Tyka MD, and Fleishman SJ (2017) Principles for computational design of binding antibodies. P Natl Acad Sci Usa 114, 10900–10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Hu X, Hu X, Wang H, Wang H, Ke H, Ke H, Kuhlman B, and Kuhlman B (2007) High-resolution design of a protein loop. Proceedings of the National Academy of Sciences 104, 17668–17673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Ferrara F, Naranjo LA, Kumar S, Gaiotto T, Mukundan H, Swanson B, and Bradbury ARM (2012) Using phage and yeast display to select hundreds of monoclonal antibodies: application to antigen 85, a tuberculosis biomarker PLoS ONE (Lenz LL, Ed.) 7, e49535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Boder ET, and Wittrup KD (2000) [25] Yeast surface display for directed evolution of protein expression, affinity, and stability, in Applications of Chimeric Genes and Hybrid Proteins - Part C: Protein-Protein Interactions and Genomics, pp 430–444. Elsevier. [DOI] [PubMed] [Google Scholar]
- (28).Mann JK, Wood JF, Stephan AF, Tzanakakis ES, Ferkey DM, and Park S (2013) Epitope-guided engineering of monobody binders for in vivo inhibition of Erk-2 signaling. ACS Chem. Biol 8, 608–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.