

Abstract
Events responsible for cardiovascular mortality and morbidity are predominantly caused by rupture of “vulnerable” atherosclerotic lesions. Vascular smooth muscle cells (VSMCs) play a key role in atherogenesis and have historically been considered beneficial for plaque stability. VSMCs constitute the main cellular component of the protective fibrous cap within lesions and are responsible for synthesising strength‐giving extracellular matrix components. However, lineage‐tracing experiments in mouse models of atherosclerosis have shown that, in addition to the fibrous cap, VSMCs also give rise to many of the cell types found within the plaque core. In particular, VSMCs generate a substantial fraction of lipid‐laden foam cells, and VSMC‐derived cells expressing markers of macrophages, osteochondrocyte, and mesenchymal stem cells have been observed within lesions. Here, we review recent studies that have changed our perspective on VSMC function in atherosclerosis and discuss how VSMCs could be targeted to increase plaque stability.
Abbreviations
- ABCA1
ATP‐binding cassette transporter
- AP‐1
activator protein‐1
- APOE
apolipoprotein E
- CArG
CC(A/T‐rich)6GG
- ECM
extracellular matrix
- H3K27me3
histone H3 lysine 27 trimethylation
- H3K4me2
histone H3 lysine 4 dimethylation
- H3K9me2/3
histone H3 lysine 9 dimethylation/trimethylation
- KLF4
Krüppel‐like factor 4
- Ldlr
LDL receptor
- MYH11
myosin heavy chain 11
- oxLDL
oxidised LDL
- Sca1
stem cell antigen 1
- SMA
smooth muscle actin
- SMMHC
smooth muscle myosin heavy chain
- SRF
serum response factor
- TCF21
transcription factor 21
- VSMC
vascular smooth muscle cell
- YFP
yellow fluorescent protein.
1. INTRODUCTION
Atherosclerosis, the leading cause of death worldwide, is a chronic and progressive inflammatory disease of large‐ to medium‐sized blood vessels (Libby, Ridker, & Hansson, 2011; Tabas, Garcia‐Cardena, & Owens, 2015; http://www.who.int/mediacentre/factsheets/fs310/en/). Atherosclerotic plaques consist of lipids and extracellular matrix (ECM) and involve several cell types, including bone marrow‐derived cells, vascular smooth muscle cells (VSMCs), and endothelial cells. The process of atherogenesis is complex and can be characterised by the following main stages (recently reviewed by Basatemur et al. (2019)). Firstly, endothelial cell damage and dysfunction stimulates the accumulation and oxidation of LDL within the vessel wall. Oxidised LDL (oxLDL) attracts monocytes from the blood into the subendothelial intima where they transform into macrophages, which ingest lipoproteins to become foam cells. The subsequent production of inflammatory mediators and cytokines stimulates VSMCs to migrate from the media to the intima where they proliferate and secrete ECM proteins. Importantly, VSMC accumulation in the intimal space (referred to as diffuse intimal thickening) occurs at sites of aberrant flow in humans already in utero and is thought to predispose for plaque development (Basatemur et al., 2019). In progressing plaques, macrophages and VSMCs undergoing cell death release lipids, which accumulate within the centre of the plaque to form the necrotic core. VSMCs are thought to migrate and proliferate to encage the necrotic core and create a fibrous cap that stabilises the plaque. Thinning of the fibrous cap in advanced plaques increases the risk of rupture, which triggers thrombus formation and subsequent clinical complications including heart attack and stroke (Libby et al., 2011; Tabas et al., 2015). In non‐lethal cases, VSMCs are thought to accumulate at the rupture site and secrete strength‐giving ECM proteins to restore the integrity of the plaque surface (Bentzon, Otsuka, Virmani, & Falk, 2014; Bentzon, Sondergaard, Kassem, & Falk, 2007; Davies, Bland, Hangartner, Angelini, & Thomas, 1989). However, this healing process may also have adverse effects such as constrictive remodelling of the vascular wall (Bentzon et al., 2014; Burke et al., 2001). It is therefore of considerable therapeutic importance to understand the mechanisms that regulate plaque stability.
Post‐mortem and clinical imaging studies have shown that vulnerable atherosclerotic plaque typically displays a thin fibrous cap, often containing micro‐calcifications, covering a lipid‐rich necrotic core, which is infiltrated by large numbers of bone marrow‐derived cells (Bennett, Sinha, & Owens, 2016; Durham, Speer, Scatena, Giachelli, & Shanahan, 2018; Shankman et al., 2015; Figure 1a). In contrast, stable lesions are thought to have a thick collagen‐rich fibrous cap covering a plaque core, which contains a high ratio of αSMA‐positive to CD68‐positive cells and possibly macro‐calcified deposits (Bennett et al., 2016; Durham et al., 2018; Shankman et al., 2015). The many cell types contributing to atherosclerotic lesions each influence plaque stability. However, an increasing body of evidence, including many genetic lineage‐tracing studies, has demonstrated that VSMCs play a substantial role in atherogenesis. This review discusses newly discovered aspects of VSMC biology, which could be targeted to detect, prevent, or treat vulnerable atherosclerotic plaques.
Figure 1.
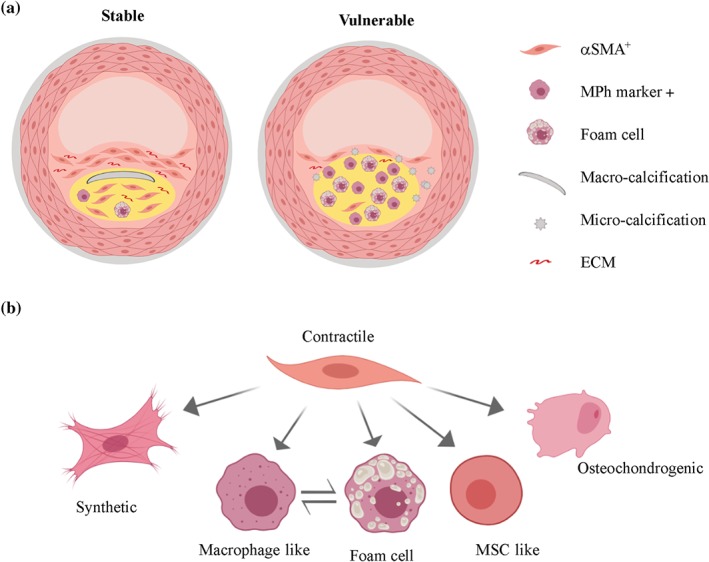
Stable versus vulnerable atherosclerotic plaque and vascular smooth muscle cell (VSMC)‐derived plaque cell phenotypes. (a) A simplified scheme showing a stable lesion with a thick collagen‐rich (extracellular matrix [ECM]) fibrous cap covering a plaque core (yellow area), which contains a high ratio of αSMA‐positive (αSMA+) cells compared with cells expressing macrophage‐associated markers (MPh‐marker +) and macro‐calcified deposits. In contrast, vulnerable plaques have a thin fibrous cap, which often contains micro‐calcified deposits, fewer cells, and less ECM. The lipid‐rich core (yellow area) of vulnerable lesions includes numerous foam cells as well as a high ratio of cells expressing macrophage‐associated markers compared with αSMA‐positive cells. Please note that details such as endothelial cells, adventitial cells, and internal and external elastic lamina are not displayed. (b) Contractile VSMCs can alter their phenotype to a more active “synthetic” state in which they up‐regulate selective gene sets important for vascular remodelling, including cytokines, chemokines, proteases, and adhesion proteins. Within plaques, VSMCs also give rise to foam cells or express markers traditionally associated with other cell types, such as macrophages, mesenchymal stem cells (MSCs), or osteochondrocytes. The relative contribution of VSMC‐derived plaque cell phenotypes in stable versus vulnerable plaque remains unknown
2. PHENOTYPIC MODULATION OF VSMCs
In the healthy blood vessel, VSMCs exhibit a low rate of proliferation and low synthetic activity and express a unique set of contractile proteins, essential for the contraction and relaxation of the vascular wall (Bennett et al., 2016). As reviewed by Owens, Kumar, and Wamhoff (2004), many VSMC‐specific genes that encode contractile proteins, including αSMA/ACTA2, Calponin1/CNN1, SM22α/TAGLN, and SMMHC/MYH11, are controlled by CC(A/T‐rich)6GG (CArG) cis‐regulatory elements in their promoter, which are bound by the widely expressed transcription factor, serum response factor (SRF). To achieve cell‐type‐specific expression of CArG‐dependent contractile VSMC genes, SRF associates with myocardin, which is only expressed in the vasculature by VSMCs (Wang et al., 2001).
Despite being a highly differentiated and specialised cell type, VSMCs retain remarkable plasticity and can alter their quiescent “contractile” phenotype to a more active “synthetic” state (Figure 1b; Chamley‐Campbell, Campbell, & Ross, 1979; Alexander & Owens, 2012). Synthetic VSMCs can re‐acquire many characteristics of the contractile phenotype, suggesting that the phenotypic switch is reversible (Aikawa et al., 1997; Christen et al., 2001; Manderson, Mosse, Safstrom, Young, & Campbell, 1989; Sottiurai et al., 1989; Thyberg, Blomgren, Hedin, & Dryjski, 1995; Thyberg, Blomgren, Roy, Tran, & Hedin, 1997). The synthetic VSMC phenotype is characterised by loss of contractile marker expression and up‐regulation of selective gene sets, including pro‐inflammatory cytokines and MMPs, leading to increased cell migration, proliferation, and secretion of pro‐inflammatory cytokines (Alexander & Owens, 2012; Clarke, Talib, Figg, & Bennett, 2010; Owens et al., 2004). Such phenotypic switching is required to maintain vascular homeostasis and regulate vascular response to injury and inflammation but can become dysregulated in disease.
3. ORIGIN OF VSMC‐LIKE CELLS WITHIN ATHEROSCLEROTIC PLAQUES
VSMCs down‐regulate contractile VSMCs genes in response to injury and inflammation, which may be linked to the reduced expression of myocardin, a key regulator of the contractile VSMC state, during plaque development (Ackers‐Johnson et al., 2015). Furthermore, contractile VSMC markers can be induced in other cell types (Gomez & Owens, 2012). For example, myofibroblasts express αSMA (Darby, Skalli, & Gabbiani, 1990), and adventitial cells have been reported to express contractile VSMC markers in vitro (Hu et al., 2004). This promiscuous expression of proposed lineage‐specific cell markers has prompted heated debate regarding the origin of VSMC‐like cells within atherosclerotic plaque.
Originally, VSMC‐like cells within plaque were suggested to be of myeloid origin (Sata et al., 2002). However, these claims have later been refuted with the emergence of genetic lineage‐tracing studies. For example, Iwata et al. (2010) transplanted bone marrow cells from mice expressing LacZ under the control of the Myh11 promoter into Apoe −/− mice and showed that while bone marrow‐derived cells contribute to atherogenesis, they do not differentiate into Myh11‐expressing cells. Several groups have suggested that adventitial cells generate contractile marker‐expressing plaque cells (Chen et al., 2013; Hu et al., 2004). In 2016, Kramann et al. demonstrated that adventitial Gli1+ MSC‐like cells can generate αSMA‐positive cells and proposed that migration of these cells into atherosclerotic lesions contributes to plaque calcification. A subpopulation of progenitor cells within healthy blood vessels that express the transcription factor TCF21 have also been described (Nurnberg et al., 2015). The authors of this study observed that cells in the media and adventitia, which are TCF21 positive prior to injury, give rise to VSMC marker‐positive cells within the fibrous cap (Nurnberg et al., 2015). It has also been hypothesised that progenitor cells within the medial layer are responsible for the VSMC‐like cells observed within plaque. For example, Myh11‐negative cells within the media were found to express multipotency‐associated factors, including Sox17, Sox10, and S100b when cultured (Tang et al., 2012). In this study, the authors used the VSMC‐specific Myh11 promoter to express a constitutively active Cre recombinase (Myh11–Cre) combined with a Rosa26–loxP–GFP recombination reporter and found an absence of GFP‐positive cells but the presence of S100b‐positive cells within the neointima following wire endothelial denudation injury to conclude that the identified cells are involved in disease‐associated cell accumulation (Tang et al., 2012). However, this observation was at odds with lineage‐tracing studies using temporally inducible recombinases (Nemenoff et al., 2011), which are key for analysis of cell fate, and the conclusions made by Tang et al. were subsequently challenged in a paper co‐authored by a number of experts in the field (Nguyen et al., 2013).
In 2013, Gomez et al. combined a tamoxifen‐inducible recombinase driven by the VSMC‐specific Myh11 promoter (Myh11–CreERt2) with the Rosa26–STOP–flox–YFP reporter (Figure 2a) for lineage tracing of VSMCs in the Apoe −/− mouse model of atherosclerosis (Gomez et al., 2013). In such experiments, animals are treated with tamoxifen to induce recombination‐mediated expression of YFP‐reporter in healthy VSMCs before being fed a high‐fat diet. Since YFP reporter expression is stably maintained, independent of Myh11 expression, this model allows tracing the fate of mature VSMC during atherosclerosis (Figure 2a). This study demonstrated that a large proportion of cells within the plaque is VSMC‐derived and that most plaque cells of VSMC origin do not express the classical contractile marker αSMA (Gomez et al., 2013). Since then, several other genetic fate mapping studies have confirmed this using transgenic mice with similar tamoxifen‐regulated, VSMC‐specific recombinase, and Cre‐dependent reporter genes to label mature VSMCs (Albarran‐Juarez, Kaur, Grimm, Offermanns, & Wettschureck, 2016; Chappell et al., 2016; Feil et al., 2014; Gomez et al., 2013; Jacobsen et al., 2017; Misra et al., 2018; Shankman et al., 2015). More recently, VSMC lineage tracing using multicolour reporters (Figure 2b) has definitively shown that atherosclerotic lesions are oligoclonal in terms of VSMC‐derived cells (Chappell et al., 2016; Feil et al., 2014; Jacobsen et al., 2017; Misra et al., 2018). Therefore, VSMC accumulation within plaque arises from proliferation of a very small number of mature yet plastic Myh11‐expressing VSMCs (Chappell et al., 2016; Feil et al., 2014; Jacobsen et al., 2017; Misra et al., 2018). It is tempting to extrapolate this oligoclonal nature of atherogenesis to human lesions, especially when considering studies on X‐chromosome inactivation patterns that suggest clonal cell expansion in human plaque (Benditt & Benditt, 1973).
Figure 2.
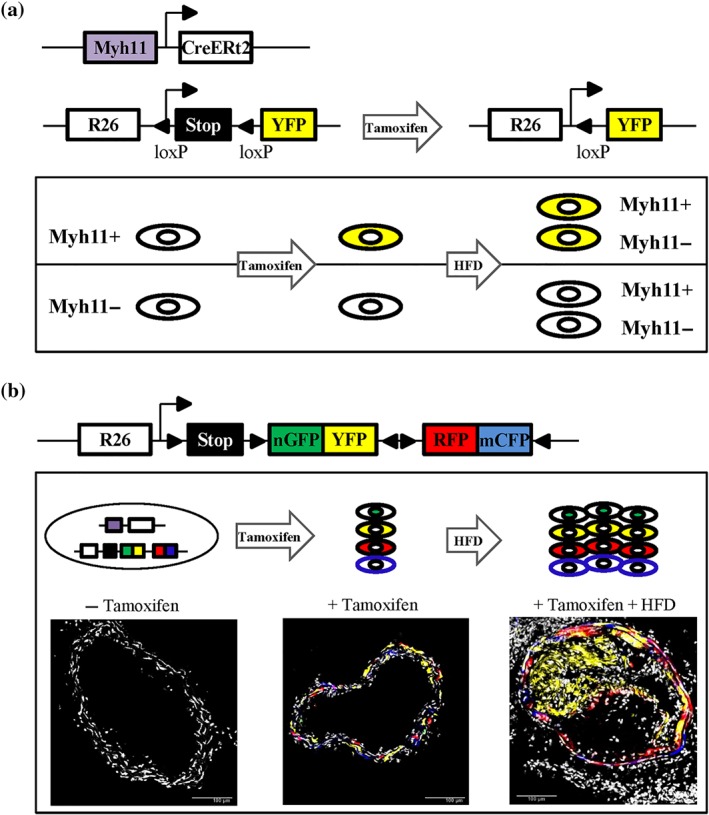
Genetic lineage labelling of vascular smooth muscle cells (VSMCs). (a) Diagram of the Myh11–CreERt2 transgene and the ROSA26–YFP reporter allele. Tamoxifen‐induced recombination at the ROSA26–YFP locus results in the expression of YFP protein, which is stably propagated within progeny after high‐fat diet (HFD)‐induced atherogenesis independent of Myh11 expression levels (b) Schematic of the ROSA26–Confetti reporter allele. Tamoxifen‐induced recombination at the ROSA26–Confetti locus results in stochastic expression of one of four fluorescent proteins (nuclear GFP, YFP, RFP, or membrane‐associated CFP), which are stably propagated within progeny after HFD‐induced atherogenesis. Confocal images show non‐labelled and Myh11‐lineage Confetti‐labelled VSMCs in carotid arteries before and after HFD‐induced atherogenesis. Scale bars are 100 μm
4. VSMC BEHAVIOUR WITHIN ATHEROSCLEROTIC PLAQUE
Genetic lineage‐tracing studies have shown that VSMCs modulate their phenotype in response to signals within the surrounding milieu (Chappell et al., 2016; Feil et al., 2014; Gomez et al., 2013; Jacobsen et al., 2017; Misra et al., 2018 ; Shankman et al., 2015). With the progression of atherosclerosis, VSMCs down‐regulate contractile proteins, adopt a more proliferative state, become more migratory, and respond to inflammatory signals (Allahverdian, Chaabane, Boukais, Francis, & Bochaton‐Piallat, 2018; Bennett et al., 2016).
In addition to adopting a “synthetic” state, VSMCs within atherosclerotic lesions can also alter their gene expression profile to resemble various other cell types (Figure 1b). For example, VSMC‐derived plaque cells have been reported to express markers that are traditionally associated with macrophages (LAMP2/MAC3, LGALS3/MAC2, and CD68; Chappell et al., 2016; Dobnikar et al., 2018; Feil et al., 2014; Misra et al., 2018; Shankman et al., 2015), mesenchymal stem cells (Sca1; Dobnikar et al., 2018; Shankman et al., 2015), and myofibroblasts (Acta2 and Pdgfrb; Misra et al., 2018; Shankman et al., 2015). A number of studies also suggest that VSMCs can acquire an osteochondrocytic transcriptional repertoire (Alp, Bglap, Opn, Runx2, and Bmp2) as reviewed by Durham et al. (2018). These phenotypically distinct cells of VSMC origin might influence multiple processes underlying atherosclerotic plaque stability, including lesion growth, lipid retention, inflammation, and ECM composition (Allahverdian et al., 2018; Bennett et al., 2016; Durham et al., 2018; Johnson, 2017). Therefore, it has been proposed that VSMC‐derived cells can both improve plaque stability and exacerbate plaque rupture (Allahverdian et al., 2018; Bennett et al., 2016; Durham et al., 2018; Johnson, 2017).
5. TARGETING A VSMC PROGENITOR
The existence of specialised VSMCs “response” population could explain the selective clonal VSMC expansion within plaque observed in mouse models of atherosclerosis (Chappell et al., 2016; Feil et al., 2014; Jacobsen et al., 2017; Misra et al., 2018). Several studies have reported subpopulations of VSMCs, which express multipotency‐associated genes (Cherepanova et al., 2016; Dobnikar et al., 2018; Sainz et al., 2006; Shankman et al., 2015). These observations have led to discussions regarding the similarities between atherosclerosis and cancer (DiRenzo, Owens, & Leeper, 2017). Like emerging pro‐efferocytic therapies that appear to target the root of cancer, selective elimination of hyper‐proliferative VSMC subpopulations has been proposed as a strategy to limiting atherosclerotic plaque growth (DiRenzo et al., 2017). However, studies have shown that plaque size is not a suitable surrogate for plaque stability. Rather than lesion size, the lipid content of the necrotic core, the thickness of the fibrous cap, the composition of the ECM, and the presence of calcification appear to provide a clearer indication of plaque vulnerability (Baylis, Gomez, & Owens, 2017). Targeting other aspects of VSMC‐progenitor function, such as migration, transdifferentiation, or synthetic activity, might therefore be a more feasible approach to treat vulnerable plaque.
The nature of VSMC‐progenitor populations and the mechanisms regulating their behaviour in atherogenesis is currently being investigated. Cherepanova et al. (2016) found that VSMCs express the pluripotency factor Oct4/Pou5f1. The authors show that Oct4 expression in VSMCs promotes migration and investment of VSMCs into the fibrous cap and improves lesion stability (Cherepanova et al., 2016). Therefore, it may be beneficial to encourage the survival or proliferation of Oct4‐positive VSMC‐derived cells or force expression of Oct4 in VSMCs to treat vulnerable plaque. Furthermore, a rare population of VSMCs express the multipotent progenitor marker stem cell antigen 1 (Sca1/Ly6a; Dobnikar et al., 2018; Sainz et al., 2006). Single cell transcriptomics showed that Sca1 expression in VSMCs correlates negatively with expression of contractile VSMC genes and is associated with up‐regulation of genes that are induced by VSMCs in response to inflammation and growth factors (Dobnikar et al., 2018). As the proportion of VSMCs expressing Sca1 is up‐regulated upon exposure to stimuli known to induce phenotypic switching (Dobnikar et al., 2018), it is tempting to hypothesise that Sca1 expression might mark a primed VSMC state that can readily respond to environmental cues (Dobnikar et al., 2018). In support of this hypothesis, Majesky et al. (2017) used lineage tracing to show that differentiated VSMCs generate a subpopulation of Sca1‐positive cells in the adventitia, which could transdifferentiate into several cell types in vitro, including macrophages. The generation of VSMC‐derived Sca1‐positive cells was dependent on the induction of the pluripotency factor KLF4 (Majesky et al., 2017), which has previously been implicated in the regulation of VSMC plasticity.
KLF4 negatively regulates VSMC contractility by interacting with SRF to repress myocardin‐induced activation of contractile VSMC genes (He et al., 2015; Liu et al., 2005; Liu, Sinha, & Owens, 2003). In addition, KLF4 has been shown to repress contractile VSMC gene expression by binding to the TGF‐β control element within their promoter, blocking the recruitment of activating complexes (Liu et al., 2003; Guo & Chen, 2012). Interestingly, VSMC‐specific conditional knockout of Klf4 in a mouse model of atherosclerosis partially suppressed macrophage‐like cell conversions and significantly reduced VSMC proliferation and apoptosis resulting in a thicker fibrous cap (Shankman et al., 2015). These results therefore suggest that KLF4 could be targeted to block VSMC transdifferentiation to a macrophage‐like state and possibly inhibit VSMCs‐derived foam cell accumulation.
6. TARGETING VSMC CONTRIBUTION TO THE FIBROUS CAP
It has long been recognised that atherosclerotic plaque stability depends on the thickness and composition of the fibrous cap and the lineage‐tracing studies described above have shown that the fibrous cap predominately contains cells derived from VSMCs. These VSMC‐derived cap cells are thought to be the primary source of collagen within the fibrous cap, providing mechanical tensile strength and resistance to rupture (Adiguzel, Ahmad, Franco, & Bendeck, 2009). Consistent with these findings, in vitro studies demonstrated that cultured VSMCs produce collagens in response to pro‐inflammatory stimuli such as IL‐1β (Adiguzel et al., 2009; Amento, Ehsani, Palmer, & Libby, 1991). Therefore, promoting VSMC collagen production may reduce cardiovascular events caused by plaque rupture. Knockout of Col15a specifically in VSMCs has been reported to reduce proliferation and result in smaller lesions, which lack a VSMC and ECM‐rich fibrous cap (Durgin et al., 2017). However, this study investigated plaque development rather than the regression or stability of established plaque. More research is required to investigate whether expression of specific ECM proteins by VSMCs similarly affects stability in established or healing plaque.
VSMCs also secrete proteases that degrade components of the ECM, including MMPs (Johnson, 2017). MMP2 is constitutively expressed in VSMCs (Newby, 2005), and several other MMPs exhibit enhanced expression within VSMCs in diseased blood vessels (Choudhary et al., 2006). VSMCs from human atherosclerotic plaque shoulder regions and areas of foam cell accumulation display increased MMP3, MMP9, and MMP12 activity compared with their medial counterparts (Galis, Sukhova, Lark, & Libby, 1994; Muller et al., 2014). This increased protease activity corresponds to regions containing higher levels of pro‐inflammatory cytokines released from necrotic VSMCs and macrophages (Galis, Sukhova, et al., 1994; Muller et al., 2014). Indeed, pro‐inflammatory cytokine‐induced expression of MMP genes has be observed in vitro (Galis et al., 1994).
MMPs play complex roles in late stage plaque and, as reviewed by Johnson, can both stabilise plaques and promote rupture (Johnson, 2017). MMPs affect plaque stability directly by degrading major components of the ECM, thereby weakening cap stability. For example, a selective MMP12 inhibitor has been shown to slow atherosclerotic plaque rupture in Apoe −/− mice (Johnson et al., 2011). However, matrix degradation also has indirect effects via modulating the migration and proliferation of VSMCs and infiltration of inflammatory cells into tissue, which incidentally affect the stability of atherosclerotic plaque (Johnson, 2017). Indeed, MMP activity is thought to stabilise atherosclerotic plaques by increasing contractile VSMC migration and proliferation. For instance, loss of either Mmp2 or Mmp9 in Apoe‐deficient animals reduces lesion stability, with plaques containing fewer VSMCs than macrophages compared with Apoe single knockout control animals (Johnson, George, Newby, & Jackson, 2005; Kuzuya et al., 2006). Due to the dual effect of MMPs on plaque stability and rupture, the development of broad spectrum MMP inhibitors to treat vascular disease has been problematic (Galis & Khatri, 2002; Newby, 2012). Given the effect of selective MMP12 inhibition, specific targeting of the activity or expression of individual proteases might be a more viable approach (Johnson, 2017) to stabilise established or healing plaque.
7. TARGETING VSMC‐DERIVED MACROPHAGE‐LIKE AND FOAM CELLS
VSMC‐derived cells not only influence the structural integrity of the fibrous cap but also substantially contribute to the generation of the plaque core. Genetic lineage‐tracing studies have shown that a large proportion of VSMC‐derived cells within the plaque express macrophage markers including CD68 (54%), Mac2 (62%), and Mac3 (Albarran‐Juarez et al., 2016; Chappell et al., 2016; Feil et al., 2014; Shankman et al., 2015). Like macrophages, some VSMC‐derived plaque cells actively participate in lipid ingestion and processing. Indeed, 81% of VSMC‐derived cells in atherosclerotic plaque take up oxLDL to form foam cells (Feil et al., 2014), and up to 75% of all foam cells within lesions of Apoe−/− animals are VSMC derived (Wang et al., 2019). Furthermore, cholesterol loading of VSMCs in vitro induces macrophage‐related gene expression (e.g., Cd68 and Lgals3/Mac2; Rong, Shapiro, Trogan, & Fisher, 2003), and VSMC treatment with oxLDL up‐regulates the expression of ACAT1, an intracellular enzyme that breaks down excess free cholesterol to facilitate storage in cytoplasmic lipid droplets (Yin et al., 2014). Although this process helps clearing atherogenic lipoprotein complexes from the vessel wall, excessive oxLDL uptake has deleterious effects (Maguire, Pearce, & Xiao, 2018). Lipid‐rich foam cells secrete a variety of pro‐inflammatory mediators, elicit pro‐apoptotic pathways, and attenuate clearance of dying cells leading to increased growth and destabilisation of the necrotic core (Maguire et al., 2018).
Importantly, transdifferentiation of VSMCs to a macrophage‐like state has also been observed in human atherosclerotic lesions (Allahverdian, Chehroudi, McManus, Abraham, & Francis, 2014; Chappell et al., 2016; Feil et al., 2014; Shankman et al., 2015). In human coronary artery plaques, 18% of CD68+ cells were marked by dimethylation of histone H3 at lysine 4 (H3K4me2) within the MYH11 promoter (Shankman et al., 2015), an epigenetic mark retained by VSMCs even after loss of the contractile state (Gomez et al., 2013). Interestingly, a large proportion of plaque cells marked by the VSMC lineage label (mouse) or αSMA (human) showed evidence of lipid ingestion (Feil et al., 2014). Moreover, Allahverdian et al. (2014) found that 50% of foam cell populations in human atherosclerotic lesions express αSMA but not CD45, suggesting that they are generated from VSMCs, although the origin of these cells remains to be verified. These αSMA‐positive foam cells exhibit lower levels of the cholesterol exporter ABCA1 compared with CD45‐positive cells in late stage plaque (Allahverdian et al., 2014). With these observations in mind, it is tempting to speculate that VSMC‐derived foam cells contribute significantly to lipid retention within the necrotic core in human lesions.
Perhaps blocking VSMC transdifferentiation to a macrophage‐like state would slow foam cell accumulation. However, it is important to remember that macrophages are also a very heterogeneous cell type and contribute to numerous processes in addition to foam cell formation (Gibson, Domingues, & Vieira, 2018). For example, M2 macrophage subpopulations are known to stimulate remodelling and anti‐inflammatory responses and hence promote plaque stability (Gibson et al., 2018; Maguire et al., 2018). It will be important to test whether VSMCs expressing macrophage markers also adopt such athero‐protective properties in order to understand how these cells affect plaque stability.
8. TARGETING VSMC‐DERIVED OSTEOCHONDROGENIC CELLS
As reviewed by Durham et al. (2018), arterial calcification is caused in part by transdifferentiation of VSMCs within the vessel wall. Inflammation, apoptosis, and oxidative stress are all thought to drive VSMC differentiation to an osteochondrogenic state and stimulate calcification (Durham et al., 2018). Lineage tracing of SM22α‐expressing cells (Tagln–Cre) in the Apoe −/− murine model of atherosclerosis revealed that ~80% of osteochondrogenic‐like (Runx2/Cbfa1+) cells and 98% of chondrocyte‐like (Sox9 + Col2a1+) cells within plaque are VSMC‐derived cells that have lost contractile VSMC marker gene expression (Naik et al., 2012). The location of these osteochondrogenic/chondrocyte‐like VSMC‐derived cells was consistent with areas of calcification within the fibrous cap and core observed in human lesions (Hutcheson et al., 2016; Otsuka, Sakakura, Yahagi, Joner, & Virmani, 2014). In support of these findings, single‐cell RNA sequencing of VSMC lineage cells (Myh11–CreERt2) from atherosclerotic plaque in Apoe −/− mice found that a subset of VSMC lineage cells express chondrocytic genes (Sox9, Ibsp, and Chad), consistent with a calcifying phenotype (Dobnikar et al., 2018). Together, these observations suggest a positive role for VSMCs in promoting vascular calcification. Indeed, deficiency of pro‐osteogenic transcription factors Msx1 and Msx2 in VSMCs within atherosclerotic Ldlr −/− mice reduced vascular calcification (Cheng et al., 2014). Moreover, VSMC‐specific deletion of Runx2 attenuated vascular osteochondrogenesis and calcification without influencing lipid metabolism, monocyte/macrophage recruitment, or atherosclerotic lesion size (Lin et al., 2016). This finding implies that the factors regulating vascular calcification are distinct from those that govern lipid storage.
Like many processes underlying atherogenesis, the effects of calcification on plaque stability are context dependent. Whereas micro‐calcifications, particularly of the fibrous cap, are associated with greater risk of plaque rupture (Kelly‐Arnold et al., 2013), macro‐calcification is widely considered to increase plaque stability (Imoto et al., 2005; Lin et al., 2006; Wong, Thavornpattanapong, Cheung, Sun, & Tu, 2012). Macrophage‐derived pro‐inflammatory cytokines such as TNF‐α, IL‐1β, IL‐6, and oncostatin M have been shown to promote VSMC calcification (Ceneri et al., 2017; Parhami, Basseri, Hwang, Tintut, & Demer, 2002; Shioi et al., 2002; Tintut et al., 2002). Furthermore, molecular imaging studies have identified a link between the presence of inflammation and calcification (Aikawa et al., 2007; Dweck et al., 2016). It has therefore been hypothesised that VSMCs undergo early stages of osteogenic differentiation in inflammatory plaques (Shanahan, 2007; New & Aikawa, 2011; Pugliese, Iacobini, Blasetti Fantauzzi, & Menini, 2015). Moreover, inflammatory signals that induce VSMC apoptosis promote matrix calcification, primarily through the release of the calcifying membrane‐bound matrix vesicles, which act as nucleation sites of calcification (Kapustin et al., 2011; Proudfoot et al., 2000). Together, these processes are thought to produce micro‐calcified deposits detrimental to the structural integrity of the plaque (Pugliese et al., 2015; Shanahan, 2007; Shioi & Ikari, 2018). However, on the resolution of inflammation, macroscopic calcium deposition is proposed to be facilitated through induction of osteoblastic differentiation and maturation of VSMCs (Pugliese et al., 2015; Shanahan, 2007; Shioi & Ikari, 2018). Macro‐calcification is associated with organised structures similar to that observed in authentic bone tissue and is therefore thought to stabilise atherosclerotic lesions (Shanahan, 2007; Pugliese et al., 2015; Shioi & Ikari, 2018).
Consequently, it is not clear how simply preventing VSMC conversion to an osteochondrocytic cell state will affect the risk of plaque rupture. However, the presence of micro‐calcification can instead be used to detect vulnerable plaques. Non‐invasive detection of micro‐calcification, as opposed to macro‐calcification, within high‐risk plaques is presently possible using 18F‐sodium fluoride PET (Irkle et al., 2015). This clinical imaging platform could therefore be used to test the efficacy of pharmacological therapies targeting vulnerable plaque.
9. TARGETING VSMC INFLAMMATORY ACTIVATION
Many experimental and clinical studies have associated inflammation with atherogenesis and increased risk of cardiovascular events (Libby, 2017). Indeed, C‐reactive protein blood concentration, an inflammatory biomarker, improves the prediction of cardiovascular events beyond traditional risk algorithms alone (Ridker, 2016).
Vascular inflammation involves bidirectional interaction between resident vascular cells and inflammatory cells, which is governed in part by pro‐inflammatory cytokines. Similar to other cell types within the plaque, VSMCs can both produce pro‐inflammatory cytokines and respond to those secreted from other cells by activating NF‐κB, AP‐1, JAK–STAT, and SMAD signalling pathways (Schober, 2008; Sprague & Khalil, 2009). IL‐1β, a key cytokine in vascular inflammation, is known to induce VSMCs to switch from a contractile to a synthetic phenotype, stimulating NF‐κB‐dependent transcription of cytokines (e.g., IL‐6), chemokines (e.g., CCL2), and MMPs (Nagase, Visse, & Murphy, 2006; Lim & Park, 2014). IL‐6 has been observed to up‐regulate VSMC migration, proliferation, and vascular calcification while attenuating VSMC contraction (Kurozumi et al., 2016; Lee et al., 2016; Watanabe et al., 2004). Also, IL‐6 treatment of cultured VSMCs activates the JAK–STAT pathway, which induces the expression of CCL2 (Watanabe et al., 2004). CCL2 plays a major role in the recruitment of monocytes and T cells to the vessel wall and has been shown to stimulate VSMC migration and proliferation (Schober, 2008; Selzman et al., 2002).
Many inflammation‐responsive genes, including those induced by IL‐1β, are under NF‐κB transcriptional control (Chistiakov, Melnichenko, Grechko, Myasoedova, & Orekhov, 2018). Activated NF‐κB transcription factors have been observed in VSMC‐like nuclei within the intima of human atherosclerotic lesions (Bourcier, Sukhova, & Libby, 1997). Furthermore, selective inhibition of NF‐κB in VSMCs attenuated loss of the contractile state and reduced neointima formation after vascular injury (Yoshida, Yamashita, Horimai, & Hayashi, 2013), highlighting a key role of NF‐κB in regulating VSMC phenotype. Many studies have also shown that NF‐κB plays a direct role in regulating the expression of contractile VSMC genes. Activation of the NF‐κB signalling pathway in VSMCs results in binding of the NF‐κB–p65 subunit to the MYOCD promoter, decreasing the expression of myocardin and myocardin‐dependent contractile genes (e.g., αSMA/ACTA2, SM22α/TAGLN, and SMMHC/MYH11; Tang et al., 2008; Yoshida et al., 2013; Singh & Zheng, 2014). Together, these studies suggest that NF‐κB inhibition, which specifically affects VSMCs could be a potential therapeutic target.
Alternatively, plaque stability could also be promoted by directly inhibiting VSMC exposure to inflammatory cytokines. Promisingly, the Canakinumab Anti‐Inflammatory Thrombosis Outcomes Study trial showed that reducing inflammation by treatment with canakinumab, an IL‐1β‐neutralising monoclonal antibody, significantly lowered the rate of cardiovascular events compared with placebo (Ridker et al., 2017). However, IL‐1β antibody treatment of Apoe‐deficient mice where VSMCs were labelled by a tamoxifen‐inducible Myh11‐driven Cre recombinase prior to inducing advanced atherosclerosis by a high‐fat diet promoted multiple characteristics of unstable plaque (Gomez, Baylis, Durgin, & Newman, 2018), such as a 40% decrease in contractile VSMC content, a 30% decrease in collagen content, and a 50% increase in M2 (Arg1+) macrophage content within the fibrous cap (Gomez et al., 2018). This study also used VSMC‐specific genetic deletion of Il1r1 combined with VSMC lineage tracing before feeding mice a high‐fat diet to demonstrate that IL‐1β signalling is required for VSMC investment into lesions and the fibrous cap (Gomez et al., 2018).
Like IL‐1β, the role of many other cytokines in VSMC behaviour remains unclear (Lim & Park, 2014). For example, Singh and Zheng (2014) report dual regulation of myocardin expression by TNF‐α in VSMCs. The authors show that, in cultured VSMCs, TNF‐α activates the NF‐κB signalling pathway and decreases the expression of myocardin and myocardin‐dependent genes by direct binding of NF‐κB (p65) to the MYOCD promoter (Singh & Zheng, 2014). In contrast, TNF‐α treatment of cultured VSMCs overexpressing myocardin significantly potentiates the expression of myocardin and CArG box‐containing contractile VSMC genes by stabilising myocardin mRNA via the NF‐κB and MAPK pathway (Singh & Zheng, 2014). These findings suggests that the effect of TNF‐α on myocardin activity depends on the VSMC phenotypic state. Therefore, blocking VSMC exposure to inflammatory stimuli or inhibiting the VSMC response to such stimuli may promote plaque stability or rupture in a context‐dependent manner.
10. EPIGENETIC TARGETS
As discussed, much is known regarding the signals that regulate VSMCs within atherosclerotic plaque. However, it is becoming increasingly clear that it is also important to understand the molecular mechanisms underlying VSMC behaviour at the epigenetic level. Recent studies have demonstrated that histone modifications, DNA methylation, and microRNAs all influence how VSMCs contribute to plaque (Alexander & Owens, 2012).
Interestingly, VSMCs within atherosclerotic lesions exhibit perturbed levels of histone methylation, including H3K27me3 (Greißel et al., 2015; Wierda et al., 2015) and H3K9me2/3 (Chen et al., 2017; Greißel et al., 2015). Additionally, VSMCs from diabetic patients display reduced levels of H3K9me2 compared with non‐diabetic controls, which suggests that dysregulation of H3K9me2 might underlie the vascular complications associated with diabetes (Chen et al., 2017; Villeneuve et al., 2008). Several studies also demonstrate that DNA methylation regulates VSMC behaviour in atherosclerotic plaque. Hiltunen et al. (2002) were the first to report DNA hypomethylation in advanced human atherosclerotic lesions. Azechi, Sato, Sudo, and Wachi (2014) observed that inhibiting DNA methyltransferases with 5‐aza‐2′‐deoxycytidine potentiates inorganic phosphate‐induced mineralisation in human aortic VSMCs, possibly through demethylation of the alkaline phosphatase promoter. However, it is important to consider local as well as global levels of histone and DNA modifications. For example, the DNA demethylase TET2 positively regulates the expression of SRF and contractile genes, including MYOCD and MYH11, in human VSMCs (Liu et al., 2013). Furthermore, TET2 expression levels are inversely correlated with severity of atherosclerosis in patients, and knock‐down of TET2 in mouse exacerbates vascular response to injury (Liu et al., 2013). Targeting the enzymes that regulate these de novo changes could be a promising strategy to inhibit the expression of inflammation‐responsive VSMC genes associated with plaque instability.
There is a growing body of evidence demonstrating that microRNAs regulate VSMC phenotype (Lu, Thavarajah, Gu, Cai, & Xu, 2018). For example, cultured VSMCs have been shown to switch to a macrophage‐like state after cholesterol loading by down‐regulating the microRNA (miR)‐143/145–myocardin axis (Vengrenyuk et al., 2015), a key pathway that is essential for maintaining the contractile VSMC state. Maintaining the expression of myocardin or miR‐143/145 prevented and reversed these phenotypic changes induced by cholesterol loading (Vengrenyuk et al., 2015). Manipulation of miRNA activity in vulnerable plaque could therefore be considered as a potential therapeutic strategy. As unstable plaque typically displays thin fibrous caps with a high ratio of macrophage‐like to VSMC‐like cells (Kolodgie et al., 2004), addition of miR‐143/145 mimics to block VSMC transdifferentiation to a macrophage‐like state could perhaps be used to stabilise plaque. Multiple miRNA‐based agents have now moved into the clinic to treat a range of diseases (Rupaimoole & Slack, 2017), suggesting that this might be a feasible strategy.
11. CONCLUSIONS
In recent years, our understanding of VSMCS within atherosclerotic plaque has changed dramatically. Historically, VSMCs were thought to be entirely beneficial by forming the fibrous cap, protecting the plaque from rupture. However, numerous genetic lineage mapping studies have definitively shown a wide‐ranging plasticity of VSMCs and suggested that these cells can modulate their behaviour in response to a variety of stimuli. Consequently, we now know that VSMCs have complex roles within atherosclerotic lesions and may act to both promote plaque stability and rupture depending on the context. To develop efficient therapeutic strategies to limit cardiovascular risk, additional knowledge about how specific VSMC‐derived cell types function in mature plaque is therefore needed. Additionally, mechanistic insight into the regulation of VSMC plasticity is required to enable specific interventions. Additionally, many murine studies focus on prevention of atherosclerosis in young healthy animals rather than regression or treatment of established plaque. As discussed by Baylis et al. (2017), there is therefore a need for implementing preclinical murine models of regression or treatment of established plaque that better mimic therapeutic intervention in humans to study VSMC function.
11.1. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGEMENTS
J.L.H. and H.F.J. are supported by the British Heart Foundation (FS/15/62/32032, RM/13/3/30159, RE/13/6/30180, and CH/20000003).
Harman JL, Jørgensen HF. The role of smooth muscle cells in plaque stability: Therapeutic targeting potential. Br J Pharmacol. 2019;176:3741–3753. 10.1111/bph.14779
REFERENCES
- Ackers‐Johnson, M. , Talasila, A. , Sage, A. P. , Long, X. , Bot, I. , Morrell, N. W. , … Sinha, S. (2015). Myocardin regulates vascular smooth muscle cell inflammatory activation and disease. Arteriosclerosis, Thrombosis, and Vascular Biology, 35(4), 817–828. 10.1161/ATVBAHA.114.305218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adiguzel, E. , Ahmad, P. J. , Franco, C. , & Bendeck, M. P. (2009). Collagens in the progression and complications of atherosclerosis. Vascular Medicine (London, England), 14(1), 73–89. 10.1177/1358863X08094801 [DOI] [PubMed] [Google Scholar]
- Aikawa, E. , Nahrendorf, M. , Figueiredo, J. L. , Swirski, F. K. , Shtatland, T. , Kohler, R. H. , … Weissleder, R. (2007). Osteogenesis associates with inflammation in early‐stage atherosclerosis evaluated by molecular imaging in vivo. Circulation, 116(24), 2841–2850. 10.1161/CIRCULATIONAHA.107.732867 [DOI] [PubMed] [Google Scholar]
- Aikawa, M. , Sakomura, Y. , Ueda, M. , Kimura, K. , Manabe, I. , Ishiwata, S. , … Nagai, R. (1997). Redifferentiation of smooth muscle cells after coronary angioplasty determined via myosin heavy chain expression. Circulation, 96(1), 82–90. 10.1161/01.CIR.96.1.82 [DOI] [PubMed] [Google Scholar]
- Albarran‐Juarez, J. , Kaur, H. , Grimm, M. , Offermanns, S. , & Wettschureck, N. (2016). Lineage tracing of cells involved in atherosclerosis. Atherosclerosis, 251, 445–453. 10.1016/j.atherosclerosis.2016.06.012 [DOI] [PubMed] [Google Scholar]
- Alexander, M. R. , & Owens, G. K. (2012). Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annual Review of Physiology, 74, 13–40. 10.1146/annurev-physiol-012110-142315 [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allahverdian, S. , Chaabane, C. , Boukais, K. , Francis, G. A. , & Bochaton‐Piallat, M. L. (2018). Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovascular Research, 114(4), 540–550. 10.1093/cvr/cvy022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allahverdian, S. , Chehroudi, A. C. , McManus, B. M. , Abraham, T. , & Francis, G. A. (2014). Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage‐like cells in human atherosclerosis. Circulation, 129(15), 1551–1559. 10.1161/CIRCULATIONAHA.113.005015 [DOI] [PubMed] [Google Scholar]
- Amento, E. P. , Ehsani, N. , Palmer, H. , & Libby, P. (1991). Cytokines and growth factors positively and negatively regulate interstitial collagen gene expression in human vascular smooth muscle cells. Arteriosclerosis and Thrombosis: A Journal of Vascular Biology/American Heart Association, 11(5), 1223–1230. 10.1161/01.ATV.11.5.1223 [DOI] [PubMed] [Google Scholar]
- Azechi, T. , Sato, F. , Sudo, R. , & Wachi, H. (2014). 5‐Aza‐2′‐deoxycytidine, a DNA methyltransferase inhibitor, facilitates the inorganic phosphorus‐induced mineralization of vascular smooth muscle cells. Journal of Atherosclerosis and Thrombosis, 21(5), 463–476. 10.5551/jat.20818 [DOI] [PubMed] [Google Scholar]
- Basatemur, G. L. , Jørgensen, H. F. , Clarke, M. C. H. , Bennett, M. R. , & Mallat, Z. (2019). Vascular smooth muscle cells in atherosclerosis. Nature Reviews Cardiology. 10.1038/s41569-019-0227-9 [DOI] [PubMed] [Google Scholar]
- Baylis, R. A. , Gomez, D. , & Owens, G. K. (2017). Shifting the focus of preclinical, murine atherosclerosis studies from prevention to late‐stage intervention. Circulation Research, 120(5), 775–777. 10.1161/CIRCRESAHA.116.310101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benditt, E. P. , & Benditt, J. M. (1973). Evidence for a monoclonal origin of human atherosclerotic plaques. Proceedings of the National Academy of Sciences of the United States of America, 70(6), 1753–1756. 10.1073/pnas.70.6.1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, M. R. , Sinha, S. , & Owens, G. K. (2016). Vascular smooth muscle cells in atherosclerosis. Circulation Research, 118(4), 692–702. 10.1161/CIRCRESAHA.115.306361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentzon, J. F. , Otsuka, F. , Virmani, R. , & Falk, E. (2014). Mechanisms of plaque formation and rupture. Circulation Research, 114(12), 1852–1866. 10.1161/CIRCRESAHA.114.302721 [DOI] [PubMed] [Google Scholar]
- Bentzon, J. F. , Sondergaard, C. S. , Kassem, M. , & Falk, E. (2007). Smooth muscle cells healing atherosclerotic plaque disruptions are of local, not blood, origin in apolipoprotein E knockout mice. Circulation, 116(18), 2053–2061. 10.1161/CIRCULATIONAHA.107.722355 [DOI] [PubMed] [Google Scholar]
- Bourcier, T. , Sukhova, G. , & Libby, P. (1997). The nuclear factor κ‐B signaling pathway participates in dysregulation of vascular smooth muscle cells in vitro and in human atherosclerosis. The Journal of Biological Chemistry, 272(25), 15817–15824. 10.1074/jbc.272.25.15817 [DOI] [PubMed] [Google Scholar]
- Burke, A. P. , Kolodgie, F. D. , Farb, A. , Weber, D. K. , Malcom, G. T. , Smialek, J. , & Virmani, R. (2001). Healed plaque ruptures and sudden coronary death: Evidence that subclinical rupture has a role in plaque progression. Circulation, 103(7), 934–940. 10.1161/01.CIR.103.7.934 [DOI] [PubMed] [Google Scholar]
- Ceneri, N. , Zhao, L. , Young, B. D. , Healy, A. , Coskun, S. , Vasavada, H. , … Morrison, A. R. (2017). Rac2 modulates atherosclerotic calcification by regulating macrophage interleukin‐1β production. Arteriosclerosis, Thrombosis, and Vascular Biology, 37(2), 328–340. 10.1161/ATVBAHA.116.308507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamley‐Campbell, J. , Campbell, G. R. , & Ross, R. (1979). The smooth muscle cell in culture. Physiological Reviews, 59(1), 1–61. 10.1152/physrev.1979.59.1.1 [DOI] [PubMed] [Google Scholar]
- Chappell, J. , Harman, J. L. , Narasimhan, V. M. , Yu, H. , Foote, K. , Simons, B. D. , … Jørgensen, H. F. (2016). Extensive proliferation of a subset of differentiated, yet plastic, medial vascular smooth muscle cells contribute to neointimal formation in mouse injury and atherosclerosis models. Circulation Research, 119(12), 1313–1323. 10.1161/CIRCRESAHA.116.309799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Zhang, J. , Yang, J. , Xu, L. , Hu, Q. , Xu, C. , … Jiang, H. (2017). Histone demethylase KDM3a, a novel regulator of vascular smooth muscle cells, controls vascular neointimal hyperplasia in diabetic rats. Atherosclerosis, 257, 152–163. 10.1016/j.atherosclerosis.2016.12.007 [DOI] [PubMed] [Google Scholar]
- Chen, Y. , Wong, M. M. , Campagnolo, P. , Simpson, R. , Winkler, B. , Margariti, A. , … Xu, Q. (2013). Adventitial stem cells in vein grafts display multilineage potential that contributes to neointimal formation. Arteriosclerosis, Thrombosis, and Vascular Biology, 33(8), 1844–1851. 10.1161/ATVBAHA.113.300902 [DOI] [PubMed] [Google Scholar]
- Cheng, S.‐L. , Behrmann, A. , Shao, J.‐S. , Ramachandran, B. , Krchma, K. , Bello Arredondo, Y. , … Towler, D. A. (2014). Targeted reduction of vascular Msx1 and Msx2 mitigates arteriosclerotic calcification and aortic stiffness in LDLR‐deficient mice fed diabetogenic diets. Diabetes, 63(12), 4326–4337. 10.2337/db14-0326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanova, O. A. , Gomez, D. , Shankman, L. S. , Swiatlowska, P. , Williams, J. , Sarmento, O. F. , … Owens, G. K. (2016). Activation of the pluripotency factor OCT4 in smooth muscle cells is atheroprotective. Nature Medicine, 22(6), 657–665. 10.1038/nm.4109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chistiakov, D. A. , Melnichenko, A. A. , Grechko, A. V. , Myasoedova, V. A. , & Orekhov, A. N. (2018). Potential of anti‐inflammatory agents for treatment of atherosclerosis. Experimental and Molecular Pathology, 104(2), 114–124. 10.1016/j.yexmp.2018.01.008 [DOI] [PubMed] [Google Scholar]
- Choudhary, S. , Higgins, C. L. , Chen, I. Y. , Reardon, M. , Lawrie, G. , Vick, G. W. 3rd , … Morrisett, J. D. (2006). Quantitation and localization of matrix metalloproteinases and their inhibitors in human carotid endarterectomy tissues. Arteriosclerosis, Thrombosis, and Vascular Biology, 26(10), 2351–2358. 10.1161/01.ATV.0000239461.87113.0b [DOI] [PubMed] [Google Scholar]
- Christen, T. , Verin, V. , Bochaton‐Piallat, M. , Popowski, Y. , Ramaekers, F. , Debruyne, P. , … Gabbiani, G. (2001). Mechanisms of neointima formation and remodeling in the porcine coronary artery. Circulation, 103(6), 882–888. 10.1161/01.CIR.103.6.882 [DOI] [PubMed] [Google Scholar]
- Clarke, M. C. , Talib, S. , Figg, N. L. , & Bennett, M. R. (2010). Vascular smooth muscle cell apoptosis induces interleukin‐1‐directed inflammation: Effects of hyperlipidemia‐mediated inhibition of phagocytosis. Circulation Research, 106(2), 363–372. 10.1161/CIRCRESAHA.109.208389 [DOI] [PubMed] [Google Scholar]
- Darby, I. , Skalli, O. , & Gabbiani, G. (1990). α‐Smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing. Laboratory Investigation; a Journal of Technical Methods and Pathology, 63(1), 21–29. [PubMed] [Google Scholar]
- Davies, M. J. , Bland, J. M. , Hangartner, J. R. , Angelini, A. , & Thomas, A. C. (1989). Factors influencing the presence or absence of acute coronary artery thrombi in sudden ischaemic death. European Heart Journal, 10(3), 203–208. 10.1093/oxfordjournals.eurheartj.a059467 [DOI] [PubMed] [Google Scholar]
- DiRenzo, D. , Owens, G. K. , & Leeper, N. J. (2017). “Attack of the clones”: Commonalities between cancer and atherosclerosis. Circulation Research, 120(4), 624–626. 10.1161/CIRCRESAHA.116.310091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobnikar, L. , Taylor, A. , Chappell, J. , Oldach, P. , Harman, J. , Oerton, E. , … Jørgensen, H. F. (2018). Disease‐relevant transcriptional signatures identified in individual smooth muscle cells from healthy mouse vessels. Nature Communications, 9, 4567 10.1038/s41467-018-06891-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durgin, B. G. , Cherepanova, O. A. , Gomez, D. , Karaoli, T. , Alencar, G. F. , Butcher, J. T. , … Connelly, J. J. (2017). Smooth muscle cell‐specific deletion of Col15a1 unexpectedly leads to impaired development of advanced atherosclerotic lesions. American Journal of Physiology. Heart and Circulatory Physiology, 312(5), H943–h958. 10.1152/ajpheart.00029.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham, A. L. , Speer, M. Y. , Scatena, M. , Giachelli, C. M. , & Shanahan, C. M. (2018). Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovascular Research, 114(4), 590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dweck, M. R. , Aikawa, E. , Newby, D. E. , Tarkin, J. M. , Rudd, J. H. , Narula, J. , & Fayad, Z. A. (2016). Noninvasive molecular imaging of disease activity in atherosclerosis. Circulation Research, 119(2), 330–340. 10.1161/CIRCRESAHA.116.307971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil, S. , Fehrenbacher, B. , Lukowski, R. , Essmann, F. , Schulze‐Osthoff, K. , Schaller, M. , & Feil, R. (2014). Transdifferentiation of vascular smooth muscle cells to macrophage‐like cells during atherogenesis. Circulation Research, 115(7), 662–667. 10.1161/CIRCRESAHA.115.304634 [DOI] [PubMed] [Google Scholar]
- Galis, Z. S. , & Khatri, J. J. (2002). Matrix metalloproteinases in vascular remodeling and atherogenesis: The good, the bad, and the ugly. Circulation Research, 90(3), 251–262. 10.1161/res.90.3.251 [DOI] [PubMed] [Google Scholar]
- Galis, Z. S. , Muszynski, M. , Sukhova, G. K. , Simon‐Morrissey, E. , Unemori, E. N. , Lark, M. W. , … Libby, P. (1994). Cytokine‐stimulated human vascular smooth muscle cells synthesize a complement of enzymes required for extracellular matrix digestion. Circulation Research, 75(1), 181–189. 10.1161/01.RES.75.1.181 [DOI] [PubMed] [Google Scholar]
- Galis, Z. S. , Sukhova, G. K. , Lark, M. W. , & Libby, P. (1994). Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. The Journal of Clinical Investigation, 94(6), 2493–2503. 10.1172/JCI117619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson, M. S. , Domingues, N. , & Vieira, O. V. (2018). Lipid and non‐lipid factors affecting macrophage dysfunction and inflammation in atherosclerosis. Frontiers in Physiology, 9, 654–654. 10.3389/fphys.2018.00654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez, D. , Baylis, R. A. , Durgin, B. G. , & Newman, A. A. C. (2018). Interleukin‐1β has atheroprotective effects in advanced atherosclerotic lesions of mice. Nature Medicine, 24(9), 1418–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez, D. , & Owens, G. K. (2012). Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovascular Research, 95(2), 156–164. 10.1093/cvr/cvs115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez, D. , Shankman, L. S. , Nguyen, A. T. , & Owens, G. K. (2013). Detection of histone modifications at specific gene loci in single cells in histological sections. Nature Methods, 10(2), 171–177. 10.1038/nmeth.2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greißel, A. , Culmes, M. , Napieralski, R. , Wagner, E. , Gebhard, H. , Schmitt, M. , … Pelisek, J. (2015). Alternation of histone and DNA methylation in human atherosclerotic carotid plaques. Thrombosis and Haemostasis, 114(2), 390–402. [DOI] [PubMed] [Google Scholar]
- Guo, X. , & Chen, S.‐Y. (2012). Transforming growth factor‐β and smooth muscle differentiation. World Journal of Biological Chemistry, 3(3), 41–52. 10.4331/wjbc.v3.i3.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, M. , Zheng, B. , Zhang, Y. , Zhang, X. H. , Wang, C. , Yang, Z. , … Wen, J. K. (2015). KLF4 mediates the link between TGF‐β1‐induced gene transcription and H3 acetylation in vascular smooth muscle cells. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology, 29(9), 4059–4070. 10.1096/fj.15-272658 [DOI] [PubMed] [Google Scholar]
- Hiltunen, M. O. , Turunen, M. P. , Hakkinen, T. P. , Rutanen, J. , Hedman, M. , Makinen, K. , … Ylä‐Herttuala, S. (2002). DNA hypomethylation and methyltransferase expression in atherosclerotic lesions. Vascular Medicine (London, England), 7(1), 5–11. 10.1191/1358863x02vm418oa [DOI] [PubMed] [Google Scholar]
- Hu, Y. , Zhang, Z. , Torsney, E. , Afzal, A. R. , Davison, F. , Metzler, B. , & Xu, Q. (2004). Abundant progenitor cells in the adventitia contribute to atherosclerosis of vein grafts in ApoE‐deficient mice. The Journal of Clinical Investigation, 113(9), 1258–1265. 10.1172/JCI19628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutcheson, J. D. , Goettsch, C. , Bertazzo, S. , Maldonado, N. , Ruiz, J. L. , Goh, W. , … Aikawa, E. (2016). Genesis and growth of extracellular‐vesicle‐derived microcalcification in atherosclerotic plaques. Nature Materials, 15(3), 335–343. https://doi.org/10.1038/nmat4519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imoto, K. , Hiro, T. , Fujii, T. , Murashige, A. , Fukumoto, Y. , Hashimoto, G. , … Matsuzaki, M. (2005). Longitudinal structural determinants of atherosclerotic plaque vulnerability: A computational analysis of stress distribution using vessel models and three‐dimensional intravascular ultrasound imaging. Journal of the American College of Cardiology, 46(8), 1507–1515. 10.1016/j.jacc.2005.06.069 [DOI] [PubMed] [Google Scholar]
- Irkle, A. , Vesey, A. T. , Lewis, D. Y. , Skepper, J. N. , Bird, J. L. , Dweck, M. R. , … Davenport, A. P. (2015). Identifying active vascular microcalcification by 18F‐sodium fluoride positron emission tomography. Nature Communications, 6, 7495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata, H. , Manabe, I. , Fujiu, K. , Yamamoto, T. , Takeda, N. , Eguchi, K. , … Nagai, R. (2010). Bone marrow‐derived cells contribute to vascular inflammation but do not differentiate into smooth muscle cell lineages. Circulation, 122(20), 2048–2057. 10.1161/CIRCULATIONAHA.110.965202 [DOI] [PubMed] [Google Scholar]
- Jacobsen, K. , Lund, M. B. , Shim, J. , Gunnersen, S. , Fuchtbauer, E. M. , Kjolby, M. , … Bentzon, J. F. (2017). Diverse cellular architecture of atherosclerotic plaque derives from clonal expansion of a few medial SMCs. JCI Insight, 2(19). 10.1172/jci.insight.95890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, J. L. (2017). Metalloproteinases in atherosclerosis. European Journal of Pharmacology, 816, 93–106. 10.1016/j.ejphar.2017.09.007 [DOI] [PubMed] [Google Scholar]
- Johnson, J. L. , Devel, L. , Czarny, B. , George, S. J. , Jackson, C. L. , Rogakos, V. , … Dive, V. (2011). A selective matrix metalloproteinase‐12 inhibitor retards atherosclerotic plaque development in apolipoprotein E‐knockout mice. Arteriosclerosis, Thrombosis, and Vascular Biology, 31(3), 528–535. 10.1161/ATVBAHA.110.219147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, J. L. , George, S. J. , Newby, A. C. , & Jackson, C. L. (2005). Divergent effects of matrix metalloproteinases 3, 7, 9, and 12 on atherosclerotic plaque stability in mouse brachiocephalic arteries. Proceedings of the National Academy of Sciences of the United States of America, 102(43), 15575–15580. 10.1073/pnas.0506201102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapustin, A. N. , Davies, J. D. , Reynolds, J. L. , McNair, R. , Jones, G. T. , Sidibe, A. , … Shanahan, C. M. (2011). Calcium regulates key components of vascular smooth muscle cell‐derived matrix vesicles to enhance mineralization. Circulation Research, 109(1), e1–e12. 10.1161/CIRCRESAHA.110.238808 [DOI] [PubMed] [Google Scholar]
- Kelly‐Arnold, A. , Maldonado, N. , Laudier, D. , Aikawa, E. , Cardoso, L. , & Weinbaum, S. (2013). Revised microcalcification hypothesis for fibrous cap rupture in human coronary arteries. Proceedings of the National Academy of Sciences of the United States of America, 110(26), 10741–10746. 10.1073/pnas.1308814110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodgie, F. D. , Virmani, R. , Burke, A. P. , Farb, A. , Weber, D. K. , Kutys, R. , … Gold, H. K. (2004). Pathologic assessment of the vulnerable human coronary plaque. Heart (British Cardiac Society), 90(12), 1385–1391. 10.1136/hrt.2004.041798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramann, R. , Goettsch, C. , Wongboonsin, J. , Iwata, H. , Schneider, R.K. , Kuppe, C ., … Humphreys, B.D. (2016). Adventitial MSC-like cells are progenitors of vascular smooth muscle cells and drive vascular calcification in chronic kidney disease. Cell Stem Cell. 19, 628–642. 10.1016/j.stem.2016.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurozumi, A. , Nakano, K. , Yamagata, K. , Okada, Y. , Nakayamada, S. , & Tanaka, Y. (2016). IL‐6/STAT3 pathway is critically involved in vascular calcification via histone modification of the RUNX2 promoter in vascular smooth muscle cells. Bone Abstracts, 5, 425. [Google Scholar]
- Kuzuya, M. , Nakamura, K. , Sasaki, T. , Cheng, X. W. , Itohara, S. , & Iguchi, A. (2006). Effect of MMP‐2 deficiency on atherosclerotic lesion formation in apoE‐deficient mice. Arteriosclerosis, Thrombosis, and Vascular Biology, 26(5), 1120–1125. 10.1161/01.ATV.0000218496.60097.e0 [DOI] [PubMed] [Google Scholar]
- Lee, G.‐L. , Wu, J.‐Y. , Tsai, C.‐S. , Lin, C.‐Y. , Tsai, Y.‐T. , Lin, C.‐S. , … Kuo, C. C. (2016). TLR4‐activated MAPK–IL‐6 axis regulates vascular smooth muscle cell function. International Journal of Molecular Sciences, 17(9), 1394 10.3390/ijms17091394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby, P. (2017). Interleukin‐1 beta as a target for atherosclerosis therapy: Biological basis of CANTOS and beyond. Journal of the American College of Cardiology, 70(18), 2278–2289. 10.1016/j.jacc.2017.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby, P. , Ridker, P. M. , & Hansson, G. K. (2011). Progress and challenges in translating the biology of atherosclerosis. Nature, 473(7347), 317–325. 10.1038/nature10146 [DOI] [PubMed] [Google Scholar]
- Lim, S. , & Park, S. (2014). Role of vascular smooth muscle cell in the inflammation of atherosclerosis. BMB Reports, 47(1), 1–7. 10.5483/BMBRep.2014.47.1.285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, M. E. , Chen, T. M. , Wallingford, M. C. , Nguyen, N. B. , Yamada, S. , Sawangmake, C. , … Giachelli, C. M. (2016). Runx2 deletion in smooth muscle cells inhibits vascular osteochondrogenesis and calcification but not atherosclerotic lesion formation. Cardiovascular Research, 112(2), 606–616. 10.1093/cvr/cvw205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, T. C. , Tintut, Y. , Lyman, A. , Mack, W. , Demer, L. L. , & Hsiai, T. K. (2006). Mechanical response of a calcified plaque model to fluid shear force. Annals of Biomedical Engineering, 34(10), 1535–1541. 10.1007/s10439-006-9182-9 [DOI] [PubMed] [Google Scholar]
- Liu, R. , Jin, Y. , Tang, W. H. , Qin, L. , Zhang, X. , Tellides, G. , … Martin, K. A. (2013). Ten‐eleven translocation‐2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation, 128(18), 2047–2057. 10.1161/CIRCULATIONAHA.113.002887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , Sinha, S. , McDonald, O. G. , Shang, Y. , Hoofnagle, M. H. , & Owens, G. K. (2005). Kruppel‐like factor 4 abrogates myocardin‐induced activation of smooth muscle gene expression. The Journal of Biological Chemistry, 280(10), 9719–9727. 10.1074/jbc.M412862200 [DOI] [PubMed] [Google Scholar]
- Liu, Y. , Sinha, S. , & Owens, G. (2003). A transforming growth factor‐beta control element required for SM α‐actin expression in vivo also partially mediates GKLF‐dependent transcriptional repression. The Journal of Biological Chemistry, 278(48), 48004–48011. 10.1074/jbc.M301902200 [DOI] [PubMed] [Google Scholar]
- Lu, Y. , Thavarajah, T. , Gu, W. , Cai, J. , & Xu, Q. (2018). Impact of miRNA in atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology, 38(9), e159–e170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire, E. M. , Pearce, S. W. A. , & Xiao, Q. (2018). Foam cell formation: A new target for fighting atherosclerosis and cardiovascular disease. Vascular Pharmacology, 112, 54–71. [DOI] [PubMed] [Google Scholar]
- Majesky, M. W. , Horita, H. , Ostriker, A. , Lu, S. , Regan, J. N. , Bagchi, A. , … Weiser‐Evans, M. C. M. (2017). Differentiated smooth muscle cells generate a subpopulation of resident vascular progenitor cells in the adventitia regulated by KLF4. Circulation Research, 120(2), 296–311. 10.1161/CIRCRESAHA.116.309322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manderson, J. A. , Mosse, P. R. , Safstrom, J. A. , Young, S. B. , & Campbell, G. R. (1989). Balloon catheter injury to rabbit carotid artery. I. Changes in smooth muscle phenotype. Arteriosclerosis (Dallas, Tex.), 9(3), 289–298. [DOI] [PubMed] [Google Scholar]
- Misra, A. , Feng, Z. , Chandran, R. R. , Kabir, I. , Rotllan, N. , Aryal, B. , … Greif, D. M. (2018). Integrin beta3 regulates clonality and fate of smooth muscle‐derived atherosclerotic plaque cells. Nature Communications, 9(1), 2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, A. , Kramer, S. D. , Meletta, R. , Beck, K. , Selivanova, S. V. , Rancic, Z. , … Ametamey, S. M. (2014). Gene expression levels of matrix metalloproteinases in human atherosclerotic plaques and evaluation of radiolabeled inhibitors as imaging agents for plaque vulnerability. Nuclear Medicine and Biology, 41(7), 562–569. 10.1016/j.nucmedbio.2014.04.085 [DOI] [PubMed] [Google Scholar]
- Nagase, H. , Visse, R. , & Murphy, G. (2006). Structure and function of matrix metalloproteinases and TIMPs. Cardiovascular Research, 69(3), 562–573. 10.1016/j.cardiores.2005.12.002 [DOI] [PubMed] [Google Scholar]
- Naik, V. , Leaf, E. M. , Hu, J. H. , Yang, H. Y. , Nguyen, N. B. , Giachelli, C. M. , & Speer, M. Y. (2012). Sources of cells that contribute to atherosclerotic intimal calcification: An in vivo genetic fate mapping study. Cardiovascular Research, 94(3), 545–554. 10.1093/cvr/cvs126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemenoff, R. A. , Horita, H. , Ostriker, A. C. , Furgeson, S. B. , Simpson, P. A. , VanPutten, V. , … Weiser‐Evans, M. C. M. (2011). SDF‐1α induction in mature smooth muscle cells by inactivation of PTEN is a critical mediator of exacerbated injury‐induced neointima formation. Arteriosclerosis, Thrombosis, and Vascular Biology, 31(6), 1300–1308. 10.1161/ATVBAHA.111.223701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- New, S. E. , & Aikawa, E. (2011). Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circulation Research, 108(11), 1381–1391. 10.1161/CIRCRESAHA.110.234146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newby, A. C. (2005). Dual role of matrix metalloproteinases (matrixins) in intimal thickening and atherosclerotic plaque rupture. Physiological Reviews, 85(1), 1–31. 10.1152/physrev.00048.2003 [DOI] [PubMed] [Google Scholar]
- Newby, A. C. (2012). Matrix metalloproteinase inhibition therapy for vascular diseases. Vascular Pharmacology, 56(5–6), 232–244. 10.1016/j.vph.2012.01.007 [DOI] [PubMed] [Google Scholar]
- Nguyen, A. T. , Gomez, D. , Bell, R. D. , Campbell, J. H. , Clowes, A. W. , Gabbiani, G. , … Owens, G. K. (2013). Smooth muscle cell plasticity: Fact or fiction? Circulation Research, 112(1), 17–22. 10.1161/CIRCRESAHA.112.281048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurnberg, S. T. , Cheng, K. , Raiesdana, A. , Kundu, R. , Miller, C. L. , Kim, J. B. , … Quertermous, T. (2015). Coronary artery disease associated transcription factor TCF21 regulates smooth muscle precursor cells that contribute to the fibrous cap. PLoS Genetics, 11(5), e1005155 10.1371/journal.pgen.1005155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka, F. , Sakakura, K. , Yahagi, K. , Joner, M. , & Virmani, R. (2014). Has our understanding of calcification in human coronary atherosclerosis progressed? Arteriosclerosis, Thrombosis, and Vascular Biology, 34(4), 724–736. 10.1161/ATVBAHA.113.302642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens, G. K. , Kumar, M. S. , & Wamhoff, B. R. (2004). Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiological Reviews, 84(3), 767–801. 10.1152/physrev.00041.2003 [DOI] [PubMed] [Google Scholar]
- Parhami, F. , Basseri, B. , Hwang, J. , Tintut, Y. , & Demer, L. L. (2002). High‐density lipoprotein regulates calcification of vascular cells. Circulation Research, 91(7), 570–576. 10.1161/01.RES.0000036607.05037.DA [DOI] [PubMed] [Google Scholar]
- Proudfoot, D. , Skepper, J. N. , Hegyi, L. , Bennett, M. R. , Shanahan, C. M. , & Weissberg, P. L. (2000). Apoptosis regulates human vascular calcification in vitro: Evidence for initiation of vascular calcification by apoptotic bodies. Circulation Research, 87(11), 1055–1062. 10.1161/01.RES.87.11.1055 [DOI] [PubMed] [Google Scholar]
- Pugliese, G. , Iacobini, C. , Blasetti Fantauzzi, C. , & Menini, S. (2015). The dark and bright side of atherosclerotic calcification. Atherosclerosis, 238(2), 220–230. 10.1016/j.atherosclerosis.2014.12.011 [DOI] [PubMed] [Google Scholar]
- Ridker, P. M. (2016). A test in context: High‐sensitivity C‐reactive protein. Journal of the American College of Cardiology, 67(6), 712–723. 10.1016/j.jacc.2015.11.037 [DOI] [PubMed] [Google Scholar]
- Ridker, P. M. , Everett, B. M. , Thuren, T. , MacFadyen, J. G. , Chang, W. H. , Ballantyne, C. , … CANTOS Trial Group (2017). Antiinflammatory therapy with canakinumab for atherosclerotic disease. The New England Journal of Medicine, 377(12), 1119–1131. 10.1056/NEJMoa1707914 [DOI] [PubMed] [Google Scholar]
- Rong, J. X. , Shapiro, M. , Trogan, E. , & Fisher, E. A. (2003). Transdifferentiation of mouse aortic smooth muscle cells to a macrophage‐like state after cholesterol loading. Proceedings of the National Academy of Sciences of the United States of America, 100(23), 13531–13536. 10.1073/pnas.1735526100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupaimoole, R. , & Slack, F. J. (2017). MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nature Reviews Drug Discovery, 16, 203–222. 10.1038/nrd.2016.246 [DOI] [PubMed] [Google Scholar]
- Sainz, J. , Al Haj Zen, A. , Caligiuri, G. , Demerens, C. , Urbain, D. , Lemitre, M. , & Lafont, A. (2006). Isolation of “side population” progenitor cells from healthy arteries of adult mice. Arteriosclerosis, Thrombosis, and Vascular Biology, 26(2), 281–286. 10.1161/01.ATV.0000197793.83391.91 [DOI] [PubMed] [Google Scholar]
- Sata, M. , Saiura, A. , Kunisato, A. , Tojo, A. , Okada, S. , Tokuhisa, T. , … Nagai, R. (2002). Hematopoietic stem cells differentiate into vascular cells that participate in the pathogenesis of atherosclerosis. Nature Medicine, 8(4), 403–409. 10.1038/nm0402-403 [DOI] [PubMed] [Google Scholar]
- Schober, A. (2008). Chemokines in vascular dysfunction and remodeling. Arteriosclerosis, Thrombosis, and Vascular Biology, 28(11), 1950–1959. 10.1161/ATVBAHA.107.161224 [DOI] [PubMed] [Google Scholar]
- Selzman, C. H. , Miller, S. A. , Zimmerman, M. A. , Gamboni‐Robertson, F. , Harken, A. H. , & Banerjee, A. (2002). Monocyte chemotactic protein‐1 directly induces human vascular smooth muscle proliferation. American Journal of Physiology‐Heart and Circulatory Physiology, 283(4), H1455–H1461. [DOI] [PubMed] [Google Scholar]
- Shanahan, C. M. (2007). Inflammation ushers in calcification: A cycle of damage and protection? Circulation, 116(24), 2782–2785. 10.1161/CIRCULATIONAHA.107.749655 [DOI] [PubMed] [Google Scholar]
- Shankman, L. S. , Gomez, D. , Cherepanova, O. A. , Salmon, M. , Alencar, G. F. , Haskins, R. M. , … Owens, G. K. (2015). KLF4‐dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nature Medicine, 21(6), 628–637. 10.1038/nm.3866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shioi, A. , & Ikari, Y. (2018). Plaque calcification during atherosclerosis progression and regression. Journal of Atherosclerosis and Thrombosis, 25(4), 294–303. 10.5551/jat.RV17020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shioi, A. , Katagi, M. , Okuno, Y. , Mori, K. , Jono, S. , Koyama, H. , & Nishizawa, Y. (2002). Induction of bone‐type alkaline phosphatase in human vascular smooth muscle cells: Roles of tumor necrosis factor‐α and oncostatin M derived from macrophages. Circulation Research, 91(1), 9–16. 10.1161/01.RES.0000026421.61398.F2 [DOI] [PubMed] [Google Scholar]
- Singh, P. , & Zheng, X. L. (2014). Dual regulation of myocardin expression by tumor necrosis factor‐α in vascular smooth muscle cells. PLoS ONE, 9(11), e112120 10.1371/journal.pone.0112120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottiurai, V. S. , Yao, J. S. , Batson, R. C. , Sue, S. L. , Jones, R. , & Nakamura, Y. A. (1989). Distal anastomotic intimal hyperplasia: Histopathologic character and biogenesis. Annals of Vascular Surgery, 3(1), 26–33. 10.1016/S0890-5096(06)62381-9 [DOI] [PubMed] [Google Scholar]
- Sprague, A. H. , & Khalil, R. A. (2009). Inflammatory cytokines in vascular dysfunction and vascular disease. Biochemical Pharmacology, 78(6), 539–552. 10.1016/j.bcp.2009.04.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas, I. , Garcia‐Cardena, G. , & Owens, G. K. (2015). Recent insights into the cellular biology of atherosclerosis. The Journal of Cell Biology, 209(1), 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, R. H. , Zheng, X. L. , Callis, T. E. , Stansfield, W. E. , He, J. , Baldwin, A. S. , … Selzman, C. H. (2008). Myocardin inhibits cellular proliferation by inhibiting NF‐κB (p65)‐dependent cell cycle progression. Proceedings of the National Academy of Sciences of the United States of America, 105(9), 3362–3367. 10.1073/pnas.0705842105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, Z. , Wang, A. , Yuan, F. , Yan, Z. , Liu, B. , Chu, J. S. , … Li, S. (2012). Differentiation of multipotent vascular stem cells contributes to vascular diseases. Nature Communications, 3, 875 10.1038/ncomms1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyberg, J. , Blomgren, K. , Hedin, U. , & Dryjski, M. (1995). Phenotypic modulation of smooth muscle cells during the formation of neointimal thickenings in the rat carotid artery after balloon injury: An electron‐microscopic and stereological study. Cell and Tissue Research, 281(3), 421–433. 10.1007/BF00417860 [DOI] [PubMed] [Google Scholar]
- Thyberg, J. , Blomgren, K. , Roy, J. , Tran, P. K. , & Hedin, U. (1997). Phenotypic modulation of smooth muscle cells after arterial injury is associated with changes in the distribution of laminin and fibronectin. The Journal of Histochemistry and Cytochemistry: Official Journal of the Histochemistry Society, 45(6), 837–846. 10.1177/002215549704500608 [DOI] [PubMed] [Google Scholar]
- Tintut, Y. , Patel, J. , Territo, M. , Saini, T. , Parhami, F. , & Demer, L. L. (2002). Monocyte/macrophage regulation of vascular calcification in vitro. Circulation, 105(5), 650–655. 10.1161/hc0502.102969 [DOI] [PubMed] [Google Scholar]
- Vengrenyuk, Y. , Nishi, H. , Long, X. , Ouimet, M. , Savji, N. , Martinez, F. O. , … Fisher, E. A. (2015). Cholesterol loading reprograms the microRNA‐143/145–myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage‐like phenotype. Arteriosclerosis, Thrombosis, and Vascular Biology, 35(3), 535–546. 10.1161/ATVBAHA.114.304029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeneuve, L. M. , Reddy, M. A. , Lanting, L. L. , Wang, M. , Meng, L. , & Natarajan, R. (2008). Epigenetic histone H3 lysine 9 methylation in metabolic memory and inflammatory phenotype of vascular smooth muscle cells in diabetes. Proceedings of the National Academy of Sciences of the United States of America, 105(26), 9047–9052. 10.1073/pnas.0803623105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, D. , Chang, P. S. , Wang, Z. , Sutherland, L. , Richardson, J. A. , Small, E. , … Olson, E. N. (2001). Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell, 105(7), 851–862. 10.1016/S0092-8674(01)00404-4 [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Dubland, J. A. , Allahverdian, S. , Asonye, E. , Sahin, B. , Jaw, J. E. , … Francis, G. A. (2019). Smooth muscle cells contribute the majority of foam cells in ApoE (apolipoprotein E)‐deficient mouse atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology, 39(5), 876–887. 10.1161/ATVBAHA.119.312434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe, S. , Mu, W. , Kahn, A. , Jing, N. , Li, J. H. , Lan, H. Y. , … Johnson, R. J. (2004). Role of JAK/STAT pathway in IL‐6‐induced activation of vascular smooth muscle cells. American Journal of Nephrology, 24(4), 387–392. 10.1159/000079706 [DOI] [PubMed] [Google Scholar]
- Wierda, R. J. , Rietveld, I. M. , van Eggermond, M. C. , Belien, J. A. , van Zwet, E. W. , Lindeman, J. H. , & van den Elsen, P. J. (2015). Global histone H3 lysine 27 triple methylation levels are reduced in vessels with advanced atherosclerotic plaques. Life Sciences, 129, 3–9. 10.1016/j.lfs.2014.10.010 [DOI] [PubMed] [Google Scholar]
- Wong, K. K. , Thavornpattanapong, P. , Cheung, S. C. , Sun, Z. , & Tu, J. (2012). Effect of calcification on the mechanical stability of plaque based on a three‐dimensional carotid bifurcation model. BMC Cardiovascular Disorders, 12, 7 10.1186/1471-2261-12-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, Y. W. , Liao, S. Q. , Zhang, M. J. , Liu, Y. , Li, B. H. , Zhou, Y. , … Zhang, L. L. (2014). TLR4‐mediated inflammation promotes foam cell formation of vascular smooth muscle cell by upregulating ACAT1 expression. Cell Death & Disease, 5(12), e1574–e1574. 10.1038/cddis.2014.535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida, T. , Yamashita, M. , Horimai, C. , & Hayashi, M. (2013). Smooth muscle‐selective inhibition of nuclear factor‐κB attenuates smooth muscle phenotypic switching and neointima formation following vascular injury. Journal of the American Heart Association, 2(3), e000230. [DOI] [PMC free article] [PubMed] [Google Scholar]