

Abstract
Hyperkalaemic periodic paralysis, paramyotonia congenita, and potassium-aggravated myotonia are three autosomal dominant skeletal muscle disorders linked to the SCN4A gene encoding the α-subunit of the human voltage-sensitive sodium channel. To date, ∼20 point mutations causing these disorders have been described. We have identified a new point mutation, in the SCN4A gene, in a family with a hyperkalaemic periodic paralysis phenotype. This mutation predicts an isoleucine-to-phenylalanine substitution at position 1495 located in the transmembrane segment S5 in the fourth homologous domain of the human α-subunit sodium channel. Introduction of the I1495F mutation into the wild-type channels disrupted the macroscopic current inactivation decay and shifted both steady-state activation and inactivation to the hyperpolarizing direction. The recovery from fast inactivation was slowed, and there was no effect on channel deactivation. Additionally, a significant enhancement of slow inactivation was observed in the I1495F mutation. In contrast, the T704M mutation, a hyperkalaemic periodic paralysis mutation located in the cytoplasmic interface of the S5 segment of the second domain, also shifted activation in the hyperpolarizing direction but had little effect on fast inactivation and dramatically impaired slow inactivation. These results, showing that the I1495F and T704M hyperkalaemic periodic paralysis mutations both have profound effects on channel activation and fast–slow inactivation, suggest that the S5 segment maybe in a location where fast and slow inactivation converge.
Keywords: Na channel, SCN4A, disorders, activation, slow inactivation, expression
Naturally occurring mutations are a powerful tool to study the relationship between protein structure and function. They have been widely used in the past decade to study integral membrane proteins for which crystal structures are difficult to obtain. This has been successfully applied to the functional analysis of the voltage-gated sodium channels. The sodium channel α-subunit is 260 kDa and associates with small β-subunits in muscle and brain. According to sodium channel models, the α-subunit is organized in four homologous domains (I–IV), and each domain is represented by six membrane spanning segments (S1–S6) (Noda et al., 1984). Two fragments located between segments S5 and S6 (SS1–SS2) are believed to form the pore region.
Over 20 single mutations affecting the human sodium channel geneSCN4A (encoding the human skeletal muscle isoform) have been linked to three autosomal dominant disorders: hyperkalaemic periodic paralysis (HyperKPP) (Ptácek et al., 1991; Rojas et al., 1991), paramyotonia congenita (PC) (Ptácek et al., 1992; McClatchey et al., 1992), and potassium-aggravated myotonia (PAM) (Lerche et al., 1993; Ptácek et al., 1994). All channel domains carry at least one disease-causing mutation, and all transmembrane segments seem to be targeted, except the membrane spanning segments S2 and S5 (for review, see Bulman, 1997). Many of these mutations have been overexpressed inXenopus oocytes or in mammalian heterologous systems. These studies demonstrated the existence of many defects in channel function that may underlie either muscle hyperexcitability or inexcitability, such as the presence of sustained inward current, shift in steady-state inactivation or activation, alteration of channel deactivation, modification of channel recovery from fast inactivation, and impairment of slow inactivation.
Five point mutations have been reported to affect the human sodium channel gene SCN4A causing HyperKPP: T704M (Cannon and Strittmatter, 1993; Cummins et al., 1993; Yang et al., 1994; Cummins and Sigworth, 1996; Hayward et al., 1997), V781I (Baquero et al., 1995), A1156T (Yang et al., 1994), M1360V (Wagner et al., 1997), and M1592V (Hayward et al., 1997). A recent study of the V781I mutation in human embryonic kidney (HEK) 293 cells suggested that this mutation may be a benign polymorphism (Green et al., 1997). All of these HyperKPP mutations show a common feature; they are localized at the intracellular membrane interface. HyperKPP-causing mutations have not been found in the extracellular loops or in the middle of any membrane spanning domain to date. We report here a novel missense mutation in the SCN4A in a patient with HyperKPP, causing the amino acid change from isoleucine to phenylalanine at position 1495 located within the putative transmembrane segment S5 of domain IV. We have also performed additional comparative experiments on the most common HyperKPP mutation (T704M), located at the intracellular interface on the S5 segment of the sodium channel.
This is the first report of a naturally occurring mutation in the S5 segment, and naturally occurring mutations in this segment may allow a better understanding of the disease and define the role of segment S5 in sodium channel function.
MATERIALS AND METHODS
Identification of a HyperKPP patient. The patient was examined by one of the authors (R.T.). The proband underwent a physical examination, a potassium-loading test, and electromyography. All human tissue samples used in this project were obtained with the approval of the Institutional Review Board at the University of Utah Health Sciences Center.
Genetic analysis. Genomic DNA was extracted from peripheral blood leukocytes and amplified using PCR as described previously (Ptácek et al., 1992). Single strand conformation polymorphism (SSCP) analysis was performed on amplified DNA using a protocol described previously (Ptácek et al., 1992). The primers used were as follows: 5′-CCTCCTCCTCTTCCTGGTCAT-3′ (Nb forward) and 5′-GGGCTCGCTGCTCTCCTCTGT-3′ (Nb reverse).
Cell culture and transfection. HEK 293 cells were grown in DMEM supplemented with penicillin, streptomycin, and 10% fetal calf serum (Life Technologies, Gaithersburg, MD) and maintained at 37°C with 5% CO2. The calcium–phosphate precipitation technique (Graham and Van Der Eb, 1973) was used for both transient and stable transfections. HEK 293 cells were grown on a 35 mm culture dish (Corning, Corning, NY) and transfected at 30% confluence. Approximately 15 hr after transfection, the media was changed. Stably expressing cell lines were produced using the expression vector pRc-CMV encoding either wild-type (WT) human skeletal muscle voltage-gated sodium channel or the corresponding I1495F mutant construct (Bendahhou et al., 1995).
Construction of hSkM1-I1495F and hSkM1-T704M. The hSkM1-I1495F was constructed using the megaprimer PCR method of site-directed mutagenesis (Sarkar and Sommer, 1990). Wild-type cDNA (hSkM1) was used as a template for the first round of PCR to generate two megaprimers (1.3 kb). The reaction was primed with an oligo-nucleotide encoding a region corresponding to nucleotides (nt) 3736–3755 of the full-length channel sequence 5′-GGCTCAATGTCAAGGTCAAC-3′ and the oligo encoding sequence (nt 4550–4569) 5′-TGGAGTAGGTGAACATGACC-3′ (reverse). The first round PCR reaction protocol was as follows: 2 min at 94°C for one cycle; 20 sec at 94°C, 20 sec at 60°C, and 1 min at 75°C for 30 cycles; and 5 min at 72°C for one cycle.
The second megaprimer set was generated using oligo-nucleotides (nt 4550–4570) 5′-GGTCATGTTCACCTACTCCA-3′ and (nt 5079–5098) 5′-TGGGCAAGTCCAGTGTGATG-3′ (reverse). The first round PCR reaction protocol for this primer set was as follows: 2 min at 94°C for one cycle; 20 sec at 94°C, 20 sec at 50°C, and 1 min at 75°C for 30 cycles; and 5 min at 72°C for one cycle.
The primer contained a nucleotide substitution T4561C (indicated above by an italic letter), which results in the amino acid substitution I1495F. The second round of PCR was primed using first round products (megaprimers) and the primers 5′-GGCTCAATGTCAAGGTCAAC-3′ (forward) and 5′-TGGGCAAGTCCAGTGTGATG-3′ (reverse). The following protocol was used for the second round PCR: 2 min at 94°C for one cycle; 20 min at 94°C, 20 sec at 54°C, and 90 sec at 72°C for 30 cycles; and 5 min at 72°C for one cycle. A 1.3 kb product was isolated from a 1% agarose gel using the Qiagen (Hilden, Germany) gel extraction kit, cut with SseI restriction enzyme and ligated to the appropriately cut fragment of wild-type hSkM1 (the pRc-CMV-hSkM1 construct was initially cut with SseI enzyme to separate a 10 kb band from the 1.3 kb wild-type band) to construct the full-length mutated channel hSkM1-I1495F.
All PCR reactions were performed in DNA Engine Tetrad (MJ Research, Watertown, MA). DNA sequencing was performed using either Applied Biosystems (Foster City, CA) dRhodamine dye terminators or Applied Biosystems Prism BigDye Terminators and cycle sequencing withTaq FS DNA polymerase. DNA sequence was collected and analyzed on an Applied Biosystems Prism 377 automated DNA sequencer. All primers were synthesized on a 394 automated DNA synthesizer (Applied Biosystems) following standard ABI procedures.
The hSkM1-T704M construct was engineered as described previously (Yang et al., 1994)
Electrophysiology. Recordings were conducted in the whole-cell configuration (Hamill et al., 1981) at room temperature (22°C) using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA) and an EPC-9 amplifier. Data acquisition and analysis were performed with pClamp6 (Axon Instruments) or Pulse and Pulsefit (Heka Elektronik, Lambrecht/Pfalz, Germany), and Igor Pro (WaveMetrics Inc., Lake Owego, OR) programs. Pipettes (Kimax; Kimble Glass Inc.) of 0.8–1.5 MΩ were prepared using a Sutter (Novato, CA) P-87 puller and were heat-polished before use. The pipette tip potential was adjusted to zero for each experiment. To minimize space-clamp problems caused by the electrical coupling between HEK 293 cells, only isolated cells with a soma size of 10–30 μm were selected for recordings. Cells were not considered for analysis if the initial seal resistance was <5 GΩ or if the cells had high leakage currents (holding current of >0.1 nA at −80 mV), membrane blebs, or an access resistance >3 MΩ. Linear leak subtraction, based on the resistance estimates from 4–6 hyperpolarizing pulses applied before the depolarizing test potential, was used for all voltage-clamp recordings.
Pulse protocols. Steady-state activation was studied by measuring the peak sodium conductance (GNa) during a 25 msec test pulse to various test potentials from −120 mV holding voltage.GNa was calculated fromGNa =INa/(V −Vrev), where INais the peak sodium current during the test depolarization (V), andVrev is the sodium reversal potential. Data were normalized to maximum peak conductance (Gmax) and fit to a two-state Boltzmann distribution:GNa/Gmax = (1 + exp[ze(V −V0.5)/kT])−1, where V0.5 is the potential for half-maximal activation, k is the Boltzmann constant, z is the apparent gating charge, T the absolute temperature, andkT/e = 25 mV at 22°C.
To study steady-state fast inactivation, cells were held at prepulse potentials ranging from −130 or −120 to +20 mV for 200 msec and then subjected to a 0 mV test pulse for 25 msec. Normalized peak currents were plotted versus prepulse potentials, and curves were fitted by the Boltzmann function: I/Imax= (1 + exp[ze(V −V0.5)/kT])−1, where Imax is the current recorded at 0 mV after the most hyperpolarizing prepulse, and k, T,z, and e are the same as described above.
For the development of fast inactivation, the holding potential was −100 mV. A series of prepulses ranging from −80 to −50 mV for increasing durations were applied before a test pulse at −20 mV for 20 msec to determine the fraction of current inactivated during the prepulse.
Recovery from fast inactivation was studied by prepulsing the cells to −20 mV for 20 msec to inactivate all of the current, and then recovery potentials from −140 to −60 mV for increasing recovery durations were applied before the test pulse to −20 mV (20 msec) to assay the fraction of current recovered. Normalized peak current amplitudes were plotted and fitted to a single-exponential function (a0 + a1exp(−t/t)), and the time constant was determined.
Steady-state slow inactivation. The first step consists of holding the cells at potentials ranging from −130 to 10 mV in 10 mV steps for 50 sec. A 30 msec recovery pulse to −100 mV and a 20 msec test pulse to −10 mV were given before the holding potential was incremented again. The holding potential was incremented by 10 mV immediately after each recovery pulse–test pulse sequence. We term this protocol sequential because the channels are not allowed to recover from slow inactivation for each test pulse; the expectation is that the sequential changes in holding potential will mimic changes in holding potential of longer durations. The short hyperpolarizing recovery pulses can be used to remove fast inactivation just before a test pulse to measure slow inactivation processes (Simoncini and Stühmer, 1987). The peak current elicited by the test pulse to −10 mV was plotted as a fraction of the maximum current. Steady-state slow inactivation data were fitted with a Boltzmann function as described for fast inactivation.
Slow inactivation recovery. Cells were held at −20 mV for at least 15 min to allow slow inactivation to reach steady-state equilibrium. Short recovery times were monitored with individual recovery pulses. Once every 50 sec, the cells were allowed to recover at −100 mV for increasing durations (from 1 to 500 msec), followed by a 20 msec test pulse to −10 mV. After the last short recovery pulse sequence, the cells were held at −20 mV for an additional 50 sec, after which the holding potential was switched to −100 mV and recovery was monitored in a continuous recording, with −10 mV test pulses at 0.5 sec intervals for 10 sec, then at 5 sec intervals for 110 sec, and finally at 15 sec intervals for 5 min. This protocol allowed us to monitor a complete recovery profile to >7 min.
Twenty-four hours after plating stable lines or 36 hr after transfection, culture dishes containing cells were transferred to the stage of an inverted microscope. The culture media was removed and replaced with a bathing solution containing (in mm): 140 NaCl, 4 MgCl2, 2 CaCl2, and 10 Na-HEPES, pH 7.3. The internal pipette solution was (in mm): 130 CsCl, 4 MgCl2, 2.5 EGTA, 5 NaCl, and 10 HEPES, pH 7.3. We have not observed significant differences in the sodium current properties when CsF is used instead of CsCl.
Values represent means ± SEM. Statistical data were obtained using t test.
RESULTS
Evaluation of patient
The patient is a 48-yr-old man who initially noted episodic weakness induced by rest after exercise. The attacks had onset at age 16 and are not affected by cold temperatures or dietary changes. His episodes of weakness were associated with cramping pain in his muscles. No symptoms suggestive of myotonia were reported; he has no paradoxical eye closure myotonia or lid lag myotonia. Similarly, no percussion or action myotonia is noted in his limbs. He has developed progressive fixed weakness in his proximal lower extremities, and significant wasting in his quadriceps muscles. In addition to the striking atrophy of his quadriceps and hamstrings, he had significant calf hypertrophy, as well as well developed upper extremity musculature. The level of creatine kinase was elevated at 523 IU/l (normal is 0–243 IU/l). The patient evaluation included an electromyogram, which showed no evidence of myotonic discharges. The patient was challenged with a sugar-free potassium chloride solution. This resulted in a peak potassium level (6.1 mmol/l; normal is 3.4–4.7 mmol/l) ∼90 min after ingestion, with a concomitant loss of strength noted in his grip, as well as more dramatically in his hip flexors and knee extensors.
The patient was treated variously over the years with acetazolamide, thiazide diuretics, or tocainide. None of these pharmacological interventions prevented the attacks. Although both parents are deceased and their affected status is not known, he has an affected older brother and nephew.
Identification of nucleotide alterations in the patient DNA
DNA from the patient was amplified using primers for exon 24 as described in Materials and Methods. An aberrant SSCP conformer was noted in this patient and his brother. Sequence analysis of the aberrant conformer revealed a T4561C mutation (data not shown) of theSCN4A gene that predicts an isoleucine-to-phenylalanine change at position 1495 in the human skeletal muscle sodium channel α-subunit. This mutation was absent in 106 normal unrelated individuals. This amino acid is located in the membrane spanning segment S5 of domain IV and is perfectly conserved among all voltage-gated sodium channels sequenced to date fromDrosophila to human (Fig.1).
Fig. 1.

Alignment of the segment IV–S5 of the sodium channel. Segment S5 of domain IV deduced amino acid sequence of the voltage-gated sodium channel from human skeletal muscle (hSkM1), human cardiac muscle (hSkM2), rat brain type II (RII), eel electroplax, squid giant axon (Squid), TTX resistant from dorsal root ganglia (DRG), jellyfish, and Drosophilapara, along with isoleucine 1495-to-phenylalanine substitution. This figure shows that isoleucine 1495 is well conserved among voltage-gated sodium channels.
Fast inactivation
A common feature of the majority of the naturally occurring mutations causing HyperKPP or PC is a sustained or noninactivating sodium current. To check for the presence of this component in the hSkM1-I1495F channels, cells were first held at −120 mV and then depolarized to different potentials for 25 msec. Figure2 illustrates the behavior of wild-type channels (Fig. 2A) and the hSkM1-I1495F (Fig.2B) expressed in HEK 293 cells, in response to increasing depolarizing pulses. The mutant channels reveal an additional sustained sodium current. This is better illustrated in Figure 2C in which two normalized current traces (at −10 mV) from wild-type and I1495F channels are superimposed to show the defect in the fast inactivation decay. Increasing the test pulse duration to 100 msec also shows the presence of this prolonged inward sodium flow (Fig. 2D). However, when currents were recorded 10 min after rupturing into the cell, the sustained inward current was absent (data not shown). This may indicate that the prolonged inward current seen during the first minutes of recording is subject to modulation. Whatever the reason, early recordings indicate that the S5 segment may be involved in channel inactivation.
Fig. 2.

Effect of I1495F mutation on macroscopic currents. Cells were held at −120 mV, and inward sodium currents were elicited by 10 mV voltage steps from −80 to 20 mV for WT (A) and I1495F (B).C shows normalized current traces from WT and I1495F mutant recorded at −10 mV. D, An example of superimposed current recordings from WT and I1495F channels using the same pulse protocol as in A–C for a 100 msec duration.
The inactivation phase was fitted with a single exponential at different voltages. A single-exponential function was sufficient for fitting wild-type currents, whereas a two-exponential function was necessary to obtain an accurate fit of sustained I1495F currents (Fig.3A). The HyperKPP current decay exhibits two components: one that is fast with a time constant slightly faster than WT throughout the range of potentials tested, and the second decaying with a slow time constant representing the sustained channels shown in the traces in Figure 2, Cand D (Fig. 2B–D).
Fig. 3.
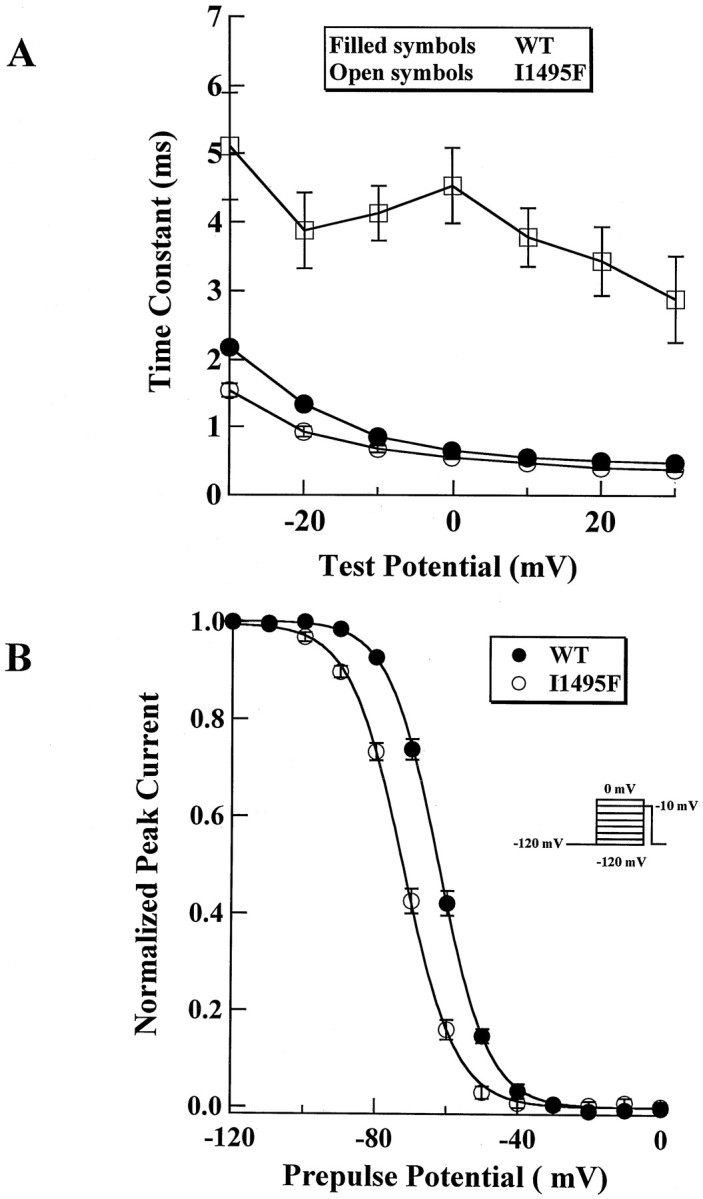
Inactivation kinetics of I1495F. A, Time constant of fast inactivation for WT (filled circles, n = 39) and I1495F (open circles, n = 14) showing an additional slow component (open squares,n = 14) for the HyperKPP mutant channels.B, Steady-state fast inactivation. HEK 293 cells were subjected to a protocol described in Materials and Methods to generate the inactivation curves for WT (filled circles,n = 43) and I1495F (open circles,n = 31).
A sustained sodium current could be the result of an increase in the “window current” caused by a positive shift of the steady-state inactivation, a negative shift of the activation curve, or both. To test for changes in steady-state fast inactivation, cells were held over a range of potentials for 200 msec, which allows the channels to undergo fast inactivation without allowing the channels to slowly inactivate, and then depolarized to a test pulse (0 mV) to measure the fraction of channels that did not fast inactivate. Figure 3Bshows that the mutant channels inactivate at more hyperpolarizing potentials than the wild type. The midpoint of inactivation was shifted by −10 mV (WT, V0.5 = −62.1 ± 4.9 mV; n = 43; I1495F, V0.5 = −72.3 ± 4.3 mV; n = 31), and this significant shift (p < 0.001) was not transient. This hyperpolarizing shift of the inactivation curve has not been reported in a pure HyperKPP mutant but was rather seen in mutations occurring at position 1448, R1448P, C, H (Chahine et al., 1994; Yang et al., 1994), or S (S. Bendahhou, T. R. Cummins, H. Kwiecinski, S. G. Waxman, and L. J. Ptácek, unpublished observations), causing PC. However, there is no significant change (p = 0.1) in the slope of the inactivation curve for I1495F channels (WT,z = 3.9 ± 0.7; n = 26; I1495F,z = 3.7 ± 0.5; n = 30) as reported for the different substitutions at position 1448 of the human skeletal muscle sodium channel.
Steady-state activation
As mentioned above, a sustained inward sodium current could result from a negative shift in the voltage dependence of the activation. Figure 4A shows the current–voltage relationship for WT and I1495F channels. The I1495F channels activate at more negative potentials. This shift is better seen by transforming the current–voltage relationship to a conductance–voltage curve (Fig. 4B). The steady-state activation is significantly shifted (p < 0.001) to the left for the hSkM1-I1495F by ∼8 mV (WT, V0.5 = −17.7 ± 3.0 mV;n = 47; I1495F, V0.5 = −25.5 ± 3.7 mV; n = 31). There was no significant change (p = 0.05) in the apparent gating charge between the two constructs (WT, z = 4.2 ± 0.6; n = 47; I1495F, z = 4.0 ± 0.4; n = 31). In addition to its role in channel inactivation (indicated by the presence of sustained current flow and the shift in the inactivation curve), our data also suggest an involvement of the S5 segment of domain IV in channel activation.
Fig. 4.

WT and I1495F activation parameters. The pulse protocol for constructing the current–voltage relationship is described in Materials and Methods. A, The current–voltage curves for WT (filled circles,n = 47) and I1495F (open circles,n = 31) were normalized for a better appreciation of the shift between the two relations. B, Currents were converted to conductance, and fit was made according to the Boltzmann function described in Materials and Methods. Symbols are the same as in A.
Deactivation, development, and recovery from fast inactivation in I1495F
A defect in channel deactivation may favor prolonged action potentials, as shown in a model of muscle membrane excitability (Featherstone et al., 1998). To examine this, tail currents, resulting from a brief 40 mV pulse preceding a command potential ranging from −120 to 0 mV, were recorded, and traces were fitted with a single-exponential function. As shown in Figure5, kinetics of deactivation of the I1495F channels are not different from WT channels. However, when testing for recovery from fast inactivation, the fraction of channels recovered after a prolonged −80 mV conditioning pulse was smaller for I1495F channels than for the WT channels (Fig. 5B), suggesting a slower recovery from fast inactivation for the mutant channels. The time constants for slow recovery from fast inactivation are illustrated in Figure 5C, which plots the time constant at each potential tested. However, development of fast inactivation seems to be enhanced at negative voltages in the I1495F channels, indicating that closed-state inactivation is enhanced in the I1495F mutation.
Fig. 5.
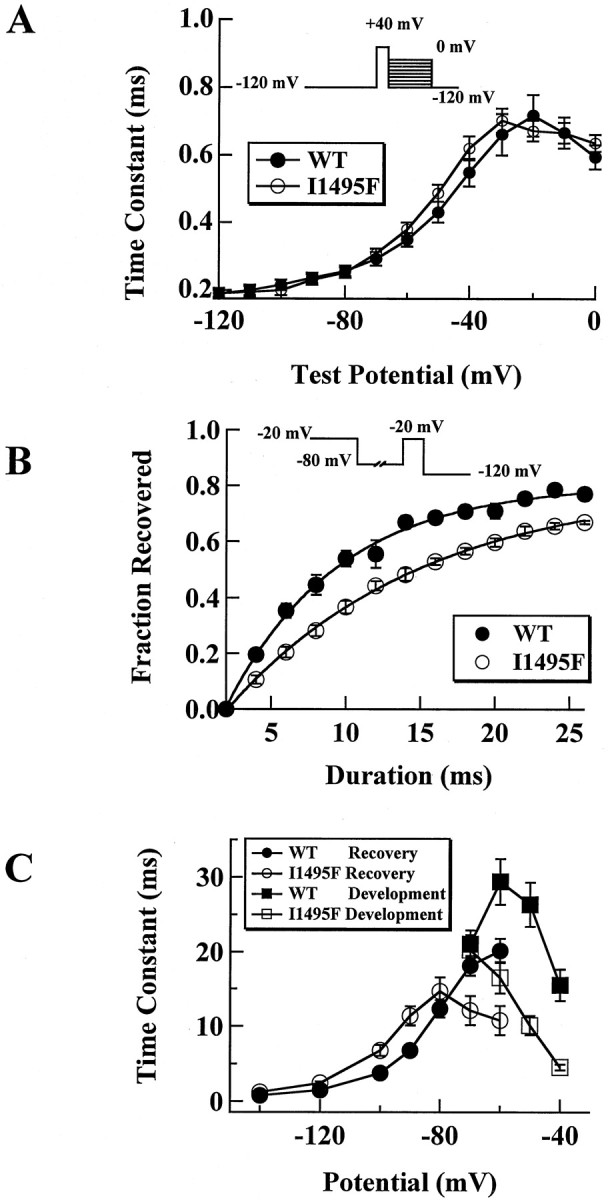
Deactivation, development, and recovery from inactivation for I1495F. A, Tail currents were elicited by a 0.5 msec test pulse to 40 mV, followed by repolarization pulses ranging from −120 to 0 mV. Resulting currents were fitted by a single-exponential decay and expressed as function of the voltage for WT (filled circles, n = 20) and for I1495F (open circles, n = 20). B, To assess the rate of recovery from fast inactivation, the following protocol was applied to HEK 293 cells: channels were fast inactivated during 200 msec using 0 mV voltage pulse, then they were allowed to recover at different voltages (−80 mV test pulse is shown as an example) for an increasing time, and finally a 0 mV test pulse was applied to test for the fraction of the channels recovered. Peak currents obtained using the test pulse were normalized to the peak current obtained during the inactivating pulse for WT (filled circles, n = 14) and I1495F (open circles, n = 26).C, Data for development of inactivation (squares) and recovery from fast inactivation (circles) were fitted to a single-exponential function and plotted as a function of the development–recovery pulse voltage. The averaged time constants from WT (filled symbols, n = 10) and I1495F (open symbols, n = 7) are shown.
I1495F enhances slow inactivation
It has been proposed that a disruption of slow inactivation is also necessary to account for the extended duration of paralysis seen in patients with HyperKPP (Ruff, 1994). Subsequently, it was demonstrated that the rT698M mutation does indeed impair slow inactivation of the rat SkM1 sodium channel (Cummins and Sigworth, 1996). Slow inactivation is functionally distinct from fast inactivation. The time constant for entering and leaving the slow inactivated state is on the order of tens of seconds rather than milliseconds as for fast inactivation (Ruff et al., 1988). Results from I1495F slow inactivation experiments are shown in Figure6. Our data show that steady-state slow inactivation (s∞) is not impaired for I1495F channels as reported for the rat T698M mutation (Cummins and Sigworth, 1996), but in contrast, is enhanced with the I1495F mutant (WT,V0.5 = −61.6 ± 2.8 mV;z = 2.7 ± 0.2; n = 8; I1495F,V0.5 = −76.7 ± 2.6 mV;z = 1.8 ± 0.1; n = 8) (Fig.6A). Both midpoint and apparent gating charge are significantly different (p < 0.002). However, development of slow inactivation at −20 mV (Fig. 6B) and recovery from slow inactivation at −100 mV (Fig. 6C) do not seem to be significantly altered by the I1495F mutation.
Fig. 6.
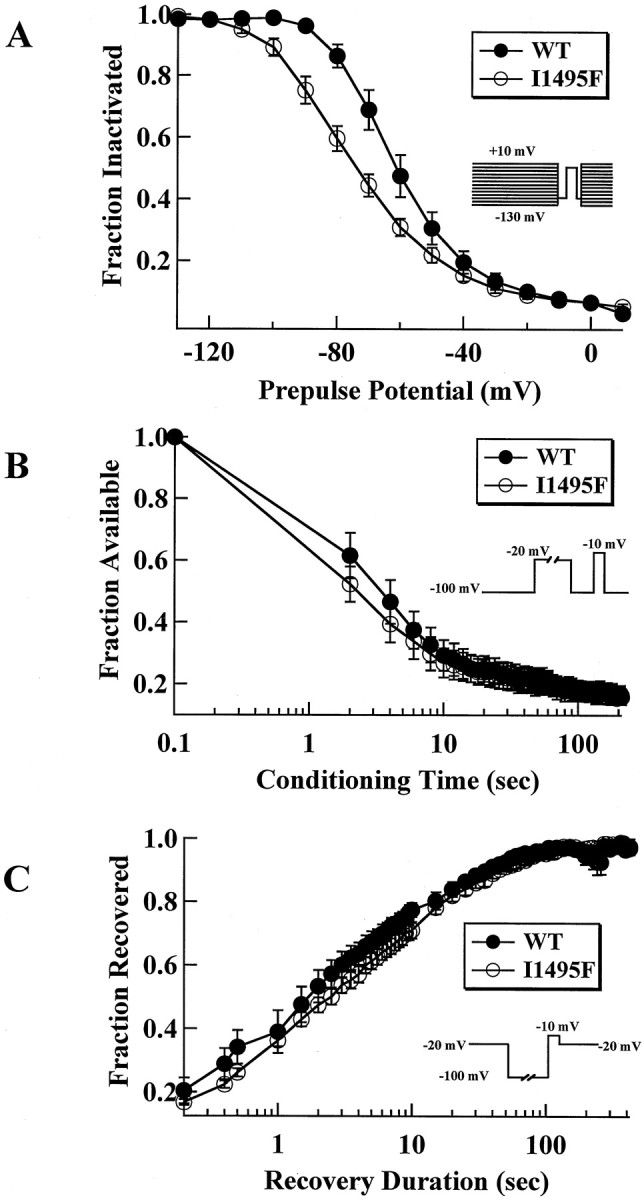
I1495F enhances slow inactivation.A, Steady-state slow inactivation in hSkM1 WT (filled circles, n = 8) and I1495F (open circles, n = 8) after 50 sec conditioning pulse ranging from −130 to 10 mV. Band C show development of slow inactivation and recovery from slow inactivation, respectively, as a function of time for WT (filled circles, n = 7) and I1495F (open circles, n = 7) channels. Protocols are as described in Materials and Methods.
The T704M mutation
The T704M is the most common HyperKPP mutation and is located at the intracellular interface on the S5 segment of domain II. Although the corresponding mutation has been extensively studied in the rat clone (rT698M; Cannon and Strittmatter, 1993; Cummins et al., 1993;Cummins and Sigworth, 1996; Hayward et al., 1997), the T704M mutation in the human clone has not been fully characterized (Yang et al., 1994). For example, it is not known whether the T704M mutation alters deactivation or slow inactivation of human skeletal muscle sodium channels. Hence, we were interested in the comparison of the T704M and I1495F mutations in the human background and in the same expression system.
Fast inactivating sodium currents are observed in cells expressing either the WT (Fig. 7A) or the T704M channels (Fig. 7B). The voltage dependence of activation and conductance was significantly different (p < 0.005) between WT (V0.5 = −19.0 ± 1.7 mV;n = 28) and T704M (V0.5 = −26.9 ± 1.8 mV; n = 26) in HEK 293 cells (Fig.7C,D). The apparent gating charge was also significantly different (p < 0.005) between WT (z = 3.7 ± 0.1; n = 15) and T704M (z = 3.0 ± 0.2; n = 13) channels. The shift in activation is similar to what has been reported previously for T704M channels (Yang et al., 1994) and for rT698M channels (Cummins et al., 1993), and to the shift in activation caused by the I1495F mutation. It has been proposed that the HyperKPP mutations increase the probability that the channels gate in a noninactivating mode, increasing the size of the sustained current throughout the activation range (Cannon and Strittmatter, 1993). To examine this, we measured the sustained current at the end of 40 msec depolarizations. At 0 mV, where window currents are not expected to contribute to the sustained current, we did not find any difference between the size of the sustained currents in WT (0.4 ± 0.8% of peak; n= 8) and T704M (0.3 ± 0.7% of peak; n = 10) cells. In contrast, at −50 mV, where window currents can contribute to the persistent current, the sustained currents were larger in T704M cells (0.5 ± 0.2% of peak; n = 10) than in WT cells (0.1 ± 0.2% of peak; n = 8). We found no difference (p = 0.3) between steady-state fast inactivation of WT (−70.5 ± 1.2; z = 4.0 ± 0.1; n = 31) and T704M (−73.6 ± 1.1;z = 3.6 ± 0.1; n = 29) channels (Fig. 7D). We were careful to measure steady-state fast inactivation at the same time point in the recording for both WT and T704M channels. We have shown previously that there is no difference between rat WT and rat T698M channels in terms of the voltage dependence of steady-state fast inactivation (Cummins et al., 1993;Cummins and Sigworth, 1996).
Fig. 7.

Effect of the T704M mutation on hSk6M1 currents. Family of traces from representative HEK 293 cells expressing either WT (A) or T704M (B) channels. The currents were elicited by 40 msec test pulses to various potentials from −60 to 30 mV. Cells were held at −100 mV. C, Peak current–voltage relationship for WT (filled squares, n = 8) and T704M (open squares, n = 7). D, The steady-state fast inactivation curves for WT (filled circles, n = 12) and T704M (open circles, n = 10) channels with 500 msec inactivating prepulses are shown. Cells were held at prepulse potentials over the range of −130 to 10 mV before a test pulse to −10 mV for 20 msec. Current is plotted as a fraction of peak current. Currents were converted to conductance, from the current–voltage curve shown in Figure 7C, for WT (filled squares, n = 8) and T704M (open squares, n = 7), and fit was made according to the Boltzmann function.
We also compared tail current kinetics of WT and T704M under the same conditions as those used for the I1495F. We found that the time constant of deactivation was slower in the T704M than in the WT channels (Fig.8A,B), in contrast to the I1495F mutant in which deactivation was not affected. This suggests that the T704M mutation alters the transition between the closed and open states. Our data on the recovery from fast inactivation show that the T704M recovers from fast inactivation in a manner similar to the I1495F mutant. However, development of fast closed-state inactivation seems to be unaffected by the T704M mutation (Fig. 8C), whereas the I1495F exhibits a faster development of closed-state inactivation.
Fig. 8.
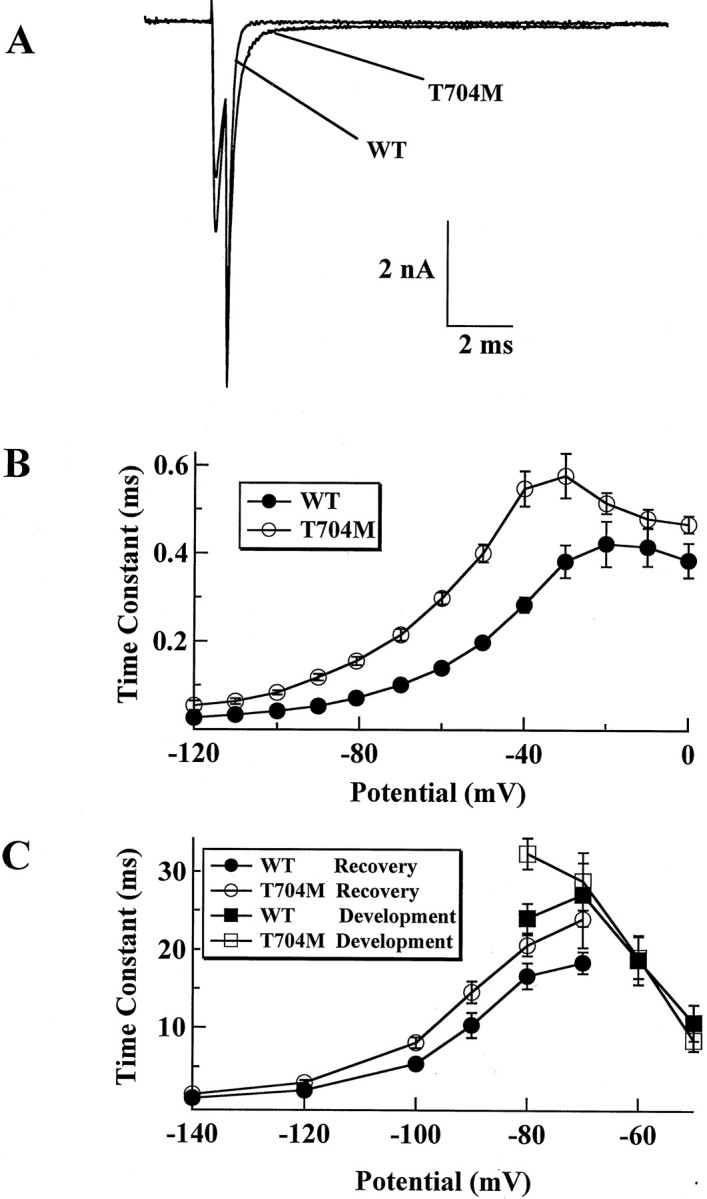
Deactivation, development, and recovery from fast inactivation for the T704M. The protocols used for the T704M are the same as those described for the I1495F mutation in Figure 5.A, Tail current traces obtained from WT and T704M channels at −70 mV. B, Time constants of tail current as shown in A were plotted as a function of the test potential for WT (filled circles,n = 5) and T704M (open circles,n = 6). C, Development of fast inactivation (squares) and recovery from fast inactivation (circles) for WT (filled symbols, n = 8) and T704M (open symbols, n = 7) were obtained as described in Figure 5.
Because the rat T698M and the I1495F mutations have altered slow inactivation and because the T704M is located at the intracellular interface of the S5 segment of domain II, we tested whether slow inactivation of human skeletal muscle sodium channels was affected by the T704M mutation. Results from slow inactivation experiments are shown in Figure 9. Although most of the WT current is slow inactivated after 5 min at 0 mV (Fig.9A), >50% of the T704M current remains available for activation under the same conditions (Fig. 9B). We measured the voltage dependence of slow inactivation using 50 sec sequential depolarizations (Fig. 9C). We observed a significant impairment of slow inactivation for T704M channels compared with WT channels. For WT channels, slow inactivation is nearly complete at 0 mV, with 6 ± 3% (n = 8) of the current available for activation after the 30 msec recovery pulse. In contrast, at 0 mV, 50 ± 10% (n = 7) of the T704M current is available for activation after the 30 msec recovery pulse.
Fig. 9.

Slow inactivation is impaired in T704M cells. Changes in peak sodium current for representative WT (A) and T704M (B) cells in response to changes in the holding potential. Cells were allowed to stabilize for 20 min at −80 mV before beginning the test protocol. The protocol involved holding the cell at a specific potential for 5 min, while once every 15 sec the membrane potential was stepped to −100 mV for 30 msec and then to 0 mV for 20 msec. The peak current elicited by the test pulse to 0 mV is plotted. C, The steady-state slow inactivation curves for WT (filled circles,n = 8) and T704M (open circles,n = 7) cells are shown. D, Normalized Na current in representative WT (filled circles, n = 5) and T704M (open circles, n = 6) cells recovering from slow inactivation. The time axis is logarithmic. The recovery protocol required ∼45 min to complete and had two phases: short recovery times were obtained with individual recovery pulses, long recovery times were obtained in a continuous recording. E, Development of slow inactivation is shown to be slower for the T704M (open circles, n = 5) than for the WT channels (filled circles, n = 5). Cells were held at −100 mV for an increasing conditioning time. Details of the protocol are described in Materials and Methods.
We also measured the time course for recovery from slow inactivation. The fraction of current was measured for recovery times ranging from 1 msec to >400 sec, as has been described previously (Cummins and Sigworth, 1996). The T704M channels recovered from slow inactivation much faster than did the WT channels (Fig. 9D). With a recovery time of 100 msec, only 11 ± 4% (n = 5) of the WT current recovers, whereas 66 ± 9% (n = 5) of T704M current recovers. The initial rate of recovery for the T704M channels is similar to that for the recovery from fast inactivation. Figure 9E shows the time course for the development of slow inactivation for WT and T704M channels. At −20 mV, the time constant was significantly (p < 0.05) smaller for WT channels (3.9 ± 0.7 sec; n = 5) than for T704M channels (12.0 ± 2.9 sec; n = 5). These data show that the T704M mutation disrupts slow inactivation in human SkM1 channels in an identical manner to the effect of the rT698M mutation on rat SkM1 channels but distinct from the I1495F mutation in which slow inactivation was particularly enhanced.
DISCUSSION
Over 20 point mutations in the SCN4A gene causing either HyperKPP, PC, or PAM have been described. The mutations are dispersed throughout the channel. However, three locations may be considered as “hot spots” according to the number of patients carrying mutations: the interdomain III-IV, cytoplasmic membrane interface, and segment S4 in domain IV. Few patients were reported with mutations in the transmembrane segments. No disease-causing mutation in segments S2 or S5 has been reported to date.
In the present study, we report a new family with the HyperKPP phenotype, which was unusual in that it was associated with muscle cramp and no myotonia was seen clinically or electrophysiologically. Scanning the sodium channel gene in the patient’s DNA with SSCP analysis and sequencing revealed a T-to-C transition at position 4561 of the SCN4A gene (in exon 24), resulting in the amino acid change isoleucine 1495 to phenylalanine located in the S5 segment of domain IV of the sodium channel α-subunit. This is the first disease-causing mutation located in this transmembrane segment. Expression of the I1495F mutation in HEK 293 cells revealed alterations in channel function that could account for the onset of HyperKPP in our patient. Comparing I1495F and T704M mutations revealed a prominent role of the S5 segment in channel activation, as well as in fast and slow inactivation.
Macroscopic inward sodium current from cells expressing I1495F showed that this substitution results in a sustained current at a wide range of physiological voltages. A two-exponential function was needed to fit the decay phase of the mutant macroscopic current, whereas a single exponential was sufficient for WT current decay fitting. The slowly decaying component measured in I1495F suggests that the S5 segment of domain IV is involved in channel inactivation. This behavior is surprising because I1495F is embedded in the membrane, presumably far from the intracellular membrane interface and also far from the III-IV linker, a strong candidate for the fast inactivation gate of the sodium channel (Stühmer et al., 1989; West et al., 1992). This mutation may have indirectly disrupted a structure directly involved in channel inactivation. Indeed, Mitrovic et al. (1996) and McPhee et al. (1998)reported recently an involvement of the cytoplasmic linker IV/S4-S5 in channel fast inactivation. In another study, a similar conclusion was reached by scanning the linker IV/S4-S5 of the hSkM1 by cysteine mutagenesis (Lerche et al., 1997).
We did not observe large sustained currents in T704M channels at 0 mV but only at more negative voltages at which window currents might occur. Although initially we do observe larger sustained currents in I1495F channels, these currents appear to run down, and after 10 min in the whole-cell configuration, we do not find any difference in the size of WT and I1495F sustained currents. This may indicate that sustained currents in skeletal muscle sodium channels can be modulated, and some of the disease-causing mutations could possibly affect this modulation. Recently, it has been shown that G-proteins modulate sustained currents in brain sodium channels (Ma et al., 1997).
Inactivation, especially at negative potentials, is significantly enhanced in the I1495F construct but not affected in T704M; this is indicated by a 10 mV hyperpolarizing shift in the steady-state fast inactivation of I1495F compared with WT and the faster development of inactivation at negative voltages (−80 to −60 mV, Fig.5C). This is the first HyperKPP mutation causing a negative shift in the voltage dependence of channel availability and conductance. However, the M1360V mutation responsible for HyperKPP and PC (Wagner et al., 1997) causes a −13 mV shift in channel availability only. The activation curve was not changed by the M1360V mutation, in contrast to the I1495F and T704M mutations that shifted the voltage dependence of activation toward negative potentials. Cummins et al. (1993) found a similar result for the rat T698M mutation (equivalent to the human T704M causing HyperKPP). A −12 mV shift in the voltage dependence of activation was reported to underlie the increase in the sustained sodium flow, mainly by increasing the window current between −75 and −40 mV, a voltage range in which sustained current is observed in HyperKPP muscle fibers.
The similarity observed here between the I1495F and T704M mutations in the activation curve may suggest a role of the S5 segments (at least in domain II and IV) in channel activation. However, although myotonia is prominent in patients with the T704M mutation, the patient with the I1495F mutation did not exhibit myotonia. This suggests that enhancement of activation is not sufficient to cause myotonia. Interestingly, another mutation in the S4–S5 linker of domain II, I693T, also shifts activation in the negative direction and induces weakness but apparently not myotonia (Plassart-Schiess et al., 1998).
Defective deactivation may well account for sodium channel disease, as reported in our own study on the R1448S mutation (Bendahhou, Cummins, Kwiecinski, Waxman, and Ptácek, unpublished observations) and studies from other laboratories (Richmond et al., 1997;Featherstone et al., 1998). These studies together have shown that a defect in sodium channel deactivation, along with altered inactivation, may lead to a destabilization of membrane repolarization resulting in prolonged action potentials. Whereas the T704M mutation significantly (p < 0.001) slowed deactivation, the I1495F mutation had much less of an effect compared with WT. We investigated also the rate of recovery from fast inactivation in WT, I1495F, and T704M. Whereas the I1495F has a slower recovery rate than WT (most prominent at −80 mV), the T704M mutation did not significantly alter the rate of recovery from fast inactivation.
It has been proposed that both fast and slow inactivation must be disrupted to trigger sustained currents in HyperKPP patients (Ruff, 1994). We found that slow inactivation was differentially disrupted in the I1495F and T704M mutants. Slow inactivation appears to be incomplete in T704M channels; after prolonged depolarizations lasting 20 min or more, more than one-third of T704M channels fail to become slow inactivated and are available for activation after a brief (e.g., 30 msec) hyperpolarization. This disruption of slow inactivation, in combination with enhanced window currents, are expected to result in sustained currents, such as those observed in the muscle of patients with HyperKPP. In the I1495F channels, in contrast, slow inactivation is dramatically enhanced; this is shown by the left shift in the steady-state slow inactivation. This is the first disease-causing mutation shown to enhance sodium channel slow inactivation. However,Wang and Wang (1997) have shown in site-directed mutagenesis that a mutation in S6 segment of domain I can cause enhancement in slow inactivation of the rat muscle homolog. An enhancement of slow inactivation could contribute to weakness in this patient.
Because the patient with the I1495F mutation did not exhibit myotonia, our data on slow inactivation suggests that disruption of slow inactivation could be crucial to the development of myotonia in HyperKPP patients. However, slow inactivation does not appear to be impaired by PC mutations associated with myotonia (Hayward et al., 1997; Richmond et al., 1997).
The HyperKPP mutation described here is the first substitution occurring in the putative membrane spanning segment IV–S5. This segment has not previously been shown to be involved in either the activation or inactivation process of the sodium channel. Mutations in the S5 segments and in the S4–S5 linker have already been shown to alter gating of voltage-sensitive potassium channels (Zagotta and Aldrich, 1990; McCormack et al., 1991; Kirsch et al., 1993; Holmgren et al., 1996; Shieh et al., 1997). An involvement of the S5 segments of the sodium channels in both activation and inactivation may well explain the mutant channel behavior in HyperKPP.
Recent studies have focused on the role of the S4–S5 and S3–S4 linkers of domain IV in sodium channel inactivation. This was shown by either studying disease-causing mutations, scanning mutagenesis (McPhee et al., 1998), cysteine scanning mutagenesis (Lerche et al., 1997), or by peptide insertion (Dib-Hajj et al., 1997). On one hand, the S3–S4 linker of domain IV may affect the coupling between activation and inactivation by altering the movement of S4 segment in the membrane (Chahine et al., 1994; Dib-Hajj et al., 1997). The S4–S5 linker, on the other hand, is a good candidate for interacting with the Ile-Phe-Met motif of the inactivation gate (Lerche et al., 1997;McPhee et al., 1998). These studies concluded that the loop S4–S5 plays a prominent role in sodium channel fast inactivation but gave no indication of the role of the S5 segment in the inactivation process. We hypothesize, based on our data and those reported for the role of the loop S4–S5 and the S4 segment in channel inactivation, that the membrane spanning segment S5 of domain IV interacts closely with the activation mechanism.
It has been proposed that paralysis can still occur in HyperKPP without impaired slow inactivation (Cannon, 1994). However, it was predicted that, without disruption of slow inactivation, there would be an asynchronous cycling of the muscle fibers between excitable and inexcitable states during the episodes of paralysis. In contrast to most patients with HyperKPP who typically have painless episodes of paralysis, the patient with the I1495F mutation experienced crampy pain during the attacks of weakness. Therefore, we propose that intermittent activation of affected muscle fibers during attacks of weakness might contribute to the crampy pain associated with the I1495F mutation. Because the I1495F mutation enhances slow inactivation, our results demonstrate that the manifestation of HyperKPP does not necessarily require disruption of slow inactivation.
Footnotes
This work was supported by a Muscular Dystrophy Association grant and by the Paralyzed Veterans of America and Eastern Paralyzed Veterans Association. We are grateful to Dr. F. Sigworth for supporting some of the work on the T704M mutation. We also thank the DNA Sequencing Facility at the University of Utah, supported in part by National Cancer Institute Grant #5P30CA42014. We thank Drs. M. Sanguinetti, J. Richmond, and D. Featherstone for critical reading of this manuscript.
Drs. Bendahhou and Cummins have contributed equally to this work.
Correspondence should be addressed to Louis J. Ptácek, Howard Hughes Medical Institute, Eccles Institute of Human Genetics, University of Utah, Building 533, Room 4425, Salt Lake City, UT 84112.
REFERENCES
- 1.Baquero JL, Ayala RA, Wang JZ, Curless RG, Feero WG, Hoffman EP, Ebeid MR. Hyperkalemic periodic paralysis with cardiac dysrhythmia: a novel sodium channel mutation. Ann Neurol. 1995;37:408–411. doi: 10.1002/ana.410370320. [DOI] [PubMed] [Google Scholar]
- 2.Bendahhou S, Cummins TR, Potts JF, Tong J, Agnew WS. Serine-1321-independent regulation of the μ1 adult skeletal muscle Na+ channel by protein kinase C. Proc Natl Acad Sci USA. 1995;92:12003–12007. doi: 10.1073/pnas.92.26.12003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bulman DE. Phenotype variation and newcomers in ion channel disorders. Hum Mol Genet. 1997;10:1679–1685. doi: 10.1093/hmg/6.10.1679. [DOI] [PubMed] [Google Scholar]
- 4.Cannon SC. Slow sodium channel inactivation need not be disrupted in the pathogenesis of myotonia and periodic paralysis. Biophys J. 1994;66:543–545. [Google Scholar]
- 5.Cannon SC, Strittmatter SM. Functional expression of sodium channel mutations identified in families with periodic paralysis. Neuron. 1993;10:317–326. doi: 10.1016/0896-6273(93)90321-h. [DOI] [PubMed] [Google Scholar]
- 6.Chahine M, George AL, Jr, Zhou M, Ji S, Sun WJ, Barchi RL, Horn R. Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron. 1994;12:281–294. doi: 10.1016/0896-6273(94)90271-2. [DOI] [PubMed] [Google Scholar]
- 7.Cummins TR, Sigworth FJ. Impaired slow inactivation in mutant sodium channels. Biophys J. 1996;71:227–236. doi: 10.1016/S0006-3495(96)79219-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cummins TR, Zhou J, Sigworth FJ, Ukomadu C, Stephan M, Ptácek LJ, Agnew WS. Functional consequences of a Na+ channel mutation causing hyperkalemic periodic paralysis. Neuron. 1993;10:667–678. doi: 10.1016/0896-6273(93)90168-q. [DOI] [PubMed] [Google Scholar]
- 9.Dib-Hajj SD, Ishikawa K, Cummins TR, Waxman SG. Insertion of a SNS-specific tetrapeptide in S3–S4 linker of D4 accelerates recovery from inactivation of skeletal muscle voltage-gated Na channel μ1 in HEK 293 cells. FEBS Lett. 1997;416:11–14. doi: 10.1016/s0014-5793(97)01154-x. [DOI] [PubMed] [Google Scholar]
- 10.Featherstone DE, Fujimoto E, Ruben PC. A defect in skeletal muscle sodium channel deactivation exacerbates hyperexcitability in human paramyotonia congenita. J Physiol (Lond) 1998;506:627–638. doi: 10.1111/j.1469-7793.1998.627bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graham FL, Van Der Eb AJ. A new technique for assay for infectivity of human adenovirus 5 DNA. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- 12.Green DS, Hayward LJ, George AL, Jr, Cannon SC. A proposed mutation, Val781Ile, associated with hyperkalemic periodic paralysis and cardiac dysrhythmia is a benign polymorphism. Ann Neurol. 1997;42:253–256. doi: 10.1002/ana.410420219. [DOI] [PubMed] [Google Scholar]
- 13.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 14.Hayward LJ, Brown RH, Jr, Cannon SC. Slow inactivation differs among mutant Na channels associated with myotonia and periodic paralysis. Biophys J. 1997;72:1204–1219. doi: 10.1016/S0006-3495(97)78768-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holmgren M, Jurman ME, Yellen G. N-type inactivation and the S4–S5 region of the Shaker K+ channel. J Gen Physiol. 1996;108:195–206. doi: 10.1085/jgp.108.3.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kirsch GE, Shieh CC, Drewe JA, Vener DF, Brown AM. Segmental exchanges define 4-aminopyridine binding and the inner mouth of K+ pores. Neuron. 1993;11:503–512. doi: 10.1016/0896-6273(93)90154-j. [DOI] [PubMed] [Google Scholar]
- 17.Lerche H, Heine R, Pika U, George AL, Jr, Mitrovic N, Browatzki M, Weiss T, Rivet BM, Franke C, Lomonaco M, Lehmann-Horn F. Human sodium channel myotonia: slowed channel inactivation due to substitutions for a glycine within the III-IV linker. J Physiol (Lond) 1993;470:13–22. doi: 10.1113/jphysiol.1993.sp019843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lerche H, Peter W, Fleischhauer R, Pika-Hartlaub U, Malina T, Mitrovic N, Lehmann-Horn F. Role in fast inactivation of the IV/S4–S5 loop of the human muscle Na+ channel probed by cysteine mutagenesis. J Physiol (Lond) 1997;505:345–352. doi: 10.1111/j.1469-7793.1997.345bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma JY, Catterall WA, Scheuer T. Persistent sodium currents through brain sodium channels induced by G protein βγ subunits. Neuron. 1997;19:443–452. doi: 10.1016/s0896-6273(00)80952-6. [DOI] [PubMed] [Google Scholar]
- 20.McClatchey AI, McKenna-Yasek D, Cros D, Worthen HG, Kuncl RW, Desilva SM, Cornblath DR, Gusella JF, Brown RH. Novel mutations in families with unusual and variable disorders of skeletal muscle sodium channel. Nat Genet. 1992;2:148–152. doi: 10.1038/ng1092-148. [DOI] [PubMed] [Google Scholar]
- 21.McCormack K, Tanouye MA, Iverson LE, Lin JW, Ramaswami M, McCormack T, Campanelli JT, Mathew MK, Rudy B. A role for hydrophobic residues in the voltage-dependent gating of Shaker K+ channels. Proc Natl Acad Sci USA. 1991;88:2931–2935. doi: 10.1073/pnas.88.7.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McPhee JC, Ragsdale DS, Scheuer T, Catterall WA. A critical role for the S4–S5 intracellular loop in domain IV of the sodium channel α-subunit in fast inactivation. J Biol Chem. 1998;273:1121–1129. doi: 10.1074/jbc.273.2.1121. [DOI] [PubMed] [Google Scholar]
- 23.Mitrovic N, Lerche H, Heine R, Fleischhauer R, Pika HU, Hartlaub U, George AL, Jr, Lehmann-Horn F. Role in fast inactivation of conserved amino acids in the IV/S4–S5 loop of the human muscle Na+ channel. Neurosci Lett. 1996;214:9–12. doi: 10.1016/0304-3940(96)12866-4. [DOI] [PubMed] [Google Scholar]
- 24.Noda M, Shimizu S, Tanabe T, Takai T, Kayano T, Ikeda T, Takahashi H, Nakayama H, Kanaoka Y, Minamino N, Kangawa K, Matsuo H, Raftery MA, Hirose T, Inayama S, Hayashida H, Miyata T, Numa S. Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature. 1984;312:121–127. doi: 10.1038/312121a0. [DOI] [PubMed] [Google Scholar]
- 25.Plassart-Schiess E, Lhuillier L, George AL, Jr, Fontaine B, Tabti N. Functional expression of the I693T Na+ channel mutation associated with paramyotonia congenita in a human cell line. J Physiol (Lond) 1998;507:721–727. doi: 10.1111/j.1469-7793.1998.721bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ptácek LJ, George AL, Jr, Griggs RC, Tawil R, Kallen RG, Barchi RL, Robertson M, Leppert MF. Identification of a mutation in the gene causing hyperkalemic periodic paralysis. Cell. 1991;67:1021–1027. doi: 10.1016/0092-8674(91)90374-8. [DOI] [PubMed] [Google Scholar]
- 27.Ptácek LJ, George AL, Jr, Barchi RL, Griggs RC, Riggs JE, Robertson M, Leppert MF. Mutations in an S4 segment of the adult skeletal muscle sodium channel cause paramyotonia congenita. Neuron. 1992;8:891–897. doi: 10.1016/0896-6273(92)90203-p. [DOI] [PubMed] [Google Scholar]
- 28.Ptácek LJ, Tawil R, Griggs RC, Meola G, McManis P, Barohn RJ, Mendell JR, Harris C, Spitzer R, Santiago F, Leppert MF. Sodium channel mutations in acetazolamide-responsive myotonia congenita, paramyotonia congenita, and hyperkalemic periodic paralysis. Neurology. 1994;44:1500–1503. doi: 10.1212/wnl.44.8.1500. [DOI] [PubMed] [Google Scholar]
- 29.Richmond JE, VanDeCarr D, Featherstone DE, George AL, Jr, Ruben PC. Defective fast inactivation recovery and deactivation account for sodium channel myotonia in the I1160V mutant. Biophys J. 1997;73:1896–1903. doi: 10.1016/S0006-3495(97)78220-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rojas CV, Wang J, Schwartz LS, Hoffman EP, Powell BR, Brown RH. A Met-to-Val mutation in the skeletal muscle Na+ channel α-subunit in hyperkalaemic periodic paralysis. Nature. 1991;354:387–389. doi: 10.1038/354387a0. [DOI] [PubMed] [Google Scholar]
- 31.Ruff RL. Slow Na+ channel inactivation must be disrupted to evoke prolonged depolarization-induced paralysis. Biophys J. 1994;66:542–545. doi: 10.1016/s0006-3495(94)80807-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruff RL, Simoncini L, Stühmer W. Slow sodium channel inactivation in mammalian muscle: a possible role in regulating excitability. Muscle Nerve. 1988;11:502–510. doi: 10.1002/mus.880110514. [DOI] [PubMed] [Google Scholar]
- 33.Sarkar G, Sommer SS. The “megaprimer” method of site-directed mutagenesis. Biotechniques. 1990;8:404–407. [PubMed] [Google Scholar]
- 34.Shieh CC, Klemic KG, Kirsch GE. Role of transmembrane segment S5 on gating of voltage-dependent K+ channels. J Gen Physiol. 1997;109:767–778. doi: 10.1085/jgp.109.6.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simoncini L, Stühmer W. Slow sodium channel inactivation in rat fast-twitch muscle. J Physiol (Lond) 1987;383:327–337. doi: 10.1113/jphysiol.1987.sp016411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stühmer W, Conti F, Suzuki H, Wang X, Noda M, Yahagi N, Kudo H, Numa S. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989;339:597–603. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- 37.Wagner S, Lerche H, Mitrovic N, Heine R, George AL, Jr, Lehmann-Horn F. A novel sodium channel mutation causing a hyperkalemic paralytic and paramyotonic syndrome with variable clinical expressivity. Neurology. 1997;49:1018–1025. doi: 10.1212/wnl.49.4.1018. [DOI] [PubMed] [Google Scholar]
- 38.Wang SY, Wang GK. A mutation in segment I-S6 alters slow inactivation of sodium channels. Biophys J. 1997;72:1633–1640. doi: 10.1016/S0006-3495(97)78809-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na+-channel inactivation. Proc Natl Acad Sci USA. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang N, Ji S, Zhou M, Ptácek LJ, Barchi RL, Horn R, George AL., Jr Sodium channel mutations in paramyotonia congenita exhibit similar biophysical phenotypes in vitro. Proc Natl Acad Sci USA. 1994;91:12785–12789. doi: 10.1073/pnas.91.26.12785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zagotta WN, Aldrich RW. Alterations in activation gating of single Shaker A-type potassium channels by the Sh5 mutation. J Neurosci. 1990;10:1799–1810. doi: 10.1523/JNEUROSCI.10-06-01799.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]