

Abstract
Cancer vaccines targeting patient-specific tumor neoantigens have recently emerged as a promising component of the rapidly expanding immunotherapeutic armamentarium. However, neoantigenic peptides typically elicit weak CD8+ T cell responses, and so there is a need for universally applicable vaccine delivery strategies to enhance the immunogenicity of these peptides. Ideally, such vaccines could also be rapidly fabricated using chemically synthesized peptide antigens customized to an individual patient. Here we describe a strategy for simple and rapid packaging of peptide antigens into pH-responsive nanoparticles with endosomal escape activity. Electrostatically-stabilized polyplex nanoparticles (nanoplexes) can be assembled instantaneously by mixing decalysine-modified antigenic peptides and poly(propylacrylic acid) (pPAA), a polyanion with pH-dependent, membrane destabilizing activity. These nanoplexes increase and prolong antigen uptake and presentation on MHC-I (major histocompatibility complex class I) molecules expressed by dendritic cells, resulting in enhanced activation of CD8+ T cells. Using an intranasal immunization route, nanoplex vaccines inhibit formation of lung metastases in a murine melanoma model. Additionally, nanoplex vaccines strongly synergized with the adjuvant α-galactosylceramide (α-GalCer) in stimulating robust CD8+ T cell responses, significantly increasing survival time in mice with established melanoma tumors. Collectively, these findings demonstrate that peptide/pPAA nanoplexes offer a facile and versatile platform for enhancing CD8+ T cell responses to peptide antigens, with potential to complement ongoing advancements in the development of neoantigen-targeted cancer vaccines.
Keywords: peptide antigen, nanoparticle, endosomal escape, antigen cross-presentation, vaccines, cancer immunotherapy
Graphical Abstract
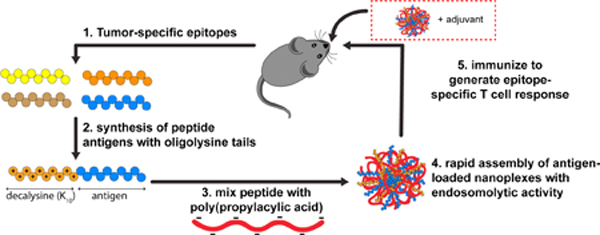
1. Introduction
Cancer vaccines have long held promise as an immunotherapeutic modality, but their clinical efficacy over the past several decades has been largely disappointing [1]. However, the discovery that effective anti-tumor immunity is typically mediated by tumor-infiltrating CD8+ T cells that recognize neoantigens [2–7] – peptides generated by tumor-specific mutations and presented on major histocompatibility complex class I (MHC-I) molecules expressed by cancer cells – has rejuvenated interest in cancer vaccines that target patient-specific mutanomes [8, 9]. With advances in exome sequencing, epitope prediction algorithms, and immunopeptidomic techniques, patient-specific peptide neoantigens can now be identified, synthesized, and administered in a personalized therapeutic cancer vaccine [10–12]. Indeed, this emergent cancer vaccine paradigm has sparked a new wave of clinical trials with promising early results [13, 14].
While synthetic peptide vaccines offer practical advantages over cell and biologic vector-based vaccine approaches, including lower cost, increased safety, and rapid and scalable methods for GMP production [15], peptide antigens are typically weakly immunogenic when delivered alone, and are particularly inept at generating CD8+ T cell responses [16]. The weak immunogenicity of peptide antigens can largely be ascribed to several interconnected delivery barriers, including poor cellular uptake by antigen presenting cells, inadequate activation of innate immunity, and, for induction of robust CD8+ T cell responses, low and/or transient antigen presentation on MHC-I by dendritic cells (DCs) [17, 18]. To achieve class I antigen presentation, administered antigen must be endocytosed by specialized cross-presenting DC subsets or delivered to the classical cytosolic MHC-I antigen processing pathway [19, 20]. However, the predominant fate of soluble endocytosed antigen is degradation in endolysosomes and presentation on MHC-II, with minimal or inefficient presentation on MHC-I [21, 22]. To circumvent this challenge, a variety of synthetic particle- based antigen delivery systems, including polymeric nano- and microparticles, liposomes, and inorganic nanoparticles, have been developed to enhance CD8+ T cell responses to protein- and peptide-based vaccines with varying degrees of efficacy in pre-clinical animal models [21, 23, 24] An additional design consideration important for neoantigen-targeted vaccines is that the vast majority of neoantigens are unique for an individual patient, and cancer vaccination regimens typically comprise multiple prime and boost administrations with pools of peptide antigens [11]. For example, in a recent landmark phase I clinical trial of personalized peptide vaccines, Wu and colleagues immunized melanoma patients with pools of 13–20 peptides using a series of five priming and two booster vaccinations [13]. Time is of the essence for patients with advanced disease who are receiving personalized cancer vaccines, and the time between tissue acquisition and vaccine administration can take several months [13]. Therefore, particle-based delivery systems for personalized neoantigen vaccines should ideally be amendable to rapid and scalable “on demand” formulation [25, 26], a shortcoming for the majority of particulate vaccine formulations, which often require labor intensive, time consuming, and/or non-scalable processing and manufacturing steps. In light of these challenges, we sought to complement ongoing advancements in personalized neoantigen-targeted vaccines through the development of a facile and versatile platform for enhancing CD8+ T cell responses to peptide antigens.
While most particulate vaccines have leveraged physicochemical (e.g., size, surface chemistry) or formulation properties (e.g., co-encapsulation of an adjuvant) to harness endogenous mechanisms of cross-presentation by specific DC subsets [22], another promising strategy has been to design antigen carriers that exploit endosomal escape mechanisms to “short circuit” endo/lysosomal antigen trafficking and actively enhance delivery of antigenic cargo to the cytosol for processing via the classical MHC-I presentation pathway [27–30]. One such carrier with pH-dependent membrane destabilizing activity is poly(propyl acrylic acid) (pPAA), a linear amphiphilic polyanion with a pKa of 6.7 that has been previously explored as a carrier to enhance cytosolic delivery of protein antigens and peptide therapeutics [31–35]. Inspired by the simplicity, speed, and scalability achieved via electrostatic self-assembly of polyplexes used commonly in nucleic acid-based vaccines [36, 37], we postulated that peptide antigen-loaded nanoparticles with endosome-destabilizing activity could be generated through facile mixing of polyanionic pPAA and peptide antigens synthesized with a cationic oligolysine tail (Fig. 1a,b).
Figure 1. Fabrication and characterization of poly(propylacrylic acid)/peptide nanoplexes for enhancing MHC-I antigen presentation.

(a) Assembly of antigen-loaded nanoplexes via simple and rapid mixing of decalysine-modified antigenic peptides and pPAA, which generates electrostatically-stabilized nanoparticles. (b) Schematic representation of nanoplexes promoting cytosolic antigen delivery via endosomal escape, resulting in enhanced levels of antigen presentation on class I major histocompatibility complex (MHC-I). (c) Horizontal, native PAGE of TAMRA-labeled K10OVA and mixtures of K10OVA with pPAA at various COOH:NH2 ratios. White box outlines lanes for gel loading. (d) Z-average diameter of indicated formulations as measured by dynamic light scattering (DLS). (e) Representative size distribution (volume average) measured by DLS of soluble pPAA, soluble K10OVA, or K10OVA/pPAA polyplex generated at 2:1 COOH:NH2. (f) Transmission electron micrograph of K10OVA/pPAA polyplex generated at 2:1 COOH:NH2. (g) Erythrocyte lysis assay demonstrating pH-dependent membrane destabilizing activity of pPAA and K10OVA/pPAA polyplex. (h) ξ-potential of K10OVA/pPAA polyplexes as a function of pH.
Here, we demonstrate feasibility and provide an initial evaluation of this versatile “mix and go” approach for generating nanoparticles that enhance cytosolic delivery and MHC-I presentation of peptide antigens. Polyplex nanoparticles (nanoplexes) are formed instantaneously upon mixing decalysine-modified peptides and pPAA, resulting in enhanced and sustained MHC-I antigen presentation by DCs and increased CD8+ T cell activation in vitro. Using an intranasal immunization route, polymer-peptide nanoplexes also dramatically enhanced antigen-specific CD8+ T cell responses in the lung when combined with the vaccine adjuvant α-galactosylceramide (α-GalCer). Finally, using a model peptide antigen, we demonstrate nanoplex vaccine formulations can prevent lung colonization and inhibit tumor growth in murine melanoma models. Collectively, these studies provide the foundation for a simple, rapid, and more universally applicable strategy for enhancing cellular immunity to peptide antigens. This offers potential as a platform technology for enhancing the efficacy of personalized neoantigen-targeted cancer vaccines.
2. Materials and Methods
Materials
Peptides containing the mouse MHC-I (H-2Kb)-restricted ovalbumin (OVA) epitope OVA257–264 (SIINFEKL) modified with a cationic oligolysine (Kn) tail (K10OVA: K10QLESIINFEKL and K5OVA: K5QLESIINFEKL) were synthesized and HPLC purified by EZBiolab (Carmel, IN). K10OVA labeled with TAMRA at the N-terminus (TAMRA-K10OVA) was purchased from Biomatik (Wilmington, DE). The purity of all peptides was above 95% as certified by HPLC and MS analyses. SIINFEKL peptide was purchased from Invivogen.
Synthesis of poly(propylacrylic acid)
Propylacrylic acid (PAA) was synthesized as described previously using diethyl propylmalonate (Sigma Aldrich) as the precursor [38]. Poly(PAA) was synthesized by reversible addition-fragmentation chain transfer (RAFT) polymerization as previously described [34]. Briefly, 4-cyano-4-(ethylsulfanylthiocarbonyl)sulfanylpentanoic acid (Boron Molecular, Raleigh, NC) was used as the chain transfer agent (CTA), azobisisobutyronitrile (AIBN) was used as the initiator (I), and a monomer to CTA to initiator ratio of 175:1:1 was used. The reaction was performed in bulk under a nitrogen atmosphere for 72 h at 60°C. Following polymerization, the reaction mixture was dissolved in acetone and purified by dialysis (3 kDa MWCO) against deionized water, followed by lyophilization. Samples were dissolved in deuterated dimethyl sulfoxide (DMSO-d6) (Sigma-Aldrich) and 1NMR spectroscopy (Bruker AV400) was used to validate polymer purity and quantify the degree of polymerization (DP = 82) based on monomer conversion (47%). A 1NMR spectrum of the purified pPAA used in these investigations is provided in Supporting Information (Fig. S1). All purified polymers were prepared as 10 mg/mL stock in PBS (pH 7.4.) for further use.
Preparation and characterization of polyplex nanoparticles
KnOVA/pPAA polyplex nanoparticles were prepared by mixing peptide and polymer stock solutions at various negative to positive charge ratios (N:P) in PBS at room temperature, where N:P was defined as the ratio between carboxyl groups on pPAA and the number of lysine residues in the oligolysine tail. The hydrodynamic diameter and ξ-potential of resultant particles were measured using a Malvern Zetasizer Nano ZS (Malvern, UK). Particle size and morphology were also characterized using transmission electron microscopy (TEM). Nanoparticles were stained with 2% methylamine tungstate solution (Ted Pella, Inc.) and images were collected with a FEI Tecnai Osiris TEM (FEI, USA). A gel retardation assay was used to assess the efficiency of peptide complexation at different charge ratios. Nanoplexes were prepared with TAMRA-labeled K10OVA to allow for band visualization, and free peptide and complexes prepared at various charge ratios were loaded into lanes of a horizontal 15% polyacrylamide gel and run at 80 V for 6 h. Gels were imaged (Gel Doc EZ, Bio-Rad) to assess migration of TAMRA-labeled K10OVA.
Erythrocyte hemolysis assay
The ability of free peptide, free polymer, and polyplex nanoparticles to disrupt lipid bilayer membranes in a pH-dependent manner was assessed by a hemolysis assay, as previously described [28]. Human blood from anonymous donors was obtained from Vanderbilt Technologies for Advanced Genomics (VANTAGE). Red blood cells (RBCs) were incubated with peptide, polymer, or polyplex nanoparticles at 5 μg/ml pPAA (2.5 μM) in 100 mM sodium phosphate buffer (supplemented with 150 mM NaCl) in the pH range of the endosomal processing pathway (7.4, 7.0, 6.6, 6.2, and 5.8). Extent of cell lysis (i.e., hemolytic activity) was determined spectrophotometrically by measuring the amount of hemoglobin released (abs = 541 nm) and normalized to a 100% lysis control (1% Triton X-100). Samples were run in quadruplicate.
Cell culture
The mouse dendritic cell line DC2.4 (H-2Kb-positive) was kindly provided by K. Rock (University of Massachusetts Medical School) and cultured in RPMI 1640 (Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco), 2 mM L-glutamine, 100 U/mL penicillin/100 μg/mL streptomycin (Gibco), 50 μM 2-mercaptoethanol (Gibco), 1×nonessential amino acids (Cellgro), and 10 mM HEPES (Invitrogen). B3Z T hybridoma, a lacZ-inducible T cell hybridoma specific for the SIINFEKL-H-2Kb complex, were a generous gift from Nilabh Shastri (UC Berkeley) and cultured in RPMI 1640 supplemented with 10% FBS, 100 U/mL penicillin/100 μg/mL streptomycin, 50 μM 2-mercaptoethanol, and 1 mM sodium pyruvate (Gibco). B16-OVA cells [39] were generously provided by Amanda Lund (Oregon Health Sciences University) cultured in DMEM (Gibco) supplemented with 2 mM L-glutamine, 10% FBS, 100 U/mL penicillin/100 μg/mL streptomycin, and 2.5 mg/mL G418. RAW264.7 cells (ATCC) were cultured in DMEM (Gibco) supplemented with 2 mM L-glutamine, 10% FBS, 100 U/mL penicillin/100 μg/mL streptomycin, and 2.5 mg/mL. All cell types were grown in a humidified atmosphere with 5% CO2 at 37°C. Viability of DC2.4 and RAW264.7 cells was evaluated using MTS (CellTiter 96, Promega).
Uptake and retention of peptide in dendritic ceils
DC2.4 cells were plated at 2×105 cells/well in a 12-well plate and allowed to adhere overnight. Cells were washed and medium was replaced by fresh FBS-free medium containing TAMRA-K10OVA or TAMRA-K10OVA/pPAA polyplexes at a charge ratio of 2:1 and a peptide concentration of 10 μM peptide. After 4 h of incubation, cells were washed twice with PBS, followed by incubation in complete medium for 0, 1, 2, 4, 8, 12, 18 or 24 h. At each time point, the cells were collected via treatment with 0.05% trypsin-EDTA and re-suspended in PBS containing 2% FBS for flow cytometric analysis using a Guava easyCyte™ 8HT flow cytometer (EMD Millipore). Data were analyzed using FlowJo software (version 10; Tree Star, Inc.). For fluorescent microscopy, cells were prepared as described above and images were gathered on a Leica DMi8 microscope using the Leica Application Suite X Software.
Flow cytometric analysis of MHC-I antigen presentation
DC2.4 cells were plated at 2×105 cells/well in 12-well plate and allowed to adhere overnight. Cells were washed and medium was replaced by fresh FBS-free medium containing K10OVA, K10OVA/pPAA polyplexes at a charge ratio of 2:1, or SIINFEKL peptide at a concentration of 10 μM. After 4 h of incubation, cells were washed twice with PBS followed by incubation in complete medium for 0, 1, 2, 4, 8, 12, 18 or 24 h. At each time point, the cells were collected, resuspended in PBS containing 2% FBS, and incubated with anti-mouse CD16/32 antibody (TruStain FcX™; Biolegend) at 4°C for 10 min, followed by staining with PE-conjugated SIINFEKL/H-2Kb-reactive monoclonal antibody (clone 25-D1.16; eBioscience) for 20 min. After staining, cells were washed twice in PBS containing 2% FBS, and the relative levels of SIINFEKL/H-2Kb presentation was measured by flow cytometry (Guava easyCyte™ 8HT; EMD Millipore).
For the protease and transport inhibition study, DC2.4 cells were cultured as described above. Thirty minutes before adding K10OVA/pPAA polyplexes at 10 μM peptide, cells were treated with 10 μM lactacystin (Sigma-Aldrich), 100 nM epoxomicin (Sigma-Aldrich), 10 μg/mL Brefeldin A (Sigma-Aldrich), 15 μM leucinethiol (Sigma-Aldrich), or left untreated. Following incubation with K10OVA/pPAA polyplexes and inhibitors for 4 h, DC2.4 cells were washed twice with PBS and incubated in complete medium for 0, 4, or 24 h. Cells were processed and stained for flow cytometric quantification of SIINFEKL/H-2Kb presentation as described above.
In vitro cross-presentation assay
To assess cross-presentation of antigen by DCs to CD8+ T cells, DC2.4 cells were incubated with K10OVA, K10OVA/pPAA polyplexes or SIINFEKL at 1–50 μM peptide as described above. After 4 h of incubation, cells were carefully washed 3x in Dulbecco’s PBS (DPBS), followed by addition of 1×105 B3Z CD8+ hybridoma T cells, induce produce β-galactosidase expression upon TCR recognition of SIINFEKL epitope presented on H-2Kb. After 22 hours of co-culture in complete medium, cells were pelleted via centrifugation (7 min, ~500 rcf), medium was carefully aspirated, and 150 μl of CPRG/lysis buffer (0.15 mM chlorophenol red-β-d-galactopyranoside (CalBiochem), 0.1% Triton-X 100, 9 mM MgCl, 100 μM mercaptoethanol) was added. Plates were incubated at 37°C in the dark for 90 min, and the absorbance of released chlorophenol red was measured at 570 nm using a plate reader (Biotek Synergy HT) [28, 40, 41].
Intranasal immunization
Male C57BL/6 mice (6–8 weeks old) were purchased from The Jackson Laboratory (Bar Harbor, ME) and maintained at the animal facilities of Vanderbilt University Medical Center under conventional conditions. All animal experiments were approved by the Vanderbilt University Institutional Animal Care and Use Committee (IACUC). Free K10OVA or K10OVA/pPAA polyplexes were freshly prepared at a peptide concentration of 250 μM in PBS from 0.2 μm filter sterilized stocks of pPAA and peptide. To prepare adjuvanted formulations, α-GalCer (Funakoshi) was mixed into peptide or polyplex solutions from a 1 mg/ml stock in DMSO to a final concentration of 12.5 μg/mL. Mice were anesthetized with ketamine/xylazine (10 mg/mL ketamine hydrochloride injection, Vedco; 1 mg/mL xylazine hydrochloride injection; Vanderbilt University Pharmacy) by intraperitoneal (IP) injection (100 μL anesthesia/18 g mice). Groups of anesthetized mice were immunized intranasally (IN) on days 0 and 14 with formulations containing 50 μg peptide and/or 47 μg pPAA with or without 1 μg α-GalCer. Vaccine formulations were delivered through the nostrils into the lungs at 80 μL/mouse. Animals were monitored daily for weight loss and signs of distress, and all mice were euthanized on day 23 by CO2 asphyxiation.
Analysis of SIINFEKL-specific CD8+ T cell response
On day 23, mice were euthanized with CO2 and lungs were harvested and processed as previously described [42]. In brief, organs were passed through a 70 μm cell strainer and were lysed with ACK lysis buffer (Gibco) to form single cell suspensions. Prior to processing, lung samples were minced with a scalpel and incubated for 1 h at 37°C in complete RPMI medium supplemented with 2 mg/mL collagenase (Sigma-Aldrich) and 50 nM dasatinib (LC Laboratories). Single cell suspensions from lungs were stained at 4°C with anti-B220-FITC (BD Biosciences), anti-CD4-FITC (BD Biosciences), anti-CD11b-FITC (Tonbo Biosciences), anti-CD11c-FITC (Tonbo Biosciences), anti-CD8α-PacificBlue (BD Biosciences), and 1.5 μg/mL PE-labeled OVA257–264 (SIINFEKL)-H-2Kb tetramer prepared according to a previously reported procedure [43]. After 1 h, cells were rinsed with PBS supplemented with 2% FBS and 50 nm dasatinib and stained with propidium iodide (BD Biosciences) to discriminate live from dead cells. Flow cytometry (BD LSRII) was used to determine the frequency of SIINFEKL-specific CD8+ T cells (tetramer-positive, CD8-positive population) and all data were analyzed using FlowJo software.
Tumor Studies
To evaluate the ability of vaccine formulations to prevent lung colonization by tumor cells, male C57BL/6 mice (7 weeks old) were immunized IN as described above. Seven days after booster immunization, mice were intravenously (IV) challenged with 2 × 105 B16-OVA melanoma cells in 200 μL sterile PBS via a caudal vein injection. Mice were euthanized with CO2 3.5–4 weeks post-challenge, lungs were excised and formalin fixed, and visible surface metastases were enumerated.
To evaluate the ability of vaccine formulations to inhibit growth of established tumors, 5 × 104 B16-OVA cells in 100 μL sterile PBS were injected subcutaneously (SC) in the right flank of 7-week old male C57BL/6 mice. At 3 and 13 days post-injection, mice were administered vaccine formulations IN as described above. Tumor volume was measured every other day via caliper measurements, and tumor volume calculated using the following equation . [44] Animals were euthanized when tumor volume reached 1500 mm3.
Statistical Analysis
Statistical analysis performed are indicated in the figure legends. All statistical analyses were performed by using GraphPad Prism software, version 7.0.
3. Results and Discussion
Assembly of pPAA/peptide polyplex nanoparticles-
To facilitate electrostatic complexation between peptide antigen and poly(propylacrylic acid) (pPAA; 9.4 kDa), a synthetic long peptide [16] containing the MHC-I-restricted epitope, SIINFEKL, from the model antigen ovalbumin (OVA) was designed with a cationic N-terminal decalysine tail (K10) and a three amino acid spacer (QLE) to support intracellular peptidase cleavage [45, 46]. This peptide, K10(QLE)SIINFEKL (hereinafter K10OVA) was mixed with pPAA over a range of charge ratios (defined as the ratio of COOH in pPAA to NH2 groups in K10 tail). After complexation, particle size was evaluated via dynamic light scattering (DLS) and peptide complexation efficiency via native PAGE. At charge ratios between 16:1 and 1:1, complete complexation of peptide was achieved, as evidenced by abrogated migration of cationic peptide towards the anode or reversed migration to the cathode due to electrostatic interaction with pPAA (Fig. 1c). However, stable nanoplexes <100 nm diameter were only observed at charge ratios of 4:1 and 2:1 (Fig. 1d); charge ratios of 1:1 and 1:2 resulted in micron-scale aggregates that quickly precipitated, while complexation at 16:1 and 8:1 resulted in only modest increases in apparent particle size (~5–10 nm by DLS) and could not be confirmed to generate nanoparticles via transmission electron microscopy (TEM; data not shown). Similarly, complexation of analogous peptides synthesized with a K5 tail at a 2:1 charge ratio failed to form nanoscale polyplexes (Fig. S2), suggesting that a critical oligolysine tail length may be necessary for efficient particle assembly. To obtain a suitable particle size (Z-average = 78 nm) and polydispersity index (0.41±0.01) for nanoparticle vaccination, a charge ratio of 2:1 was utilized for generating nanoplexes for further analysis and investigation. TEM characterization of nanoplexes confirmed the assembly of polyplex nanoparticles with a size distribution consistent with DLS data (Fig. 1e, f). Additionally, zeta potential measurements revealed a negative surface charge at neutral pH (Fig. 1h), suggesting neutralization and sequestration of K10 tails with colloidal stability imparted by negatively charged pPAA chains localized on the particle surface. In the presence of 10% serum, particle size increased slightly but remained stable for at least 3.5 h (Fig. S3).
The ability of pPAA to mediate endosomal escape arises from its ability to undergo a switch from a hydrophilic soluble conformation at physiologic pH to a hydrophobic and membrane interactive state upon protonation of carboxylic acid groups within acidic environments. Because electrostatic complexation neutralizes a fraction of the carboxylate groups and alters chain conformation, we sought to verify that pPAA/peptide nanoplexes retained pH-dependent, membrane-destabilizing activities. Using a red blood cell hemolysis assay that has been shown to correlate strongly with endosomal escape [47], nanoplexes demonstrated hemolytic activity at endosomal pH of 6.2–5.8 and negligible activity at higher pH values of 6.6–7.4 (Fig. 1g). This is also consistent with zeta potential measurements (Fig. 1h) demonstrating a steady increase in surface charge of polyplexes from −22 mV at pH 7.4 to 0 mV at pH 5.8.
Therefore, simple mixing of pPAA with decalysine-modified peptides enables instantaneous assembly of antigen-loaded nanoparticles with pH-responsive, membrane-destabilizing activity. In principle, this approach can be extended to virtually any decalysine-modified peptide as well as pools of modified peptides, though further investigation into the dependence on the epitope composition and properties (e.g., charge, hydrophobicity) on nanoplex formation is necessary to validate the generalizability of this approach. Additionally, while K5 modified peptides failed to form polyplexes, the optimal number, density, and type of basic amino acid residues in the cationic peptide tail remain to be investigated.
Nanoplexes enhance antigen uptake and intracellular retention-
The weak immunogenicity of peptide antigens is in part attributable to low cellular uptake, rapid exocytosis, and/or degradation [22], and so we investigated the capacity of nanoplexes to increase antigen uptake and prolong intracellular retention. To evaluate this, a pulse-chase study was performed in which DC2.4 cells, a murine dendritic cell line, were treated with TAMRA-labeled soluble K10OVA or K10OVA/pPAA nanoplexes for 4 h, followed by washing and quantification of peptide uptake using flow cytometry. Despite having a negative zeta potential, nanoplexes enhanced peptide uptake ~4.5-fold over free peptide (Fig. 2a). The free peptide was nonetheless internalized to a considerable extent, likely due to its cationic properties. Uptake of K10OVA/pPAA nanoplexes was reduced slightly in the presence of 10% serum, but nonetheless remained significantly higher than free K10OVA (Fig. S4). Additionally, incubation at 4°C, where endocytosis is inhibited, nearly completed abrogated fluorescent intensity (Fig. S5), indicating that the vast majority of fluorescent peptide was internalized versus cell-surface associated. This was further corroborated by fluorescent microscopy (Fig. S6), which demonstrated significantly higher levels of intracellular TAMRA-labeled K10OVA with nanoplexes relative to free peptide. Upon washing of cells, the change in median fluorescence intensity (MFI) relative to the initial MFI measurement after pulsing with peptide, defined here as the retention ratio, was determined using flow cytometry. Free peptide was rapidly cleared, with >50% of internalized peptide lost within 2 h and completely eliminated within a day, whereas assembly of the peptide into nanoplexes significantly extended intracellular antigen retention, with >20% of internalized peptide remaining after 24 h (Fig. 2b). This is consistent with previous reports demonstrating that pPAA, and other endosomolytic polymers, can enhance intracellular retention of associated cargo, likely through a combination of reduced endosomal recycling, stalling of endosomal trafficking, and sustained endosomal release into the cytosol [32, 33, 47].
Figure 2. Nanoplexes enhance uptake and prolong intracellular antigen retention.

(a) Representative flow cytometry histograms of DC2.4 cells treated for 4 h with TAMRA-labeled K10OVA or K10OVA/pPAA polyplexes at a concentration of 10 μM K10OVA. (b) Change in median fluorescence intensity (MFI) at indicated time points relative to that measured after initial 4 h incubation with TAMRA-labeled K10OVA or nanoplexes. ***p<0.001 between K10OVA and K10OVA/pPAA at all time points between 4 and 24 h by two-tailed t-test.
Nanoplexes enhance and sustain MHC-I antigen presentation-
Both the magnitude and duration of antigen presentation by APCs play an important role in generating CD8+ T cell responses [16, 48–50]. Therefore, we investigated whether the enhanced intracellular uptake and endosomolytic properties of the polyplexes could enhance and prolong antigen presentation on MHC-I. DC2.4 cells were pulsed with indicated antigen formulations for 4 h, followed by flow cytometric quantification of class I antigen presentation using an antibody against the H-2Kb/SIINFEKL complex (Fig. 3a). Nanoplexes significantly increased antigen presentation relative to free K10OVA while also sustaining enhanced antigen presentation for 24 h. Pulsing of DC2.4 cells with nanoplexes in the presence of serum did not appear to have a significant effect on initial levels SIINFEKL presentation, though resulted in reduced levels after 24 h, potentially due to reduced intracellular uptake (Fig. S7). While incubation with the exact epitope SIINFEKL, which does not require intracellular processing and can directly bind MHC-I on the cell surface, initially resulted in ~4-fold higher levels of antigen presentation relative to nanoplexes, SIINFEKL presentation rapidly declined. Within 8 h, SIINFEKL presentation levels fell below that achieved with nanoplexes and was undetectable by 18 h, whereas nanoplexes enhanced antigen presentation up to 24 h. These findings are consistent with the increased antigen retention achieved using nanoplexes, and demonstrate the importance of maintaining an intracellular reservoir for sustained antigen presentation.
Figure 3. pPAA/peptide nanoplexes enhance and prolong antigen presentation on class I MHC.

(a) Representative flow cytometry histograms (left) and MFI over time of DC2.4 cells treated for 4 h with K10OVA, K10OVA/pPAA polyplexes, or the exact class I epitope SIINFEKL at 10 μM peptide, followed by washing, culture for time indicated, and staining with an antibody (25-D1.16) specific to the SIINFEKL/H-2Kb complex. ****p<0.0001 by one-way ANOVA with Tukey post-hoc analysis for multiple comparison. Statistical comparison between K10OVA/pPAA and SIINFEKL are shown; K10OVA/pPAA is significantly lower than SIINFEKL between 0 and 4 h and significantly higher between 8 and 24h. (b) Thirty minutes prior to 4 h treatment with K10OVA/pPAA polyplexes at 10 μM peptide, DC2.4 cells were treated with the indicated inhibitor of antigen processing or presentation followed by staining with 25-D1.16 and quantification of relative levels of SIINFEKL presentation by flow cytometry. Treatment with brefeldin A and leucinethiol inhibited antigen presentation. Dashed line represents baseline MFI of untreated DC2.4 cell; ****p<0.001, **p<0.01 relative to no inhibitor group. (c) DC2.4 were treated with the indicated formulation for 4 h at 10 μM peptide and subsequently co-cultured for 24 h with B3Z hybridoma T cells. ****p<0.001 relative to all other groups by one-way ANOVA with Tukey post-hoc test. (d) B3Z T cell response to DC2.4 cells treated with indicated formulation at different peptide concentrations. Statistical significance between K10OVA/pPAA and SIINFEKL are shown; **p<0.01, ****p<0.001 by one-way ANOVA with Tukey post-hoc test.
To gain insight into the mechanism of antigen processing, DC2.4 cells were pre-treated with epoxomicin and lactacystin, proteasome inhibitors; leucinethiol, a leucine aminopeptidase; and brefeldin A, which inhibits transport of membrane proteins – including assembled p/MHC-I complexes – from the endoplasmic reticulum to the cell membrane (Fig. 3b). Brefeldin A almost completely prevented antigen presentation, suggesting a requirement for intracellular loading of peptide onto MHC-I. Proteasome inhibition did not decrease antigen presentation, whereas leucinethiol inhibited presentation by ~30%, indicating that K10OVA processing is mediated endoplasmic reticulum-associated aminopeptidases associated with antigen processing (ERAPs) and not by proteasomal cleavage. The incomplete inhibition of SIINFEKL presentation in response to treatment with leucinethiol likely indicates that additional intracellular proteases (e.g., insulin-regulated aminopeptidase; IRAP) [51] are involved in trimming of peptides delivered using nanoplexes. However, it should be noted the degree of pPAA-mediated antigen presentation and the mechanism of antigen processing may be different for primary dendritic cells or between dendritic cell subsets, and further investigation is necessary to elucidate this.
We next sought to demonstrate whether the ability of nanoplexes to increase and prolong antigen presentation would result in improved CD8+ T cell activation. This was evaluated using a co-culture model comprising DC2.4 cells and a B3Z T cell hybridoma that produces β-galactosidase upon recognition of SIINFEKL/H-2Kb (Fig. 3c). As expected, nanoplexes enhanced B3Z T cell activation ~2.5-fold over soluble K10OVA and modestly but significantly over SIINFEKL. Additionally, a mixture of pPAA and K5OVA, which did not assemble into particles (Fig. S2), did not enhance the B3Z T cell response, reinforcing the importance of oligolysine length and assembly of polyplexes in enhancing peptide presentation on MHC-I. We further compared B3Z T cell responses at peptide concentrations ranging from 1–50 μM, and found that nanoplexes consistently enhanced B3Z T cell activation relative to free K10OVA (Fig. 3d). Additionally, at concentrations above 10 μm, nanoplexes improved T cell activation relative to soluble SIINFEKL, despite increased initial levels of antigen presentation. This may be a consequence of the prolonged antigen presentation achieved with nanoplexes, resulting in increased probability and/or stability of pMHC/TCR interactions during DC-T cell co-culture [49]. This could also reflect a higher affinity between peptide/MHC complexes associated with intracellular processing of synthetic long peptides relative to extracellular epitope binding, which can be low affinity with potential to induce tolerance instead of immunity [52].
Pulmonary delivery of peptide/pPAA nanoplexes enhances CD8+ T cell responses-
The lungs are one of the most common sites of metastasis for many cancer types [53]. However, most cancer vaccines are delivered intramuscularly or SC, injection routes that often fail to elicit pulmonary T cell responses optimal for eliminating lung metastases [54, 55]. Hence, we investigated the ability of IN administered nanoplex vaccines to generate antigen-specific CD8+ T cell responses in the lung. Mice were administered soluble K10OVA or K10OVA/pPAA nanoplexes IN on day 0, boosted IN on day 14, and p/MHC-I tetramer staining was used to evaluate SIINFEKL-specific CD8+ T cell responses in the lungs on day 23 (Fig. 4). Nanoplex vaccines were well tolerated, as no weight loss or adverse effects were observed (Fig. S8). This outcome is consistent with the low cytotoxicity of nanoplexes observed in vitro (Fig. S9). IN administration with nanoplexes resulted in a modest antigen-specific CD8+ T cell responses in the lung (~2% SIINFEKL-specific CD8+ T cells), whereas free K10OVA failed to elicit a response above the limit of detection (~0.1%). Notably, nanoplexes were generated immediately prior to administration (5–10 min before immunization) by simply mixing pre-made, filter-sterilized stock solutions of K10OVA and pPAA at the appropriate ratio and concentration, a testament to the ease and speed with which nanoplex vaccines can be fabricated.
Figure 4. Intranasal administration of nanoplex vaccines augments antigen-specific CD8+ T cell response in the lungs.

(a) Mice were immunized intranasally (IN) on d0 and 14 and peptide/MHC tetramer staining was used to quantify the SIINFEKL-specific CD8+ T cell response on d23. (b) Representative flow cytometry dot-plots and (c) quantification of the frequency of SIINFEKL-specific CD8+ T cells in the lung elicited by immunization with the indicated formulation. ****p<0.0001 by one-way ANOVA with Tukey post-hoc analysis for multiple comparison.
In addition to class I antigen presentation by DCs, generating a robust CD8+ T cell response also requires engagement of costimulatory molecules and cytokine signaling that provide context to T cell priming and enhance T cell activation and effector function [17]. We therefore postulated that CD8+ T cell responses using nanoplex vaccines could be further augmented through addition of an immunostimulatory adjuvant [56]. To evaluate this, we mixed nanoplexes with α-galactosylceramide (α-GalCer), a glycolipid adjuvant that has been widely explored for applications in cancer immunotherapy and mucosal vaccination, in part owing to its ability to potently stimulate CD8+ T cell responses in the absence of CD4+ T cell help.[42, 54, 57] Combining nanoplexes with α-GalCer resulted in an ~10-fold increase in the CD8+ T cell response in the lungs relative to the non-adjuvanted nanoplex (18% vs. 2%). Interestingly, the addition of α-GalCer to soluble K10OVA elicited a nearly identical response as non-adjuvanted nanoplexes, a further indication of the ability of the nanoplex platform to enhance immunogenicity of peptide antigens. The dramatic enhancement in CD8+ T cell response achieved by combining nanoplexes with α-GalCer suggests synergy between delivery systems that enhance and sustain MHC-I antigen presentation and molecular adjuvants that stimulate Th1-biased inflammatory responses. Whether similar or greater enhancement in cellular immunity can be achieved through addition of other immunostimulatory adjuvants (e.g., TLR agonists) remains to be investigated. Furthermore, there is significant evidence demonstrating that co-loading of antigen and adjuvant into a common particle can significantly enhance immune responses [28, 58, 59]. While this possibility was not explored herein, the nanoplex approach offers potential for generation of ternary electrostatic complexes between oligolysine-modified peptides, pPAA, and immunostimulatory nucleic acids (e.g., CpG, poly(I:C)). The development of nanoplexes for dual-delivery peptide and nucleic acid adjuvants may further augment CD8+ T cell responses and anti-tumor efficacy and will be the focus of future investigations.
Nanoplex vaccines protect against lung metastasis and inhibit tumor growth-
As a demonstration of T cell effector function, we next evaluated the ability of nanoplex vaccines to protect against lung metastases following IV administration of B16 murine melanoma cells expressing OVA protein as a model antigen (B16-OVA; Fig. 5a) [39]. Immunization with K10OVA/pPAA nanoplexes significantly reduced the number of surface lung metastases relative to soluble K10OVA, which failed to provide significant protection relative to mice receiving only PBS (Fig. 5b,c). This is consistent with the weak immunogenicity of peptide antigens when delivered alone and further supports the ability of nanoplexes to elicit functional effector CD8+ T cell responses in the absence of an exogeneous adjuvant and without incorporation of an MHC class II (MHC-II) restricted epitope for generating CD4+ T cell help.
Figure 5. Nanoplex vaccines enhance anti-tumor immunity.

(a) Mice were immunized IN with indicated formulations on d0 and d14, followed by intravenous challenge with ovalbumin (OVA)-expressing B16 melanoma cells (B16-OVA) and quantification of surface lung colonies. (b) Representative images (scale bar = 0.5 cm) and (c) enumeration of surface lung metastases of mice immunized with indicated formulation. *p<0.05, ****p<0.0001 by one-way ANOVA with Tukey post-hoc analysis for multiple comparison. (d) To evaluate efficacy in a therapeutic setting, mice were inoculated with B16-OVA cells subcutaneously (SC) 3 days prior to IN administration of indicated formulations. (e) Kaplan-Meier curves (****p<0.0001 vs. all other groups by Mantel-Cox test) and (f) survival time are shown comparing the indicated vaccine formulations, determined based on a tumor volume of 1500 mm3. *p<0.05, **p<0.01,****p<0.0001 by one-way ANOVA with Tukey post-hoc test.
Though the lung is common site of metastasis for many cancers, motivating our investigation into a pulmonary delivery route, an effective cancer vaccine would optimally inhibit tumor growth at other sites as well. We therefore further evaluated the ability of IN administered nanoplexes to inhibit growth of a subcutaneous B16-OVA tumor established three days prior to vaccine administration. IN immunization of non-adjuvanted nanoplexes was unable to significantly inhibit tumor growth or extend survival in this model, reflecting an insufficient number and/or functionality of peripheral CD8+ T cells for treatment of established distal disease. The addition of α-GalCer to nanoplexes resulted in a moderate, but statistically insignificant, reduction in the number of surface lung tumor nodules (Fig. 5c), but a significant reduction in tumor growth (Fig. S10) and improvement in overall survival of mice with established B16-OVA tumors relative to all other formulations tested (Fig. 5e,f).
Collectively, these data demonstrate the ability of the peptide/pPAA nanoplex vaccine platform to enhance the immunogenicity of peptide antigens, potentially in synergy with co-administered molecular adjuvants, resulting in more effective anti-tumor immunity, particularly in the lung when administered IN. In these proof-of-concept investigations, we have explored only the use of a model MHC-I restricted epitope (SIINFEKL) and, therefore, it will be important in future work to validate that this approach can be used to enhance the immunogenicity of neoantigenic peptides, such as the established M27 and M30 epitopes generated by B16.F10 melanoma cells [60]. Additionally, increasing evidence suggests that many neoantigens are MHC-II epitopes, which can stimulate anti-tumor CD4+ T cell responses that support CD8+ T cells and/or have direct anti-tumor effects [60, 61]. While the cytosolic delivery of antigen is typically associated with enhanced MHC-I presentation, a cohort of antigen likely remains in endo/lysosomal compartments [27, 33]. This, combined with the increased cellular uptake and prolonged intracellular retention achieved using nanoplex vaccines, may also bolster MHC-II presentation and CD4+ T cell responses. Indeed, increased CD4+ T cell responses have been previously noted using endosomolytic antigen carriers.[28] Therefore, evaluating and optimizing the ability of nanoplexes to increase the immunogenicity of both MHC-I and MHC-II restricted epitopes will be an important next step in the development of this approach for personalized cancer vaccination. Likewise, combining cancer vaccines with other immunotherapeutic modalities that reverse immunosuppression in the tumor microenvironment [62], in particular CTLA-4 and PD-1/PD-L1 checkpoint blockade, has significantly enhanced therapeutic efficacy, and so exploring such combinations in the context of nanoplex vaccines merits future investigation.
5. Conclusion
The recent revival of cancer vaccines as a promising strategy for personalized immunotherapy has generated a need for strategies to enhance the immunogenicity of peptide neoantigens. While a wide diversity of particle-based antigen delivery platforms have been explored, the vast majority are not readily amenable to rapid and facile loading of neoantigenic peptides that must be custom synthesized for each patient. Here, we describe a versatile “mix and go” approach for instantaneous fabrication of peptide antigen-loaded, endosomolytic nanoparticulate polyplexes via facile mixing of oligolysine modified peptide antigens and poly(propylacrylic acid). This work builds upon and corroborates previous studies utilizing pPAA and other endosome-destabilizing polymers to achieve cytosolic delivery and enhanced MHC-I presentation of covalently linked antigens; however, it is distinguished by the simplicity and speed with which antigen-loaded, endosomolytic nanoparticles can be assembled via electrostatic interactions. Relative to soluble peptides, nanoplex vaccines enhance intracellular antigen uptake and retention, increase and prolong MHC-I antigen presentation, elicit CD8+ T cell responses that can protect against lung metastasis, and can synergize with molecular adjuvants to further enhance cellular immunity and anti-tumor efficacy. Therefore, peptide/pPAA nanoplexes offer a versatile, scalable, and more universally applicable delivery platform for enhancing immunogenicity of peptide antigens with potential to complement continued advancements in individualized, neoantigen-targeted vaccines for cancer immunotherapy.
Supplementary Material
Acknowledgments
We gratefully acknowledge Prof. Kenneth Rock (University of Massachusetts Medical School) for providing DC2.4 cells, Prof. Nilabh Shastri (UC Berkeley) for providing B3Z T cells, and Prof. Amanda Lund (Oregon Health Sciences University) for providing B16-OVA cells. We also acknowledge the core facilities of the Vanderbilt Institute of Nanoscale Sciences and Engineering (VINSE) and for use dynamic light scattering and TEM instruments, Prof. Marjan Rafat (Vanderbilt University) for assistance with fluorescent microscopy, the VUMC Flow Cytometry Shared Resource, supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404), for the usage of BD 3-laser LSRII flow cytometery. This research was supported by grants from the National Institutes of Health 5R21AI121626 (JTW), the National Science Foundation CBET-1554623 (JTW), the Vanderbilt University Discovery Grant Program (JTW, SJ), the State Scholarship Fund by China Scholarship Council 201406245022 (FQ), and a Veteran’s Administration Merit Award BX001444 (SJ). This research was supported by a grant from Susan G. Komen® PDF18377947 (SS), and also supported by a Stand Up To Cancer Innovative Research Grant, Grant Number SU2C-AACR-IRG 20-17 (JTW). Stand Up To Cancer is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C.
Footnotes
Data Availability
The raw/processed data required to reproduce these findings cannot be shared at this time due to technical or time limitations.
References
- [1].Romero P, Banchereau J, Bhardwaj N, Cockett M, Disis ML, Dranoff G, Gilboa E, Hammond SA, Hershberg R, Korman AJ, Kvistborg P, Melief C, Mellman I, Palucka AK, Redchenko I, Robins H, Sallusto F, Schenkelberg T, Schoenberger S, Sosman J, Tureci O, Van den Eynde B, Koff W, Coukos G, The Human Vaccines Project: A roadmap for cancer vaccine development, Sci Transl Med 8(334) (2016) 334ps9. [DOI] [PubMed] [Google Scholar]
- [2].McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, Watkins TB, Shafi S, Murugaesu N, Mitter R, Akarca AU, Linares J, Marafioti T, Henry JY, Van Allen EM, Miao D, Schilling B, Schadendorf D, Garraway LA, Makarov V, Rizvi NA, Snyder A, Hellmann MD, Merghoub T, Wolchok JD, Shukla SA, Wu CJ, Peggs KS, Chan TA, Hadrup SR, Quezada SA, Swanton C, Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade, Science 351(6280) (2016) 1463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, Zappasodi R, Maeda Y, Sander C, Garon EB, Merghoub T, Wolchok JD, Schumacher TN, Chan TA, Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer, Science 348(6230) (2015) 124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, van Dijk LJ, Behjati S, Hilkmann H, El Atmioui D, Nieuwland M, Stratton MR, Kerkhoven RM, Kesmir C, Haanen JB, Kvistborg P, Schumacher TN, Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma, J Clin Oncol 31(32) (2013) e439–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E, Samuels Y, Rosenberg SA, Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells, Nat Med 19(6) (2013) 747–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, Hollmann TJ, Bruggeman C, Kannan K, Li Y, Elipenahli C, Liu C, Harbison CT, Wang L, Ribas A, Wolchok JD, Chan TA, Genetic basis for clinical response to CTLA-4 blockade in melanoma, N Engl J Med 371(23) (2014) 2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, Mulder GE, Toebes M, Vesely MD, Lam SS, Korman AJ, Allison JP, Freeman GJ, Sharpe AH, Pearce EL, Schumacher TN, Aebersold R, Rammensee HG, Melief CJ, Mardis ER, Gillanders WE, Artyomov MN, Schreiber RD, Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens, Nature 515(7528) (2014) 577–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Vormehr M, Diken M, Boegel S, Kreiter S, Tureci O, Sahin U, Mutanome directed cancer immunotherapy, Curr Opin Immunol 39 (2016) 14–22. [DOI] [PubMed] [Google Scholar]
- [9].Gubin MM, Artyomov MN, Mardis ER, Schreiber RD, Tumor neoantigens: building a framework for personalized cancer immunotherapy, J Clin Invest 125(9) (2015) 3413–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sahin U, Tureci O, Personalized vaccines for cancer immunotherapy, Science 359(6382) (2018) 1355–1360. [DOI] [PubMed] [Google Scholar]
- [11].Hu Z, Ott PA, Wu CJ, Towards personalized, tumour-specific, therapeutic vaccines for cancer, Nat Rev Immunol 18(3) (2018) 168–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Schumacher TN, Schreiber RD, Neoantigens in cancer immunotherapy, Science 348(6230) (2015) 69–74. [DOI] [PubMed] [Google Scholar]
- [13].Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, Chen C, Olive O, Carter TA, Li S, Lieb DJ, Eisenhaure T, Gjini E, Stevens J, Lane WJ, Javeri I, Nellaiappan K, Salazar AM, Daley H, Seaman M, Buchbinder EI, Yoon CH, Harden M, Lennon N, Gabriel S, Rodig SJ, Barouch DH, Aster JC, Getz G, Wucherpfennig K, Neuberg D, Ritz J, Lander ES, Fritsch EF, Hacohen N, Wu CJ, An immunogenic personal neoantigen vaccine for patients with melanoma, Nature 547(7662) (2017) 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, Bukur V, Tadmor AD, Luxemburger U, Schrors B, Omokoko T, Vormehr M, Albrecht C, Paruzynski A, Kuhn AN, Buck J, Heesch S, Schreeb KH, Muller F, Ortseifer I, Vogler I, Godehardt E, Attig S, Rae R, Breitkreuz A, Tolliver C, Suchan M, Martic G, Hohberger A, Sorn P, Diekmann J, Ciesla J, Waksmann O, Bruck AK, Witt M, Zillgen M, Rothermel A, Kasemann B, Langer D, Bolte S, Diken M, Kreiter S, Nemecek R, Gebhardt C, Grabbe S, Holler C, Utikal J, Huber C, Loquai C, Tureci O, Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer, Nature 547(7662) (2017) 222–226. [DOI] [PubMed] [Google Scholar]
- [15].Hacohen N, Fritsch EF, Carter TA, Lander ES, Wu CJ, Getting personal with neoantigen-based therapeutic cancer vaccines, Cancer Immunol Res 1(1) (2013) 11–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Melief CJM, van der Burg SH, Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines, Nature Reviews Cancer 8(5) (2008) 351–360. [DOI] [PubMed] [Google Scholar]
- [17].Yewdell JW, Designing CD8+ T cell vaccines: it’s not rocket science (yet), Current Opinion in Immunology 22(3) (2010) 402–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJ, Vaccines for established cancer: overcoming the challenges posed by immune evasion, Nat Rev Cancer 16(4) (2016) 219–33. [DOI] [PubMed] [Google Scholar]
- [19].Foged C, Hansen J, Agger EM, License to kill: Formulation requirements for optimal priming of CD8(+) CTL responses with particulate vaccine delivery systems., European journal of pharmaceutical sciences : official journal of the European Federation for Pharmaceutical Sciences 45(4) (2012) 482–491. [DOI] [PubMed] [Google Scholar]
- [20].Amigorena S, Savina A, Intracellular mechanisms of antigen cross presentation in dendritic cells, Current Opinion in Immunology 22(1) (2010) 109–117. [DOI] [PubMed] [Google Scholar]
- [21].Hubbell JA, Thomas SN, Swartz MA, Materials engineering for immunomodulation, Nature 462(7272) (2009) 449–460. [DOI] [PubMed] [Google Scholar]
- [22].De Temmerman M-L, Rejman J, Demeester J, Irvine DJ, Gander B, De Smedt SC, Particulate vaccines: on the quest for optimal delivery and immune response, Drug Discovery Today 16(13–14) (2011) 569–582. [DOI] [PubMed] [Google Scholar]
- [23].Moon JJ, Huang B, Irvine DJ, Engineering Nano- and Microparticles to Tune Immunity, Advanced Materials 24(28) (2012) 3724–3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mehta NK, Moynihan KD, Irvine DJ, Engineering New Approaches to Cancer Vaccines, Cancer Immunol Res 3(8) (2015) 836–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Li AW, Sobral MC, Badrinath S, Choi Y, Graveline A, Stafford AG, Weaver JC, Dellacherie MO, Shih TY, Ali OA, Kim J, Wucherpfennig KW, Mooney DJ, A facile approach to enhance antigen response for personalized cancer vaccination, Nat Mater (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ, Designer vaccine nanodiscs for personalized cancer immunotherapy, Nat Mater 16(4) (2017) 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Keller S, Wilson JT, Patilea GI, Kern HB, Convertine AJ, Stayton PS, Neutral polymer micelle carriers with pH-responsive, endosome-releasing activity modulate antigen trafficking to enhance CD8(+) T cell responses., Journal of Controlled Release 191 (2014) 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wilson JT, Keller S, Manganiello MJ, Cheng C, Lee C-C, Opara C, Convertine A, Stayton PS, pH-Responsive Nanoparticle Vaccines for Dual-Delivery of Antigens and Immunostimulatory Oligonucleotides., ACS Nano 7(5) (2013) 3912–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wilson JT, Postma A, Keller S, Convertine AJ, Moad G, Rizzardo E, Meagher L, Chiefari J, Stayton PS, Enhancement of MHC-I Antigen Presentation via Architectural Control of pH-Responsive, Endosomolytic Polymer Nanoparticles., The AAPS journal 17(2) (2014) 358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Morishita M, Takahashi Y, Nishikawa M, Ariizumi R, Takakura Y, Enhanced Class I Tumor Antigen Presentation via Cytosolic Delivery of Exosomal Cargos by Tumor-Cell-Derived Exosomes Displaying a pH-Sensitive Fusogenic Peptide, Mol Pharm 14(11) (2017) 4079–4086. [DOI] [PubMed] [Google Scholar]
- [31].Foster S, Duvall CL, Crownover EF, Hoffman AS, Stayton PS, Intracellular Delivery of a Protein Antigen with an Endosomal-Releasing Polymer Enhances CD8 T-Cell Production and Prophylactic Vaccine Efficacy, Bioconjugate Chemistry 21(12) (2010) 2205–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Berguig GY, Convertine AJ, Shi J, Palanca-Wessels MC, Duvall CL, Pun SH, Press OW, Stayton PS, Intracellular Delivery and Trafficking Dynamics of a Lymphoma-Targeting Antibody-Polymer Conjugate., Molecular Pharmaceutics (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Evans BC, Hocking KM, Kilchrist KV, Wise ES, Brophy CM, Duvall CL, Endosomolytic Nano-Polyplex Platform Technology for Cytosolic Peptide Delivery To Inhibit Pathological Vasoconstriction, ACS Nano 9(6) (2015) 5893–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Evans BC, Hocking KM, Osgood MJ, Voskresensky I, Dmowska J, Kilchrist KV, Brophy CM, Duvall CL, MK2 inhibitory peptide delivered in nanopolyplexes prevents vascular graft intimal hyperplasia, Sci Transl Med 7(291) (2015) 291ra95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yang L, Bracho-Sanchez E, Fernando LP, Lewis JS, Carstens MR, Duvall CL, Keselowsky BG, Poly(2-propylacrylic acid)/poly(lactic-co-glycolic acid) blend microparticles as a targeted antigen delivery system to direct either CD4(+) or CD8(+) T cell activation, Bioeng Transl Med 2(2) (2017) 202–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Demoulins T, Ebensen T, Schulze K, Englezou PC, Pelliccia M, Guzman CA, Ruggli N, McCullough KC, Self-replicating RNA vaccine functionality modulated by fine-tuning of polyplex delivery vehicle structure, J Control Release 266 (2017) 256–271. [DOI] [PubMed] [Google Scholar]
- [37].Cui L, Osada K, Imaizumi A, Kataoka K, Nakano K, Feasibility of a subcutaneously administered block/homo-mixed polyplex micelle as a carrier for DNA vaccination in a mouse tumor model, J Control Release 206 (2015) 220–31. [DOI] [PubMed] [Google Scholar]
- [38].Ferrito MT, Tirrell DA, Poly(2-ethylacrylic acid), Macromolecular Syntheses 11 (1992) 59–62. [Google Scholar]
- [39].Preynat-Seauve O, Contassot E, Schuler P, Piguet V, French LE, Huard B, Extralymphatic tumors prepare draining lymph nodes to invasion via a T-cell cross-tolerance process, Cancer Res 67(10) (2007) 5009–16. [DOI] [PubMed] [Google Scholar]
- [40].Karttunen J, Sanderson S, Shastri N, Detection of rare antigen-presenting cells by the lacZ T-cell activation assay suggests an expression cloning strategy for T-cell antigens, Proceedings of the National Academy of Sciences (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Rosalia RA, Quakkelaar ED, Redeker A, Khan S, Camps M, Drijfhout JW, Silva AL, Jiskoot W, van Hall T, van Veelen PA, Janssen G, Franken K, Cruz LJ, Tromp A, Oostendorp J, van der Burg SH, Ossendorp F, Melief CJ, Dendritic cells process synthetic long peptides better than whole protein, improving antigen presentation and T-cell activation, Eur J Immunol 43(10) (2013) 2554–65. [DOI] [PubMed] [Google Scholar]
- [42].Gilchuk P, Knight FC, Wilson JT, Joyce S, Eliciting Epitope-Specific CD8+ T Cell Response by Immunization with Microbial Protein Antigens Formulated with alpha-Galactosylceramide: Theory, Practice, and Protocols, Methods Mol Biol 1494 (2017) 321–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rodenko B, Toebes M, Hadrup SR, van Esch WJ, Molenaar AM, Schumacher TN, Ovaa H, Generation of peptide-MHC class I complexes through UV-mediated ligand exchange, Nat Protoc 1(3) (2006) 1120–32. [DOI] [PubMed] [Google Scholar]
- [44].Tomayko MM, Reynolds CP, Determination of subcutaneous tumor size in athymic (nude) mice, Cancer Chemother Pharmacol 24(3) (1989) 148–54. [DOI] [PubMed] [Google Scholar]
- [45].Hirosue S, Kourtis IC, van der Vlies AJ, Hubbell JA, Swartz MA, Antigen delivery to dendritic cells by poly(propylene sulfide) nanoparticles with disulfide conjugated peptides: Cross-presentation and T cell activation, Vaccine 28(50) (2010) 7897–7906. [DOI] [PubMed] [Google Scholar]
- [46].York IA, Chang SC, Saric T, Keys JA, Favreau JM, Goldberg AL, Rock KL, The ER aminopeptidase ERAP1 enhances or limits antigen presentation by trimming epitopes to 8–9 residues, Nat Immunol 3(12) (2002) 1177–84. [DOI] [PubMed] [Google Scholar]
- [47].Kilchrist KV, Evans BC, Brophy CM, Duvall CL, Mechanism of Enhanced Cellular Uptake and Cytosolic Retention of MK2 Inhibitory Peptide Nano-polyplexes, Cell Mol Bioeng 9(3) (2016) 368–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Obst R, van Santen HM, Melamed R, Kamphorst AO, Benoist C, Mathis D, Sustained antigen presentation can promote an immunogenic T cell response, like dendritic cell activation, Proc Natl Acad Sci U S A 104(39) (2007) 15460–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Iezzi G, Karjalainen K, Lanzavecchia A, The duration of antigenic stimulation determines the fate of naive and effector T cells, Immunity 8(1) (1998) 89–95. [DOI] [PubMed] [Google Scholar]
- [50].Remakus S, Ma X, Tang L, Xu RH, Knudson C, Melo-Silva CR, Rubio D, Kuo YM, Andrews A, Sigal LJ, Cutting Edge: Protection by Antiviral Memory CD8 T Cells Requires Rapidly Produced Antigen in Large Amounts, J Immunol (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Saveanu L, Carroll O, Weimershaus M, Guermonprez P, Firat E, Lindo V, Greer F, Davoust J, Kratzer R, Keller SR, Niedermann G, van Endert P, IRAP identifies an endosomal compartment required for MHC class I cross-presentation, Science 325(5937) (2009) 213–7. [DOI] [PubMed] [Google Scholar]
- [52].Bijker MS, van den Eeden SJF, Franken KL, Melief CJM, Offringa R, van der Burg SH, CD8+ CTL priming by exact peptide epitopes in incomplete Freund's adjuvant induces a vanishing CTL response, whereas long peptides induce sustained CTL reactivity., The Journal of Immunology 179(8) (2007) 5033–5040. [DOI] [PubMed] [Google Scholar]
- [53].Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, Psaila B, Kaplan RN, Bromberg JF, Kang Y, Bissell MJ, Cox TR, Giaccia AJ, Erler JT, Hiratsuka S, Ghajar CM, Lyden D, Pre-metastatic niches: organ-specific homes for metastases, Nat Rev Cancer 17(5) (2017) 302–317. [DOI] [PubMed] [Google Scholar]
- [54].Nizard M, Roussel H, Diniz MO, Karaki S, Tran T, Voron T, Dransart E, Sandoval F, Riquet M, Rance B, Marcheteau E, Fabre E, Mandavit M, Terme M, Blanc C, Escudie JB, Gibault L, Barthes FLP, Granier C, Ferreira LCS, Badoual C, Johannes L, Tartour E, Induction of resident memory T cells enhances the efficacy of cancer vaccine, Nat Commun 8 (2017) 15221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Li AV, Moon JJ, Abraham W, Suh H, Elkhader J, Seidman MA, Yen M, Im E-J, Foley MH, Barouch DH, Irvine DJ, Generation of effector memory T cell-based mucosal and systemic immunity with pulmonary nanoparticle vaccination., Science Translational Medicine 5(204) (2013) 204ra130–204ra130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Gouttefangeas C, Rammensee HG, Personalized cancer vaccines: adjuvants are important, too, Cancer Immunol Immunother (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Cerundolo V, Silk JD, Masri SH, Salio M, Harnessing invariant NKT cells in vaccination strategies, Nat Rev Immunol 9(1) (2009) 28–38. [DOI] [PubMed] [Google Scholar]
- [58].Schlosser E, Mueller M, Fischer S, Basta S, Busch DH, Gander B, Groettrup M, TLR ligands and antigen need to be coencapsulated into the same biodegradable microsphere for the generation of potent cytotoxic T lymphocyte responses, Vaccine 26(13) (2008) 1626–1637. [DOI] [PubMed] [Google Scholar]
- [59].Moon JJ, Suh H, Bershteyn A, Stephan MT, Liu H, Huang B, Sohail M, Luo S, Um S.Ho, Khant H, Goodwin JT, Ramos J, Chiu W, Irvine DJ, Interbilayer-crosslinked multilamellar vesicles as synthetic vaccines for potent humoral and cellular immune responses, Nature Materials 10(3) (2011) 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, Boegel S, Schrors B, Vascotto F, Castle JC, Tadmor AD, Schoenberger SP, Huber C, Tureci O, Sahin U, Mutant MHC class II epitopes drive therapeutic immune responses to cancer, Nature 520(7549) (2015) 692–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, Behjati S, Velds A, Hilkmann H, Atmioui DE, Visser M, Stratton MR, Haanen JB, Spits H, van der Burg SH, Schumacher TN, High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma, Nat Med 21(1) (2015) 81–5. [DOI] [PubMed] [Google Scholar]
- [62].Melief CJ, van Hall T, Arens R, Ossendorp F, van der Burg SH, Therapeutic cancer vaccines, J Clin Invest 125(9) (2015) 3401–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.