

Abstract
Opioids increase the firing of dopamine cells in the ventral tegmental area by presynaptic inhibition of GABA release. This report describes an acute presynaptic inhibition of GABAB-mediated IPSPs by μ- and κ-opioid receptors and the effects of withdrawal from chronic morphine treatment on the release of GABA at this synapse. In slices taken from morphine-treated guinea pigs after washing out the morphine (withdrawn slices), a low concentration of a μ receptor agonist increased, rather than decreased, the amplitude of the GABAB IPSP. In withdrawn slices, after blocking A1-adenosine receptors with 8-cyclopentyl-1,3-dipropylxantine, μ-opioid receptor activation inhibited the IPSP at all concentrations and increased the maximal inhibition. In addition, during withdrawal, there was a tonic increase in adenosine tone that was further increased by forskolin or D1-dopamine receptor activation, suggesting that metabolism of cAMP was the source of adenosine. The results indicate that during acute morphine withdrawal, there was an upregulation of the basal level of an opioid-sensitive adenylyl cyclase. Inhibition of this basal activity by opioids had two effects. First, a decrease in the formation of cAMP that decreased adenosine tone. This effect predominated at low μ receptor occupancy and increased the amplitude of the IPSP. Higher agonist concentrations inhibited transmitter release by both kinase-dependent and -independent pathways. This study indicates that the consequences of the morphine-induced upregulation of the cAMP cascade on synaptic transmission are dependent on the makeup of receptors and second messenger pathways present on any given terminal.
Keywords: adenosine, cAMP, μ-opioid receptor, κ-opioid receptor, GABA, tolerance, dependence
The ventral tegmental area (VTA) is a heterogeneous population of neurons that plays a role in endogenous reward and is affected by many drugs of abuse (Bozarth and Wise, 1981;Wise, 1987). Electrophysiological studies of substantia nigra pars compacta and the ventral tegmental area in both rats and guinea pigs have identified neurons that fall into two or three categories (Lacey et al., 1989; Johnson and North, 1992b; Cameron et al., 1997). Two groups of neurons are directly hyperpolarized by opioids. The best characterized are presumed GABAergic interneurons (Johnson and North, 1992a). The second group of cells are distinct in that they are hyperpolarized by opioids, 5-HT and dopamine (Cameron et al., 1997). The third group are the dopamine cells. The membrane potential of dopamine cells is not directly affected by opioids, but presynaptic inhibition of GABA release onto these cells increases the firing frequency through disinhibition (Gysling and Wang, 1983; Johnson and North, 1992a). Although separate terminals are thought to mediate GABAA and GABAB synaptic potentials (Johnson et al., 1992; Cameron and Williams, 1993), both are inhibited by opioids (Johnson and North, 1992a). The first aim of this study was to characterize the acute presynaptic effects of opioids on synaptic transmission mediated by GABAB receptors.
Acutely, opioids are known to inhibit adenylyl cyclase activity, and one of the best characterized consequences of chronic morphine treatment is the upregulation of the adenylyl cyclase cascade despite the continued presence of agonist (Sharma et al., 1975; Law et al., 1982; Avidor-Reiss et al., 1996, 1997; Johnson and Fleming, 1989). After removal of morphine, adenylyl cyclase activity increases above normal and is thought to be an important cellular component of withdrawal. The cAMP cascade is known to have potent effects on transmitter release at both excitatory and inhibitory synapses in the CNS (Cameron and Williams, 1993; Chavez-Noriega and Stevens, 1994;Salin et al., 1996; Bonci and Williams, 1997; Chen and Regehr, 1997;Chavis et al., 1998; Chieng and Williams, 1998). During withdrawal from morphine, a cAMP-dependent increase in GABAA-mediated synaptic transmission has been observed in several sites, including the VTA (Bonci and Williams, 1997), periaqueductal gray (PAG) (Ingram et al., 1998), nucleus accumbens (Chieng and Williams, 1998), and dorsal raphe (Jolas et al., 1998). The release of GABA that mediates GABAB IPSPs in the VTA is known to be regulated by D1-dopamine receptors through activation of adenylyl cyclase (Cameron and Williams, 1993; Bonci and Williams, 1996). This study examines the interaction of adenylyl cyclase and opioids on the regulation of this GABAB IPSP during acute morphine withdrawal.
MATERIALS AND METHODS
Intracellular recordings were made from dopamine neurons in horizontal slices of guinea pig midbrain. Preparation of slices has been described previously (Cameron and Williams, 1993). In brief, male guinea pigs (300–400 gm) were anesthetized with halothane and killed. The midbrain was sliced (300 μm) in the horizontal plane using a vibratome. Slices containing the VTA were stored in morphine-free solution for at least 1 hr before being placed in the recording chamber and superfused (1.5 ml/min) with oxygenated artificial CSF (ACSF) at 34 ± 0.5°C. The ACSF was of the following composition (in mm): NaCl 126, KCl 2.5, NaH2PO41.2, MgCl2 1.2, CaCl2 2.4, glucose 11, and NaHCO3 21.4, saturated with 95% O2 and 5% CO2, pH 7.4. Recordings were made with KCl-filled (2 m) glass microelectrodes (40–60 MΩ) using standard techniques. Identification of dopamine cells was made based on the physiological properties that have been characterized previously (Lacey et al., 1989; Johnson and North, 1992b), including the presence of a regular spontaneous firing activity, a relaxation in hyperpolarizing electrotonic potentials mediated by the activation of Ih and the GABAB IPSP. Bipolar tungsten-stimulating electrodes were placed within the VTA. Neurons were maintained at a membrane potential of −60 to −65 mV by injecting hyperpolarizing current (10–20 pA), and synaptic potentials were evoked with a train of stimuli (500 μsec at 70 Hz, 10 stimuli, 60 sec interval) ranging from 0.5–1.5 mA delivered using a constant current stimulation unit. The stimulus intensity was adjusted such that the initial amplitude of the IPSP recorded in all experiments was between 12 and 18 mV. By adjusting the initial amplitude of the IPSP, all drug-induced effects, presented as a percentage change from control, can be compared directly.
All drugs were applied by superfusion. The GABAB synaptic potential was pharmacologically isolated by using a superfusion medium containing 2-amino-5-phosphonopentanoic acid (AP-5; 100 μm), 6-cyano-2,3,-dihydroxy-7-nitro-quinoxaline (CNQX; 10 μm) or 6-nitro-7-sulfamoybenzo-quinoxaline-2,3-dione disodium (NBQX; 5 μm), picrotoxin (100 μm), strychnine (1 μm), and eticlopride (100 nm) to block NMDA, AMPA, GABAA, glycine, and dopamine D2-mediated synaptic potentials, respectively. There was no effect of this solution on the membrane potential. Recently, a metabotropic glutamate receptor (mGluR)-mediated IPSP was observed in dopamine cells that had an overlapping time course with the GABAB IPSP (Fiorillo and Williams, 1998). That IPSP was blocked by mGluR antagonists [S-α-methyl-3-carboxyphenylalanine (MCPG)], and the underlying potassium conductance was selectively blocked by apamin. Except where otherwise stated, apamin (100 nm) or MCPG (1 mm) were contained in the superfusion medium. GABAB IPSPs were blocked by the GABAB receptor antagonist CGP35348 (100 μm).
Morphine base pellets were obtained from the National Institute on Drug Abuse (Bethesda, MD); DAMGO, (5α,7α,8α)-(+)-N-methyl-N-[7-(pyrrolidinyl)-1-oxaspiro [4,5]dec-8-yl]-benzeneacetamide (U69593), AP-5, picrotoxin, baclofen, strychnine, forskolin, 1,9-dideoxyforskolin, and staurosporin were obtained from Sigma (St Louis, MO). CNQX, eticlopride, 8-cyclopentyl-1,3-dipropylxantine (DPCPX), SKF82958, SCH23390, [d-Pen]2[d-Pen]5enkephalin (DPDPE), and naloxone were obtained from Research Biochemicals (Natick, MA). NBQX was obtained from Tocris Cookson (St Louis, MO). CGP35348 was a gift from Novartis. Results in the text and figures are presented as the mean ± SEM. The percent change is presented in all bar graphs [(IPSP amplitude in the presence of drug)/(IPSP amplitude in control) * 100] − 100. The average of five IPSPs was taken in control just before adding the drug and again after the effects of the drug had reached steady state (5–20 min). The difference in response to each concentration of drug between groups was compared using the Mann–Whitney U test. A pvalue <0.05 indicated statistical significance. To estimate the EC50 and maximal response, concentration–response curves were fit with a least squares regression using the logistic equation. The statistical analysis of the interaction between μ and κ receptor agonists was performed with an ANOVA with a Fisher’spost hoc test. y
Male guinea pigs (200–250 gm) were anesthetized with ketamine–xylazine cocktail, and morphine pellets (75 mg morphine base each) were implanted subcutaneously, one on day 1 and two on days 3 and 5. Experiments were done 7–10 d after the start of treatment (2–5 d after the last set of pellets). We and others have used this protocol to induce tolerance and dependence in both rats and guinea pigs (Chieng and Christie, 1995; Bonci and Williams, 1997). Slices were washed extensively in morphine-free ACSF solution for at least 1 hr before any experiments.
RESULTS
μ- and κ-subtype opioid receptors inhibit GABAB IPSPs
Intracellular recordings were made from dopamine cells, and the effects of opioids were examined on GABAB IPSPs. IPSPs were isolated pharmacologically (see Materials and Methods). Initial experiments were performed without separating the mGluR and GABAB IPSPs, and the mixed IPSPs were termed “slow IPSP”. Superfusion with the μ-opioid agonist DAMGO (10 μm) or the κ-opioid agonist U69593 (10 μm) depressed the amplitude of the slow IPSP (Fig.1). The ∂-opioid agonist DPDPE (1–10 μm) had no effect on the slow IPSP (data not shown). The EC50 for DAMGO on the slow IPSP was 365 ± 127 nm with a maximum inhibition of 46 ± 5% (n = 8; Fig. 1A). The κ agonist U69593 was more potent, having an EC50 of 42 ± 1 nm and a maximum inhibition of 56 ± 6% (n = 6; Fig. 1B). In experiments in which the GABAB IPSP was isolated by adding apamin (100 nm) to the superfusion solution, the dose–response curves to DAMGO and U69593 were similar to experiments examining the slow IPSP (Fig. 1). None of the opioid agonists or antagonists had an effect on the membrane potential.
Fig. 1.

Both μ- and κ-opioid receptors decrease the slow IPSP and the GABAB IPSP. Concentration–response curves for DAMGO (A) and U69593 (B) measuring both the slow IPSP and the GABAB IPSP. The insets in Aand B are examples of slow IPSPs. There are two superimposed traces in each showing the inhibition of DAMGO (10 μm) in A and U69593 (10 μm) in B.
μ- and κ-mediated inhibition are additive
Superfusion with a mixture of DAMGO and U69593 caused a greater inhibition (80–90%) than either agonist when applied alone (40–60%;p < 0.001; ANOVA, F = 24, df = 4; Fig. 2). Both agonists were applied at a concentration that maximally depressed the amplitude of the slow IPSP and/or the GABAB IPSP (DAMGO, 10 μm; U69593, 10 μm). The κ-opioid antagonist nor-binaltorphimine dihydrochloride (nor-BNI; 100 nm) decreased the inhibition of the slow IPSP by DAMGO and U69593 to 42 ± 12%, similar to the inhibition caused by DAMGO alone (37 ± 5%). The μ-opioid antagonist CTAP (1 μm) did not significantly reduce the inhibition by the combination of DAMGO and U69593 (from 75 ± 4 to 62 ± 6%; p = 0.07). This may result from the necessity to use a concentration of CTAP that was selective for μ receptors in the presence of a saturating concentration of DAMGO. The fact that the inhibition of the slow IPSP by DAMGO was smaller than the inhibition by U69593 (Fig. 2) also limited the interpretation of this experiment. Taken together, however, the results indicate that both μ- and κ-subtype receptors cause inhibition of GABA release. Because the inhibition caused by maximally effective concentrations of these agonists add, it appears that these agonists may not share a common effector. This could be taken to indicate that there are two groups of GABA-releasing terminals or that μ and κ receptors act by separate cellular mechanisms.
Fig. 2.
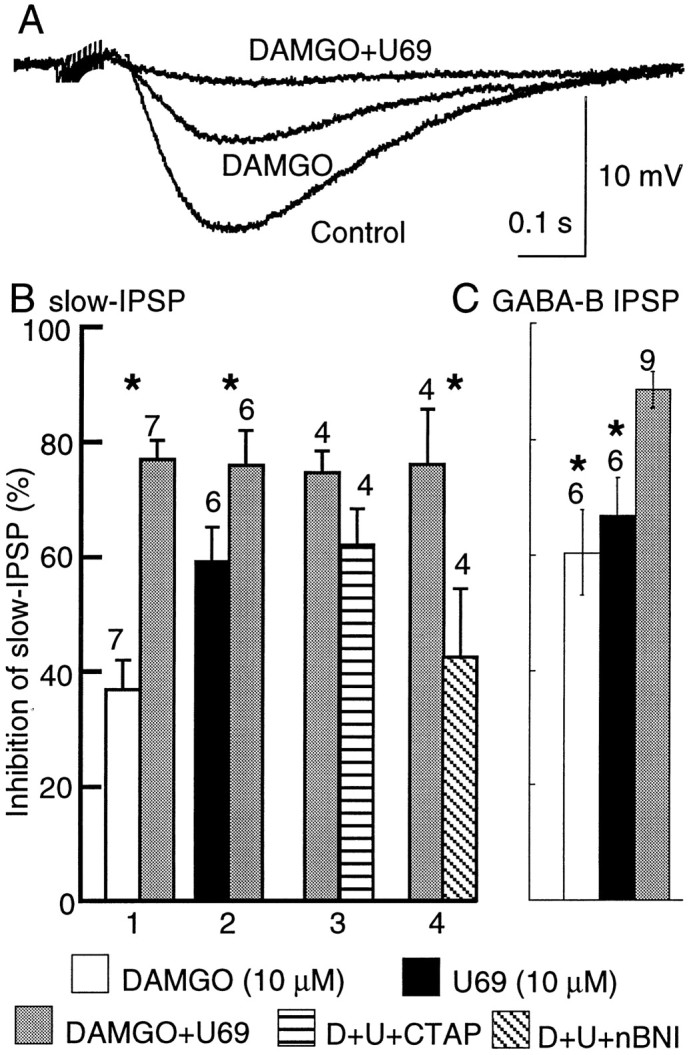
The inhibition caused by DAMGO alone (10 μm), U69593 alone (10 μm), or a mixture of each (DAMGO+U69) indicate that the inhibition caused by the combination of the two agonists is greater than the maximal inhibition caused by either one alone. This additivity was observed for both the slow IPSP and the isolated GABABIPSP. A illustrates three superimposed slow IPSPs showing the additive inhibition. B, Summarized data measuring the slow IPSP. 1 indicates an experiment in which DAMGO was applied first, and U69593 was added to the DAMGO solution. 2 is the reverse experiment in which U69593 was applied first, and DAMGO was added to that solution.3 summarizes experiments in which CTAP (1 μm) was added to the DAMGO and U69593 solution to block μ receptors, and 4 summarizes the same experiment in which nor-BNI (100 nm) was used to block κ receptors. The inhibition caused by DAMGO plus U69593 plus CTAP was the same as that induced by U96593 alone (D+U+CTAP). On the other hand, the inhibition caused by DAMGO plus U69593 plus nor-BNI was similar to that caused by DAMGO alone (D+U+nBNI).C shows that the same additive effects of DAMGO and U69593 occur when measuring the isolated GABAB IPSP. Theasterisks above the bars indicate statistical significance (p < 0.01).
Two effects of opioids during morphine withdrawal
The effect of acute withdrawal on the regulation of GABAB IPSPs was examined initially by comparing the inhibition caused by DAMGO in control and withdrawn slices. The concentration–response curves to DAMGO were the same in control and morphine-withdrawn slices except at a single concentration (10 nm, Fig. 3). DAMGO (10 nm) had no effect on the GABAB IPSP in control slices but increased the IPSP in morphine-withdrawn slices (control, −1 ± 2%, n = 9; morphine-withdrawn, 9 ± 2%, n = 8; Mann–Whitney U test,p = 0.05). It was not possible to fit the DAMGO concentration–response curve with a logistic equation in withdrawn slices because of the increase in the IPSP at 10 nm DAMGO. It was therefore not possible to make a reasonable estimate of the EC50.
Fig. 3.

DAMGO had two effects on the GABABIPSP in withdrawn slices; an increase in GABA release at low concentration and a decrease in GABA release at higher concentrations. The traces at the top are examples of IPSPs in cells from a control (left) and withdrawn (right) slices. Three traces are superimposed in each of the two examples: the IPSP in control, after superfusion with 10 nm DAMGO, and after 10 μm DAMGO. The inhibition caused by DAMGO (10 μm) is ∼50% in each example, whereas DAMGO (10 nm) caused an increase in the IPSP in the withdrawn slice. Below are DAMGO concentration–response curves from control and withdrawn slices. The only point where there is a significant difference is at DAMGO 10 nm.
The inhibition of the slow IPSP by the κ agonist U69593 was not different from the control at any concentration (Fig. 1,control; EC50, 37 ± 4 nm; maximum inhibition, 57 ± 1%, n = 6; withdrawn EC50, 45 ± 7 nm; maximum inhibition, 45 ± 4%, n = 5; data not shown). In addition, the inhibition of the slow IPSP by the 5-HT-1 agonist, 5-carboxamidotryptamine was also not different in control and withdrawn slices. The EC50 was 2.5 ± 0.9 nm in control and 3.3 ± 1.0 nm in withdrawn slices, and the maximum inhibition was 66 ± 4% and 66 ± 5% in the two groups. The selective increase in the IPSP caused by DAMGO (10 nm) may be taken to indicate that μ and κ agonists act through separate second messenger pathways (see Fig. 7).
Fig. 7.
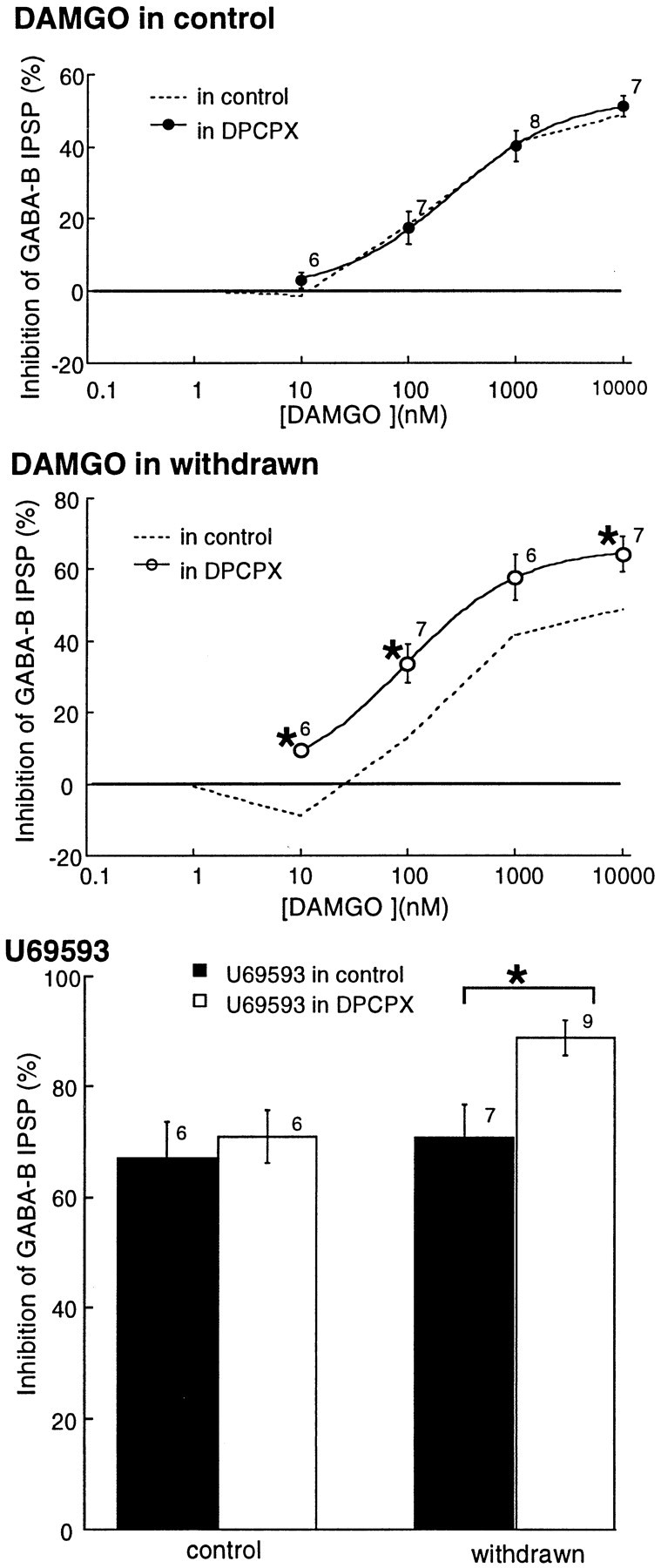
Blockade of A1 adenosine receptors with DPCPX increased the sensitivity and maximal inhibition caused by DAMGO and U69593 in withdrawn slices. A, The concentration–response curve to DAMGO in control slices in the absence (dashed line; data from Fig. 1) and in the presence (solid circles) of DPCPX. B shows the change in the DAMGO concentration–response curve caused by DPCPX in withdrawn slices. The DAMGO inhibition was greater and occurred at lower concentrations in the presence of DPCPX (1 μm;open circles). The dashed line is before treatment with DPCPX (data taken from Fig. 1). Part C is a bar graph showing the effect of DPCPX on the inhibition mediated by U69593 (1 μm). In withdrawn slices, the inhibition was significantly larger in the presence of DPCPX.
The upregulation of adenylyl cyclase may mediate the μ receptor-induced increase in IPSP
One potential mechanism that could account for the DAMGO-induced increase in GABAB IPSP observed in withdrawn slices is through an opioid-sensitive decrease in adenosine tone. This mechanism requires that during withdrawal, the upregulation of adenylyl cyclase results in an increase in extracellular adenosine. An increase in extracellular adenosine after activation of adenylyl cyclase with forskolin has been observed in several sites (Brundege et al., 1997;Chieng and Williams, 1998), including the VTA (Bonci and Williams, 1996). The increased adenosine tone was dependent on the transport and metabolism of cAMP in the extracellular space, as has been described previously (Barber and Butcher, 1981; Rosenberg and Ditchter, 1989;Henderson and Strauss, 1991; Rosenberg et al., 1994). A selective inhibition of adenylyl cyclase by a low concentration of DAMGO would decrease the production of cAMP, reduce the extracellular level of adenosine, and remove inhibition of GABA release mediated by adenosine. The following experiments were aimed at testing this hypothesis. All experiments were performed on the GABAB IPSP isolated with apamin (100 nm).
Adenosine tone is augmented during withdrawal
Adenosine tone was determined by measuring the increase in the amplitude of the GABAB IPSP caused by the adenosine A1 antagonist DPCPX (1 μm). The amplitude of the IPSP was increased by DPCPX in control and withdrawn slices, however the increase was significantly larger in withdrawn slices (Fig.4A, control, 5 ± 1%, n = 7; morphine-withdrawn, 16 ± 2%,n = 8; Mann–Whitney U test,p < 0.002). An increase in the sensitivity of adenosine receptors was not responsible for the augmented response to DPCPX in drug-treated animals because the inhibition of the GABAB IPSP caused by an exogenously applied A1 agonist, N6-cyclopentyladenosine (N6-CPA), was not changed (Fig.4B; control, 67 ± 6%, n = 9; morphine-withdrawn, 65 ± 4%, n = 7; Mann–Whitney U test, p > 0.05). There was no effect of either DPCPX or N6-CPA on the membrane potential (data not shown). Thus, as was found during acute withdrawal in the nucleus accumbens (Chieng and Williams, 1998) and after long-term withdrawal in the VTA (Bonci and Williams, 1996), adenosine tone was elevated.
Fig. 4.

Adenosine tone was increased in withdrawn slices.A shows examples of IPSPs in cells from control (left) and withdrawn (right) slices. Two traces are superimposed in each of the examples: the IPSP before (control) and after treatment of the slice with DPCPX. The IPSP in the withdrawn slice was increased by DPCPX. InB the increase in the IPSP in control and withdrawn slices is summarized. C shows that the inhibition caused by a maximally effective concentration of the A1 adenosine agonist N6-CPA (1 μm) has the same effect in control and withdrawn slices.
Activation of adenylyl cyclase and adenosine tone
Direct activation of adenylyl cyclase with forskolin (10 μm) augmented the GABAB IPSP in control slices (Fig. 5; control, 26 ± 3%,n = 8) but had no effect on the amplitude of the IPSP in morphine-withdrawn slices (morphine-withdrawn, −4 ± 3%,n = 6). The inactive forskolin analog dideoxyforskolin (10 μm) did not affect the GABAB IPSP (0.1 ± 2%; n = 4). The interaction between the activation of adenylyl cyclase and adenosine receptors was investigated by determining adenosine tone through the increase in the IPSP caused by blockade of adenosine receptors with DPCPX (1 μm). In the presence of DPCPX, forskolin (10 μm) augmented the GABAB IPSP in both groups (Fig. 5B; control, 37 ± 5%, n = 7; morphine-withdrawn, 41 ± 4%, n = 7). These observations are consistent with those made after long-term withdrawal from morphine or cocaine in the VTA and suggest that during acute morphine withdrawal, activation of adenylyl cyclase indirectly increases extracellular adenosine to the extent that the IPSP amplitude is decreased through activation of A1 receptors (Bonci and Williams, 1996).
Fig. 5.
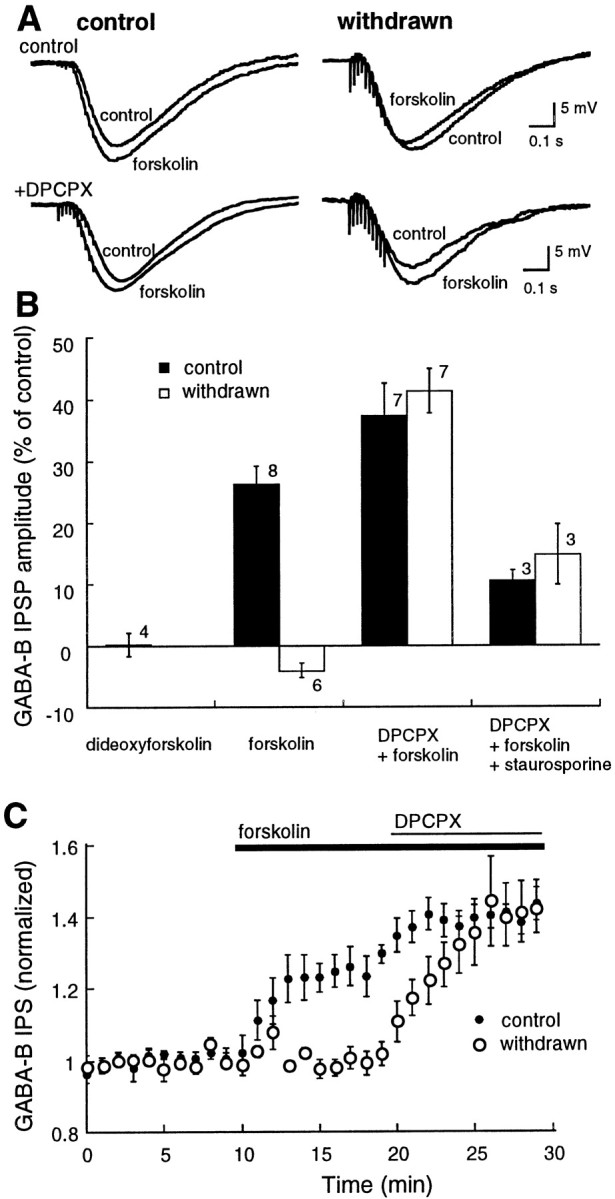
Forskolin had two effects: an increase in GABA release and an increase in adenosine tone. In withdrawn slices, the increase in adenosine tone overrides the increase in GABA release so that the amplitude of the IPSP is decreased. A shows examples of IPSPs from four different cells. In each example, two superimposed IPSPs are illustrated, the control and in the IPSP in the presence of forskolin (10 μm). The top traces are taken from a control (left) and a withdrawn (right) slice. The bottom traces are also taken from control (left) and withdrawn (right) slices, however in these experiments the slice had been treated with DPCPX (1 μm). Forskolin caused an inhibition of the GABAB IPSP in the withdrawn slice, and this inhibition was blocked by DPCPX. B is a bar graph of summarized results. Dideoxyforskolin had no effect. The inhibition of the IPSP by forskolin (forskolin) was blocked by DPCPX (DPCPX+forskolin), and the increase caused by forskolin was largely reduced by treatment with the kinase inhibitor staurosporin (1 μm;DPCPX+forskolin+staurosporin).C shows the time course of action of forskolin and DPCPX in control (solid circles) and withdrawn (open circles) slices.
The increase in IPSP caused by the combination of DPCPX and forskolin was significantly reduced by the nonselective protein kinase inhibitor staurosporin (10 μm; Fig. 5B; control, 10 ± 2%, n = 3; morphine-withdrawn, 15 ± 5%,n = 3). Thus, the augmentation of the IPSP by forskolin was mediated by an increase in kinase activity.
One concern was that forskolin could have a postsynaptic interaction with GABAB receptors because forskolin always caused a small depolarization of the membrane potential (2–5 mV). The effect of forskolin on the hyperpolarization caused by the GABABagonist baclofen (1–3 μm) was tested to determine whether activation of adenylyl cyclase could have a postsynaptic action. Forskolin (10 μm) increased the hyperpolarization induced by baclofen in cells from both control and withdrawn slices (control, 31 ± 8%, n = 6; morphine-withdrawn, 28 ± 6%, n = 6, data not shown). Dideoxyforskolin (10 μm) had no effect (0.7 ± 2%;n = 4). The result with forskolin could suggest a potential postsynaptic interaction, however, this interaction is not affected by morphine withdrawal.
D1-dopamine receptors, adenylyl cyclase, and acute withdrawal
Activation of D1 dopamine receptors increases the GABAB IPSP through a cAMP-dependent pathway (Cameron and Williams, 1993). The role of receptor-mediated activation of adenylyl cyclase during withdrawal was examined. The dopamine D1 agonist SKF82958 (1 μm) produced an increase in the GABAB IPSP that was completely blocked by the D1 receptor antagonist SCH23390 (1 μm; Fig.6A). The augmentation of the GABAB IPSP by SKF82958 was significantly less in morphine-withdrawn slices than controls (Fig. 6B; control, 26 ± 4%, n = 8; morphine-withdrawn, 4 ± 4%, n = 7; Mann–Whitney U test,p < 0.01). In the presence of DPCPX, SKF82958 (1 μm) augmented the GABAB IPSP to the same extent in both control and withdrawn slices (Fig. 6C; control, 30 ± 4%, n = 7; morphine-withdrawn, 29 ± 3%, n = 6). Thus, after blocking A1 adenosine receptors, the increase in transmitter release resulting from D1 receptor stimulation was not different in control and withdrawn slices.
Fig. 6.

D1-dopamine receptors have two effects in withdrawn slices: increased adenosine tone and increased GABA release. The top shows examples of IPSPs from four different cells. In each example, three superimposed IPSPs are illustrated: the control, in the presence of SKF82958 (1 μm), and after treatment with SCH23390 (1 μm). The top traces are taken from control (left) and withdrawn (right) slices. The bottom traces are also taken from control (left) and withdrawn (right) slices, however in these experiments the slices had been treated with DPCPX (1 μm). The bar graph at the bottom shows summarized data indicating that the increase in the GABAB IPSP by SKF82958 in control slices was not changed by DPCPX, whereas in withdrawn slices it was significantly increased. Similar results were obtained with the D1 antagonist SCH23390 (1 μm). In this case, SCH23390 had no effect on the IPSP in control but significantly increased the IPSP in withdrawn slices treated with DPCPX.
In the presence of DPCPX, a larger inhibition of the IPSP was induced by the D1 antagonist SCH23390 in withdrawn slices (Fig. 6C; Mann–Whitney U test, p < 0.05). This observation was taken to indicate that the basal level of D1-dependent adenylyl cyclase was greater in morphine-withdrawn slices than in controls and could result from two possible mechanisms. Either dopamine tone was increased or there was an increased coupling efficiency between the D1 receptor and the upregulated adenylyl cyclase. In either case, the inhibition of D1 receptor-dependent adenylyl cyclase by SCH23390 had two effects resulting from a decrease in cAMP synthesis: a decrease extracellular adenosine and a decline in cAMP-dependent kinase activity. The decrease in the level of extracellular adenosine increased the IPSP, whereas inhibition of cAMP-dependent kinase decreased the IPSP. Thus, the decrease in IPSP caused by blockade of D1 receptors in the presence of DPCPX was an indication that the basal activity of the D1 receptor–cAMP pathway was greater in the morphine-withdrawn slices.
Increased opioid inhibition
To determine whether the increase in adenosine tone during morphine withdrawal affected the sensitivity to opioids, the inhibition by opioids was examined in the absence and presence of DPCPX (1 μm). In control slices, DPCPX did not change the dose–response curve to DAMGO (Fig.7A; EC50, 253 ± 28 nm; maximum inhibition, 51 ± 3%;n = 7). In morphine-withdrawn slices, however, DPCPX altered the dose–response curve in several ways. First, the concentration of DAMGO that increased the IPSP (10 nm) caused an inhibition of the IPSP. Second, the maximal inhibition caused by DAMGO was significantly increased from 49 ± 6% (n = 7) in control to 64 ± 5% (n= 7) in withdrawn slices (Mann–Whitney U test,p < 0.05). Finally, although the EC50 for DAMGO could not be determined accurately in withdrawn slices because of the biphasic effects of DAMGO, it was not remarkably different from in control slices (Fig. 3). In DPCPX however, the EC50 for DAMGO was 93 ± 4 nm, less than half that estimated from the control slices. The increased inhibition caused by DAMGO was completely antagonized by naloxone (1 μm,n = 7). Thus, during withdrawal, the efficacy and potency of DAMGO were increased.
Similar experiments were performed with a maximally effective concentration of the κ-selective agonist U69593. In the absence of DPCPX (1 μm), a maximal concentration of U69593 (10 μm) caused an inhibition of 67 ± 7% (n = 6) in control, the same as found in morphine-withdrawn slices (71 ± 6%, n = 6). In the presence of DPCPX, however, the maximum inhibition induced by U69593 was significantly greater in morphine-withdrawn slices (Fig. 7; control, 71 ± 5%, n = 7; morphine-withdrawn, 89 ± 3%, n = 9; Mann–Whitney U test,p < 0.01). Thus, rather than finding tolerance to opioids, treatment of withdrawn slices with DPCPX revealed an increase in the presynaptic inhibition caused by opioids.
Kinase dependence of opioid inhibition
The increased sensitivity to opioids in the presence of DPCPX during morphine withdrawal could be an indication of an upregulation of the cAMP-dependent cascade. An increased adenylyl cyclase activity would be expected to have two actions, an increase in adenosine tone and protein kinase A activity. To determine the role of kinase activity, the sensitivity to opioids was first examined in the absence and presence of forskolin. For these experiments, DPCPX (1 μm) was included in the superfusion solution to eliminate the effect of increased extracellular adenosine after the activation of adenylyl cyclase. In the presence of forskolin (10 μm), neither the EC50 (142 ± 11 nm in control and 192 ± 65 nm in morphine-withdrawn) nor the maximal inhibition (control, 78 ± 1%, n = 6; morphine-withdrawn, 86 ± 5%, n = 6) for DAMGO were significantly different (Fig. 8). Only one point on the concentration curve was statistically different between control and withdrawn slices; DAMGO (10 nm) caused an inhibition of 6 ± 1% in control and 17 ± 2% in withdrawn slices.
Fig. 8.
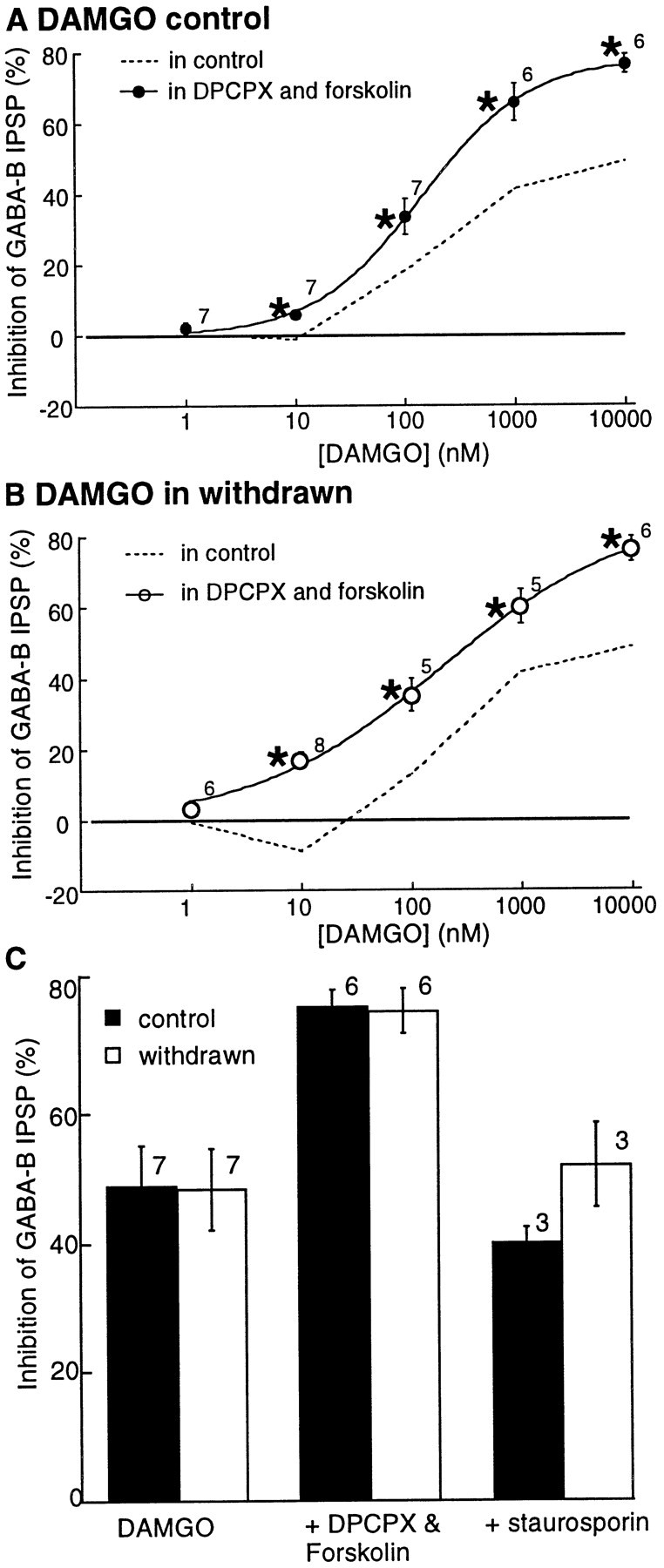
Forskolin increased the maximal inhibition caused by DAMGO in both control and withdrawn slices.A, B, Concentration–response curves to DAMGO are in the presence of both DPCPX and forskolin. Dashed lines indicate the concentration curves from control and withdrawn slices taken from Figure 2. In forskolin and DPCPX (unlike DPCPX alone) the concentration–response curves were similar in control and withdrawn slices. C indicates that the maximal DAMGO (10 μm) inhibition was increased in both tissues to the same extent by forskolin and DPCPX. Staurosporin (1 μm) decreased the augmented inhibition to a level similar to that found before treatment with forskolin and DPCPX.
The DAMGO-induced inhibition in the presence of forskolin and DPCPX was reduced by the nonselective protein kinase inhibitor staurosporin (10 μm) in both control and morphine-withdrawn slices (Fig.7; control, from 78 ± 1% to 40 ± 2%, n = 3; morphine-withdrawn, from 86 ± 5% to 52 ± 7%,n = 3). The inhibition caused by DAMGO was not completely blocked by staurosporin, indicating that presynaptic inhibition by DAMGO had both kinase-dependent and -independent mechanisms in control and morphine-withdrawn slices. The results indicate that a latent opioid-sensitive kinase-dependent inhibition can be activated by increasing the activity of adenylyl cyclase.
DISCUSSION
Acute opioid inhibition
The results of this study show that opioids act presynaptically to inhibit the GABAB IPSP recorded in dopamine cells of the VTA. Both μ and κ-subtype opioid receptors mediate this inhibition. The maximal inhibition caused by one receptor did not occlude inhibition caused by the other, suggesting that receptors are located on separate terminals or act through differing second messenger pathways. Another possibility is that μ and κ receptors are on the same terminals and act through a shared second messenger pathway, but that the maximal activation of a single receptor population alone is not capable of maximally activating the effector pathway. At the present time there is no way to distinguish these possibilities. The present results suggest that opioids can increase in dopamine cell activity by inhibition of both GABAA- (Johnson and North, 1992a) and GABAB-mediated synaptic potentials.
Morphine withdrawal, adenylyl cyclase, and adenosine
In slices taken from morphine-treated animals that were recorded under conditions of acute withdrawal (hours after removal of morphine), the presynaptic actions of opioids were affected in at least two ways, both of which suggest that the basal activity of adenylyl cyclase was increased (Fig. 9). First, there was an increase in adenosine tone that resulted in a tonic A1 receptor-mediated inhibition of release. This inhibition would functionally mimic the action of opioids during withdrawal and serve to blunt a rebound increase in transmitter release. In fact, adenosine receptor antagonists have been found to exacerbate opioid withdrawal signs (Kaplan and Sears, 1996). The increase in adenosine tone itself would be expected to be self-limiting because activation of A1 receptors is also known to inhibit adenylyl cyclase activity (Dunwiddie, 1985).
Fig. 9.

Summary of the three effects caused by an increase in adenylyl cyclase during withdrawal from chronic morphine.1, In control, μ-opioids inhibit GABA release, and D1-dopamine receptors increase GABA release. Under normal conditions, the inhibition of adenylyl cyclase by opioids does not have an influence on the inhibition of GABA release. The regulation of GABA release is changed in three ways during withdrawal from chronic morphine treatment. 2, An upregulation of adenylyl cyclase increased adenosine tone. This upregulation tonically inhibited GABA IPSPs by activation of A1 adenosine receptors. The upregulated adenylyl cyclase was dependent on D1 dopamine receptors because the D1 antagonist SCH23390 caused a larger inhibition of the IPSP after adenosine receptors were blocked. 3, A low concentration of DAMGO increased the IPSP in withdrawn slices. This increase was blocked or occluded after blockade of adenosine receptors. This observation suggests that μ-opioid receptors may couple more efficiently to the inhibition of adenylyl cyclase. Finally,4, μ-opioid receptor-mediated inhibition was augmented in withdrawn slices after blockade of adenosine receptors. After activation of adenylyl cyclase by forskolin, the inhibition by opioids was increased in both control and withdrawn slices. The results indicate that an opioid-sensitive adenylyl cyclase can be activated to the same extent in both control and withdrawn slices but that the basal activity is higher in withdrawn slices.
The increase in adenosine tone appears to be synapse-selective in that there was no evidence of an increase in adenosine tone in the VTA measuring GABAA IPSCs or glutamate EPSCs in dopamine cells (Bonci and Williams, 1997; Manzoni and Williams, 1999). In addition, there was no evidence for increased adenosine tone at GABAAsynapses in the PAG (Bagley and Christie, personal communication). There was, however, an increase in adenosine tone found in the nucleus accumbens measuring GABAA IPSCs during acute withdrawal from morphine (Chieng and Williams, 1998). Thus, it appears that the role that endogenous adenosine plays on synaptic transmission during withdrawal may be common, is synapse-specific, and is not found at all opioid-sensitive synapses.
The second effect of withdrawal at this synapse was the observation that a low concentration of DAMGO (10 nm) consistently caused an increase in the GABAB IPSP (Fig. 9). This augmentation was not observed in control. Biphasic actions of opioids that depend on the concentration of agonist applied have been reported in at least two preparations and are most evident after chronic opioid treatment (Gintzler and Xu, 1991; Cruciani et al., 1993). In the present study, both DAMGO and the adenosine antagonist DPCPX increased the IPSP in withdrawn slices. The increase in the IPSP caused by DAMGO (10 nm) was blocked or occluded by DPCPX. These results can be explained by the action of DAMGO to reduce extracellular adenosine. One important source of extracellular adenosine is thought to result from the metabolism of cAMP (Barber and Butcher, 1981; Dunwiddie, 1985;Rosenberg and Ditchter, 1989; Rosenberg et al., 1994; Brundege et al., 1997; Manzoni et al., 1998). With the activation of adenylyl cyclase, the extracellular levels of adenosine have been shown to rise in several areas, including the VTA (Bonci and Williams, 1996; Brundege et al., 1997). Given that the upregulation of adenylyl cyclase is a common effect of withdrawal from chronic morphine treatment, an increase in adenosine tone could be predicted. Agents that inhibit adenylyl cyclase, such as DAMGO would therefore indirectly decrease the level of endogenous adenosine and result in an increase in the amplitude of the IPSP.
D1 receptors, opioids, and adenylyl cyclase
D1 receptors on GABA-releasing terminals in the VTA and substantia nigra originate from projection neurons in the nucleus accumbens (Mansour et al., 1991). The nucleus accumbens is an area enriched in type V adenylyl cyclase (Glatt and Snyder, 1993; Mons and Cooper, 1995). This isoform was both acutely inhibited by opioid receptor activation and upregulated with chronic morphine treatment (Avidor-Reiss et al., 1996, 1997). The present results suggest that the same cyclase that was activated by D1 receptors is inhibited by opioid receptors. Both μ and κ-opioid receptors inhibit adenylyl cyclase (Murthy and Makhlouf, 1996), although others have found a selective inhibition of adenylyl cyclase by μ agonists and not κ agonists in slices of nucleus accumbens (Izenwasser et al., 1993). When adenylyl cyclase activity was stimulated with forskolin, transmitter release was increased, and the inhibition of the IPSP by both μ and κ receptors was augmented (Fig. 9). This result suggests that activation of either μ or κ receptors on GABA-releasing terminals in the VTA can inhibit forskolin-activated adenylyl cyclase activity and decrease GABA release. In addition, in withdrawn slices after blockade of A1 adenosine receptors, opioids caused a larger inhibition, suggesting that there was an increased basal activity of adenylyl cyclase. This increase in opioid sensitivity has been observed in both the nucleus accumbens (Chieng and Williams, 1998) and the PAG (Ingram et al., 1998) and thus appears to be a common result of the withdrawal activation of adenylyl cyclase.
Is the expression of opioid withdrawal primarily through presynaptic mechanisms?
It has been known since the early work on NG108–15 cells that the activity of adenylyl cyclase was upregulated in the continued presence of morphine (Sharma et al., 1975; Law et al., 1982; Puttfarcken et al., 1988). The rebound increase in adenylyl cyclase activity that occurred with the rapid removal of morphine became the cellular hallmark of withdrawal. The link between the increase in adenylyl cyclase activity and a cellular response measured physiologically has been difficult to identify. Although the uncoupling of opioid receptors from ion channel effectors caused by chronic morphine treatment has been demonstrated in both neurons (Christie et al., 1987) and cell lines (Kennedy and Henderson, 1991), the expected rebound after the withdrawal of morphine has been elusive.
The neurons of the locus coeruleus have been suggested as a model for the chronic actions of morphine (Nestler and Aghajanian, 1997). Despite robust tolerance to the inhibitory action of opioids (Christie et al., 1987), the primary mechanism for the increased activity of these cells during withdrawal is mediated by an increased presynaptic release of glutamate (Akaoka and Aston-Jones, 1991). The only site where the isolation and characterization of a postsynaptic current activated by acute withdrawal has been successful is a subgroup of neurons in the PAG (Chieng and Christie, 1996). It is not known if this current is dependent on cAMP.
Recent reports measuring synaptic transmission during morphine withdrawal have indicated a robust interaction with the cAMP cascade during acute withdrawal (Bonci and Williams, 1996; Chieng and Williams, 1998; Ingram et al., 1998). It appears that cAMP-dependent modulation of transmitter release may be the missing link between the increased adenylyl cyclase and physiological response. There are several synapses where the cAMP-dependent regulation of transmitter release has been well characterized (Cameron and Williams, 1993; Chavez-Noriega and Stevens, 1994; Salin et al., 1996; Chen and Regehr, 1997; Chavis et al., 1998). At each of these sites, an upregulation of cAMP caused a robust augmentation of transmitter release. Given the known interaction between chronic morphine treatment and the upregulation of the cAMP cascade, along with the increasing number of synapses where transmitter release is potently regulated by a cAMP-dependent process, the presynaptic regulation of transmitter release during opioid withdrawal may be the primary cellular target of acute opioid withdrawal.
Summary
The effects of withdrawal on the modulation of GABA release are summarized in Figure 9. At this synapse in control, both μ- and κ-opioid receptors decrease GABA release, whereas D1 receptor activation increases release. One effect of withdrawal was an increased synthesis of cAMP, which resulted in an increase in extracellular adenosine. The increased adenosine tone caused a tonic inhibition of GABA release. Activation of μ-opioid receptors with a low concentration of DAMGO selectively inhibited adenylyl cyclase, which decreased adenosine tone. The decline in adenosine tone removed the tonic inhibition, and the GABA IPSP was increased. After blockade of adenosine receptors, both μ- and κ-opioid agonists decrease GABA release by two mechanisms. One mechanism was not dependent on kinase activity and was similar in amplitude to the inhibition found in slices from untreated animals. The second, and additional, mechanism found in withdrawn slices was mediated by an inhibition of adenylyl cyclase. Thus, the upregulation of adenylyl cyclase with chronic morphine treatment has several consequences on the regulation of transmitter release.
Footnotes
This work was supported by National Institute on Drug Abuse Grants DA08163 and DA07262. We thank Drs. Brundege, Ingram, and Manzoni for comments on this work and manuscript and Dr. A. Sutter at Novartis Pharma for the gift of CGP35348.
Correspondence should be addressed to Dr. Williams, The Vollum Institute, L474, Oregon Health Sciences University, 3181 SW Sam Jackson Park Road, Portland, OR 97201.
Dr. Delfs’ present address: Department of Psychiatry, University of Pennsylvania Medical School, Philadelphia, PA 19104.
REFERENCES
- 1.Akaoka H, Aston-Jones G. Opiate withdrawal-induced hyperactivity of locus ceruleus neurons is substantially mediated by augmented excitatory amino acid input. J Neurosci. 1991;11:3830–3839. doi: 10.1523/JNEUROSCI.11-12-03830.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avidor-Reiss T, Nevo I, Pfeuffer T, Vogel Z. Chronic opioid treatment induces adenylyl cyclase V superactivation: involvement of Gβγ. J Biol Chem. 1996;271:21309–21315. doi: 10.1074/jbc.271.35.21309. [DOI] [PubMed] [Google Scholar]
- 3.Avidor-Reiss T, Nevo I, Saya D, Bayewitch M, Vogel Z. Opiate-induced adenylyl cyclase superactivation is isozyme-specific. J Biol Chem. 1997;272:5040–5047. doi: 10.1074/jbc.272.8.5040. [DOI] [PubMed] [Google Scholar]
- 4.Barber R, Butcher RW. The quantitative relationship between intracellular concentration and egress of cyclic AMP from cultured cells. Mol Pharmacol. 1981;19:38–43. [PubMed] [Google Scholar]
- 5.Bonci A, Williams JT. A common mechanism mediates long-term changes in synaptic transmission after chronic cocaine and morphine. Neuron. 1996;16:631–639. doi: 10.1016/s0896-6273(00)80082-3. [DOI] [PubMed] [Google Scholar]
- 6.Bonci A, Williams JT. Increased probability of GABA release during withdrawal from morphine. J Neurosci. 1997;17:796–803. doi: 10.1523/JNEUROSCI.17-02-00796.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bozarth MA, Wise RA. Intracranial self-administration of morphine into the ventral tegmental area in rats. Life Sci. 1981;28:551–555. doi: 10.1016/0024-3205(81)90148-x. [DOI] [PubMed] [Google Scholar]
- 8.Brundege JM, Diao L, Proctor WR, Dunwiddie TV. The role of cyclic AMP as a precursor of extracellular adenosine in the rat hippocampus. Neuropharmacology. 1997;36:1201–1210. doi: 10.1016/s0028-3908(97)00102-0. [DOI] [PubMed] [Google Scholar]
- 9.Cameron DL, Williams JT. Dopamine D1 receptors facilitate transmitter release. Nature. 1993;366:344–347. doi: 10.1038/366344a0. [DOI] [PubMed] [Google Scholar]
- 10.Cameron DL, Wessendorf MW, Williams JT. A subset of VTA neurons are inhibited by dopamine, 5-HT and opioids. Neuroscience. 1997;77:155–166. doi: 10.1016/s0306-4522(96)00444-7. [DOI] [PubMed] [Google Scholar]
- 11.Chavis P, Mollard P, Bockaert J, Manzoni O. Visualization of cyclic AMP-regulated presynaptic activity at cerebellar granule cells. Neuron. 1998;20:773–781. doi: 10.1016/s0896-6273(00)81015-6. [DOI] [PubMed] [Google Scholar]
- 12.Chavez-Noriega LE, Stevens CF. Increased transmitter release at excitatory synapses produced by direct activation of adenylate cyclase in rat hippocampal slices. J Neurosci. 1994;14:310–317. doi: 10.1523/JNEUROSCI.14-01-00310.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen C, Regehr WG. The mechanism of cAMP-mediated enhancement at a cerebellar synapse. J Neurosci. 1997;17:8687–8694. doi: 10.1523/JNEUROSCI.17-22-08687.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chieng B, Christie MJ. Lesions to terminals of noradrenergic locus coeruleus neurones do not inhibit opiate withdrawal behaviour in rats. Neurosci Lett. 1995;186:37–40. doi: 10.1016/0304-3940(95)11276-3. [DOI] [PubMed] [Google Scholar]
- 15.Chieng B, Christie MJ. Local opioid withdrawal in rat single periaqueductal gray neurons in vitro. J Neurosci. 1996;16:7128–7136. doi: 10.1523/JNEUROSCI.16-22-07128.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chieng B, Williams JT. Increased opioid inhibition of GABA release in nucleus accumbens during morphine withdrawal. J Neurosci. 1998;18:7033–7039. doi: 10.1523/JNEUROSCI.18-17-07033.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Christie MJ, Williams JT, North RA. Cellular mechanisms of opioid tolerance: studies in single brain neurons. Mol Pharmacol. 1987;32:633–638. [PubMed] [Google Scholar]
- 18.Cruciani RA, Dvorskin B, Morris SA, Crain SM, Makman MH. Direct coupling of opioid receptors to both stimulatory and inhibitory guanine nucleotide-binding proteins in F-11 neuroblastoma-sensory neurons hybrid cells. Proc Natl Acad Sci USA. 1993;90:3019–3023. doi: 10.1073/pnas.90.7.3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dunwiddie TV. The physiological role of adenosine in the central nervous system. Int Rev Neurobiol. 1985;27:63–139. doi: 10.1016/s0074-7742(08)60556-5. [DOI] [PubMed] [Google Scholar]
- 20.Fiorillo CD, Williams JT. Glutamate mediates an inhibitory postsynaptic potential in dopamine neurons. Nature. 1998;394:78–81. doi: 10.1038/27919. [DOI] [PubMed] [Google Scholar]
- 21.Glatt CE, Snyder SH. Cloning and expression of an adenylyl cyclase localized to the corpus striatum. Nature. 1993;361:536–538. doi: 10.1038/361536a0. [DOI] [PubMed] [Google Scholar]
- 22.Gintzler AR, Xu H. Different G proteins mediate the opioid inhibition or enhancement of evoked [5-methionine]enkephalin release. Proc Natl Acad Sci USA. 1991;88:4741–4745. doi: 10.1073/pnas.88.11.4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gysling K, Wang RY. Morphine-induced activation of A10 dopamine neurons in the rat. Brain Res. 1983;277:119–127. doi: 10.1016/0006-8993(83)90913-7. [DOI] [PubMed] [Google Scholar]
- 24.Henderson GB, Strauss BP. Evidence for cAMP and cholate extrusion in C6 rat glioma cells by a common anion efflux pump. J Biol Chem. 1991;266:1641–1645. [PubMed] [Google Scholar]
- 25.Ingram SL, Vaughan CW, Bagley EE, Connor M, Christie MJ. Enhanced opioid efficacy in opioid dependence is due to an additional signal transduction pathway. J Neurosci. 1998;18:10269–10276. doi: 10.1523/JNEUROSCI.18-24-10269.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Izenwasser S, Buzas B, Cox BM. Differential regulation of adenylyl cyclase activity by mu and delta opioids in rat caudate putamen and nucleus accumbens. J Pharmacol Exp Ther. 1993;267:145–152. [PubMed] [Google Scholar]
- 27.Johnson SM, Fleming WW. Mechanisms of cellular adaptive sensitivity changes: Applications to opioid tolerance and dependence. Pharmacol Rev. 1989;41:435–487. [PubMed] [Google Scholar]
- 28.Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992a;12:483–488. doi: 10.1523/JNEUROSCI.12-02-00483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson SW, North RA. Two types of neuron in the rat ventral tegmental area and their synaptic inputs. J Physiol (Lond) 1992b;450:455–468. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson SW, Mercuri NB, North RA. 5-hydroxytryptamine-1B receptors block the GABA-B synaptic potential in rat dopamine neurons. J Neurosci. 1992;12:2000–2006. doi: 10.1523/JNEUROSCI.12-05-02000.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jolas T, Gilden L, Nestler E, Aghajanian GK. Opiate upregulation of adenylyl cylase linked to GABA tone in 5-HT dorsal raphe neurons. Soc Neurosci Abstr. 1998;24:1352. [Google Scholar]
- 32.Kaplan GB, Sears MT. Adenosine receptor agonists attenuate and adenosine receptor antagonists exacerbate opiate withdrawal signs. Psychopharmacology. 1996;123:64–70. doi: 10.1007/BF02246282. [DOI] [PubMed] [Google Scholar]
- 33.Kennedy C, Henderson G. μ-opioid receptor inhibition of calcium current: Development of homologous tolerance in single SH-SY5Y cells after chronic exposure to morphine in vitro. Mol Pharmacol. 1991;40:1000–1005. [PubMed] [Google Scholar]
- 34.Lacey MG, Mercuri N, North RA. Two cell types in rat substantia nigra zona compacta distinguished by membrane properties and actions of dopamine and opioids. J Neurosci. 1989;9:1233–1241. doi: 10.1523/JNEUROSCI.09-04-01233.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Law PY, Hom DS, Loh HH. Loss of opiate receptor activity in neuroblastoma X glioma NG108–15 hybrid cells after chronic opiate treatment. Mol Pharmacol. 1982;22:1–4. [PubMed] [Google Scholar]
- 36.Mansour A, Meador-Woodruff JH, Zhou QY, Civelli O, Akil H, Watson SJ. A comparison of D1 receptor binding and mRNA in rat brain using receptor autoradiographic and in situ hybridization techniques. Neuroscience. 1991;45:359–371. doi: 10.1016/0306-4522(91)90233-e. [DOI] [PubMed] [Google Scholar]
- 37.Manzoni OJ, Williams JT (1999) Opioid inhibition of excitatory transmission in the ventral tegmental area. J Neurosci, in press.
- 38.Manzoni O, Pujalte D, Williams JT, Bockaert J. Decreased presynaptic sensitivity to adenosine after cocaine withdrawal. J Neurosci. 1998;18:7996–8002. doi: 10.1523/JNEUROSCI.18-19-07996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mons N, Cooper DMF. Adenylate cyclases: critical foci in neuronal signaling. Trends Neurosci. 1995;18:536–542. doi: 10.1016/0166-2236(95)98375-9. [DOI] [PubMed] [Google Scholar]
- 40.Murthy KS, Makhlouf GM. Opioid mu, delta, and kappa receptor-induced activation of phospholipase C-beta 3 and inhibition of adenylyl cyclase is mediated by Gi2 and G(o) in smooth muscle. Mol Pharmacol. 1996;50:870–877. [PubMed] [Google Scholar]
- 41.Nestler EJ, Aghajanian GK. Molecular and cellular basis of addiction. Science. 1997;278:58–63. doi: 10.1126/science.278.5335.58. [DOI] [PubMed] [Google Scholar]
- 42.Puttfarcken PS, Werling LL, Cox BM. Effects of chronic morphine exposure on opioid inhibition of adenylyl cyclase in 7315c cell membranes: a useful model for the study of tolerance at μ opioid receptors. Mol Pharmacol. 1988;33:520–527. [PubMed] [Google Scholar]
- 43.Rosenberg PA, Ditchter MA. Extracellular cAMP accumulation and degradation in rat cerebral cortex in dissociated cell culture. J Neurosci. 1989;9:2654–2663. doi: 10.1523/JNEUROSCI.09-08-02654.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosenberg PA, Knowles R, Knowles KP, Li YJ. β-Adrenergic receptor-mediated regulation of extracellular adenosine in cerebral cortex in culture. J Neurosci. 1994;14:2953–2965. doi: 10.1523/JNEUROSCI.14-05-02953.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salin PA, Malenka RC, Nicoll RA. Cyclic AMP mediates a presynaptic form of LTP at cerebellar parallel fiber synapses. Neuron. 1996;16:797–803. doi: 10.1016/s0896-6273(00)80099-9. [DOI] [PubMed] [Google Scholar]
- 46.Sharma SK, Klee WA, Nirenberg M. Dual regulation of adenylate cyclase accounts for narcotic dependence and tolerance. Proc Natl Acad Sci USA. 1975;72:3092–3096. doi: 10.1073/pnas.72.8.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wise RA. The role of reward pathways in the development of drug dependence. Pharmacol Ther. 1987;35:227–263. doi: 10.1016/0163-7258(87)90108-2. [DOI] [PubMed] [Google Scholar]