

Abstract
Selective inhibitors of Cryptosporidium Calcium-Dependent Protein Kinase 1 (CpCDPK1) based on the 1H-pyrazolo[3,4-d]pyrimidin-4-amine (pyrazolopyrimidine, PP) scaffold are effective in both in vitro and in vivo models of cryptosporidiosis. However, the search for distinct safety and pharmacokinetic (PK) properties has motivated our exploration of alternative scaffolds. Here, we describe a series of 7H-pyrrolo[2,3-d]pyrimidin-4-amine (pyrrolopyrimidine, PrP)-based analogs of PP CpCDPK1 inhibitors. Most of the PrP-based inhibitors described potently inhibit the CpCDPK1 enzyme, demonstrate no toxicity against mammalian cells, and block proliferation of the C. parvum parasite in the low micromolar range. Interestingly, certain substituents that show reduced CpCDPK1 potency when displayed from a PP scaffold provided notably enhanced efficacy in the context of a PrP scaffold. PK studies on these paired compounds show that some PrP analogs have distinct physiochemical properties compared with their PP counterparts. These results demonstrate that inhibitors based on a PrP scaffold are distinct therapeutic alternatives to previously developed PP inhibitors.
Keywords: Cryptosporidium parvum; Calcium-Dependent Protein Kinase 1 (CpCDPK1) inhibitors; 7H-pyrrolo[2,3-d]pyrimidin-4-amine; 1H-pyrazolo[3,4-d]pyrimidin-4-amine
Multiple studies have reported significant mortality and morbidity in children less than 2 years old due to Cryptosporidium-induced diarrhea, especially in resource limited regions of the world.1–3 Nonetheless, nitazoxanide is the only Food and Drug Administration (FDA) approved drug for treatment.1 Nitazoxanide has shown minimal efficacy in immunocompromised patients and malnourished children4 and is not approved or recommended for use in children under the age of one year.5 Hence, the public health benefit of more potent and safe treatment options cannot be overemphasized.1 Calcium dependent protein kinase 1 (CDPK1) has been reported as an essential enzyme for the survival of Cryptosporidium parasites and is an attractive molecular target for anti-Cryptosporidium drug development programs.6–8 Selective targeting of Cryptosporidium parvum CDPK1 (CpCDPK1) with “bumped kinase inhibitors” (BKIs) based on 1H-pyrazolo[3,4-d]pyrimidin-4-amine (pyrazolopyrimidine, PP) and 5-aminopyrazole-4-carboxamide (AC) chemical scaffolds blocks parasite cell proliferation in both in vitro and in vivo models of cryptosporidiosis.6–10 Partial knockdown of CpCDPK1 with small interfering RNAs (siRNAs) led to significantly decreased C. parvum parasite invasion and growth. Moreover, partial CpCDPK1 siRNA knockdown in combination with treatment of either BKIs 1294 (PP) or 1517 (AC) produced further inhibitory effects on the proliferation of C. parvum parasites.8 The observation that the EC50 values of BKIs were reduced proportionally with the extent of siRNA knockdown supports the validation of CpCDPK1 as the target of BKI mediated C. parvum growth inhibition for compound BKI-1294.
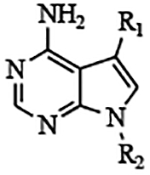
Iterative improvements for safety and efficacy of the PP scaffold inhibitors have resulted in pre-clinical leads that are advancing toward human and animal treatment of cryptosporidiosis.11–12 However, desired improvements in safety and efficacy require an array of CpCDPK1 inhibitors that have varying physiochemical properties. Therefore, we have explored how small variations in inhibitor structure affect their in vitro and in vivo properties. Surprisingly, we observed that changing the core scaffold to 7H-pyrrolo[2,3-d]pyrimidin-4-amine (pyrrolopyrimidine, PrP) while keeping previously explored substituents of the pyrazolopyrimidine scaffold (Figure 1) can lead to significant changes in biochemical, anti-parasitic, and pharmacokinetic (PK) properties in some of the derivative analogues. BKI-1649 is a previously described compound, which is based on a PrP scaffold instead of a PP scaffold, like most BKIs11–12. PrP BKI-1649 (compound 13) was shown to block the activity of CpCDPK111–12, prevent the growth and proliferation of C. parvum in vitro, and reduce oocyst shedding in a mouse model of cryptosporidiosis. In this study, we compared other PrP-based inhibitors to their PP-BKI analogues, for activity against CpCDPK1 and inhibition of C. parvum growth in cell culture and demonstrated that PrP-based compounds often retain potency against parasites and low toxicities to mammalian cells, while demonstrating distinct PK properties.
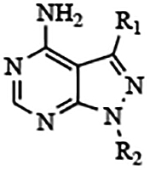
Figure 1. Pyrazolopyrimidine and pyrrolopyrimidine scaffolds.

Previously explored functional groups of the 1H-pyrazolo[3,4-d]pyrimidin-4-amine (Pyrazolopyrimidine, PP) based bumped kinase inhibitors that included both potent and non-potent inhibitors of CpCDPK1 and TgCDPK1 were used to generate analogues based on the 7H-pyrrolo[2,3-d]pyrimidin-4-amine (pyrrolopyrimidine, PrP) core. The PrP derivatives are potent inhibitors of CpCDPK1 and TgCDPK1.
PP-based inhibitors of CpCDPK1
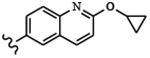
For compounds based on the PP scaffold, we previously demonstrated that analogues possessing 6-alkoxynaphthalen-2-yl groups at the R1 position in combination with branched R2 substituents linked to the scaffold through a methylene group are highly potent and selective inhibitors of TgCDPK1.13 In particular, inhibitors that contain a 2-methylpropan-2-ol group at the R2 position were found to be advantageous because they possess favorable solubility and pharmacokinetic properties and reduced hERG activity. To explore the effectiveness of inhibitors of this PP scaffold against CpCDPK1, inhibitors 6-12 were synthesized (for synthesis scheme and spectroscopic data: see Supplementary Information) from a common PP intermediate. Compounds 6-10 have a 2-methylpropan-2-ol as R2 groups at the N-1 position, and either 6-alkoxynaphthalen-2-yl or 2-alkoxyquinolin-6-yl moieties as the R1 group (C-3 position). We also explored the importance of the tertiary alcohols of inhibitors 6-10 by generating O-methylated analogs of 6 and 7–inhibitors 11 and 12.
All inhibitors were tested for their ability to inhibit recombinant CpCDPK1 in an in vitro activity assay. As counter-screens all inhibitors were tested for inhibition of the mammalian tyrosine kinase SRC–a common off-target of similar PP-based inhibitors–and for cytotoxicity against CRL-8155 (human lymphoblasts) and HepG2 (human hepatocellular carcinoma) cells. Similar to TgCDPK1, inhibitors 6-9 demonstrated highly potent inhibition (IC50s = 2–19 nM) of CpCDPK1. Interestingly, inhibitor 10, which contains a 2-difluoromethoxyquinolin-6-yl group, is also potent against CpCDPK1 (IC50 = 4 nM) but >10-fold less so against TgCDPK1. This represents a rare case of an inhibitor showing significant selectivity for CpCDPK1 over TgCDPK1. The most surprising results from this series were obtained with inhibitors 11 and 12. Despite only containing an extra methyl group at the R2 position, which is directed towards the ribose-binding pocket, inhibitors 11 and 12 are ~200–600-fold less potent against CpCDPK1 than 6 and 7. Inhibitors 6-12 showed no inhibition of SRC kinase at the highest concentration tested and no cytotoxicity against CRL-8155 and HepG2 cells.
PrP-based inhibitors of CpCDPK1
We next explored whether similar trends would be observed for the same substituents displayed from an alternative, isosteric PrP scaffold that contains only one atom difference from the parent PP. Such isosteric swaps have previously been successful in tuning the physiochemical properties of different kinase inhibitor series15 while maintaining overall potency and selectivity against the kinase being targeted. Both the PP and PrP scaffolds maintain the same hydrogen-bonding pattern with the hinge region of kinases. Furthermore, both scaffolds project substituents from the same region of the scaffold and would be expected to make the same contacts with different regions of the ATP-binding pocket. Inhibitors 13-19 were synthesized from a common PrP intermediate (for synthesis scheme, spectroscopic data see Supplementary Information).
PrP inhibitors 13, 14, and 16 demonstrate almost identical IC50s against CpCDPK1 as their PP analogues, with 15 showing a small decrease in potency relative to 8. Like inhibitor 10, 17 is selective for CpCDPK1 over TgCDPK1–demonstrating increased potency for both enzymes. Strikingly, PrP inhibitors 18 and 19 are significantly more potent against CpCDPK1 than their PP analogues, inhibitors 11 and 12. It is further noted that 18 retains activity against Cryptosporidium parasites with EC50s of <2 μM, whereas 11 and 12 have Cryptosporidium EC50s of >5 μM, correlating with the differences in activity against CpCDPK1. This is further correlative evidence that BKIs act to inhibit Cryptosporidium replication through the CpCDPK1 target.
Inhibitors 18 and 19 retain the lack of inhibition of SRC, so appear to maintain selectivity, while regaining lost activity of 11 and 12 against CpCDPK1. However, 18 loses some specificity compared with 11, when selectivity is evaluated on a kinomewide scale (see below).
We hypothesize that the significant difference in IC50 observed for some paired PP and PrP compounds is at least partially explained as a structural consequence of combining two features, the single atom difference (N→C) in their respective scaffolds (Figure 1), and the extension of the R2 substituent by replacing -OH with -OMe (compare compounds from Table 1 with Table 2).
Table 1.
Enzymatic CpCDPK1, TgCDPK1, SRC inhibition (IC50), Toxicity (CC50) against CRL-8155 and HepG2, and C. parvum cellular assay (EC50) results for pyrazolopyrimidine (PP) compounds (6-12).
 |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| BKI | Entry | R2 | R1 | IC50 (μM) | Toxicity CC50 (μM) | EC50 (μM) | Solubility (μM) | ||||
| CpCDPK1 | TgCDPK1 | SRC | CRL-8155 | HEPG2 | C. parvum | pH 2.0 | pH 6.5 | ||||
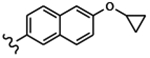
| 1561 | 6 | 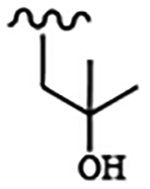 |
 |
0.0032 | 0.0020 | >10.0 | >40.0 | >40.0 | 0.464 | >100.0 | 87.1 |
| 1553 | 7 | 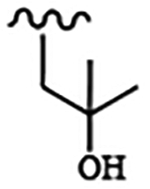 |
 |
0.0021 | 0.0010 | >10.0 | >40.0 | >40.0 | 1.63 | >95.0 | 50.0 |
| 1829 | 8 | 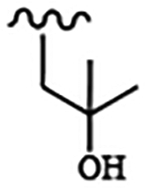 |
 |
0.019 | 0.025 | >10.0 | >80.0 | >40.0 | 0.882 | >100.0 | 98.7 |
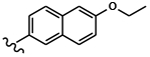
| 1547 | 9 | 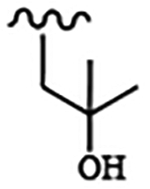 |
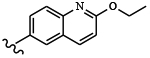 |
0.0030 | 0.0033 | >10.0 | >80.0 | >80.0 | 5.89 | >100.0 | 88.0 |
| 1830 | 10 | 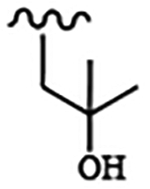 |
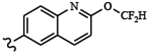 |
0.0040 | 0.058 | >10.0 | >80.0 | >80.0 | 0.286 | 95.1 | 81.0 |
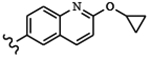
| 1676 | 11 | 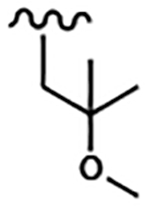 |
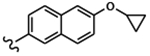 |
1.33 | 1.66 | >10.0 | >80.0 | >80.0 | 5.87 | 99.6 | 52.9 |
| 1677 | 12 | 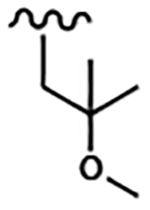 |
 |
0.62 | 0.042 | >10.0 | >80.0 | >80.0 | >20.0 | >100.0 | >100.0 |
Table 2.
Enzymatic CpCDPK1, TgCDPK1, SRC inhibition (IC50), Toxicity (CC50) against CRL-8155 and HepG2, and C. parvum cellular assay (EC50) results for pyrrolopyrimidine (PrP) compounds (13-19).
 |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| BKI | Entry | R2 | R1 | IC50 (μM) | Toxicity CC50 (μM) | EC50 (μM) | Solubility (μM) | ||||
| CpCDPK1 | TgCDPK1 | SRC | CRL-8155 | HEPG2 | C. parvum | pH 2.0 | pH 6.5 | ||||
| 1649 | 13 | 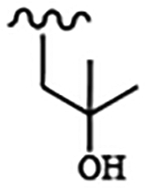 |
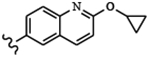 |
0.0022 | 0.0032 | >10.0 | >40.0 | >40.0 | 1.03 | >100.0 | 83.0 |
| 1660 | 14 | 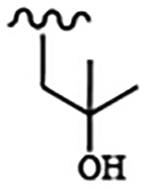 |
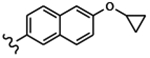 |
0.0016 | 0.0010 | >10.0 | >40.0 | >40.0 | 2.66 | 72.5 | >100.0 |
| 1811 | 15 | 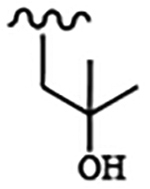 |
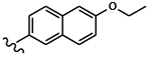 |
0.0496 | 0.0527 | >10.0 | >80.0 | >80.0 | 1.19 | >100.0 | 65.4 |
| 1812 | 16 | 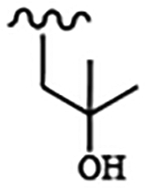 |
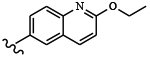 |
0.0025 | 0.011 | >10.0 | >80.0 | >80.0 | 0.52 | >100.0 | 96.5 |
| 1815 | 17 | 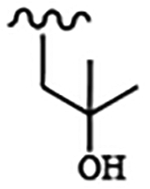 |
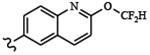 |
0.0009 | 0.013 | >10.0 | >80.0 | >80.0 | 0.67 | 98.2 | 82.2 |
| 1813 | 18 | 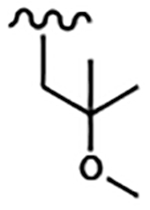 |
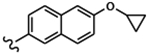 |
0.030 | 0.015 | >10.0 | >80.0 | 43.88 | 1.82 | 74.3 | >100.0 |
| 1814 | 19 | 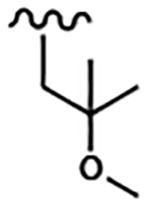 |
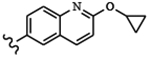 |
0.005 | 0.010 | >10.0 | >80.0 | >80.0 | 1.39 | >100.0 | >100.0 |
In many of the 50+ TgCDPK1 crystal structures obtained in-house for PP scaffold compounds, the PP nitrogen N-2 participates in a polar interaction network linking it via a primary site water to the NZ of Lys 80 and indirectly via secondary site water-water-sidechain hydrogen bonds to Glu 178, Asp 179, Asp 195 (Figure 3). These waters are necessarily labile as they are displaced by the ATP α-phosphate upon substrate binding. In previously observed crystal structures of PP compounds 6 and 7, the R2 group -OH also participates in this network via hydrogen bonding to the primary site water [ref: PDB 4tzr 6bfa]. Substitution of the PP nitrogen N-2 with a carbon in the PrP scaffold is expected to disfavor formation of such a polar interaction network, but by itself this has no observed effect on affinity (top 5 compounds in Table 2). However, a substantial change in affinity occurs when this substitution is combined with extension of R2 by an additional methyl group. Retaining the binding pose seen crystallographically for PP compounds 6 and 7 (e.g. Figures 6b, 6c of Vidadala et al [2016] (Figure 3)13 would place the methyl group in the restricted volume between the PP N-2 and the protein backbone at Gly 60, overlapping the primary water site previously observed. Such placement of the non-polar methyl would be disfavored in the presence of the polar PP N-2, consistent with the 10–100 fold worse IC50 observed for compounds 11, 12 (Table 2) compared to 7, 6 (Table 1). In contrast, placement of the methyl adjacent to the analogous PrP scaffold is not so obviously disfavored and may in fact constitute a preferred binding pose.
Figure 3. Crystal structure of the TgCDPK1∙compound 1553 (7) complex (PDB code 6bfa).

The primary water site bridging the pyrazolopyrimidine (PP) scaffold and the protein is evident in difference electron density at 3σ (green).
Human kinome profiling of 1553 (7), 1676 (11), and 1813 (18)
We next explored whether the trends observed in inhibitor potency against CDPK1 are reflected across the human kinome. To do this, we tested representative PP (7 and 11) and PrP (18) compounds in a competitive, kinobead MS-chemoproteomics assay that allows human kinome-wide profiling of inhibitors.14, 16–17 We have previously used this method to profile the selectivity of compound 7 and several other more selective BKIs.14 To explore whether O-methylation of PP compound 7 diminished its interactions with human kinases like it did with CDPK1, we profiled PP compound 11 (the ether analog of 7) in a label-free kinobead-competition experiment at a high inhibitor concentration (50 μM)–to capture even weak kinase interactors.14 Comparison of the kinases competed by 7 and 11 at 50 μM final concentration are shown in the heat map in Figure 2. At this high concentration, 7 interacts with 16 kinases, including, most prominently, PKD isoforms 1, 2 and 3. Among these, kinases that were competed with high LFQ ratios (log2 ratio >5) are ACVR1B, LIMK1, MAP5K4, PTK6, SLK, STK10, RIPK2, and TGFBR1 (Figure 2). Strikingly, 11 did not interact with any of the 180 quantified human kinases. This indicates that O-methylation abrogates interaction with the ATP-binding pocket of human kinases–even weakly–within the context of the pyrazolopyrimidine scaffold. Interestingly, like it does with Tg/CpCDPK1, the direct PrP analogue of 11 (compound 18) recovers the ability to interact–although fairly weakly–with a few human kinases. Thus, the interdependence of the scaffold and R2 substituent on potency appears to hold for several other kinases.
Figure 2. Kinome target profiling of Cp/TgCDPK1 inhibitors against 180 human protein and lipid kinases.
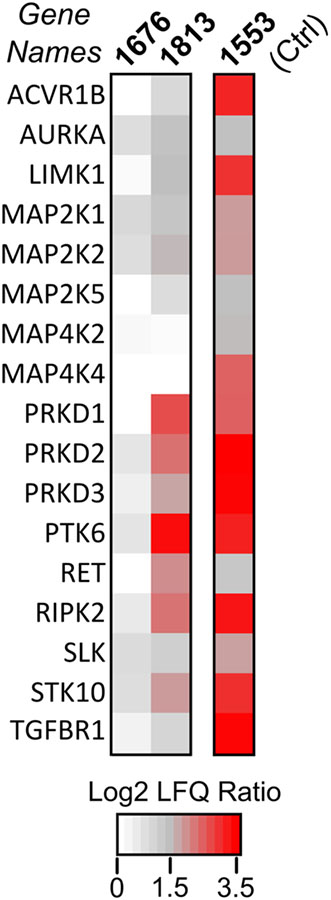
The heat map shows all putative drug−kinase interactions detected in the kinobead-competition assay at 50 Mm of PP compound 1553 (7), PP compound 1676 (11) and PrP compound 1813 (18) in the LFQ master mix. The panel’s color scale indicates the mean log2 LFQ ratios from two replicate LFQ pulldown experiments. A log2 LFQ ratio cut-off of ≥2 (i.e., 4-fold) was apply to distinguish kinase interactors from false positives.
Further characterization of PP versus PrP inhibitors
Oral pharmacokinetic (PK) analysis was performed on 3 of the 7 sets of compounds for both the PP and PrP analogs (Table 3). In two of three cases, the PrP compound showed a comparatively higher Cmax and systemic exposure (AUC) than its PP counterpart, as seen with 14 (27900 min*μmol/L) versus 7 (13700 min*μmol/L), and 16 (8500 min*μmol/L) versus 9 (770 min*μmol/L). Even when scaled down to account for 16’s increased dosage of 25 mg/kg, assuming a linear relationship, PrP compound 16 would have an estimated AUC of 3398 min*μmol/L from a 10 mg/kg dose, over 4 fold higher than its PP equivalent 9. In this regard, it is important to remember that although GI tract exposure is necessary for treatment of Cryptosporidium,11–12, 18 systemic exposure after oral dosing may be vital for treatment of immunocompromised individuals, where the infection can move out of the GI tract.19 Although direct comparative in vivo efficacy analysis of pairs of PP and PrP compounds are beyond the scope of the present study; we hypothesize that experimentally derived properties of the PrP inhibitors of CpCDPK1 are encouraging, but more importantly, provide additional distinct oral PK, safety, and efficacy properties beyond what can be observed with PP compounds alone. Thus, PrP analogs are worth further exploration in parallel with PP analogs, as this approach may allow the discovery of safe therapeutics that are potent, safe, and display appropriate systemic exposure.
Table 3.
Pharmacokinetics of plasma exposure after oral dosing of PP (top of pair) and PrP (bottom of pair) in BALB/c mice.
| Dose (mg/kg) | Tmax (min) | Cmax (μM) | AUC (min*μmol/L) | Half-life (min) | |
|---|---|---|---|---|---|
| 6 | 10 | 560 | 7.8 | 13700 | 1110 |
| 13 | 10 | 400 | 4.9 | 6400 | 900 |
| 7 | 10 | 320 | 12.8 | 13700 | 1190 |
| 14 | 10 | 480 | 24.9 | 27900 | 930 |
| 9 | 10 | 50 | 5.2 | 770 | 110 |
| 16 | 25 | 50 | 28 | 8500 | 100 |
Materials and Methods
The PP analogues were synthesized and characterized with few modification to previously reported procedures (Supplementary Material).11 The potency of the PrP analogues to inhibit recombinant CpCDPK1 and TgCDPK1 enzymes were evaluated as described.20 Expression of recombinant CpCDPK1 and TgCDPK1 protein using auto induction of E. coli strain BL21(DE3) was previously described.6 Inhibitory effects of BKIs on CpCDPK1 and TgCDPK1;20–21 and off-target inhibition of small gatekeeper human tyrosine kinase, SRC, was determined by indirect measurement of changes in initial reaction ATP concentration after phosphorylation of peptide substrate Syntide 2 (PLARTLSVAGLPGKK) and (Ac-EIYGEFKKK) (GenScript, Piscataway, NJ), respectively, via luminescence with Kinaseglo® as previously described.21 Compound inhibitory effect (EC50) on C. parvum cellular growth and proliferation in HCT-8 cells was performed with serial dilutions of test compound in 96 well plates with an initial inoculum size of 1000 oocyst per well as previously described.11–12 Mammalian cell cytotoxicity assays (CC50) of serial dilutions of each compound were performed in HepG2 or CRL-8155 cells for 48 h and growth measured using AlamarBlue® (Life Technologies, USA) assay.13, 22 Pharmacokinetic (PK) analysis of BKIs dosed at 10 or 25 mg/kg body weight in 3% ethanol/7% Tween 80/90% saline by oral gavage of female 10–12 weeks old BALB/c mice was previously described.11 Compounds were extracted from plasma samples in acetonitrile/0.1% formic acid with an internal standard and quantified by LC-MS/MS analysis. PK calculations including time at maximum concentration (Tmax), maximum concentration (Cmax), area under the curve (AUC), and half-life (Table 3) were performed using Pharsight Phoenix WinNonlin software (Certara, St. Louis, MO, USA).11–12 Solubility tests for all inhibitors were determined at pH 2 and pH 6.5 as previously described.10
Supplementary Material
Acknowledgments
The study was supported by the National Institute of Allergy and Infectious Diseases (USA) and National Institute of Child Health and Human Development of the National Institutes of Health (USA) under the award numbers R01AI089441, R01AI111341, R21AI123690, R21CA177402 and R01HD080670.
Footnotes
Animal ethics
All animal experiments conducted at the University of Washington, USA, were approved by the Institutional Animal Care and Use Committees. All animals used in this study were handled in strict accordance with practices to minimize suffering.
SUPPORTING INFORMATION AVAILABLE
One supplemental detail for synthetic procedures, analytical data is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Checkley W; White AC Jr.; Jaganath D; Arrowood MJ; Chalmers RM; Chen XM; Fayer R; Griffiths JK; Guerrant RL; Hedstrom L; Huston CD; Kotloff KL; Kang G; Mead JR; Miller M; Petri WA Jr.; Priest JW; Roos DS; Striepen B; Thompson RC; Ward HD; Van Voorhis WA; Xiao L; Zhu G; Houpt ER, A review of the global burden, novel diagnostics, therapeutics, and vaccine targets for cryptosporidium. Lancet Infect Dis 2015, 15 (1), 85–94. DOI: 10.1016/S1473-3099(14)70772-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shirley DA; Moonah SN; Kotloff KL, Burden of disease from cryptosporidiosis. Curr Opin Infect Dis 2012, 25 (5), 555–63. DOI: 10.1097/QCO.0b013e328357e569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kotloff KL; Nataro JP; Blackwelder WC; Nasrin D; Farag TH; Panchalingam S; Wu Y; Sow SO; Sur D; Breiman RF; Faruque AS; Zaidi AK; Saha D; Alonso PL; Tamboura B; Sanogo D; Onwuchekwa U; Manna B; Ramamurthy T; Kanungo S; Ochieng JB; Omore R; Oundo JO; Hossain A; Das SK; Ahmed S; Qureshi S; Quadri F; Adegbola RA; Antonio M; Hossain MJ; Akinsola A; Mandomando I; Nhampossa T; Acácio S; Biswas K; O’Reilly CE; Mintz ED; Berkeley LY; Muhsen K; Sommerfelt H; Robins-Browne RM; Levine MM, Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 2013, 382 (9888), 209–22. DOI: 10.1016/S0140-6736(13)60844-2. [DOI] [PubMed] [Google Scholar]
- 4.Sparks H; Nair G; Castellanos-Gonzalez A; White AC Jr., Treatment of Cryptosporidium: What We Know, Gaps, and the Way Forward. Curr Trop Med Rep 2015, 2 (3), 181–187. DOI: 10.1007/s40475-015-0056-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fox LM; Saravolatz LD, Nitazoxanide: a new thiazolide antiparasitic agent. Clin Infect Dis 2005, 40 (8), 1173–80. DOI: 10.1086/428839. [DOI] [PubMed] [Google Scholar]
- 6.Murphy RC; Ojo KK; Larson ET; Castellanos-Gonzalez A; Perera BG; Keyloun KR; Kim JE; Bhandari JG; Muller NR; Verlinde CL; White AC; Merritt EA; Van Voorhis WC; Maly DJ, Discovery of Potent and Selective Inhibitors of Calcium-Dependent Protein Kinase 1 (CDPK1) from C. parvum and T. gondii. Acs Med Chem Lett 2010, 1 (7), 331–335. DOI: 10.1021/ml100096t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castellanos-Gonzalez A; White AC; Ojo KK; Vidadala RS; Zhang Z; Reid MC; Fox AM; Keyloun KR; Rivas K; Irani A; Dann SM; Fan E; Maly DJ; Van Voorhis WC, A novel calcium-dependent protein kinase inhibitor as a lead compound for treating cryptosporidiosis. J Infect Dis 2013, 208 (8), 1342–8. DOI: 10.1093/infdis/jit327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Castellanos-Gonzalez A; Sparks H; Nava S; Huang W; Zhang Z; Rivas K; Hulverson MA; Barrett LK; Ojo KK; Fan E; Van Voorhis WC; White AC Jr., A Novel Calcium-Dependent Kinase Inhibitor, Bumped Kinase Inhibitor 1517, Cures Cryptosporidiosis in Immunosuppressed Mice. J Infect Dis 2016, 214 (12), 1850–1855. DOI: 10.1093/infdis/jiw481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schaefer DA; Betzer DP; Smith KD; Millman ZG; Michalski HC; Menchaca SE; Zambriski JA; Ojo KK; Hulverson MA; Arnold SL; Rivas KL; Vidadala RS; Huang W; Barrett LK; Maly DJ; Fan E; Van Voorhis WC; Riggs MW, Novel Bumped Kinase Inhibitors Are Safe and Effective Therapeutics in the Calf Clinical Model for Cryptosporidiosis. J Infect Dis 2016, 214 (12), 1856–1864. DOI: 10.1093/infdis/jiw488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang W; Choi R; Hulverson MA; Zhang Z; McCloskey MC; Schaefer DA; Whitman GR; Barrett LK; Vidadala RSR; Riggs MW; Maly DJ; Van Voorhis WC; Ojo KK; Fan E, 5-Aminopyrazole-4-Carboxamide-Based Compounds Prevent the Growth of Cryptosporidium parvum. Antimicrob Agents Chemother 2017, 61 (8). DOI: 10.1128/AAC.00020-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hulverson MA; Vinayak S; Choi R; Schaefer DA; Castellanos-Gonzalez A; Vidadala RSR; Brooks CF; Herbert GT; Betzer DP; Whitman GR; Sparks HN; Arnold SLM; Rivas KL; Barrett LK; White AC Jr.; Maly DJ; Riggs MW; Striepen B; Van Voorhis WC; Ojo KK, Bumped-Kinase Inhibitors for Cryptosporidiosis Therapy. J Infect Dis 2017, 215 (8), 1275–1284. DOI: 10.1093/infdis/jix120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hulverson MA; Choi R; Arnold SLM; Schaefer DA; Hemphill A; McCloskey MC; Betzer DP; Muller J; Vidadala RSR; Whitman GR; Rivas KL; Barrett LK; Hackman RC; Love MS; McNamara CW; Shaughnessy TK; Kondratiuk A; Kurnick M; Banfor PN; Lynch JJ; Freiberg GM; Kempf DJ; Maly DJ; Riggs MW; Ojo KK; Van Voorhis WC, Advances in bumped kinase inhibitors for human and animal therapy for cryptosporidiosis. International journal for parasitology 2017, 47 (12), 753–763. DOI: 10.1016/j.ijpara.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vidadala RS; Rivas KL; Ojo KK; Hulverson MA; Zambriski JA; Bruzual I; Schultz TL; Huang W; Zhang Z; Scheele S; DeRocher AE; Choi R; Barrett LK; Siddaramaiah LK; Hol WG; Fan E; Merritt EA; Parsons M; Freiberg G; Marsh K; Kempf DJ; Carruthers VB; Isoherranen N; Doggett JS; Van Voorhis WC; Maly DJ, Development of an Orally Available and Central Nervous System (CNS) Penetrant Toxoplasma gondii Calcium-Dependent Protein Kinase 1 (TgCDPK1) Inhibitor with Minimal Human Ether-a-go-go-Related Gene (hERG) Activity for the Treatment of Toxoplasmosis. J Med Chem 2016, 59 (13), 6531–46. DOI: 10.1021/acs.jmedchem.6b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Golkowski M; Vidadala RS; Lombard CK; Suh HW; Maly DJ; Ong SE, Kinobead and Single-Shot LC-MS Profiling Identifies Selective PKD Inhibitors. J Proteome Res 2017, 16 (3), 1216–1227. DOI: 10.1021/acs.jproteome.6b00817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meanwell NA, Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. Journal of Medicinal Chemistry 2011, 54 (8), 2529–2591. DOI: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- 16.Golkowski M; Maly DJ; Ong SE, Proteomic Profiling of Protein Kinase Inhibitor Targets by Mass Spectrometry. Methods Mol Biol 2017, 1636, 105–117. DOI: 10.1007/978-1-4939-7154-1_8. [DOI] [PubMed] [Google Scholar]
- 17.Golkowski M; Brigham JL; Perera GK; Romano GE; Maly DJ; Ong SE, Rapid profiling of protein kinase inhibitors by quantitative proteomics. Medchemcomm 2014, 5 (3), 363–369. DOI: 10.1039/C3MD00315A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arnold SLM; Choi R; Hulverson MA; Schaefer DA; Vinayak S; Vidadala RSR; McCloskey MC; Whitman GR; Huang W; Barrett LK; Ojo KK; Fan E; Maly DJ; Riggs MW; Striepen B; Van Voorhis WC, Necessity of Bumped Kinase Inhibitor Gastrointestinal Exposure in Treating Cryptosporidium Infection. J Infect Dis 2017, 216 (1), 55–63. DOI: 10.1093/infdis/jix247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verdon R; Polianski J; Grodet A; Garry L; Carbon C, Cryptosporidium parvum biliary tract infection in adult immunocompetent and immunosuppressed mice. J Med Microbiol 1998, 47 (1), 71–7. DOI: 10.1099/00222615-47-1-71. [DOI] [PubMed] [Google Scholar]
- 20.Ojo KK; Larson ET; Keyloun KR; Castaneda LJ; Derocher AE; Inampudi KK; Kim JE; Arakaki TL; Murphy RC; Zhang L; Napuli AJ; Maly DJ; Verlinde CL; Buckner FS; Parsons M; Hol WG; Merritt EA; Van Voorhis WC, Toxoplasma gondii calcium-dependent protein kinase 1 is a target for selective kinase inhibitors. Nat Struct Mol Biol 2010, 17 (5), 602–7. DOI: 10.1038/nsmb.1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keyloun KR; Reid MC; Choi R; Song Y; Fox AM; Hillesland HK; Zhang Z; Vidadala R; Merritt EA; Lau AO; Maly DJ; Fan E; Barrett LK; VAN Voorhis WC; Ojo KK, The gatekeeper residue and beyond: homologous calcium-dependent protein kinases as drug development targets for veterinarian Apicomplexa parasites. Parasitology 2014, 141 (11), 1499–509. DOI: 10.1017/S0031182014000857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang W; Hulverson MA; Zhang Z; Choi R; Hart KJ; Kennedy M; Vidadala RS; Maly DJ; Van Voorhis WC; Lindner SE; Fan E; Ojo KK, 5-Aminopyrazole-4-carboxamide analogues are selective inhibitors of Plasmodium falciparum microgametocyte exflagellation and potential malaria transmission blocking agents. Bioorg Med Chem Lett 2016, 26 (22), 5487–5491. DOI: 10.1016/j.bmcl.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.