

Abstract
The PerMM web server and database were developed for quantitative analysis and visualization of passive translocation of bioactive molecules across lipid membranes. The server is the first physics-based web tool that calculates membrane binding energies and permeability coefficients of diverse molecules through artificial and natural membranes (phospholipid bilayers, PAMPA-DS, blood–brain barrier, and Caco-2/MDCK cell membranes). It also visualizes the transmembrane translocation pathway as a sequence of translational and rotational positions of a permeant as it moves across the lipid bilayer, along with the corresponding changes in solvation energy. The server can be applied for prediction of permeability coefficients of compounds with diverse chemical scaffolds to facilitate selection and optimization of potential drug leads. The complementary PerMM database allows comparison of computationally and experimentally determined permeability coefficients for more than 500 compounds in different membrane systems. The website and database are freely accessible at https://permm.phar.umich.edu/.
Graphical Abstract
INTRODUCTION
Recent progress in combinatorial synthesis has rapidly increased the number of tangible small molecules to more than a billion, and millions of them are of pharmaceutical interest.1 Discovery of natural products from microorganisms additionally brought >23 000 valuable agents for medicine, veterinary science, and agriculture.2 Identification of possible drug leads from synthetic chemicals and natural products relies on experimental methods for high-throughput screening of these compounds against potential therapeutic targets and on the development of computational tools for prediction of drug-likeness of a compound of interest and evaluation of its pharmacokinetics profiles defined by its absorption, distribution, metabolism, and excretion (ADME).
Currently, quantitative structure–permeability relationship (QSPR) models are the primary tools for ADME optimization and selection of potential drug candidates in the early stages of drug development. These models use experimentally and computationally derived properties of organic molecules to predict their oral bioavailability, intestinal absorption, and permeability through black lipid membranes (BLM), the blood–brain barrier (BBB), the gut epithelium, or skin multilayered membranes.3–6 Statistics-based models for prediction of various ADME properties have been combined into state-of-the-art publicly or commercially available web tools, such as admetSAR,7 pkCSM,8 SwissADME,9 or QikProp (Schrödinger, LLC).
The QSPR models are usually trained on limited sets of compounds and show good performance for classes of similar molecules, but they may have a poor transferability to compounds with different molecular skeletons. Besides, QSPR models do not consider the actual process and physical mechanisms of permeation. Therefore, to rationalize key factors underlying permeation of solutes through the lipid bilayers, solubility–diffusion models of different complexity have been developed.10–12
In order to describe the thermodynamics of membrane–solute interactions and to uncover mechanisms of passive permeation across the lipid bilayer, general physics-based methods are needed. For example, MD simulations were used to describe diffusivity and free energy profiles, to define optimal orientations of molecules in membranes, and to calculate their permeability coefficients.13–16 However, all-atom MD simulations are highly computationally expensive, which hampers their application for high-throughput drug screening. Simulations of molecules in the lipid bilayer treated as a low dielectric continuum are more computationally efficient. This approach was applied in physics-based methods developed by Leung et al.,17 Swift and Amaro,18 Ferrarini et al.,19 and, recently, by Brocke et al.20
Despite the advancement of physics-based methods for modeling of permeability, they have certain limitations in calculating the absolute membrane permeability17 and, until very recently,20 were not available online. Therefore, we have developed a new physics-based computational method, PerMM, aimed at the fast and reliable modeling of passive permeability of structurally diverse compounds across artificial and natural membranes (first reported as a conference abstract21) as an extension of our PPM method.22 The development and testing of the PerMM method was described in the accompanying paper.46
In this publication, we describe implementation of the PerMM method as a public web server for modeling the transmembrane translocation pathways and permeability coefficients of diverse molecules. A complementary PerMM database is the first web resource that contains experimentally determined permeability coefficients of bioactive molecules through liposomes and black lipid membranes (BLM) collected from publications alongside with permeability coefficients through PAMPA, BBB, and Caco-2/MDCK systems compiled by Avdeef.23–25 Furthermore, the PerMM database provides for the first time the computationally simulated pathways for numerous drugs and other biologically important molecules.
PERMM METHOD
The PerMM method combines the heterogeneous solubility–diffusion theory10 and the anisotropic solvent model of the lipid bilayer characterized by transbilayer profiles of dielectric and hydrogen-bonding capacity parameters.22 PerMM calculates the membrane binding energies (ΔGbind) and the transfer energy profiles (ΔGtransf(z)) of permeants in membranes and obtains their optimal spatial positions and conformations during rotational and translational motion along the membrane normal. The membrane-bound state of a permeant is defined as its conformation and the spatial position in membrane with the lowest transfer energy from water. The integration of free energy profile over the permeation pathway allows evaluation of permeability coefficients of molecules through artificial (BLM) and natural membranes (BBB and Caco-2/MDCK) based on the following equations (all details are provided in the accompanying paper46):
| (1) |
| (2) |
| (3) |
| (4) |
where K(z) are the local partition coefficients, ASA is the accessible surface area to account for the size-dependence of the diffusivity, is the integral of the Gibbs free energy of a molecule over the hydrophobic thickness d (30 Å) of the dioleoyl-phosphatidylcholine (DOPC) bilayer. The dependency of the partition coefficient K(z) on the Gibbs free energy (ΔGtransf(z)) of a solute along the membrane normal is calculated as
| (5) |
The integral of the transbilayer energy profile (eq 1) is calculated in the interval from −15 to +15 Å distance from the membrane center with the step of 1 Å.
PERMM DATABASE
The PerMM database (https://permm.phar.umich.edu/) was created for the following reasons: (1) to allow comparison of permeability parameters measured for the same compounds in different experimental systems; (2) to facilitate development and testing of computational methods; and (3) to provide access to coordinate files and results of calculations by our PerMM method for each included molecule.
Database Content.
The database contains a set of 506 organic molecules and FDA-approved drugs with molecular weights ranging from 17 to 1202 Da (Figure S1) with experimentally measured and PerMM-calculated permeability coefficients. The molecules are divided into different chemical classes (organic acids, amino acids, nucleotides, lipids, alkaloids, polyketides, macrolides, etc.) and to groups with different ionization properties.
Experimental permeability data were collected from the literature for assays performed in artificial and natural membrane systems, including phospholipid bilayers (BLM and liposomes), parallel artificial membrane permeability assay (PAMPA), BBB, and Caco-2 (colon adenocarcinoma cell line) and MDCK (Mardin–Darby canine kidney) cells. The data set of experimental permeability coefficients includes 132 data points for phospholipid bilayers (obtained by Xiang and Anderson,11,26,27 Walter and Gutknecht,28–30 and others) and 1808 data points for PAMPA, BBB, and Caco-2/MDCK assays that were collected and processed by Avdeef23–25 (see notes for Figure S1). These data cover a wide range of experimental values spanning 9 orders of magnitude for the neutral states (from −7.5 to 1.5) and 6 orders of magnitude for species ionized in water (from −13 to −7).
The permeation pathways, membrane binding energies, and permeability coefficients of all compounds were calculated at physiological conditions (at pH = 7.4 and 298 K), i.e., taking into account the deionization costs of acids, bases, and zwitterions. The permeability coefficients are provided for ionized (log Pm−7.4 at pH 7.4) and un-ionized (log P0) species through BLM and PAMPA-DS and for un-ionized species through BBB and Caco-2/MDCK cells (i.e., intrinsic permeability coefficients, log P0). The database also offers information about influx and efflux transporters, including the classification of compounds as substrates, inducers, or inhibitors. These data were collected from the Human Metabolome Database,31 DrugBank,32 and Metrabase.33
Three types of downloadable coordinate files (in pdb format) are available for each molecule: (a) a set of source conformations which can be used as input for the PerMM web server; (b) the permeation pathway, i.e., multiple models of the molecule representing optimal conformers and orientations of the molecule in different positions along the bilayer normal; and (c) the lowest-energy membrane-bound state of the molecule.
Database Access.
Access to data is provided through a menu on the website that allows browsing of the content for each compound individually using classification on chemical classes and groups or based on experimental systems for permeability measurement. Dynamic pages are generated for every compound, chemical class, charge group, and type of experimental permeability data. To facilitate retrieval and analysis of data, web pages are organized as sortable lists and tables. Pages for each individual molecule display physicochemical properties of the molecule (pKa, MW), experimentally measured and predicted permeability coefficients in different membrane systems, brief annotation, and links to external web resources, such as PubChem,34 DrugBank,32 KEGG,35 ChEBI,36 the Human Metabolome Database (HMDB),31 the Protein Data Bank (PDB),37 and Wikipedia. Static 2D images of molecules were produced by PubChem or ChemDraw (PerkinElmer Informatics, Inc.). Dynamic images of 3D structures of molecules bound to the lipid bilayer can be viewed by Jmol. Translational and rotational motions of molecules along the translocation pathway can be viewed using GLmol. The individual tables can be downloaded in csv format.
PERMM WEB SERVER
The database website provides access to the web server (https://permm.phar.umich.edu/server) (Figure S2). The web server implements our PerMM method for calculation of the membrane binding energies of molecules, energy barriers along the membrane normal, and permeability coefficients for different membrane systems.
Input.
The input includes (1) 3D structure(s) of a compound with all hydrogens in the pdb format; (2) experimental conditions (T K, pH); (3) the choice of the optimization method for calculating energy barriers along the membrane normal (pathway); and (4) two options for calculating permeability coefficients of ionizable compounds.
The 3D structures of organic molecules can be obtained from the PerMM database, the Protein Data Bank (PDB),37 the Cambridge Structural Database,38 DrugBank,32 or PubChem.34 Files in the structure data format (sdf) should be converted into the pdb format, for example, by using PyMol (https://pymol.org/2/). 3D coordinates for molecules not found in public databases can be generated by molecular modeling software, such as QUANTA (BIOVIA-Accerlys Inc.), Marvin Suite (ChemAxon), Chem3D (PerkinElmer Informatics, Inc.), or similar ones, with subsequent local energy minimization. Multiple conformers of FDA-approved drugs and various organic molecules can be downloaded from PubChem3D39 or generated by molecular modeling software, such as Frog2.40 pKa values of ionizable groups should be added to the coordinate file of a compound as REMARK PKA record (see server instruction for details). The required pKa values can be taken from public databases or calculated by publicly or commercially available software.41 Dipole moments of different chemical groups are automatically defined by PerMM using a library of dipole moments that was compiled based on the previously published tabulations.42,43 However, a user can also use dipole moments of specific groups from the literature and include them manually in the source coordinate file.
User-Defined Options.
Two different optimization methods are included for calculating the lowest transfer energy pathway of the molecule along the membrane normal: drag method and global rotational optimization. In drag method,44 the transfer energy is locally minimized with respect to rotational variables of the molecule in every z + Δz point of the transmembrane pathway, starting from the optimal rotational orientation calculated in the previous point z. This method is preferred, because the global rotational optimization effectively nullifies the energy barrier for flip-flop of the molecule in the middle of membrane.
The user can define an option for the ionization state. To estimate the actual membrane permeability coefficients of bases and acids through the DOPC bilayer that depend on pH (i.e., log Pm−7.4 at pH = 7.4), the user must select option 1, where the deionization energy is included. The user can also select option 2 to obtain intrinsic permeability coefficients (log P0) for neutral molecules or the uncharged forms of ionizable compounds through BLM, BBB, and Caco-2/MDCK systems.
Output.
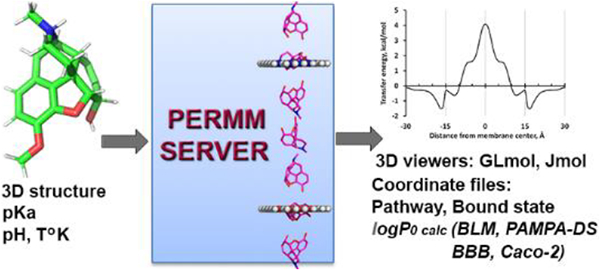
The output includes (1) the graphical representation of the calculated transfer energy profile across the DOPC bilayer, ΔGtranf(z); (2) the membrane binding energy, optimal conformation and orientation of the molecule in the membrane-bound state; (3) the permeability coefficients through the DOPC bilayer (BLM), BBB, and Caco-2/MDCK cells; (4) interactive 3D images of the molecule along the permeation pathway and in the membrane-bound state; and (5) the downloadable coordinate file that provides multiple positions of the molecule during its movement across the lipid bilayer (Figure 1).
Figure 1.

Output of the PerMM server that displays results of calculation of codeine in membranes. Graphical representation of the calculated transfer energy profile (ΔGtransf(z)), the membrane binding energy (ΔGbind = −1.55 kcal/mol), the membrane permeability coefficient at pH 7 through the DOPC bilayer , and downloadable coordinate files (A); interactive 3D images of codeine in membrane by GLmol (B) and Jmol (C).
Performance.
While testing the PerMM method, we found a good correlation (R2 of 0.88 and rmse of 1.15 log units) between experimental and calculated permeability coefficients of 78 compounds (log P0 for 58 un-ionized and log Pm for 20 ionized species) studied in BLM or liposomes. The prediction of intrinsic permeability coefficients (log P0) was less precise for PAMPA and natural membrane systems: the correlation coefficient between experimental and predicted data for PAMPA-DS (280 compounds), BBB (182 compounds), and Caco-2/MDCK cell membranes (165 compounds) demonstrated R2 values of 0.75, 0.69, and 0.52 and rmse values of 1.59, 0.87, and 0.89 log units, respectively. The estimated threshold for BBB-permeable drugs was , i.e., compounds are presumably BBB-permeable (CNS-active) if their calculated intrinsic permeability coefficients were higher than the threshold and they are not substrates for efflux transporters.
The accuracy of the calculation of the membrane binding energy (the lowest transfer energy along the translocation pathway) was assessed for all compounds from the database whose binding energies were experimentally determined. These energies were reproduced with the correlation coefficient R2 of 0.63 and rmse of 0.96 kcal/mol (Figure S3).
The calculation for a single molecule requires 1–4 min (single CPU), depending on the number of conformations. The results of calculations are displayed on a web page and can be received via email.
IMPLEMENTATION
The PerMM database was developed using the Ruby on Rails server-side web application framework and the PostgreSQL database management system for the back-end and React JavaScript library for building user interfaces for the front-end (Figures S4 and S5). The database is hosted on the Heroku Cloud platform with assets (images, PDB files, server results) stored on the Google Cloud platform. Firebase hosting was used for the front-end of the website.
The PerMM web server was developed using Python3 with the Flask framework and an executable script that runs the PerMM program. The software is running in the Yottabyte Research Cloud environment provided by the Advanced Computing Research Services at the University of Michigan. The virtual server that hosts our programs and web applications is equipped with Gunicorn, a Python WSGI HTTP Server for UNIX.
COMPARISON WITH OTHER WEB TOOLS
The PerMM server is the first web-based tool that calculates membrane binding energies of organic molecules, permeability coefficients through BLM, PAMPA-DS, BBB, and Caco-2/MDCK cell membranes, and their transbilayer free energy profiles. On the other hand, state-of-the-art public web tools, such as admetSAR,7 pkCSM,8 and SwissADME,9 as well as commercial packages, calculate a number of ADME properties not considered here.
A comparison of permeability coefficients calculated by our and statistics-based server demonstrates that PerMM has better performance than both pkCSM8 and admetSAR7 for prediction of Caco-2 permeability but shows rather similar performance as admetSAR7 for prediction of BBB permeability for the set of 14 natural product-derived drugs with large sizes (MW > 400 Da) (Figure S6). Quantitative comparison with SwissADME9 is not possible, as this server shows only a binary classification for GI absorption (high and low) and BBB permeation (yes or no). PerMM also significantly out-performed QikProp for Caco-2 permeability prediction during testing on 44 compounds, as was demonstrated in the accompanying paper.46 A comparison with MemDrugPerm operating with HDGB and DHDGB-based implicit membrane models and partial charges from the AM1-BCC force field20 for PAMPA-DS permeability prediction of 58 compounds demonstrates much better accuracy of our PerMM method, which yielded R2 of 0.67 and rmse of 1.51 log units, as compared to R2 of 0.41 and 0.46 and rmse values of 3.83 and 1.90 log units obtained by the MemDrugPerm method with both models, respectively (Figure S7).
CONCLUSIONS
Here we present the public web server that implements the physics-based computational method for fast evaluation of passive permeability of drug-like molecules of different size and scaffolds across artificial and natural membranes. For artificial membranes, the server calculates intrinsic and membrane (at specified pH) permeability coefficients of neutral and ionizable molecules and allows visualization of permeant movement across the DOPC bilayer. The server application for natural membranes is limited to reproducing intrinsic permeability of drugs through BBB and Caco-2/MDCK cells rather than their measurable effective permeability in vivo. The underlying method is approximate, as it does not account for the mechanical properties of lipid bilayers or the influence of lipid composition on membrane permeability. As a future direction, we envision to address these issues and to include distinct polarity profiles for diverse artificial and natural membranes.45 The method can be extended to assess permeation through the lipid bilayer of larger molecules, such as cell-penetrating peptides.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Dr. Avdeef for the provided sets of membrane binding and permeability data of drugs and numerous organic molecules that were experimentally obtained in PAMPA, BBB, and Caco-2/MDCK assays.
Funding
NIH R21DA040752 (to A.L.L. and I.D.P.).
ABBREVIATIONS
- ADME
absorption, distribution, metabolism and excretion
- BBB
blood–brain barrier
- BLM
black lipid membranes
- Caco-2
colon adenocarcinoma cell line
- CSD
Cambridge Structural Database
- DHDGB
dynamic heterogeneous dielectric generalized Born
- FDA
Food and Drug Administration
- DOPC
dioleoyl-phosphatidylcholine
- HDGB
heterogeneous dielectric generalized Born
- MD
molecular dynamics
- MDCK
Madin–Darby canine kidney cell line
- NP
natural product
- PAMPA
parallel artificial membrane permeability assay
- PAMPA-DS
PAMPA double-sink
- PC
phosphatidylcholine
- PDB
Protein Data Bank
- QSPR
quantitative structure–permeability relationship
- rmse
root-mean-square error
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jcim.9b00225.
Description of the database and web server, deployment and activity diagrams, method assessment, and comparison with other web servers (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Ursu O; Rayan A; Goldblum A; Oprea TI Understanding drug-likeness. WIREs Comput. Mol. Sci 2011, 1, 760–781. [Google Scholar]
- (2).Katz L; Baltz RH Natural product discovery: past, present, and future. J. Ind. Microbiol. Biotechnol 2016, 43, 155–176. [DOI] [PubMed] [Google Scholar]
- (3).Nitsche JM; Kasting GB Permeability of fluid-phase phospholipid bilayers: assessment and useful correlations for permeability screening and other applications. J. Pharm. Sci 2013, 102, 2005–2032. [DOI] [PubMed] [Google Scholar]
- (4).Subirats X; Munoz-Pascual L; Abraham MH; Roses M Revisiting blood-brain barrier: A chromatographic approach. J. Pharm. Biomed. Anal 2017, 145, 98–109. [DOI] [PubMed] [Google Scholar]
- (5).Pham-The H; Cabrera-Perez MA; Nam NH; Castillo-Garit JA; Rasulev B; Le-Thi-Thu H; Casanola-Martin GM In Silico assessment of ADME properties: advances in Caco-2 cell monolayer permeability modeling. Curr. Top. Med. Chem 2019, 18, 2209–2229. [DOI] [PubMed] [Google Scholar]
- (6).Alves VM; Muratov E; Fourches D; Strickland J; Kleinstreuer N; Andrade CH; Tropsha A Predicting chemically-induced skin reactions. Part II: QSAR models of skin permeability and the relationships between skin permeability and skin sensitization. Toxicol. Appl. Pharmacol 2015, 284, 273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Cheng F; Li W; Zhou Y; Shen J; Wu Z; Liu G; Lee PW; Tang Y admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model 2012, 52, 3099–3105. [DOI] [PubMed] [Google Scholar]
- (8).Pires DE; Blundell TL; Ascher DB pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem 2015, 58, 4066–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Daina A; Michielin O; Zoete V SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep 2017, 7, 42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Diamond JM; Katz Y Interpretation of nonelectrolyte partition coefficients between dimyristoyl lecithin and water. J. Membr. Biol 1974, 17, 121–154. [DOI] [PubMed] [Google Scholar]
- (11).Xiang TX; Xu YH; Anderson BD The barrier domain for solute permeation varies with lipid bilayer phase structure. J. Membr. Biol 1998, 165, 77–90. [DOI] [PubMed] [Google Scholar]
- (12).Oren I; Fleishman SJ; Kessel A; Ben-Tal N Free diffusion of steroid hormones across biomembranes: a simplex search with implicit solvent model calculations. Biophys. J 2004, 87, 768–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bemporad D; Essex JW; Luttmann C Permeation of small molecules through a lipid bilayer: A computer simulation study. J. Phys. Chem. B 2004, 108, 4875–4884. [Google Scholar]
- (14).Dickson CJ; Hornak V; Pearlstein RA; Duca JS Structure-kinetic relationships of passive membrane permeation from multiscale modeling. J. Am. Chem. Soc 2017, 139, 442–452. [DOI] [PubMed] [Google Scholar]
- (15).Palaiokostas M; Ding W; Shahane G; Orsi M Effects of lipid composition on membrane permeation. Soft Matter 2018, 14, 8496–8508. [DOI] [PubMed] [Google Scholar]
- (16).Shinoda W Permeability across lipid membranes. Biochim. Biophys. Acta, Biomembr 2016, 1858, 2254–2265. [DOI] [PubMed] [Google Scholar]
- (17).Leung SS; Mijalkovic J; Borrelli K; Jacobson MP Testing physical models of passive membrane permeation. J. Chem. Inf. Model 2012, 52, 1621–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Swift RV; Amaro RE Back to the future: can physical models of passive membrane permeability help reduce drug candidate attrition and move us beyond QSPR? Chem. Biol. Drug Des 2013, 81, 61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Parisio G; Stocchero M; Ferrarini A Passive membrane permeability: beyond the standard solubility-diffusion model. J. Chem. Theory Comput 2013, 9, 5236–5246. [DOI] [PubMed] [Google Scholar]
- (20).Brocke SA; Degen A; MacKerell AD; Dutagaci B; Feig M Prediction of membrane permeation of drug molecules by combining an implicit membrane model with machine learning. J. Chem. Inf. Model 2019, 59, 1147–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Pogozheva ID; Mosberg HI; Lomize A PerMM: web server and database for prediction of membrane permeability and translocation pathways of molecules. Biophys. J 2018, 114, 343a–344a.29401432 [Google Scholar]
- (22).Lomize AL; Pogozheva ID; Mosberg HI Anisotropic solvent model of the lipid bilayer. 2. Energetics of insertion of small molecules, peptides, and proteins in membranes. J. Chem. Inf. Model 2011, 51, 930–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Avdeef A Permeability: Blood–Brain Barrier In Absorption and Drug Development; John Wiley & Sons, Inc., Hoboken, NJ, 2012; pp 575–680. [Google Scholar]
- (24).Avdeef A Permeability: Caco-2/MDCK In Absorption and Drug Development; John Wiley & Sons, Inc., Hoboken, NJ, 2012; pp 499–574. [Google Scholar]
- (25).Avdeef A Permeability—PAMPA In Absorption and Drug Development; John Wiley & Sons, Inc., Hoboken, NJ, 2012; pp 319–498. [Google Scholar]
- (26).Xiang TX; Anderson BD The relationship between permeant size and permeability in lipid bilayer membranes. J. Membr. Biol 1994, 140, 111–122. [DOI] [PubMed] [Google Scholar]
- (27).Cao Y; Xiang TX; Anderson BD Development of structure-lipid bilayer permeability relationships for peptide-like small organic molecules. Mol. Pharmaceutics 2008, 5, 371–88. [DOI] [PubMed] [Google Scholar]
- (28).Gutknecht J; Walter A Histamine, theophylline and tryptamine transport through lipid bilayer membranes. Biochim. Biophys. Acta, Biomembr 1981, 649, 149–154. [DOI] [PubMed] [Google Scholar]
- (29).Walter A; Gutknecht J Monocarboxylic acid permeation through lipid bilayer membranes. J. Membr. Biol 1984, 77, 255–264. [DOI] [PubMed] [Google Scholar]
- (30).Gutknecht J Aspirin, acetaminophen and proton transport through phospholipid bilayers and mitochondrial membranes. Mol. Cell. Biochem 1992, 114, 3–8. [DOI] [PubMed] [Google Scholar]
- (31).Wishart DS; Jewison T; Guo AC; Wilson M; Knox C; Liu Y; Djoumbou Y; Mandal R; Aziat F; Dong E; Bouatra S; Sinelnikov I; Arndt D; Xia J; Liu P; Yallou F; Bjorndahl T; Perez-Pineiro R; Eisner R; Allen F; Neveu V; Greiner R; Scalbert A HMDB 3.0–The Human Metabolome Database in 2013. Nucleic Acids Res 2012, 41, D801–D807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Law V; Knox C; Djoumbou Y; Jewison T; Guo AC; Liu Y; Maciejewski A; Arndt D; Wilson M; Neveu V; Tang A; Gabriel G; Ly C; Adamjee S; Dame ZT; Han B; Zhou Y; Wishart DS DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res 2014, 42, D1091–D1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Mak L; Marcus D; Howlett A; Yarova G; Duchateau G; Klaffke W; Bender A; Glen RC Metrabase: a cheminformatics and bioinformatics database for small molecule transporter data analysis and (Q)SAR modeling. J. Cheminform 2015, 7, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wang YL; Xiao JW; Suzek TO; Zhang J; Wang JY; Bryant SH PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res 2009, 37, W623–W633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Kanehisa M; Furumichi M; Tanabe M; Sato Y; Morishima K KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 2017, 45, D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Degtyarenko K; De Matos P; Ennis M; Hastings J; Zbinden M; McNaught A; Alcantara R; Darsow M; Guedj M; Ashburner M ChEBI: a database and ontology for chemical entities of biological interest. Nucleic Acids Res 2007, 36, D344–D350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Rose PW; Prlic A; Bi C; Bluhm WF; Christie CH; Dutta S; Green RK; Goodsell DS; Westbrook JD; Woo J; Young J; Zardecki C; Berman HM; Bourne PE; Burley SK The RCSB Protein Data Bank: views of structural biology for basic and applied research and education. Nucleic Acids Res 2015, 43, D345–D356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Groom CR; Bruno IJ; Lightfoot MP; Ward SC The Cambridge Structural Database. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater 2016, 72, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Bolton EE; Chen J; Kim S; Han L; He S; Shi W; Simonyan V; Sun Y; Thiessen PA; Wang J; Yu B; Zhang J; Bryant SH PubChem3D: a new resource for scientists. J. Cheminform 2011, 3, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Miteva MA; Guyon F; Tuffery P Frog2: Efficient 3D conformation ensemble generator for small compounds. Nucleic Acids Res 2010, 38, W622–W627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Manchester J; Walkup G; Rivin O; You Z Evaluation of pKa estimation methods on 211 druglike compounds. J. Chem. Inf. Model 2010, 50, 565–571. [DOI] [PubMed] [Google Scholar]
- (42).Lien EJ; Guo ZR; Li RL; Su CT Use of dipolemoment as a parameter in drug receptor interaction and quantitative structure activity relationship studies. J. Pharm. Sci 1982, 71, 641–655. [DOI] [PubMed] [Google Scholar]
- (43).Li WY; Guo ZR; Lien EJ Examination of the interrelationship between aliphatic group dipole-moment and polar substituent constants. J. Pharm. Sci 1984, 73, 553–558. [DOI] [PubMed] [Google Scholar]
- (44).Henkelman G; Johannesson G; Jónsson H Methods for finding saddle points and minimum energy paths In Theoretical Methods in Condensed Phase Chemistry, Schwartz SD, Ed.; Springer: Dordrecht, The Netherlands, 2002; pp 269–302. [Google Scholar]
- (45).Pogozheva ID; Tristram-Nagle S; Mosberg HI; Lomize AL Structural adaptations of proteins to different biological membranes. Biochim. Biophys. Acta, Biomembr 2013, 1828, 2592–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Lomize AL; Pogozheva ID A physics-based method for modeling passive membrane permeability and translocation pathways of bioactive molecules. J. Chem. Inf. Model 2019, DOI: 10.1021/acs.jcim.9b00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.