

Abstract
SMAD4 pathogenic variants cause juvenile polyposis (JPS) and hereditary hemorrhagic telangiectasia (HHT), and 40% of affected individuals also have thoracic aortic disease. At the same time, SMAD4 pathogenic variants have not been reported in thoracic aortic disease families without JPS-HHT. A SMAD4 heterozygous variant, c.290G>T, p.(Arg97Leu), not present in population databases and predicted to be damaging to protein function, was identified in a family with thoracic aortic disease and no evidence of HHT or JPS. Cellular studies revealed that the SMAD4 p.(Arg97Leu) alteration increased SMAD4 ubiquitination and 26S proteasome-mediated protein degradation. Smooth muscle cells (SMCs) infected with lentivirus expressing the SMAD4 p.(Arg97Leu) variant demonstrated reduced contractile protein gene expression when compared to that of wild-type SMAD4. In addition, two rare variants were identified in individuals with early age of onset of thoracic aortic dissection. These results suggest that SMAD4 rare missense variants can lead to thoracic aortic disease in individuals who do not have JPS or HHT.
Subject terms: Medical genetics, Genetics research
Introduction
The natural history of aneurysms involving the aortic root and/or ascending aorta is to asymptomatically enlarge over time until an acute tear in the intimal and medial layers leads to an ascending aortic dissection [1–3]. A predisposition for thoracic aortic aneurysms and dissections can be inherited in an autosomal dominant manner in families and may or may not be associated with syndromic features, termed heritable thoracic aortic disease (HTAD). Rare pathogenic variants that are highly likely to cause HTAD have been identified in genes encoding proteins involved in canonical TGF-β signaling (TGFBR2, TGFBR1, TGFB2, TGFB3, and SMAD3) [4–9]. TGF-β signaling is initiated when the cytokine binds to the TGF-β type II receptor on the cell surface, which recruits and phosphorylates the TGF-β type I receptor. The active type I receptor phosphorylates SMAD2 and SMAD3 (Mothers Against Decapentaplegic Homolog 2 and 3 (MIM 601366 and MIM 603109), respectively) at the C-terminus, which forms a complex with SMAD4 and is translocated to the nucleus to alter gene transcription. Pathogenic SMAD4 variants cause juvenile polyposis (JPS) and hereditary hemorrhagic telangiectasia (HHT), and 40% of affected individuals have thoracic aortic disease [10–12], but SMAD4 variants have not been reported in thoracic aortic disease families without JPS–HHT. We report here a family with HTAD segregating with a SMAD4 missense variant that disrupts SMAD4 stability. Furthermore, additional rare variants in SMAD4 were identified in patients with early age onset of sporadic thoracic aortic dissections (ESTAD).
Patients and methods
Patients
DNA samples were collected from affected individuals and family members after obtaining informed consent and human subject research approval from all participating institutions, including the University of Texas Health Science Center at Houston and Baylor College of Medicine. Clinical data were obtained from medical records, and phenotypic features were assessed in the SMAD4 variant carriers by a clinical geneticist. Additional methods are available in the online Supplementary material.
Results
Identification of SMAD4 pathogenic variants in thoracic aortic disease patients
Exome-sequencing data from probands and family members of 223 unrelated HTAD families (Supplementary Table 1), defined as two or more members with thoracic aortic disease, were analyzed for rare heterozygous variants as previously described [13]. One SMAD4 (NM_005359.5) variant, c.290G>T p.(Arg97Leu)(ClinVar SCV000804195.1), was identified in the proband and affected brother of family TAA281; Sanger sequencing validated the variant and confirmed it was inherited from proband’s affected father (Fig. 1a). No other rare variants in known HTAD genes were identified [14]. This variant is absent in the gnomAD database and has a CADD score of 33 [15] (Supplementary Table 2). The proband presented with an ascending aortic dissection at the age of 24 years and underwent initial ascending aortic repair, and subsequent aortic root and valve replacement at the age of 34 years. Postoperative CT imaging showed an aberrant right subclavian artery dilated at the origin and markedly a tortuous distal thoracic and abdominal aorta. Clinical examination by a geneticist was remarkable for mild degree of esotropia, uvula with groove but not bifid, mild scoliosis, and joint laxity. She died of ovarian cancer at the age of 44 years. Her brother also presented with an ascending aortic dissection at the age of 41 years and had a bicuspid aortic valve. Their father was diagnosed with an ascending aortic aneurysm at the age of 75 years and underwent an ascending aortic replacement. Complete medical records from the proband and her father did not identify any features of JPS, HHT, or Myhre syndrome.
Fig. 1.
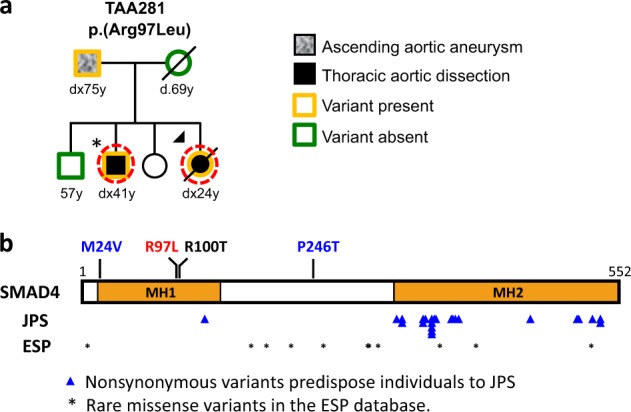
SMAD4 rare variants identified in a family with heritable thoracic aortic disease. a Pedigree of TAA281 with SMAD4 p.(Arg97Leu) variant. The legend indicates the disease status and genotypes of the family members. Arrow points to the proband. Asterisk indicates the presence of a bicuspid aortic valve. The age at diagnosis of aortic aneurysm or dissection (‘‘dx’’) and age at death (‘‘d’’) are shown in years. A dashed circle around a symbol indicates individuals whose DNA was used for exome sequencing. b Schematic representation of the SMAD4 domains and variants. The SMAD4 rare missense variant identified in TAA281 is shown in red, the somatic variant identified in pancreatic cancer in black, and missense variants identified in individuals with early dissections are in blue above the protein diagram. The blue triangles indicate the location of SMAD4 missense variants identified in patients with JPS or JPS–HHT. Asterisks indicate variants identified in the NHLBI ESP database
Analysis of exome-sequencing data from 355 individuals with aortic dissections ≤56 years of age with no family history or syndromic features (ESTAD, Supplementary Table 1) identified two additional rare nonsynonymous SMAD4 variants, a c.70A>G p.(Met24Val)(ClinVar SCV000804196.1) and c.736C>A, p.(Pro246Thr)(ClinVar SCV000804197.1) [16]; these variants were validated by Sanger sequencing. These variants have minor allele frequencies <5.0 × 10–4 in the gnomAD database and CADD scores of >15 (Supplementary Table 1). Variant p.(Met24Val) was identified in a patient with a type A aortic dissection at the age of 37 and p.(Pro246Thr) was identified in a patient with a type A aortic dissection at 54 years old; these individuals did not have features of HHT or JPS. Individual with the SMAD4 p.(Pro246Thr) variant did not have any rare variants in known HTAD genes, whereas the individual with the SMAD4 p.(Met24Val) variant also had rare variants in MYH11 c3281C>T, p.(Ala1094Val) and TGFBR1 c.457G>A, p.(Val153Ile) (Supplementary Table 3).
SMAD4 p.(Arg97Leu) alters protein stability and decreases TGFβ signaling
SMAD4 has two Mad Homology domains, MH1 and MH2, and a region linking these domains (Fig. 1b). The SMAD4 p.(Arg97Leu) variant is located in the MH1 domain of SMAD4 (Fig. 1b) and is immediately adjacent to a previously identified somatic SMAD4 variant, p.(Arg100Thr), found in a pancreatic cancer patient and previously shown to decrease SMAD4 stability [17]. Given the proximity of Arg97 to Arg100, we hypothesized that the p.(Arg97Leu) variant affects SMAD4 protein stability in a similar way [18]. To test this hypothesis, lentiviruses of control vector, Flag-tagged wild type (WT) and Leu97 were infected in HEK293T cells and an immortalized smooth muscle cell (SMC) line (SMC11023) [19]. Quantitative RT-PCR (qPCR) showed similar levels of SMAD4 WT and Leu97 transcripts in both cell lines (Fig. 2a), but immunoblot analyses demonstrated significantly reduced levels of SMAD4 Leu97 compared to WT protein, suggesting that the SMAD4 Leu97 variant reduced the stability of the protein (Fig. 2b). After treating the HEK293T cells expressing SMAD4 WT or Leu97 with a protein synthesis inhibitor (cycloheximide) for up to 8 h, half of the SMAD4 Leu97 protein was degraded within 8 h, while the level of WT protein was unchanged (Fig. 2c). Similar analyses of SMAD4 Val24 and Thr246 indicated that these variants did not lead to increased degradation of the protein (Supplementary Fig. 1).
Fig. 2.

SMAD4 Leu97 reduces protein stability through increased binding to SKP2. a Lentiviruses of control vector and Flag-tagged WT and Leu97 SMAD4 were infected in HEK293T and SMC11023 followed by real-time qPCR with GAPDH as internal control. SMAD4 WT and Leu97 have similar expression of mRNA. b In these same cells, the protein level of SMAD4 Leu97 was significantly lower than that of SMAD4 WT. The average protein levels over triplicate experiments are shown on the graph. c SMAD4 Leu97 variant protein is unstable after cycloheximide treatment. HEK293T cells expressing SMAD4 WT and Leu97 variant were treated with cycloheximide for up to 8 h followed by immunoblot analyses with anti-Flag antibodies. The levels of SMAD4 and GAPDH protein were quantified using immunoblot assay. d MG132 partially rescued the degradation of SMAD4 Leu97 variant. HEK293T cells expressing SMAD4 WT and Leu97 variant were treated with MG132, for 8 h. A representative experiment is shown on the left and quantification of three individual experiments are shown at the right. e Leu97 variant increased the ability of SMAD4 binding to HA-SKP2. HEK293T cells were co-transfected with Myc-SMAD4 and HA-SKP2 followed by immunoprecipitation with Myc beads. The SMAD4-bound SKP2 was detected with anti-HA antibodies. f Ubiquitination of SMAD4 Leu97 is increased compared to WT protein. HEK293T cells were co-transfected with His-SMAD4, Flag-SKP2, and HA-ubiquitin followed by immunoprecipitation with Ni-NTA agarose beads under denatured conditions. The ubiquitination of Smad4 was detected with anti-HA antibodies. Three individual experiments were performed and one representative result is shown. Asterisks indicate a p value <0.05
The ubiquitination and degradation of the SMAD4 variant p.(Arg100Thr) identified in cancer cells is augmented through the ubiquitin E3 ligase SKP2-mediated 26S proteasome pathway [17, 18]. To determine if a similar pathway is degrading the SMAD4 p.(Arg97Leu), HEK293T cells expressing Flag-tagged WT or Leu97 SMAD4 were treated with a 26S proteasome inhibitor (MG132) for 8 h. Results showed that MG132 slightly increased the levels for Leu97 SMAD4, suggesting that the mutant SMAD4 degradation is partially mediated through the 26S proteasome (Fig. 2d). To test whether SMAD4 Leu97 binds the E3 ubiquitin ligase SKP2 more efficiently, HEK293T cells were co-transfected with HA-tagged SKP2 and Myc-tagged SMAD4 WT or Leu97, followed by immunoprecipitation using anti-Myc antibodies. Subsequent immunoblot analyses using anti-HA antibodies showed increased SKP2 binding to SMAD4 Leu97 compared to WT (Fig. 2e). To test whether SMAD4 Leu97 mutant increases its ubiquitination by SKP2-mediated 26S proteasome, His-tagged SMAD4 and HA-tagged ubiquitin, with and without Flag-tagged SKP2 were co-transfected in HEK293T cells, followed by immunoprecipitation with nickle beads to pull down the SMAD4 under denatured conditions and immunoblot analyses done with both anti-Flag and anti-HA antibodies. The results showed that the ubiquitination of SMAD4 Leu97 was increased compared to WT at baseline and further increased when SKP2 was co-expressed (Fig. 2f). Thus, these results indicate that the SMAD4 Leu97 variant increases binding to SKP2, leading to increased ubiquitination and 26S proteosomal degradation.
With TGFβ activation of the type I and II receptors, SMAD4 forms a complex with SMAD2/3 and translocates into nucleus to increase the expression of genes by binding to conserved SMAD binding elements (SBE) in promoter regions. To test if any of the identified SMAD4 rare variants disrupt TGFβ signaling, these variants were expressed with a 3TP-Lux plasmid which contains an SBE driving luciferase expression with TGFβ and a SMAD2 construct in COS7 cells. The results show that the luciferase activity increases in response to TGFβ1 treatment in cells expressing increasing amounts of WT SMAD4, and similar increased activity was found when SMAD4 Val24 and Thr246 were expressed (Fig. 3a). In contrast, luciferase activity was minimally increased when the SMAD4 Leu97 was expressed. Cyclin-dependent kinase 4 inhibitor B (p15INK4B) has an SBE in its promoter region and is a well-characterized early response gene of TGFβ signaling. SMCs expressing SMAD4 WT and Leu97 variant were exposed to TGFβ1 for 4 h, and RNA harvested for qPCR analyses. The induction of the expression of p15INK4B was greater in SMCs overexpressing WT SMAD4 than in cells expressing Leu97 SMAD4 variant (Fig. 3b).
Fig. 3.

SMAD4 Leu97 variant reduces TGFβ signaling. a The effect of SMAD4 rare variants on the TGFβ was assessed in COS7 cells signaling. The SMAD4 activity of Leu97 alteration to induce 3TP luciferase activity was reduced compared to WT. COS7 cells were co-transfected with 3TP-lux plasmid and plasmids expressing WT or rare variants in SMAD4, followed by TGFβ1 treatment for 24 h. The 3TP luciferase activities were measured with renilla luciferase as internal control. The enhancement of luciferase activity in response to TGFβ1 treatment in SMAD4 Leu97 was significantly lower than that of SMAD4 WT, Val24, Thr246. b Immortalized SMCs (SMC11023) expressing control vector, SMAD4 WT or SMAD4 Leu97 were treated with TGFβ1 for 4 h and the expression of p15INK4b was measured by real-time qPCR and with GAPDH as internal control. c, d SMAD4 Leu97 variant reduced TGFβ1-induced smooth muscle specific gene expression and protein levels. SMC11023 cells infected with control vector, Flag-tagged SMAD4 WT or Leu97 variant were treated with TGFβ1 for 48 h followed by Western blot (c) and real-time qPCR (d). Asterisks indicate a p value <0.05 when compared to the SMAD4 WT
TGFβ signaling drives SMC differentiation, which is defined by high levels of SMC contractile genes, including smooth muscle α-actin (ACTA2) and calponin (CNN1). Previous studies have shown that SMCs explanted from Smad4-deficient mice have decreased expression of SMC differentiation markers, including Acta2 [20]. To test whether the SMAD4 Leu97 variant alters the expression of these genes, SMAD4 WT and Leu97 variants were expressed in human immortalized SMCs and then exposed to TGFβ1 for 48 h. The result showed that the expression and protein levels of ACTA2 and CNN1 were increased in SMCs expressing WT SMAD4 with exposure to TGFβ1, but not increased in SMCs expressing SMAD4 Leu97 (Fig. 3c, d). Thus, the SMAD4 Leu97 variant reduces SMC differentiation with exposure to TGFβ1.
Discussion
We demonstrate here that a rare heterozygous missense variant in the MH1 domain of SMAD4 leads to an unstable protein and segregates with thoracic aortic disease in the absence of JPS–HHT in a HTAD family. The thoracic aortic disease in this family first presented as either a type A dissection or aneurysm and had a variable age of onset. Pathogenic heterozygous variants in SMAD4 lead to a spectrum of syndromes, including JPS, JPS with HHT, and Myhre syndrome [11, 12, 21]. SMAD4 variants associated with JPS or JPS–HHT are predicted to result in haploinsufficiency or are missense variants located in the MH2 domain of SMAD4 (Fig. 1b). Functional studies show that these missense SMAD4 variants reduce bone morphogenetic protein (BMP) signaling but to a lesser extent than predicted for haploinsufficiency [22]. Pathogenic variants in SMAD4 that lead to Myhre syndrome alter Arg496 or Ile500 in the MH2 domain and increase the stability of SMAD4 by decreasing its ubiquitination and degradation, and thus most likely increase signaling through SMAD4 [21]. In contrast, SMAD4 p.(Arg97Leu) reduces the stability of SMAD4 protein by increasing binding to the ubiquitin E3 ligase SKP2, which decreases TGFβ signaling and SMC differentiation. Based on the decreased stability, this SMAD4 variant should also cause JPS and HHT. It may be that the cells altered in JPS and HHT do not degrade SMAD4 using SKP2, and thus the mutant SMAD4 would not be degraded more rapidly than WT in these cells.
SMAD4 variants, p.(Met24Val) and p.(Pro246Thr), were identified in patients with early onset sporadic thoracic aortic disease. Cell biology studies of these variants indicated that they did not affect SMAD4 stability or TGFβ signaling (Fig. 3a; Supplementary Fig. 1). Futhermore, one individual with SMAD4 variant p.(Met24Val) has additional variants of uncertain significance in MYH11 and TGFBR1, known genes that lead to HTAD. These SMAD4 variants did not disrupt SMAD4 stability or function and therefore are less likely to predispose to thoracic aortic disease. At the same time, we cannot exclude the possibility that the variants altered a function of SMAD4 that was not assayed in this study.
In summary, this study provides evidence that a rare SMAD4 missense variant predisposes individuals to thoracic aortic disease in the absence of JPS and HHT. We also identified rare SMAD4 variants in patients with sporadic thoracic aortic diseases but based on our assessment, these variants did not alter protein function. Additional studies are needed to validate and determine the frequency of SMAD4 variants leading to thoracic aortic disease in the absence of features of JPS and HHT.
Supplementary information
Acknowledgements
This work is supported by National Heart, Lung and Blood Institute (RO1 HL109942) and the John Ritter Foundation to Dr. Milewicz, National Human Genome Research Institute (1U54HG006493) to Drs. Nickerson and Bamshad.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Xue-Yan Duan, Dong-chuan Guo
Supplementary information
The online version of this article (10.1038/s41431-019-0357-x) contains supplementary material, which is available to authorized users.
References
- 1.Howard DP, Banerjee A, Fairhead JF, Perkins J, Silver LE, Rothwell PM. Population-based study of incidence and outcome of acute aortic dissection and premorbid risk factor control: 10-year results from the Oxford Vascular Study. Circulation. 2013;127:2031–7. doi: 10.1161/CIRCULATIONAHA.112.000483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prakash SK, Haden-Pinneri K, Milewicz DM. Susceptibility to acute thoracic aortic dissections in patients dying outside the hospital: an autopsy study. Am Heart J. 2011;162:474–9. doi: 10.1016/j.ahj.2011.06.020. [DOI] [PubMed] [Google Scholar]
- 3.Hiratzka LF, Bakris GL, Beckman JA, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation. 2010;121:e266–369. doi: 10.1161/CIR.0b013e3181d4739e. [DOI] [PubMed] [Google Scholar]
- 4.Mizuguchi T, Collod-Beroud G, Akiyama T, et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet. 2004;36:855–60. doi: 10.1038/ng1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loeys BL, Schwarze U, Holm T, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006;355:788–98. doi: 10.1056/NEJMoa055695. [DOI] [PubMed] [Google Scholar]
- 6.Boileau C, Guo DC, Hanna N, et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat Genet. 2012;44:916–21. doi: 10.1038/ng.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van de Laar IM, Oldenburg RA, Pals G, Roos-Hesselink JW, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet. 2011;43:121–6. doi: 10.1038/ng.744. [DOI] [PubMed] [Google Scholar]
- 8.Regalado ES, Guo DC, Villamizar C, et al. Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circ Res. 2011;109:680–6. doi: 10.1161/CIRCRESAHA.111.248161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andrabi S, Bekheirnia MR, Robbins-Furman P, Lewis RA, Prior TW, Potocki L. SMAD4 mutation segregating in a family with juvenile polyposis, aortopathy, and mitral valve dysfunction. Am J Med Genet A. 2011;155A:1165–9. doi: 10.1002/ajmg.a.33968. [DOI] [PubMed] [Google Scholar]
- 11.Teekakirikul P, Milewicz DM, Miller DT, et al. Thoracic aortic disease in two patients with juvenile polyposis syndrome and SMAD4 mutations. Am J Med Genet A. 2013;161:185–91. doi: 10.1002/ajmg.a.35659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heald B, Rigelsky C, Moran R, et al. Prevalence of thoracic aortopathy in patients with juvenile Polyposis Syndrome-Hereditary Hemorrhagic Telangiectasia due to SMAD4. Am J Med Genet A. 2015;167A:1758–62. doi: 10.1002/ajmg.a.37093. [DOI] [PubMed] [Google Scholar]
- 13.Guo DC, Regalado ES, Gong L, et al. LOX mutations predispose to thoracic aortic aneurysms and dissections. Circ Res. 2016;118:928–34. doi: 10.1161/CIRCRESAHA.115.307130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Renard M, Francis C, Ghosh R, et al. Clinical validity of genes for heritable thoracic aortic aneurysm and dissection. J Am Coll Cardiol. 2018;72:605–15. doi: 10.1016/j.jacc.2018.04.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo DC, Hostetler EM, Fan Y, et al. Heritable thoracic aortic disease genes in sporadic aortic dissection. J Am Coll Cardiol. 2017;70:2728–30. doi: 10.1016/j.jacc.2017.09.1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang M, Liang YY, Wrighton K, et al. Ubiquitination and proteolysis of cancer-derived Smad4 mutants by SCFSkp2. Mol Cell Biol. 2004;24:7524–37. doi: 10.1128/MCB.24.17.7524-7537.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu J, Attisano L. Mutations in the tumor suppressors Smad2 and Smad4 inactivate transforming growth factor beta signaling by targeting Smads to the ubiquitin-proteasome pathway. Proc Natl Acad Sci USA. 2000;97:4820–5. doi: 10.1073/pnas.97.9.4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo DC, Duan XY, Regalado ES, et al. Loss-of-function mutations in YY1AP1 lead to grange syndrome and a fibromuscular dysplasia-like vascular disease. Am J Hum Genet. 2017;100:21–30. doi: 10.1016/j.ajhg.2016.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mao X, Debenedittis P, Sun Y, et al. Vascular smooth muscle cell Smad4 gene is important for mouse vascular development. Arterioscler Thromb Vasc Biol. 2012;32:2171–7. doi: 10.1161/ATVBAHA.112.253872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Le Goff Carine, Mahaut Clémentine, Abhyankar Avinash, Le Goff Wilfried, Serre Valérie, Afenjar Alexandra, Destrée Anne, di Rocco Maja, Héron Delphine, Jacquemont Sébastien, Marlin Sandrine, Simon Marleen, Tolmie John, Verloes Alain, Casanova Jean-Laurent, Munnich Arnold, Cormier-Daire Valérie. Mutations at a single codon in Mad homology 2 domain of SMAD4 cause Myhre syndrome. Nature Genetics. 2011;44(1):85–88. doi: 10.1038/ng.1016. [DOI] [PubMed] [Google Scholar]
- 22.Carr Jennifer C., Dahdaleh Fadi S., Wang Donghong, Howe James R. Germline Mutations in SMAD4 Disrupt Bone Morphogenetic Protein Signaling. Journal of Surgical Research. 2012;174(2):211–214. doi: 10.1016/j.jss.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.