

Self-resonant microhelix provides sensitivity for X-band electron paramagnetic resonance on protein crystals with <27-nl volume.
Abstract
Electron paramagnetic resonance (EPR) spectroscopy on protein single crystals is the ultimate method for determining the electronic structure of paramagnetic intermediates at the active site of an enzyme and relating the magnetic tensor to a molecular structure. However, crystals of dimensions typical for protein crystallography (0.05 to 0.3mm) provide insufficient signal intensity. In this work, we present a microwave self-resonant microhelix for nanoliter samples that can be implemented in a commercial X-band (9.5 GHz) EPR spectrometer. The self-resonant microhelix provides a measured signal-to-noise improvement up to a factor of 28 with respect to commercial EPR resonators. This work opens up the possibility to use advanced EPR techniques for studying protein single crystals of dimensions typical for x-ray crystallography. The technique is demonstrated by EPR experiments on single crystal [FeFe]-hydrogenase (Clostridium pasteurianum; CpI) with dimensions of 0.3 mm by 0.1 mm by 0.1 mm, yielding a proposed g-tensor orientation of the Hox state.
INTRODUCTION
The catalytic cycle of redox enzymes often contains paramagnetic intermediates, and electron paramagnetic resonance (EPR) spectroscopy is the method of choice used to study these occurrences. Through EPR experiments, information on the electronic and geometrical structure of the active site can be obtained. For typical EPR experiments on proteins, a frozen solution of 0.1 to 1 mM concentration is prepared and placed in a microwave cavity. Standard sample volumes at X-band (nominally 9.5 GHz) are in the 200 μl range. However, frozen solution EPR experiments only allow the determination of the principal values of magnetic interactions at an active site and, thus, provide only a limited view of the electronic structure.
To resolve the full-tensor magnetic interaction parameters, such as g-, zero-field, hyperfine, and quadrupole tensors, single-crystal EPR experiments must be performed. In combination with x-ray crystallography, the magnetic interaction tensors obtained with EPR experiments can be directly related to the protein geometry to help identify and better understand the catalytic mechanism of the enzyme (1, 2). Despite its usefulness, single-crystal EPR is rarely applied to protein systems because of challenges in growing crystals of sufficient quality and volume for these experiments. Many protein crystals used in x-ray crystallography are of dimensions in the 0.05-0.3 mm range and, hence, are too small to be studied using commercial EPR instrumentation. Crystallization methods, such as macroseeding (3), have the potential to increase the volume of the crystals, but these techniques are difficult to implement.
To study magnetic interactions of the paramagnetic center with the surrounding nuclear spins, pulse and double-resonance EPR experiments are required. However, these experiments require even higher sensitivity. Unlike nuclear magnetic resonance (NMR), where all nuclei are excited and contribute to the NMR signal, double-resonance experiments, such as electron spin echo envelope modulation (ESEEM), hyperfine sublevel correlation (HYSCORE), and electron nuclear double resonance (ENDOR), probe only the nuclei that are magnetically coupled to the paramagnetic center. Extending these experiments to single crystals provides not only the magnitude of the hyperfine and quadrupole tensors of ligand nuclear spins that interact with the paramagnetic centers but also the associated angles relative to the active site of an enzyme. These interacting nuclei are either naturally abundant, such as 1H and 14N, or the catalytic cofactors can be enriched with nuclei such as 2H, 13C, 15N, and 57Fe, for further analysis of magnetic interaction tensors with respect to the first ligand sphere. Furthermore, the same interaction tensors can be calculated from the molecular structure using quantum chemical calculations (4). These experimentally determined spectroscopic parameters can, therefore, be used to verify the adequacy of the level of theory, which, in turn, gives confidence to the predicted electronic and geometric structure of the involved intermediates and transition states in the whole catalytic cycle. The groundwork for understanding the inner workings of enzymes lies in collecting as much accurate spectroscopic information as possible, including other spectroscopic and structural methods (optical and vibrational spectroscopy, Mößbauer, x-ray spectroscopy, and diffraction). Every experiment contributes to the total picture and ultimately leads to a fundamental understanding of the catalytic mechanism of these enzymes.
Currently, volume-limited crystals can only be studied using W-band (94 GHz) (or higher) EPR in a single-mode resonator (5) or, for continuous-wave experiments, a Fabry-Pérot (6) resonator. These high-frequency EPR spectrometers are not widely available, and high-frequency conditions are usually unfavorable for pulse experiments such as ESEEM or HYSCORE, which provide the best response when the nuclear Zeeman and hyperfine/quadrupole interactions are of the same order of magnitude (7). Instead, at higher frequencies, where the ESEEM effect becomes increasingly difficult to detect, ENDOR- and ELDOR-detected NMR experiments are performed to obtain hyperfine and quadrupole information.
To improve the sensitivity for studying single crystals using EPR on readily available spectrometers, typically at X-band, one must abandon the microwave cavity design and move to small-volume resonators based on lumped circuits in the microwave frequency range. This allows the reduction of the sample volume by one order of magnitude, from 200 to 20 μl using a loop-gap resonator (LGR) (8). Further reductions can be achieved by incorporating materials with a high dielectric constant in a standard resonator to reduce the active volume down to 1 μl (9). For protein single crystals, one must reduce the volume even further (less than 0.03 μl), which requires radical new approaches.
Here, we combine the concept of a self-resonant microhelix, shown in Fig. 1A, with that of a planar microcoupler. The coupling structure on the printed circuit board is a resonant structure, which drives the self-resonant microhelix placed in the center of the coupling loop (10), shown in Fig. 1B. The microhelix geometry offers notable advantages in that the microwave field homogeneity is strongly improved along with volume sensitivity for small samples compared to other microresonator geometries, such as the planar microresonator (PMR) (11–13). The microwave characteristics of the self-resonant microhelix are optimal for pulse and continuous-wave experiments requiring very little microwave power, and the microhelix assembly is easily matched and tuned over a variety of samples and temperatures.
Fig. 1. The self-resonant microhelix.
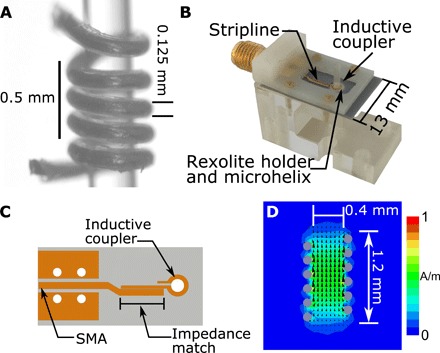
(A) A fabricated five-turn microhelix wrapped around a 0.4-mm–outer diameter capillary. During fabrication, the microhelix is tightly wound around a 0.4-mm drill bit and glued inside a Rexolite cylinder. The drill bit is removed, and the glue is allowed to dry for several days. The microhelix assembly is placed in (B) a coupling and support assembly, which includes a planar microcoupler. (C) The planar microcoupler consists of a stripline impedance match to an inductive coupling loop. SMA, SubMiniature version A. (D) Finite-element modeling simulations of the microwave magnetic field, normalized to input power, at 9.5 GHz show an active region of good magnetic field homogeneity over a 0.8-mm height. The measured microwave magnetic field of 3.2 G/W1/2 corresponds to a 20-ns π/2 pulse at approximately 20 mW. Dimensions of the microhelix, where the self-resonance is determined by the capacitance formed between each turn and the inductance of the windings, are shown. The frequency can be tuned during fabrication by the number of turns, the pitch of the turns, or the inner diameter. The microwave characteristics of the fabricated microhelix can be found in table S1.
Single-crystal EPR for metalloenzyme research
Nature has evolved enzymes with various metallic cofactors (metalloenzymes) to efficiently catalyze a broad variety of chemical reactions (14). These enzymes mostly use first-row transition metals to perform their catalytic functions. One of the main challenges is to fully understand these enzymatic mechanisms and provide a basis for cheap, robust, and highly active molecular catalysts designed for practical applications, e.g., in the field of energy conversion and storage (15). The ultimate goal is to alleviate the requirement of noble metals, such as platinum, that limit the scalability of current technology. This important biophysical and biochemical research seeks metalloenzyme-based and metalloenzyme-inspired systems as an interesting route to advance toward the future of clean energy and efficient energy storage.
For hydrogenases, specifically [NiFe]-hydrogenase, the single-crystal EPR strategy has been very successful (2). The active site of this enzyme harbors a [NiFe] binuclear cluster in which the iron carries two cyanide (CN−) and one carbon monoxide (CO) ligand. The metals are bridged by two cysteine thiols, and the nickel center is further coordinated by two cysteine thiolate side groups. The paramagnetic states all originate from the nickel center, while the iron center remains Fe(II) during the catalytic cycle (16). The open-coordination site between the two metals can be occupied by an oxygen species leading to the inactive oxidized states or a hydride, which is the key intermediate in the catalytic cycle (2). For all these species, the g-tensor magnitude and orientation were determined and analyzed in terms of ligand-field theory and verified using quantum chemical calculations, providing a fundamental insight into the electronic structure and the dependence on the first ligand sphere (2). The [NiFe]-hydrogenase crystals in these studies were relatively large (2 mm by 0.5 mm by 0.5 mm), enabling measurements in standard X-band probeheads with a measuring time of 2 to 3 hours per angle, stepping over 180°. However, even with the large crystal volume, ESEEM and HYSCORE experiments on [NiFe]-hydrogenase were only published in frozen solution with volumes of 50 to 200 μl and experimental times upward of 2 hours for each experiment (2).
Similar experiments were anticipated for [FeFe]-hydrogenase, which has a much higher activity and exhibits a different catalytic mechanism compared to [NiFe]-hydrogenase. The [FeFe]-hydrogenase active site carries a [4Fe-4S] cluster linked via a cysteine ligand connecting a [2Fe] site. The [2Fe] cluster contains an iron atom proximal (Fep) and one distal (Fed) to the [4Fe-4S] cluster. Each iron carries a cyanide (CN−) and one carbon monoxide (CO) ligand, and the two irons share a bridging carbon monoxide. In addition, the two iron atoms are bridged by an azapropane-dithiolate ligand (ADT ligand). The molecular structure of the [FeFe]-hydrogenase active site, known as the H-cluster, can be found in Fig. 2. The whole active site has a total of six iron atoms at various redox states in the catalytic cycle (17). Unfortunately, the crystals obtained from [FeFe]-hydrogenase are much smaller than those available from [NiFe]-hydrogenase with dimensions less than 0.3 mm. Frozen solution EPR on [FeFe]-hydrogenase has provided a lot of information on the binuclear active site, such as the discovery of a nitrogen in the dithiolate bridge (ADT ligand) (18). However, the full g-tensor and the hyperfine tensor, including angular information, of the active site are still elusive. Therefore, there is continuing interest in developing EPR instrumentation, specifically microresonators, at 9.5 and 35 GHz, X- and Q-band, respectively, optimized for metalloenzyme research. Furthermore, other potentially interesting proteins are rarely studied in single crystals, and doing so would contain a wealth of information on the electronic structure and enzymatic function (14).
Fig. 2. The molecular structure of the [FeFe]-hydrogenase active site, the H-cluster.
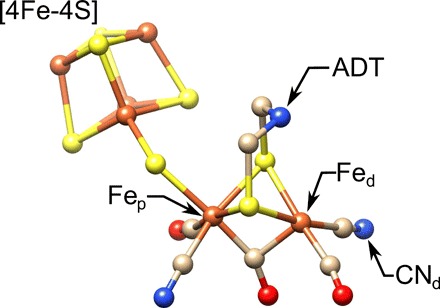
Highlighted are the proximal and distal irons, Fep and Fed, respectively, the cyanide ligand (), and the ADT ligand. S, yellow; Fe, orange; N, blue; C, tan; O, red. Structure is from Protein Data Bank (PDB) ID 4XDC.
We report here an EPR crystal rotational study of a [FeFe]-hydrogenase in the active oxidized state (Hox) from Clostridium pasteurianum (CpI) with crystal dimensions of 0.3 mm by 0.1 mm by 0.1 mm using the self-resonant microhelix. The excellent signal-to-noise ratio has also allowed for advanced pulse EPR experiments to be performed. These data demonstrate the utility of the microhelix in studying protein single crystals at volumes relevant for x-ray crystallography.
Theory and design
We focus on the development of resonant structures to increase EPR absolute spin sensitivity at X-band without the application of commercial bridge modifications, which require technical expertise not commonly available in EPR laboratories. We start with the well-established EPR signal analysis by Feher (19), which states that the continuous-wave EPR signals for a critically coupled resonator on a reflection bridge is proportional to
| (1) |
where χ is the magnetic susceptibility, Q is the Q-value of the cavity, P is the incident power, and η is the filling factor. The Q-value is defined as the ratio of the stored energy in the resonator to the power loss in the sample and coupled resonator, whereas the filling factor is defined as the ratio of the circularly polarized microwave magnetic field stored energy perpendicular to the static magnetic field that gives rise to EPR transitions and the complete magnetic field stored energy in the geometry
| (2) |
where B1 is the microwave magnetic field in all space (V) and B1r is one component of the clockwise (or counterclockwise) rotational component of the linear B1 field perpendicular to the static magnetic field in the sample volume (Vs) (19, 20).
From Eq. 1, we can derive a metric that encompasses the filling factor η and the Q-value, called the resonator efficiency (8). The resonator efficiency (or conversion factor) is defined as
| (3) |
where Pl is the power loss in the system, including sample. However, Λ assumes that the microwave magnetic field is uniform and does not take into account nonuniformity of the field over the sample. Experimentally, the microwave magnetic field is distributed throughout the sample, and the average of Λ is measured. We can define the resonator efficiency average as
| (4) |
The resonator efficiency average is proportional to the signal and, for a fixed sample volume, is proportional to the absolute spin sensitivity.
From Eqs. 1 and 4, we can outline the criteria needed to increase the sensitivity for fixed sample volume EPR experiments. We choose the target dimensions of 0.3 mm by 0.3 mm by 0.3 mm (27 nl) as a maximum sample size. These dimensions are a reasonable size for protein single crystals used in x-ray crystallography diffraction, and any increase in EPR sensitivity would benefit single crystals of smaller dimensions. To maximize sensitivity, one must increase the ηQ product, as shown in Eq. 1. However, there are practical limitations that must be considered while designing a resonator.
For instance, cavity resonators, such as the Bruker Super High-Q probehead, are not sufficiently sensitive for extremely small samples since the very small filling factor η is not compensated by the high Q0-value, resulting in an overall poor EPR signal (21). Further increase of the Q0-value, by superconducting materials, is detrimental for pulse experiments where the available bandwidth of the resonator is inversely reduced. The resonator bandwidth filters the excitation pulses as well as the EPR signal, distorting the spectrum. The trade-off required for advanced pulse experiments results in the design parameters of a large filling factor η while keeping the bandwidth around 100 MHz (Q0-value of 190 at 9.5 GHz).
Therefore, one way to maximize ηQ, and consequently Λave, is to reduce the size of the resonant structure relative to the sample volume, increasing the filling factor while maintaining a homogeneous magnetic field profile. The challenge arises because of the potential increase of the losses in the system can degrade the Q0-value more than the increase in the filling factor. Two common methods to reduce the size of a resonant geometry are to use either dielectric resonators to reduce the wavelength and, consequently, the size of the resonator or LGRs to reduce the cutoff frequency of a waveguide by introducing protrusions to create regions of inductive loops and capacitive gaps. Dielectric resonator and LGR geometries with commercially available dimensions are illustrated in fig. S1, and the microwave characteristics are tabulated in table S1. However, in order to study sample volumes less than 0.03 μl, further resonator reduction strategies are needed.
Limitations to minimizing LGR geometries stem from an increase in ohmic losses due to a reduction in the gap spacing to maintain a constant resonant frequency as the sample-loop radius is reduced. In practice, this has put a limit on the Λave obtainable to less than 1 mT/W1/2 for X-band. Further EPR signal improvement is possible using dielectric resonators by increasing the dielectric permittivity, and dielectric resonators with permittivity up to 80 have been used for continuous-wave EPR experiments on crystals of porous materials and polymers (9, 22). However, these resonators exhibit Q0-values over 2500 that make pulse experiments problematic.
Here, we introduce a new type of resonator based on a self-resonant microhelix that is particularly useful for protein single-crystal experiments at X-band and can be used as a drop-in replacement on a standard commercial system. The self-resonant microhelix geometry, illustrated in Fig. 1, solves these challenges by providing good magnetic field homogeneity, a high efficiency parameter, an optimum Q0-value for both pulse and continuous-wave EPR experiments, straightforward impedance matching, and ease of sample placement.
Helical resonators were first introduced to EPR in the early 1960s as a method to increase the microwave magnetic field at the sample. Resonant helical geometries were affixed to one end of a shorted waveguide creating a slow-wave structure (23, 24). Coupling was achieved by direct connection to a coaxial line with a capacitive matching network or by microwave incident on the helical structure from a waveguide. The sample was placed within the helix and showed reasonable sensitivity increase and larger microwave magnetic field due to higher filling factors compared to typical cavities (25). Broadband slow-wave helical resonators were used for multifrequency experiments, where a nonresonant structure, having a Q-value close to unity, could be matched with a slide-screw tuner over an octave bandwidth (26). However, over time, they were replaced by LGRs, which achieved higher concentration sensitivity for volume-limited samples.
Recently, microcoils have gained popularity in NMR for nanoliter samples (27, 28) and for microfluidics (29). However, three characteristics differentiate our microhelix configuration from those described in the EPR (23, 24) and NMR literature: (i) The helix is self-resonant, meaning that the self-inductance of n-turns (Ltot) and self-capacitance between the loops (Ctot) resonate at a frequency determined by ω2LtotCtot = 1, where ω is the resonant frequency in radians per second. Since the geometry is self-resonant, no additional capacitors are needed. A self-resonant microhelix has lower ohmic loss, which provides a higher Q0-value than is typically feasible with microcoil geometries in NMR, where, with an NMR microcoil, a typical Q0-value is around 30 (28). With a self-resonant microhelix, the volume-to-surface ratio is maximized and a Q0-value of 300 is achievable. (ii) Unlike the slow-wave structures in (23) and (24), the helix length is much smaller than the wavelength (31.6 mm at 9.5 GHz), which increases the uniformity. In addition, the inner diameter is 0.4 mm, which increases the resonator efficiency. Therefore, the microhelix is not a slow-wave structure but an inductor at self-resonance. The microwave magnetic field profile is shown in Fig. 1D. (iii) Last, the helix is coupled to an inductive coupling loop on a printed circuit board by mutual inductance, which can be designed to minimize noise and further increase the EPR signal-to-noise ratio (10). Mutual inductance does not require a balun or additional capacitive matching networks since impedance matching can be achieved by varying the distance between the microhelix and the inductive coupling loop, simplifying coupling methods.
RESULTS AND DISCUSSION
Performance compared to commercially available and state-of-the-art microwave probes
The self-resonant microhelix geometry wound around a 0.4-mm capillary is shown in Fig. 1A. The final number of the microhelix windings is determined by the pitch of the helix, the quartz capillary sample tube (0.4 mm outer diameter and 0.3 mm inner diameter), and the surrounding Rexolite, which all affect the resonance frequency. The 6.5-turn microhelix had a resonant frequency around 9.8 GHz with sample when coupled to the printed circuit board inductive coupler. The microhelix assembly is attached to a custom insert that is compatible with commercial EPR systems. The complete structure is shown in Fig. 1B with an expanded view of the printed circuit board geometry in Fig. 1C. Comparison of the fabricated microhelix geometry with commercial (Bruker MD5W1 and Bruker MS3) and state-of-the-art microwave probes is provided in Table 1.
Table 1. Resonator EPR signal characteristics calculated and measured using a power saturation measurement of a lithium phthalocyanine point sample.
Dimensions of the resonators and further experimental details are provided in the Supplementary Materials.
| Geometry |
Unsat. signal |
Sat. signal |
Eff. η |
Q0-value Meas. |
ηQ0 | ||
| Calc. | Meas. | Calc. | Meas. | ||||
| Bruker MD5W1 | 1.0 | 1.0 | 1.0 | 1.0 | 6.12 × 10−6 | 6650 | 1.0 |
| Bruker MS3 | 1.5 | 1.2 | 1.0 | 1.0 | 79.6 × 10−6 | 600 | 1.17 |
| PMR RO6010LM | 4.4 | 1.2 | 0.9 | 1.2 | 4.63 × 10−3 | 61 | 6.94 |
| PMR sapphire | 18.6 | 13.3 | 3.9 | 3.8 | 4.63 × 10−3 | 181 | 20.59 |
| Microhelix | 35.7 | 28.2 | 6.1 | 5.7 | 6.63 × 10−3 | 220 | 35.84 |
As a comparison to the state-of-the-art microwave probes, two Ω-shaped 0.5 mm inner diameter PMRs (based on Rogers RO6010LM printed circuit board or sapphire substrates) are tested, which were fabricated by printing the microresonator geometry on a substrate using photolithographic techniques (11–13). The PMRs are considered state-of-the-art because of the significant improvement in absolute spin sensitivity compared to the best commercial probeheads available (21). However, the self-resonant microhelix introduced in this work exhibits the highest absolute sensitivity with no modification to the commercial bridge.
As described in Table 1, if the EPR signal cannot be saturated (Unsat.), a factor of approximately 28 can be achieved compared to commercially available probeheads. EPR signals that cannot be saturated are proportional to the square root of the incident microwave power; therefore, the EPR signal intensity is only limited by the amount of power available. However, most protein samples saturate readily; hence, the maximum signal that can be obtained is determined by the microwave magnetic field at the sample. When the sample is saturable (Sat.), a factor of 5.7 can be achieved. Further experimental details are provided in the Supplementary Materials and fig. S3. The procedure to calculate the relative EPR signal intensities can be found in (20). Also found in Table 1 is an effective filling factor (Eff. η) calculated by using the ratio of a “point” sample (1 nl reference volume) with the effective active volume of the resonator. The effective height of the PMR is assumed to be 1.1 mm, taken from the on-axis profile in fig. S2C. The ηQ0 product can be related to the unsaturable signal as per Eq. 1. In a pulse experiment, the signal enhancement will be proportional to the saturable signal, in this case, approximately a factor of 6.
Frozen solution EPR of photosystem II tyrosine D radical ()
Water oxidation in photosystem II takes place at the tetranuclear manganese cluster, with a redox-active tyrosine radical () as an interface to the light-induced electron transfer process (30). Symmetrically to , a long-lived tyrosine radical () exists in the second branch of the photosystem II that contains no manganese cluster. In this work, the radical is used as a standard probe because it is stable for a number of hours under ambient conditions (31) and has been well characterized using a variety of EPR techniques (5, 30). The hyperfine interactions from several protons, both on the phenyl ring and distal CH2 carbon, lead to the distinct splittings of the radical (S = 1/2). To generate the tyrosine radical () EPR signal, the photosystem II core complex samples are illuminated in ambient light and rapidly frozen.
The tyrosine D radical () of photosystem II is measured in two forms: (i) a frozen solution of photosystem II prepared by the method of Berthold, Babcock, and Yocum (BBY particles) (32) placed in a 0.3 mm inner diameter capillary and (ii) a 0.3 mm by 0.18 mm by 0.18 mm single crystal of photosystem II core complexes (33). In both photosystem II samples, the and first ligand sphere are known to be identical. These samples provide a benchmark for future work.
Shown in Fig. 3 is the radical EPR signal in an 85 nl frozen solution of photosystem II BBY particles at a temperature of 80 K using the self-resonant microhelix. A continuous-wave EPR experiment, shown in Fig. 3A, was performed using an Elexsys E580 X-band bridge by sweeping 10 mT in 1 min (4096 points) with a modulation rate of 100 kHz and an amplitude of 0.5 mT. The data were averaged 49 times for a total of 49 min at an incident power of 0.2 μW. To further improve the signal-to-noise ratio of the continuous-wave experiment, we performed a field-swept nonadiabatic rapid scan (NARS) experiment (data shown in Fig. 3B). The field-swept NARS experiment was performed on the same commercial hardware using the rapid-scan method of Möser et al. (34) and processed with the method described by Hyde et al. (35). Here, the scan rate was a sinusoidal 100 kHz field sweep at 1 mT amplitude and a field-step size of 0.05 mT. The collected real and imaginary, pure-absorption and pure-dispersion, spectra were pseudo-modulated with a 0.5 mT moving difference (MDIFF) (35) to compare to the field-modulated continuous-wave experiment. A factor of 2 in signal-to-noise improvement is obtained for the same signal acquisition time.
Fig. 3. Frozen solution EPR on an 85-nl-volume sample at X-band.
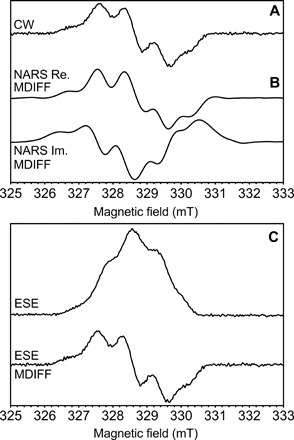
Three EPR experiments performed with a 0.4 mm inner diameter self-resonant microhelix. Shown are the (A) continuous-wave (CW), (B) real (Re.) and imaginary (Im.) nonadiabatic rapid scan (NARS), and (C) field-swept two-pulse ESE EPR experiments of the tyrosine D radical () in photosystem II with 85 nl of frozen solution sample at a temperature of 80 K. Calculated MDIFF pseudo-modulation of 0.5 mT is shown for the NARS and field-swept ESE experiments to directly compare to the continuous-wave EPR experiment. The total time for the experiments was 49, 55, and 45 min, respectively. The signal-to-noise ratio is calculated and tabulated in Table 2.
A field-swept two-pulse electron spin-echo (ESE) EPR experiment was performed on the same commercial hardware over an 8 mT sweep, shown in Fig. 3C. The field-swept ESE data were pseudo-modulated with a 0.5 mT MDIFF to compare the experiment with the field-modulated continuous-wave experiment in Fig. 3A. Only 9 mW of incident power was needed to obtain a sufficient echo using a 40 ns π/2 pulse. The signal-to-noise ratio for all three experiments was calculated and tabulated in Table 2.
Table 2. Signal-to-noise calculations for the three experiments performed on the photosystem II radical in frozen solution at a temperature of 80 K.
Approximately 1.6 × 1012 spins were calculated to be in the 85 nl that fill the microhelix. SNR, signal-to-noise ratio.
| Experiment | SNR Re. | SNR Im. | Time |
| Continuous wave | 197 | 131 | 49 min |
| NARS | 4400 | 2300 | 55 min |
| NARS (MDIFF) | 410 | 423 | – |
| ESE | 248 | – | 45 min |
| ESE (MDIFF) | 106 | – | – |
Last, a comparison of radical EPR signal between the MD5W1 dielectric resonator and the self-resonant microhelix was performed at a temperature of 80 K. A 636 nl volume sample was placed in the MD5W1 resonator and a signal-to-noise ratio of approximately 300 was measured (data shown in fig. S5). This represents a volume-normalized absolute spin sensitivity improvement of approximately 5 at similar microwave magnetic field and EPR experimental parameters. These experiments serve to show the versatility of the microhelix to perform EPR experiments on limited sample volumes (less than 85 nl) at X-band.
Single-crystal continuous-wave EPR of the tyrosine D radical () in the photosystem II core complex
Shown in Fig. 4 are continuous-wave EPR data collected at two separate angles of the photosystem II radical in a single crystal at a temperature of 80 K as a sensitivity test for the 0.4-mm–inner diameter self-resonant microhelix. The photosystem II core complex crystal had dimensions of 0.3 mm by 0.18 mm by 0.18 mm. The spectra were collected by sweeping 15 mT in 1 min (4096 points) with a modulation rate of 100 kHz and an amplitude of 0.3 mT. The data are averaged 49 times for a total time of 49 min at an incident power of 0.2 μW. Simulations using the known g-tensor and hyperfine tensors (5) were performed with an EasySpin (http://easyspin.org; 36) global fit routine to find the crystal orientation and plotted in red in Fig. 4. At X-band, the g-anisotropy of the radical is very small and is not resolved. Instead, the orientation dependence is primarily determined by the hyperfine interaction pattern of the coupled proton nuclei (5). Using only two angles, a unique fit cannot be found, but a demonstration of the features is shown. A nonspecifically bound Mn2+ signal is also present in the crystal, yielding the signals indicated by an asterisk (*).
Fig. 4. Single-crystal continuous-wave EPR of in the photosystem II core complex.
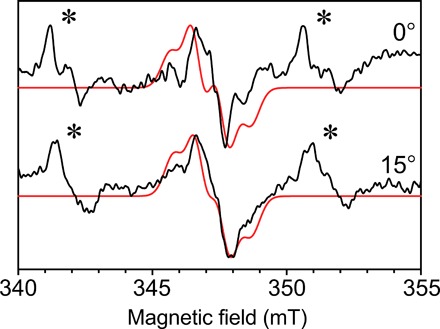
Continuous-wave EPR collected with the 0.4 mm inner diameter self-resonant microhelix at two angles of the photosystem II radical from a single crystal at a temperature of 80 K. The crystal dimensions were 0.3 mm by 0.18 mm by 0.18 mm. Shown in red is a fitted simulation with similar features. A nonspecifically bound Mn2+ signal is also present in the mother liquor of the crystal, indicated by an asterisk (∗). Each spectrum was collected in 49 min with a signal-to-noise ratio of approximately 35.
The use of photosystem II crystals as a benchmark provides a challenging system to measure. The photosystem II core complex has a molecular mass of approximately 350 kDa as a monomer, and each complex contains only one radical. With a crystal size of 0.3 mm by 0.18 mm by 0.18 mm, the size of the unit cell, and the fact that there are eight photosystem II complexes per unit cell, one can calculate approximately 8.9 × 1012 radicals to be present in the sample. This demonstrates the versatility of the microhelix to study large complexes in small crystal dimensions. A photosystem II core complex crystal can be routinely grown to dimensions of 0.3 mm but requires significant effort to increase in size. Last, the radical is easily saturable with large microwave magnetic fields, which limits the available microwave power and maximum EPR signal at a given temperature. Despite these challenges, a signal-to-noise ratio of approximately 35 could be obtained for the radical.
Pulse EPR on the H-cluster in single crystals of [FeFe]-hydrogenase
Using the photosystem II radical, we have established that the self-resonant microhelix is suitable for single-crystal protein samples. However, we now want to demonstrate that (i) a full angular g-tensor determination can be performed and that (ii) advanced pulse EPR experiments, like ESEEM and HYSCORE, are also possible in this setup. The self-resonant microhelix is optimal for these experiments because of the relatively large bandwidth (90 MHz critically coupled); an efficiency of 3.2 mT/W1/2, which corresponds to a π/2 pulse of as short as 20 ns with an incident power of only 20 mW; and the relatively homogeneous microwave magnetic field incident on the sample.
First, a field-swept two-pulse ESE EPR experiment has been performed every 5° on a protein single crystal of the [FeFe]-hydrogenase of C. pasteurianum (CpI) in the oxidized Hox state (16). Under a microscope in an anaerobic chamber, the protein crystal is drawn by capillary action into a 0.3 mm inner diameter capillary with mother liquor and cryoprotectant, centered in the microhelix, and flash-frozen. The microhelix assembly is affixed to a Bruker Flexline–compatible support assembly, placed in a precooled cryostat, and attached to the EPR bridge. The whole assembly is then rotated in 5° steps over 180° in one plane within the magnet (shown in Fig. 5). A very good signal-to-noise ratio of approximately 290 is calculated for a collection time of 8 min for each spectrum at a temperature of 15 K. The [FeFe]-hydrogenase of C. pasteurianum (CpI) has a molecular mass of 67 kDa. The unit cell has P1 21 1 symmetry with two molecules in the asymmetric unit [Protein Data Bank (PDB) ID: 4XDC], resulting in four distinct signals in the EPR spectrum. The single crystal had dimensions of approximately 0.3 mm by 0.1 mm by 0.1 mm and, on the basis of the unit cell dimensions, approximately 17 × 1012 enzymes within the crystal are calculated, each containing one active site (H-cluster, shown in Fig. 2). Each enzyme in the unit cell provides one of the four observed signals corresponding to 4.25 × 1012 spins per peak.
Fig. 5. Pulse EPR on a single crystal of the H-cluster in [FeFe]-hydrogenase.
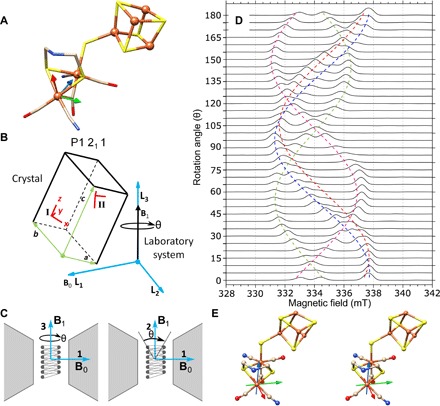
(A) The molecular structure of the [FeFe]-hydrogenase active site, the H-cluster, from PDB ID 4XDC is shown with the molecular frame located with the distal iron (Fed) as the origin. The molecular frame rotational coordinates can be found in table S2. S, yellow; Fe, orange; N, blue; C, tan; O, red. (B) The P1211 symmetry schematic relating the molecular frame (x, y, z) to the crystal frame (a, b, c) and, last, to the laboratory system frame (L1, L2, L3) is shown. The two molecular frames from the asymmetric unit are present in Site I and can be translated to Site II by crystal symmetry operations. (C) The static magnetic field (B0) is positioned along the L1 axis, while the microwave magnetic field (B1) can be either along the L2 axis or along the L3 axis. A rotation of 180° is feasible around the L3 axis, but only a partial rotation around the L2 axis is feasible because of the B1 rotating with the crystal resulting in B1 to become parallel to B0. A third partial rotation is feasible if the sample is rotated by 90° around the L2 axis. (D) Pulse EPR experiments collected with the 0.4 mm inner diameter self-resonant microhelix with a [FeFe]-hydrogenase single crystal of C. pasteurianum (CpI) in the Hox state showing collected data in one plane for a full rotation of 180° in 5° steps at a temperature of 15 K. The crystal dimensions were approximately 0.3 mm by 0.1 mm by 0.1 mm, and each spectrum was collected in 8 min with a signal-to-noise ratio of approximately 290. (E) A stereo view of the analyzed g-tensor (gx, red; gy, green; and gz, blue) is mapped on the crystal structure (PDB ID: 4XDC). For a three-dimensional (3D) view of the proposed g-tensor, see https://act-epr.org/FeFeHydrogenase.html.
From these spectra, the data can be fitted to simulations that relate the different frames of reference to each other, as defined in the EasySpin simulation package (further details in the Supplementary Materials). The [FeFe]-hydrogenase H-cluster is shown in Fig. 5A with the chosen molecular frame (details in the Supplementary Materials), and a schematic that relates the molecular frame to the laboratory system frame is shown in Fig. 5B. Here, the laboratory system frame is defined and the static magnetic field (B0) is oriented along the L1 axis, while in this work, the microwave magnetic field (B1) is along the L3 axis. The crystal frame with respect to the laboratory frame is unknown and depends on how the crystal lies within the capillary. The molecular frames of each of the four molecules in the unit cell are related to each other by the known crystal symmetry. The two molecules in the asymmetric unit are labeled Molecular-Frame AI and Molecular-Frame BI (Site I). The three axes for the molecular frames (AI and BI) were chosen on the basis of the x-ray crystal structure as (i) the axis from the distal iron (Fed) to the proximal iron (Fep); (ii) the normal of a plane calculated from the proximal iron, distal iron, and the nitrogen of the ADT ligand (Fep-Fed-N1); and (iii) the cross product of (i) into (ii). The crystal symmetry–related molecules, Molecular-Frame AII and Molecular-Frame BII from Site II, are generated by the screw axis along the b axis of the unit cell. The g-tensor frame, which gives rise to the EPR signal, is also unknown and is defined as a rotation with respect to the molecular frame. (Rotational matrices for the relationship of these frames can be found in table S2.) The proposed g-tensor frame was solved using the principal g-values obtained from the frozen solution EPR experiment in (16) (g-values: [1.999, 2.039, 2.097] corresponding to the x, y, and z axes, respectively), and the resonance roadmap is overlaid on the field-swept two-pulse ESE EPR experiment shown in Fig. 5D. Since four molecules with different (known) orientations are present in the P1211 unit cell symmetry, it can be anticipated that a rotational study in one plane might be sufficient to fully define the g-tensor axis with the molecular frame.
The preliminary proposed g-tensor orientation is plotted as a stereo view in Fig. 5E. The g-tensor (gx, red; gy, green; and gz, blue) is drawn with an origin at the distal iron (Fed) since density functional theory calculations consistently identify Fed to contain most of the spin density in the Hox state (37). In this work, we find that the g-tensor principal axis does not coincide with the molecular frame, as defined by the two iron atoms and the ADT-amine nitrogen, as suggested by Adamska-Venkatesh et al. (38). Rather, the g-tensor orientation seems to be symmetric with respect to the plane defined by the two ADT sulfurs, the distal iron atom, and the distal CN− and CO ligand. The orientation of the largest g-component (gz) dissects the plane between the distal CN− and CO ligands, while the gx and gy components tend to be roughly symmetric with respect to the plane spanned by the two iron atoms and the ADT-amine nitrogen. See fig. S6 for a direct comparison. A rotation of the molecular frame by (−142.0, −84.1, 137.6) using a ZYZ convention Euler angle produces the proposed g-frame. Rotational matrices relating the g-tensor to the molecular frame within the PDB ID 4XDC crystal structure can be found in table S2.
In the study by Adamska-Venkatesh et al. (38), the orientation of the Fed-CN axis was established with respect to the g-frame using the axial components of the 13CN hyperfine and C14N quadrupole interaction, as determined by orientation-selective 13C ENDOR and 14N HYSCORE experiments in frozen solution. The calculated direction cosines of the Fed-CN orientation within the g-frame seem to be consistent with g-frame being coincident with the molecular frame. It turns out, however, that the direction cosines of the Fed-CN axis with respect to the actual g-frame (not coinciding with the molecular frame), as determined in our current single-crystal study, are still similar to those found in the study of Adamska-Venkatesh et al. and only deviate by about 10° (see fig. S6). The information obtained from the frozen solution orientation-selective ESEEM and ENDOR experiments was underdetermined and did not allow accurate assignment of the full magnetic interaction tensors. Accurate analysis and refinement is only possible with the collection of hyperfine and quadrupole data originating from single-crystal EPR and relating the whole dataset to quantum chemical calculations.
Advanced pulse EPR on the H-cluster in single crystals of [FeFe]-hydrogenase
To illustrate the availability of more advanced experiments for single-crystal studies, a HYSCORE experiment for the 150° field-swept ESE EPR dataset was performed on each of the peaks and is plotted in Fig. 6. Each HYSCORE spectrum was collected over approximately 1 hour, using a standard four-pulse HYSCORE pulse sequence (7). To obtain information on the hyperfine and quadrupole tensors, HYSCORE or ESEEM data must be collected on at least one peak and followed through a 180° rotation to obtain the axial relationship of the hyperfine interactions. Multiple peaks can be used to overdetermine the system.
Fig. 6. Single-crystal HYSCORE EPR of the H-cluster in [FeFe]-hydrogenase.
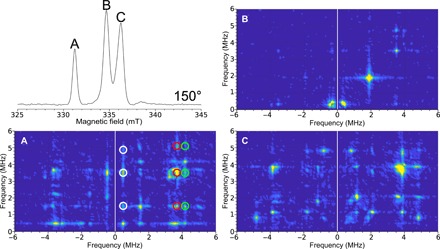
Top left: Field-swept two-pulse ESE EPR spectrum at 150°. The figure labels (A, B, and C) are representative of the spectral peaks. The HYSCORE spectra collected with the 0.4 mm inner diameter self-resonant microhelix of a [FeFe]-hydrogenase single crystal of C. pasteurianum (CpI) in the Hox state at an orientation of 150° collected at a temperature of 15 K. The 2D density representation shows correlations between the nuclear spin transitions in both projections of the electronic spin. (A) Clean HYSCORE spectrum due to the peak corresponding to only one of the EPR signals in the unit cell of the crystal. The correlated features between these transitions are indicated by the white, red, and green circles. (B) Relatively featureless HYSCORE spectrum suggests little hyperfine interaction at this orientation. (C) HYSCORE on two overlapping EPR signals representing different orientations of the enzyme molecule with respect to the magnetic field. The HYSCORE was set up using the Bruker HYSCORE wizard with the following settings: π/2, 40 ns; τ, 280 ns; and Δτ, 48 ns with 256 points each and 20 shots per point. Each HYSCORE spectrum was collected in approximately 1 hour.
In a HYSCORE experiment, the two-dimensional (2D) density representation shows correlations between the nuclear-spin transitions (mI) in both projections of the electron spin. Both the 14N nucleus (I = 1) from a distal cyanide ligand () and the secondary-amine group in the ADT ligand can potentially contribute to the HYSCORE spectrum generating three transitions per ligand for each electron-spin transition (ms) manifold for a maximum of 12 modulation frequencies. According to an earlier study on Hox in frozen solution, the features of the distal cyanide ligand spread out up to 6 MHz, while the transitions of the ADT-amine nitrogen are found between 2 and 4 MHz (38).
In the single-crystal 2D spectrum, shown in Fig. 6, six main transitions can be identified, which are assigned to the 14N of the distal CN ligand. The modulation frequencies can be grouped into two sets (0.5, 3.7, and 4.2 MHz) and (1.5, 3.5, and 5.0 MHz), each originating from a different ms manifold. The correlation features between these transitions are indicated by the white, red, and green circles and are in agreement with frozen-solution HYSCORE performed in (38). However, the current work only seeks to highlight the feasibility of these advanced EPR experiments. Future ESEEM/HYSCORE experiments will address the 14N couplings of the CN− and ADT ligand in greater detail. Possibly, this will involve selective 15N labeling as has been demonstrated before (38, 39). From these experiments, extracting the magnitude and orientation of the hyperfine and nitrogen quadrupole tensors in the molecular axis frame and relating these to the electronic structure as predicted through quantum chemical calculations are possible.
CONCLUSIONS AND OUTLOOK
An application of the self-resonant microhelix geometry and planar-coupling structure that increases the EPR absolute spin sensitivity by a factor of approximately 28 if the signal is unsaturable, and 6 if the EPR signal is able to be saturated, is presented. For saturable EPR signals, such as those found in protein samples, the self-resonant microhelix saves up to a factor of 36 in measuring time. From this gain in sensitivity, the self-resonant microhelix is well suited for EPR studies on protein single crystals with dimensions less than 0.3 mm. Because of the very high efficiency parameter of 3.2 mT/W1/2, which corresponds to a π/2 pulse of 20 ns with an incident power of 20 mW, the microhelix geometry is advantageous in extending pulse EPR to experiments that usually require costly high-powered microwave amplifiers (e.g., HYSCORE), further expanding the applicability of pulse EPR. The significantly reduced power contributes to reduced dead time and, therefore, potentially expands the use of Fourier transform EPR on systems with short relaxation rates (40). We also show that the microhelix performs well for field-swept NARS techniques because of its small size and “open” structure, which increases the continuous-wave EPR spin sensitivity further by a factor of 2 for the same experimental time. Because of the relatively large bandwidth of the microhelix (90 MHz critically coupled), this geometry is particularly well suited for frequency-swept NARS and rapid scan experiments, which further improve the signal-to-noise ratio for saturable samples (35, 34) and for the use of arbitrary-waveform generators for advanced pulse spectroscopy.
This advance in resonator design has allowed the collection of EPR data from a 0.3 mm by 0.1 mm by 0.1 mm single crystal of [FeFe]-hydrogenase in the Hox state from C. pasteurianum (CpI) at a temperature of 15 K. To our knowledge, the HYSCORE spectra collected are the first published results from a protein single crystal with dimensions less than 0.3 mm. Full g-tensor and 14N hyperfine tensor analysis of the active-site cofactor from the collected data is to follow. In addition, further studies of the hyperfine and quadrupole tensor of the [FeFe]-hydrogenase are now feasible in single-crystal experiments. These studies will provide further insight for protein engineering and artificial enzyme research for creating bio-inspired and bio-mimicking hydrogenase systems (16).
As this technology matures, further improvements to enhance the sensitivity based on new fabrication techniques and choice of other materials will be explored. Not only does the increase in sensitivity save time in EPR data measurements, but it also reduces the need for the availability of, or necessity to grow, larger crystals. Other microhelix structures, for example those made from superconducting materials have very high Q0-values and, consequently, a limited bandwidth, which makes them ideal for continuous-wave experiments. Because of the extremely high resonator efficiency, only nonsaturable samples will fully benefit from these designs. Therefore, the current microhelix geometry and fabrication provides an optimal compromise between maximum sensitivity and bandwidth for a broad range of temperatures. Specifically, the self-resonant microhelix provides the possibility to study catalytically active proteins at crystal dimensions relevant to x-ray crystallography and, hence, is a significant advancement in the field of enzyme research.
MATERIALS AND METHODS
Simulations and experiments
All microhelix and planar-coupling designs were modeled in the commercially available finite-element modeling program Ansys Electronics Desktop with HFSS (High Frequency Structure Simulator; v. 19.1) using driven mode. In driven mode, Ansys HFSS requires a coupling structure and mimics the output of a network analyzer. All designs were matched to 50 ohms with an S11 < − 35 dB. Frequency and Q-values (−3 dB) were read directly from a simulated S11 plot, and Q0-values were calculated by the known equation Q = Q0/(1 + β), where β is the reflection coefficient at the frequency of resonance (β = 1 for critically coupled). EPR signal intensity and resonator efficiency values (mT/W1/2) were calculated using Ansys HFSS (20) and tabulated. EPR experimental comparisons were performed on an Elexsys E580 X-band bridge by Bruker Biospin. Four resonators were used for this comparison. The Bruker Biospin (i) dielectric ER4118X-MD-5 W1 (MD5W1) and (ii) LGR ER4118X-MS-3 W1 (MS3; split-ring) resonators were used as comparisons with known commercial resonator geometries. Two Ω-shaped 0.5-mm–inner diameter PMR resonators were also tested. The first used (iii) Rogers 6010LM (RO6010LM printed circuit board; Rogers Corporation, Chandler, AZ, USA) substrate as per (11–13) and one with (iv) sapphire substrate. The sapphire substrate was fabricated by Technical University Ilmenau (Ilmenau, Germany). Both PMR geometries had a 0.5 mm hole through the substrate. Resonator characteristics are found in table S1.
The EPR experiments performed in this work were as follows. The continuous-wave EPR experiment measures the sample using a constant microwave power incident on the sample and slowly sweeping a quasi-static magnetic field through the resonance condition, ν = γB0, where ν is the operating frequency (nominally 9.5 GHz for X-band), γ is the gyromagnetic ratio of the spin system (γ = 2.8 MHz/G for a free electron), and B0 is the quasi-static magnetic field. The magnetic field was modulated, typically at 100 kHz, and collected using a phase-sensitive detector. Continuous wave was the standard EPR experiment. With modern digital signal processing and fast analog-to-digital converters, the continuous-wave experiment has recently been improved upon. The NARS (35, 41) and adiabatic rapid scan (34, 42) methods collect real and imaginary, pure-absorption and pure-dispersion, EPR spectra using fast quasi-static magnetic field or microwave frequency sweeps without the need for a phase-sensitive detector. Both rapid scan data can be pseudo-modulated to the conventional first-derivative EPR spectrum using an MDIFF pseudo-modulation (35). The NARS experiment uses a field sweep fast enough to overcome 1/f noise but remains in a thermal equilibrium, while adiabatic rapid scan sweeps the field fast enough to cause passage. The advantage of NARS is the signal-to-noise improvement of collecting pure-absorption EPR spectra, while adiabatic rapid scan can further improve the continuous-wave and NARS experiment by changing the effective microwave magnetic field at the sample. This allows for an increase of microwave power and, thus, increase in EPR signal for saturable signals (42). While the NARS method can be implemented on commercial bridges with no hardware changes, it does require some technical expertise (34). However, to perform adiabatic rapid scan experiments on protein samples, custom current drivers are needed to increase the swept field amplitude. For simplicity, in this work, we implemented only NARS. Other experimental parameters are as stated in the manuscript.
The field-swept two-pulse ESE experiment used a π/2 − τ − π pulse sequence (π is 80 ns) that resulted in an echo τ seconds; herein, τ is 300 ns, after the π pulse. The field was stepped, and a whole spectrum was acquired. This is the standard pulse sequence to collect an EPR spectrum (7). The HYSCORE experiment performed in this work has a π/2 − τ − π/2 − t1 − π − t2 − π/2 pulse sequence, which resulted in an echo τ seconds after the last π/2 pulse. The values for t1 and t2 were swept to form a 2D experiment at a fixed magnetic field position. Here, the magnetic field was set to one of the peaks measured in the two-pulse ESE experiment: τ is 280 ns, t1 and t2 start at 300 ns with 256 48 ns steps, and π is 80 ns.
Fabrication techniques
The microhelix was fabricated by hand winding 5 to 8 turns of 0.125 mm diameter silver wire with polytetrafluoroethylene (PTFE) coating (0.0255-mm thickness, total 0.18 mm diameter; Science Products GmbH, Hofheim, Germany) around a 0.4 mm drill bit and placed inside a Rexolite cylinder (0.8 mm inner diameter and 1.2 mm outer diameter) with a length of 10 mm. The drill bit was removed as the coil was affixed with super glue by capillary action, waiting for 1 min and blowing out the excess. The assembly was left to dry for several days.
In the current setup, 6.5 turns are necessary to obtain a helix that has a resonant frequency around 10 GHz without sample and 9.8 GHz with a 0.4 mm outer diameter and 0.3 mm inner diameter quartz capillary. The microhelix is reproducible within 500 MHz (9.3 to 9.8 GHz). Further reduction of the variability is feasible with the use of an assembly jig. The resonator withstands many freeze-thaw cycles and is quite robust.
The coupling loop was designed in Ansys HFSS and prepared for fabrication in AutoDesk Inventor Professional 2019. The printed circuit board designs were emailed to Streamline Circuit (Santa Clara, CA, USA) engineers and manufactured on a PTFE substrate. The printed circuit board was connected to the bridge by a high-frequency SubMiniature version A (SMA) end launcher (AmphenolRF, 901-10510-1). Impedance matching was achieved by moving the microhelix relative to the coupling loop until critically coupled on a network analyzer. Fine-tune matching was obtained with a slide-screw tuner at the bridge output. Bench tests of resonator characteristics—such as the frequency measurements, Q0-value, and sample frequency shifts—were performed on an Agilent 8722ES (now Keysight Technologies, Santa Rosa, CA, USA) vector network analyzer.
Sample preparation
The photosystem II complex sample from spinach was prepared by following the BBY method and placed in a 0.4 mm outer diameter capillary with a 0.3 mm inner diameter (32). The tyrosine D () radical was generated by ambient light at room temperature for a few minutes and then rapidly frozen in liquid nitrogen. The radical is well suited as a biological test sample since it has been extensively studied and the g- and hyperfine tensors are known (5). Using ultraviolet-visible (UV-VIS) spectroscopy, the number of chlorophyll molecules in the sample can be determined, and taking into account that there are approximately 250 chlorophyll molecules per photosystem II complex in spinach, the approximate amount of enzyme can be calculated. Each photosystem II complex contains one radical. For the sample prepared in this work, chlorophyll molecules (7.9 μM/ml) were measured, resulting in approximately 1.6 × 1012 radicals in the 85 nl that fill the microhelix.
In addition, the photosystem II core complex, extracted and purified from the thermophilic cyanobacterium Thermosynechococcus elongatus, was crystallized to reach a crystal size of 0.3 mm by 0.18 mm by 0.18 mm, using the method of Kern et al. (33). The crystals were gradually transferred to a cryogenic protection buffer [100 mM MES (pH 6.5), 5 mM CaCl2, 30% (w/w) glycerol, and 16% polyethylene glycol 2000]. The photosystem II core complex forms an asymmetric unit, and the crystal has a unit cell space group symmetry P21 21 21, which generates four sites per unit cell (PDB ID: 1W5C). From this, one can calculate that there are approximately 8.9 × 1012 radicals in the test crystal. The active center and the of a cyanobacterium were equivalent to that of the photosystem II from spinach.
Last, [FeFe]-hydrogenase from C. pasteurianum (CpI) was grown and crystallized to dimensions of 0.3 mm by 0.1 mm by 0.1 mm by the method of Esselborn et al. (43) under auto-oxidative conditions, i.e., without reducing agents. This leaves the enzyme in the characteristic active oxidized state (Hox), giving rise to an S = 1/2 ground state of the H-cluster. The accessory iron-sulfur clusters in the protein were oxidized and remained EPR silent (16). The [FeFe]-hydrogenase crystal has a space group symmetry P1211 with two asymmetric units in two sites per unit cell (PDB ID: 4XDC). With this information, we can calculate that there are approximately 17 × 1012 single enzymes within the crystal, with each peak corresponding to 4.25 × 1012 spins.
Supplementary Material
Acknowledgments
We thank M. Chrysina from the Max Planck Institute for Chemical Energy Conversion for guidance on photosystem II spectra and M. Reus for the photosystem II BBY prepared sample and the UV-VIS measurements used in this work. Funding: The research reported in this publication was supported by funding from the European Union Horizon 2020 Marie Skłodowska-Curie Fellowship (no. 745702; ACT-EPR, https://act-epr.org), the Max Planck Society, and Sonderforschungsbereich Sfb1078 by Humboldt Universität zu Berlin, Project A5 (R.H. and A.Z.), Cluster of Excellence-2033 RESOLV #390677874 (Deutsche Forschungsgemeinschaft, DFG), DFG Research Training Group GRK 2341 Microbial Substrate Conversion (MiCon), and Volkswagen Stiftung (Design of [FeS] cluster containing Metallo-DNAzymes [Az 93412]) (M.W. and T.H.). Author contributions: J.W.S., E.J.R., and W.L. conceived the project. J.W.S. designed and fabricated the self-resonance microhelix, executed the project, and performed the measurements. R.H. and A.Z. fabricated photosystem II crystals from T. elongatus. J.D., M.W., and T.H. fabricated [FeFe]-hydrogenase crystals from C. pasteurianum (CpI) in the Hox state. D.S., A.S., W.L., and E.J.R. oversaw the project. J.W.S., W.L., and E.J.R. wrote the manuscript. All authors discussed and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. MATLAB code and data can be found at https://act-epr.org/data. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaay1394/DC1
EPR characteristics comparison of multiple resonators
Power saturation and Λave measurements
Sensitivity comparison of the X-band microhelix to a commercial dielectric resonator
Qualitative sensitivity comparison of the X-band microhelix to high-frequency single-mode resonators
Crystal rotation and simulation
Fig. S1. Dimensions and geometry of the four resonators compared in this paper.
Fig. S2. Ansys HFSS finite-element modeling simulation of the microwave magnetic fields comparing the PMR and microhelix.
Fig. S3. Power saturation curve of LiPC using various resonators.
Fig. S4. Continuous-wave EPR of frozen solution photosystem II BBY particles performed in the Bruker MD5W1 dielectric resonator at a temperature of 80 K.
Fig. S5. Comparison of the g-tensor proposed by Adamska-Venkatesh et al. (38) and the current proposed g-tensor from this work.
Table S1. Resonator characteristics calculated and measured.
Table S2. Rotational matrices for the crystal frame with respect to the laboratory frame and the g-tensor with respect to the molecular frame.
REFERENCES AND NOTES
- 1.Bowman S. E. J., Bridwell-Rabb J., Drennan C. L., Metalloprotein crystallography: More than a structure. Acc. Chem. Res. 49, 695–702 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lubitz W., Reijerse E., van Gastel M., [NiFe] and [FeFe] hydrogenases studied by advanced magnetic resonance techniques. Chem. Rev. 107, 4331–4365 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Hussein R., Ibrahim M., Chatterjee R., Coates L., Müh F., Yachandra V. K., Yano J., Kern J., Dobbek H., Zouni A., Optimizing crystal size of photosystem II by macroseeding: Toward neutron protein crystallography. Cryst. Growth Des. 18, 85–94 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.F. Neese, Quantum chemistry and EPR parameters, in eMagRes (John Wiley & Sons, 2017), vol. 6, pp. 1–22. [Google Scholar]
- 5.Hofbauer W., Zouni A., Bittl R., Kern J., Orth P., Lendzian F., Fromme P., Witt H. T., Lubitz W., Photosystem II single crystals studied by EPR spectroscopy at 94 GHz: The tyrosine radical . Proc. Natl. Acad. Sci. U.S.A. 98, 6623–6628 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klette R., Törring J. T., Plato M., Möbius K., Boenigk B., Lubitz W., Determination of the g tensor of the primary donor cation radical in single crystals of rhodobacter sphaeroides R-26 reaction centers by 3-mm high-field EPR. J. Phys. Chem. 97, 2015–2020 (1993). [Google Scholar]
- 7.A. Schweiger, G. Jeschke, Principles of Pulse Electron Paramagnetic Resonance (Oxford Univ. Press, 2001). [Google Scholar]
- 8.J. S. Hyde, W. Froncisz, in Advanced EPR: Applications in Biology and Biochemistry, A. J. Hoff, Ed. (Elsevier, 1989), pp. 277–306. [Google Scholar]
- 9.Friedländer S., Petkov P. S., Bolling F., Kultaeva A., Böhlmann W., Ovchar O., Belous A. G., Heine T., Pöppl A., Continuous-wave single-crystal electron paramagnetic resonance of adsorption of gases to cupric ions in the Zn(II)-doped porous coordination polymer Cu2.965Zn0.035(btc)2. J. Phys. Chem. C 120, 27399–27411 (2016). [Google Scholar]
- 10.Mett R. R., Sidabras J. W., Hyde J. S., MRI surface-coil pair with strong inductive coupling. Rev. Sci. Instrum. 87, 124704 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Narkowicz R., Suter D., Stonies R., Planar microresonators for EPR experiments. J. Magn. Reson. 175, 275–284 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Narkowicz R., Suter D., Niemeyer I., Scaling of sensitivity and efficiency in planar microresonators for electron spin resonance. Rev. Sci. Instrum. 79, 084702 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Narkowicz R., Suter D., Tuner and radiation shield for planar electron paramagnetic resonance microresonators. Rev. Sci. Instrum. 86, 024701 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Holm R. H., Solomon E. I., Introduction: Bioinorganic enzymology II. Chem. Rev. 114, 3367–3368 (2014). [DOI] [PubMed] [Google Scholar]
- 15.R. Schlögl, A. Aho, M. Antonietti, S. Arndt, M. Behrens, E. Bill, A. Brandner, G. Centi, P. Claus, N. Cox, S. DeBeer, N. DeMartini, K. Doblhofer, T. Franzke, H.-J. Freund, M. Gastel, J.-D. Grunwaldt, G. Hofmann, M. Hupa, K. Kähler, E. Kunkes, J. Loosdrecht, W. Lubitz, J. Maier, D. Menzel, M. Muhler, D. Y. Murzin, F. Neese, J. W. Niemantsverdriet, N. Nilius, R. Palkovits, D. A. Pantazis, S. Perathoner, T. Petrenko, J. Rossmeisl, D. Samuelis, R. Schlögl, R. Schomäcker, F. Schüth, S. Shaikhutdinov, M. Sterrer, P. Strasser, A. Trunschke, W. R. H. Wright, S. Ye, Chemical Energy Storage (De Gruyter Textbook, De Gruyter, 2012). [Google Scholar]
- 16.Lubitz W., Ogata H., Rüdiger O., Reijerse E., Hydrogenases. Chem. Rev. 114, 4081–4148 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Sommer C., Adamska-Venkatesh A., Pawlak K., Birrell J. A., Rüdiger O., Reijerse E. J., Lubitz W., Proton coupled electronic rearrangement within the H-cluster as an essential step in the catalytic cycle of [FeFe] hydrogenases. J. Am. Chem. Soc. 139, 1440–1443 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Silakov A., Wenk B., Reijerse E., Lubitz W., 14N HYSCORE investigation of the H-cluster of [FeFe] hydrogenase: Evidence for a nitrogen in the dithiol bridge. Phys. Chem. Chem. Phys. 11, 6592–6599 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Feher G., Sensitivity considerations in microwave paramagnetic resonance absorption techniques. Bell Syst. Tech. J. 36, 449–484 (1957). [Google Scholar]
- 20.J. S. Hyde, J. W. Sidabras, R. R. Mett, in Multifrequency Electron Paramagnetic Resonance: Theory and Applications, S. K. Misra, Ed. (Wiley-VCH, 2011), pp. 244–269. [Google Scholar]
- 21.E. Reijerse, A. Savitsky, Electron paramagnetic resonance instrumentation, in eMagRes (John Wiley & Sons, 2017), vol. 6, pp. 187–206. [Google Scholar]
- 22.Friedländer S., Šimėnas M., Kobalz M., Eckold P., Ovchar O., Belous A. G., Banys J., Krautscheid H., Pöppl A., Single crystal electron paramagnetic resonance with dielectric resonators of mononuclear Cu2+ ions in a metal-organic framework containing Cu2 paddle wheel units. J. Phys. Chem. C 119, 19171–19179 (2015). [Google Scholar]
- 23.Webb R. H., Use of traveling wave helices in ESR and double resonance spectrometers. Rev. Sci. Instrum. 33, 732–737 (1962). [Google Scholar]
- 24.Collingwood J. C., White J. W., Helical resonators for spin resonance spectroscopy. J. Sci. Instrum. 44, 509 (1967). [Google Scholar]
- 25.Adkins L. R., Nolle A. W., EPR spectrometer utilizing traveling wave helix for 2 Gc region. Rev. Sci. Instrum. 37, 1404–1405 (1966). [Google Scholar]
- 26.Mahdjour H., Clark W. G., Baberschke K., High-sensitivity broadband microwave spectroscopy with small nonresonant coils. Rev. Sci. Instrum. 57, 1100–1106 (1986). [Google Scholar]
- 27.Kentgens A. P. M., Bart J., van Bentum P. J. M., Brinkmann A., van Eck E. R. H., Gardeniers J. G. E., Janssen J. W. G., Knijn P., Vasa S., Verkuijlen M. H. W., High-resolution liquid- and solid-state nuclear magnetic resonance of nanoliter sample volumes using microcoil detectors. J. Chem. Phys. 128, 052202 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Jones C. J., Larive C. K., Could smaller really be better? Current and future trends in high-resolution microcoil NMR spectroscopy. Anal. Bioanal. Chem. 402, 61–68 (2012). [DOI] [PubMed] [Google Scholar]
- 29.Mompeán M., Sánchez-Donoso R. M., de la Hoz A., Saggiomo V., Velders A. H., Gomez M. V., Pushing nuclear magnetic resonance sensitivity limits with microfluidics and photo-chemically induced dynamic nuclear polarization. Nat. Commun. 9, 108 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Styring S., Sjöholm J., Mamedov F., Two tyrosines that changed the world: Interfacing the oxidizing power of photochemistry to water splitting in photosystem II. Biochim. Biophys. Acta Bioenerg. 1817, 76–87 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Saito K., Rutherford A. W., Ishikita H., Mechanism of tyrosine D oxidation in photosystem II. Proc. Natl. Acad. Sci. U.S.A. 110, 7690–7695 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berthold D. A., Babcock G. T., Yocum C. F., A highly resolved, oxygen-evolving photosystem II preparation from spinach thylakoid membranes: EPR and electron-transport properties. FEBS Lett. 134, 231–234 (1981). [Google Scholar]
- 33.Kern J., Loll B., Lüneberg C., Di Fiore D., Biesiadka J., Irrgang K.-D., Zouni A., Purification, characterisation and crystallisation of photosystem II from Thermosynechococcus elongatus cultivated in a new type of photobioreactor. Biochim. Biophys. Acta Bioenerg. 1706, 147–157 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Möser J., Lips K., Tseytlin M., Eaton G. R., Eaton S. S., Schnegg A., Using rapid-scan EPR to improve the detection limit of quantitative EPR by more than one order of magnitude. J. Magn. Reson. 281, 17–25 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hyde J. S., Bennett B., Kittell A. W., Kowalski J. M., Sidabras J. W., Moving difference (MDIFF) non-adiabatic rapid sweep (NARS) EPR of copper(II). J. Magn. Reson. 236, 15–25 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stoll S., Schweiger A., EasySpin, a comprehensive software package for spectral simulation and analysis in epr. J. Magn. Reson. 178, 42–55 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Greco C., Silakov A., Bruschi M., Ryde U., De Gioia L., Lubitz W., Magnetic properties of [FeFe]-hydrogenases: A theoretical investigation based on extended QM and QM/MM models of the H-cluster and its surroundings. Eur. J. Inorg. Chem. 2011, 1043–1049 (2011). [Google Scholar]
- 38.Adamska-Venkatesh A., Simmons T. R., Siebel J. F., Artero V., Fontecave M., Reijerse E., Lubitz W., Artificially maturated [FeFe] hydrogenase from Chlamydomonas reinhardtii: A HYSCORE and ENDOR study of a non-natural H-cluster. Phys. Chem. Chem. Phys. 17, 5421–5430 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Adamska-Venkatesh A., Roy S., Siebel J. F., Simmons T. R., Fontecave M., Artero V., Reijerse E., Lubitz W., Spectroscopic characterization of the bridging amine in the active site of [FeFe] hydrogenase using isotopologues of the H-cluster. J. Am. Chem. Soc. 137, 12744–12747 (2015). [DOI] [PubMed] [Google Scholar]
- 40.M. K. Bowman, H. Chen, A. G. Maryasov, Fourier-transform EPR, in eMagRes (John Wiley & Sons, 2017), vol. 6, pp. 387–406. [Google Scholar]
- 41.Kittell A. W., Camenisch T. G., Ratke J. J., Sidabras J. W., Hyde J. S., Detection of undistorted continuous wave (CW) electron paramagnetic resonance (EPR) spectra with non-adiabatic rapid sweep (NARS) of the magnetic field. J. Magn. Reson. 211, 228–233 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Joshi J. P., Ballard J. R., Rinard G. A., Quine R. W., Eaton S. S., Eaton G. R., Rapid-scan EPR with triangular scans and Fourier deconvolution to recover the slow-scan spectrum. J. Magn. Reson. 175, 44–51 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Esselborn J., Muraki N., Klein K., Engelbrecht V., Metzler-Nolte N., Apfel U.-P., Hofmann E., Kurisu G., Happe T., A structural view of synthetic cofactor integration into [FeFe]-hydrogenases. Chem. Sci. 7, 959–968 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Narkowicz R., Ogata H., Reijerse E., Suter D., A cryogenic receiver for EPR. J. Magn. Reson. 237, 79–84 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Torrezan A. C., Mayer Alegre T. P., Medeiros-Ribeiro G., Microstrip resonators for electron paramagnetic resonance experiments. Rev. Sci. Instrum. 80, 075111 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Ghirri A., Bonizzoni C., Righi M., Fedele F., Timco G., Winpenny R., Affronte M., Microstrip resonators and broadband lines for X-band EPR spectroscopy of molecular nanomagnets. Appl. Magn. Reson. 46, 749–756 (2015). [Google Scholar]
- 47.Twig Y., Dikarov E., Blank A., Ultra miniature resonators for electron spin resonance: Sensitivity analysis, design and construction methods, and potential applications. Mol. Phys. 111, 2674–2682 (2013). [Google Scholar]
- 48.Twig Y., Suhovoy E., Blank A., Sensitive surface loop-gap microresonators for electron spin resonance. Rev. Sci. Instrum. 81, 104703 (2010). [DOI] [PubMed] [Google Scholar]
- 49.Walsh W. M., Rupp L. W., Enhanced ESR sensitivity using a dielectric resonator. Rev. Sci. Instrum. 57, 2278–2279 (1986). [Google Scholar]
- 50.Golovina I., Geifman I., Belous A., New ceramic EPR resonators with high dielectric permittivity. J. Magn. Reson. 195, 52–59 (2008). [DOI] [PubMed] [Google Scholar]
- 51.Liu K. J., Gast P., Moussavi M., Norby S. W., Vahidi N., Walczak T., Wu M., Swartz H. M., Lithium phthalocyanine: A probe for electron paramagnetic resonance oximetry in viable biological systems. Proc. Natl. Acad. Sci. U.S.A. 90, 5438–5442 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiang J. H., Wu D. L., Ice and water permittivities for millimeter and sub-millimeter remote sensing applications. Atmos. Sci. Lett. 5, 146–151 (2004). [Google Scholar]
- 53.A. Oppenheim, R. Schafer, J. Buck, Discrete-Time Signal Processing (Pearson Education Signal Processing Series, Pearson Education, 1999). [Google Scholar]
- 54.G. Eaton, S. Eaton, D. Barr, R. Weber, Quantitative EPR (Springer, 2010). [Google Scholar]
- 55.I. Gromov, P. Höfer, EUROMAR 2013, Crete, Greece (2013).
- 56.Teutloff C., Pudollek S., Keßen S., Broser M., Zouni A., Bittl R., Electronic structure of the tyrosine D radical and the water-splitting complex from pulsed ENDOR spectroscopy on photosystem II single crystals. Phys. Chem. Chem. Phys. 11, 6715–6726 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaay1394/DC1
EPR characteristics comparison of multiple resonators
Power saturation and Λave measurements
Sensitivity comparison of the X-band microhelix to a commercial dielectric resonator
Qualitative sensitivity comparison of the X-band microhelix to high-frequency single-mode resonators
Crystal rotation and simulation
Fig. S1. Dimensions and geometry of the four resonators compared in this paper.
Fig. S2. Ansys HFSS finite-element modeling simulation of the microwave magnetic fields comparing the PMR and microhelix.
Fig. S3. Power saturation curve of LiPC using various resonators.
Fig. S4. Continuous-wave EPR of frozen solution photosystem II BBY particles performed in the Bruker MD5W1 dielectric resonator at a temperature of 80 K.
Fig. S5. Comparison of the g-tensor proposed by Adamska-Venkatesh et al. (38) and the current proposed g-tensor from this work.
Table S1. Resonator characteristics calculated and measured.
Table S2. Rotational matrices for the crystal frame with respect to the laboratory frame and the g-tensor with respect to the molecular frame.