

Abstract
Dopamine and the neuropeptides Ala-Pro-Gly-Trp-NH2(APGWamide or APGWa) and Phe-Met-Arg-Phe-NH2 (FMRFamide or FMRFa) all activate an S-like potassium channel in the light green cells of the mollusc Lymnaea stagnalis, neuroendocrine cells that release insulin-related peptides. We studied the signaling pathways underlying the responses, the role of the G-protein βγ subunit, and the interference by phosphorylation pathways. All responses are blocked by an inhibitor of arachidonic acid (AA) release, 4-bromophenacylbromide, and by inhibitors of lipoxygenases (nordihydroguaiaretic acid and AA-861) but not by indomethacin, a cyclooxygenase inhibitor. AA and phospholipase A2 (PLA2) induced currents with similarI–V characteristics and potassium selectivity as dopamine, APGWa, and FMRFa. PLA2 occluded the response to FMRFa. We conclude that convergence of the actions of dopamine, APGWa, and FMRFa onto the S-like channel occurs at or upstream of the level of AA and that formation of lipoxygenase metabolites of AA is necessary to activate the channel. Injection of a synthetic peptide, which interferes with G-protein βγ subunits, inhibited the agonist-induced potassium current. This suggests that βγ subunits mediate the response, possibly by directly coupling to a phospholipase. Finally, the responses to dopamine, APGWa, and FMRFa were inhibited by activation of PKA and PKC, suggesting that the responses are counteracted by PKA- and PKC-dependent phosphorylation. The PLA2-activated potassium current was inhibited by 8-chlorophenylthio-cAMP but not by 12-O-tetradecanoylphorbol 13-acetate (TPA). However, TPA did inhibit the potassium current induced by irreversible activation of the G-protein using GTP-γ-S. Thus, it appears that PKA targets a site downstream of AA formation, e.g., the potassium channel, whereas PKC acts at the active G-protein or the phospholipase.
Keywords: FMRFamide, dopamine, neuropeptide, K+-current, S-current, molluscs, arachidonic acid, G-protein βγ subunits, neuron, signal transduction, convergence
G-protein-mediated activation of K+ channels involves either direct interaction of G-proteins with the channels [so-called G-protein-gated inward rectifier K+ channels (GIRKs); Wickman and Clapham, 1995] or intervention of an enzyme system and second messengers. The latter route has been well documented in molluscan systems. Thus, Phe-Met-Arg-Phe-NH2 (FMRFamide or FMRFa)-induced activation of S-channels in sensory cells of Aplysia(Piomelli et al., 1987a; Belardetti et al., 1989; Buttner et al., 1989), an S-like current in neuron B5 of Helisoma (Bahls et al., 1992) and an S-like but voltage-dependent K+-current in the caudodorsal cells ofLymnaea (Kits et al., 1997), involves lipoxygenase metabolites of arachidonic acid (AA). However, other studies suggested AA-independent activation of K+ channels. Examples are the S-current activated by the peptide myomodulin inAplysia (Critz et al., 1991) and a K+-conductance activated by dopamine, glutamate, and a muscarinic agonist in Planorbarius (Bolshakov et al., 1993).
Previously, we described the convergent activation of an S-like K+ channel by the classical transmitter dopamine and the neuropeptides Ala-Pro-Gly-Trp-NH2 (APGWamide or APGWa) and FMRFa in the light green cells (LGCs) ofLymnaea stagnalis (van Tol-Steye et al., 1997), a set of growth-regulating neurons that produce insulin-related peptides (Geraerts, 1976; Geraerts et al., 1991; Smit et al., 1988). The responses are G-protein-dependent, but the signaling pathway has not been elucidated. Here, we focus on three main questions. (1) What is the signal transduction pathway underlying the convergent activation of the K+ channel by dopamine and APGWa and FMRFa? Convergence may occur at any site between the receptors and the channel, implying that the agonists may use completely different or identical pathways. To solve this, we used pharmacological tools to interfere with signal transduction. (2) Which G-protein subunit, α or βγ, is the active mediator in these responses? Unlike for GIRKs in which Gβγ subunits induce the response (Clapham and Neer, 1997;Isomoto et al., 1997), for receptor-operated K+channels in molluscs (Sasaki and Sato, 1987; Volterra and Siegelbaum, 1988; Bolshakov et al., 1993), it is unclear whether Gα or βγ subunits are the active mediators. To address this problem, we used a peptide that specifically interacts with βγ-mediated signaling (Koch et al., 1993; Macrez et al., 1997). (3) Do modulatory pathways interact with the K+ current response? In LGCs, responses to dopamine are inhibited by cAMP. This resembles the situation in S channels in which protein kinase A (PKA)-dependent phosphorylation shuts down the channels (Shuster et al., 1985). Therefore, we studied for all three messengers the role of phosphorylation by PKA and protein kinase C (PKC) in modulation of the current responses. Also, we asked which sites in the signaling route are targeted by PKA and PKC by studying the effect of PKA and PKC activation on responses activated downstream of the receptors.
We provide evidence that channel activation by all three agonists involves lipoxygenase metabolism of AA, implying that not only FMRFa but also dopamine and APGWa, couple to the AA pathway. Gβγ subunits appear to mediate the response and presumably activate the phospholipase catalyzing AA formation. The AA pathway is modulated by phosphorylation through PKA and PKC, with each kinase apparently targeting a separate site in the signaling route.
MATERIALS AND METHODS
Animals and cells. Adult pond snails (Lymnaea stagnalis), bred in the laboratory under standard conditions (van der Steen et al., 1969), were used. LGCs were dissociated mechanically from the CNS after 45 min incubation at 37°C in HEPES-buffered saline (HBS) (see below) supplemented with 0.2% trypsin (type III; Sigma, St. Louis, MO). Dissociated cells were plated in culture dishes (Costar, Cambridge, MA) containing HBS and were left undisturbed for at least 1 hr to allow the cells to attach to the bottom of the dish. In all experiments, isolated cells (diameter of ∼60 μm) without neurites were used. Cells were always used within 7 hr after isolation.
Solutions and chemicals. CNSs and isolated cells were bathed in HBS containing (in mm): NaCl 30, NaCH3SO4 10, NaHCO3 5, KCl 1.7, CaCl2 4, MgCl2 1.5, and HEPES 10, pH 7.8 adjusted with NaOH. The same buffer was used as extracellular solution during electrophysiological recordings, unless otherwise specified. Extracellular high-K+ solution contained 20 mm KCl and 10 mm NaCl. For the remainder, it was identical to HBS. Internal pipette solution contained (in mm): KCl 29, ATPMg 2, GTP (Tris-salt) 0.1, HEPES 10, EGTA 11, and CaCl2 2.3, pH 7.4 adjusted with KOH.
Solutions of agonists and all other drugs were freshly prepared before use by dilution from stock solutions into extracellular or intracellular medium. The following agents were prepared as stocks in DMSO: 4-bromophenacylbromide (4-BPB), nordihydroguaiaretic acid (NDGA), indomethacin, and 8-chlorophenylthio-cAMP (8-cpt-cAMP) (all obtained from Sigma); 12-O-tetradecanoylphorbol 13-acetate [(TPA) also referred to as phorbol 12-myristate 13-acetate (PMA)], 4-α-phorbol 12-myristate 13-acetate (4α-PMA), staurosporin, and okadaic acid (all obtained from Research Biochemicals, Natick, MA); AA (Sigma and Research Biochemicals); and 1-(2-nitrophenyl)ethyl cAMP (NPE-caged cAMP) (Molecular Probes, Eugene, OR). AA-861 stock solutions (Biomol, Plymouth Meeting, PA) were prepared in ethanol. Stocks of 3,4-dihydroxyphenylethylamine hydrochloride (dopamine) (Sigma), APGWamide (American Peptide Co., Sunnyvale, CA), FMRFamide (Peninsula Laboratories Inc., Belmont, CA), phospholipase A2(PLA2) from bee venom (Sigma), and βγ-interacting peptide (peptide G) (Isogen Bioscience BV, Maarssen, The Netherlands) were prepared in distilled water. Stocks of cAMP (Boehringer Mannheim, Mannheim, Germany) were prepared in distilled water containing 2 mm KOH. cAMP-dependent protein kinase inhibitor 5–24 amide (wiptide) (Peninsula Laboratories Inc.) was directly dissolved in pipette medium. Peptide G is an N-acylated and C-amidated 28-mer: WKKELRDAYREAQQLVQRVPKMKNKPRS, corresponding to amino acids 643–670 from the βγ-binding sequence of β-adrenergic receptor kinase 1 (βARK1) (Koch et al., 1993). AA-containing solutions were always prepared under N2 and sonicated before use. Dopamine, APGWa, FMRFa, 8-cpt-cAMP, staurosporin, AA, and PLA2 were applied to the cell by means of a gravity-driven Y-tube system described previously (Kits et al., 1997) or by means of a Picospritzer (General Valve, Fairfield, NJ). The vital dye amaranth (0.01%) (Merck, Darmstadt, Germany) was added to visualize the application. Because of the continuous perfusion of the culture dish with extracellular solution, applied agents were washed away within seconds after application was stopped. Okadaic acid and NPE-caged cAMP were applied intracellularly via the patch electrode. Peptide G was administered by microinjection. All other solutions were administered via the pump-driven bath perfusion system, unless stated otherwise. Bath medium was completely replaced within 5 min. Transmitters were dissolved in the same solution as used for perfusion. Recordings were always started in standard extracellular solution. When high potassium or drugs were bath-perfused during an experiment, subsequent experiments were performed on a fresh dish of cells.
Electrophysiological recordings and microinjections.Whole-cell voltage-clamp recordings were made with an Axoclamp-2B or Axopatch-200B amplifier (Axon Instruments, Foster City, CA). Voltage-clamp electrodes (2–4 MΩ) were pulled from Clark GC-150T or GC-150 glass (Clark Electromedical Instruments, Reading, UK). Seal resistance was ≥1 GΩ; after disruption of the patch membrane, series resistance (<5 MΩ) was compensated for ≥70%. Sharp injection electrodes were pulled from Clark GC-150F glass; resistance was 15–20 MΩ when filled with standard pipette medium. After impalement of the cell, which was confirmed by monitoring the membrane potential using a custom-built microelectrode amplifier, injection (≤1% of the cell volume) was accomplished under visual control, using a Picospritzer.
Data-acquisition was controlled by a CED 1401 (Cambridge Electronics Design, Cambridge, UK) or a Digidata 1200 (Axon Instruments) analog-to-digital converter connected to a personal computer. Voltage-clamp software developed in our laboratory, as well as pClamp (Axon Instruments), was used. Current recordings were filtered at 100 Hz, sampled at >200 Hz, and stored on disk. This system allowed simultaneous control of applied voltage protocols, data acquisition, and drug application.
Cells were kept at a holding potential of −80 mV. Before application of agonist, a voltage step to −40 mV was made, unless stated otherwise, to enhance the driving force for potassium. I–Vrelationships were determined using ramp protocols, imposing linear voltage ramps (slope of 34 mV/sec; duration of 5 sec). Current responses to ramps under control conditions (no agonist present) were subtracted from those measured after application (10–40 sec) of agonist to obtain the current–voltage relationship of the agonist-induced current.
Error bars indicate SEM.
Flash experiments. Uncaging of NPE-caged cAMP was accomplished using a 35 S mercury arch Flashtube and Strobex model 238 power pack (Chadwick-Helmuth, El Monte, CA), delivering 200 J per flash. For these experiments, the bottoms of the Costar culture dishes, normally used to plate the cells, were replaced by glass coverslips, and an Axiovert 35 microscope with 40× plan-neofluar objective (both from Zeiss, Jena, Germany) was used.
RESULTS
Arachidonic acid pathway
Previously, we have shown that the transmitter dopamine and the neuropeptides APGWa and FMRFa activate a single type of potassium channel in the neuroendocrine LGCs of the pond snail Lymnaea stagnalis (van Tol-Steye et al., 1997). To assess whether AA is involved in activation of this potassium channel, we tested the effect of 4-BPB. 4-BPB irreversibly blocks the formation of AA by inhibiting phospholipase A2 activity or by inhibiting the combined action of phospholipase C (PLC) and diacylglycerol lipase (Blackwell and Flower, 1983; Piomelli et al., 1987a). The current responses to all three transmitters were blocked for ≥90% by 10 μm 4-BPB (Fig.1). The inhibition was obtained within 10–20 min.
Fig. 1.
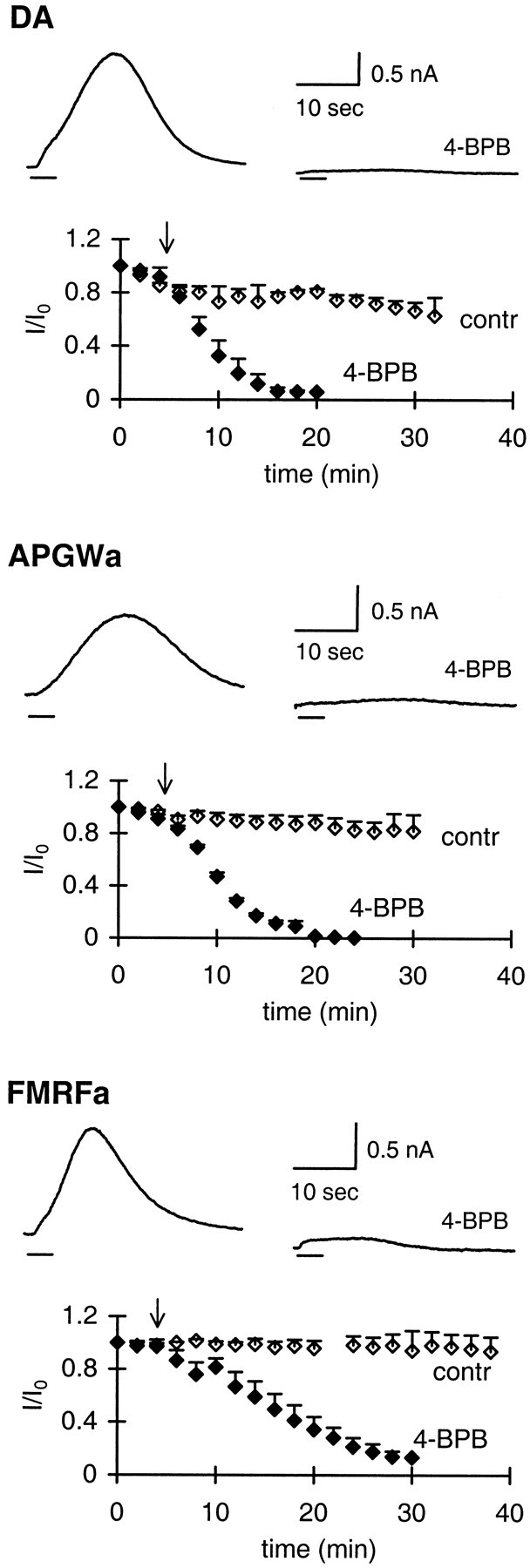
4-BPB inhibits the outward current induced by dopamine, APGWa, and FMRFa. For each agonist, the control response and the strongly reduced response in the presence of 4-BPB are shown. Responses were evoked by 2 sec applications of agonist by means of Y-tube. Graphs are plots of current amplitudes (I), normalized to the current response att = 0 (I0), against time.Arrows indicate the start of bath perfusion with 0.01% DMSO in standard medium (control, open diamonds;n = 3 for each agonist) or with 10 μm4-BPB (filled diamonds; n = 3 for each agonist; n indicates number of cells tested per agonist). SEMs are indicated only by upward error bars. Concentrations used were as follows: dopamine, 1 μm; APGWa, 7 μm; and FMRFa, 0.1 μm.
Whereas 4-BPB inhibited the responses, AA (5–50 μm) mimicked the agonist-induced effects on the LGCs (Figs.2A,3). However, the AA-evoked responses were rather variable in intensity and stability after repeated applications (2–20 sec applications, once per minute). In some cases, AA application failed to induce a response. Although we prepared all AA-containing solutions under N2and sonicated solutions before use, these problems could well be attributable to the fact that AA is poorly soluble in aqueous solutions, tends to stick to glassware and tubing (Yamada et al., 1994), and is prone to oxidation. In line with this explanation, we found that a given solution was either effective or ineffective in all cells tested. To consolidate our observation that AA is capable of mimicking the effect of dopamine, APGWa, and FMRFa on the LGCs, we stimulated AA formation in the cell by application of PLA2. Extracellular application of PLA2 (50–122.5 U/ml, corresponding to micromolar range) consistently induced a slow outward current similar to that evoked by the three agonists (Figs.2A, 3). Figure 2A shows examples of the AA- and PLA2-induced responses and the dopamine-induced potassium current.
Fig. 2.

PLA2 and AA are involved in the signaling route activated by the agonists. A, AA and PLA2 induce an outward current, similar to that induced by dopamine. Bars indicate extracellular applications of AA (5 sec, 50 μm), PLA2 (20 sec, 122.5 U/ml), and dopamine (1 sec, 1 μm). The cells were voltage clamped at −40 mV. Responses are from different cells. B, C, FMRFa occludes the response to PLA2. B, Combined application of FMRFa and PLA2 fails to show summation of the responses to FMRFa and PLA2. Average amplitudes (right) of the responses to a maximal dose of FMRFa (10 μm) alone and to the subsequently applied combination of FMRFa and PLA2 (50 U/ml) (n = 4). C, Application of FMRFa during a response to PLA2 increases the current amplitude up to the level of the response to FMRFa alone. The right panel shows the average current amplitudes to FMRFa alone and to PLA2 alone compared with the response amplitude obtained when FMRFa was applied during a PLA2 response (n = 4).
Fig. 3.

AA and PLA2 induce a current with similar I–V characteristics as the current induced by dopamine, both in standard and high extracellular [K+]o. A,I–V relationships of the current induced by AA (50 μm), PLA2 (50–122.5 U/ml), or dopamine (1 μm) were determined with the use of linear voltage ramp protocols (34 mV/sec) as described in Materials and Methods.Left panels, Standard extracellular potassium (1.7 mm; n = 3, n = 4,n = 7 for AA, PLA2, and dopamine, respectively). Right panels, High extracellular potassium (20 mm; n = 2,n = 4, n = 2 for AA, PLA2, and dopamine, respectively). B, Curves from A were normalized with respect to the current at −40 mV (1.7 mm K+) or to the peak inward current amplitude (20 mmK+).
If direct application of AA or PLA2 activates the same channels as the agonists, there should be occlusion of the agonist responses and the PLA2-evoked response. To test this, we used a saturating dose of FMRFa (10 μm) to evoke the agonist-induced response because it was shown previously that FMRFa maximally activates the population of K+ channels involved and completely occludes the responses to the other agonists (van Tol-Steye et al., 1997). This response was compared with the response to combined application of PLA2 (50 U/ml) and FMRFa (n = 4). Figure 2B shows there is no summation of the responses, i.e., that FMRFa completely occludes the response to PLA2. If FMRFa was applied during the PLA2 current response, an increase in current amplitude was observed up to the level of the response to FMRFa alone (n = 4) (Fig. 2C). This indicates that PLA2 activates the same population of channels as FMRFa but does so less effectively.
As a next test to establish that PLA2 and AA evoke the same current as dopamine, APGWa, and FMRFa, we determined the reversal potential and voltage dependence of the responses using voltage ramp protocols (Fig. 3). In standard extracellular potassium, the current induced by dopamine reversed at −82.0 ± 0.4 mV (± SEM;n = 4) and that induced by AA and PLA2 at −81.9 ± 2.3 mV (n = 3) and −83.6 ± 0.4 mV (n = 7), respectively. At high extracellular potassium, the reversal potential shifted to −23.1 ± 0.1 mV for dopamine (n = 4), −23.1 ± 1.3 mV for AA (n = 2), and −21.2 ± 1.1 mV for PLA2(n = 2). Thus, for the three agents, the shift in reversal potential caused by the change in extracellular potassium concentration amounts to ∼60 mV. This corresponds well with the value calculated by the Nernst equation for potassium (61.6 mV), indicating that the current induced by AA and PLA2, like that induced by dopamine, APGWa, and FMRFa, is carried by potassium.
The I–V relationship of the K+ channels activated by dopamine, FMRFa, and APGWa has a characteristic shape that is a result of outward rectification, accounted for by asymmetry in potassium concentrations across the membrane and a slight voltage dependence (van Tol-Steye et al., 1997). The I–Vrelationships of the AA- and PLA2-induced currents also display these properties (Fig. 3A). Outward rectification is especially evident at standard extracellular potassium. Voltage dependence is apparent at voltages below approximately −100 mV, where all conductances decreased despite the increasing driving force. This is best appreciated at 20 mm[K+]o. To permit a better comparison, normalized I–V curves are shown Figure 3B. In low [K+]o, the I–Vcurves of the dopamine-, AA-, and PLA2-induced responses overlap completely at all voltages negative to approximately −20 mV. Also in high [K+]o, the overall shape of the I–V relationships is very similar. The differences between the dopamine- and PLA2-evoked currents at voltages positive to −20 mV reflect effects of the agonist on high voltage-gated calcium channels that are not mediated by activation of PLA2 (H. van Tol-Steye and K. Kits, unpublished observations). The much stronger deviation of the AA-induced current at positive potentials is unexplained.
The data presented suggest that AA, whether applied exogenously or formed endogenously by application of PLA2, activates the same type of potassium channel as dopamine, APGWa, and FMRFa. Together with the fact that an inhibitor of AA formation potently inhibits the responses to the three transmitters, this strongly suggests that formation of AA underlies the generation of these responses.
To test whether AA itself, or one of its metabolites, is responsible for the generation of the potassium current, we tested several inhibitors of AA metabolism. NDGA, an inhibitor of the lipoxygenase pathway (Needleman et al., 1986; Yamada et al., 1994), is active inAplysia nervous tissue, with an IC50 of 3 μm (Piomelli et al., 1987a,b). In the LGCs, 15 μm NDGA inhibited the responses evoked by dopamine, APGWa, and FMRFa for ∼90% or more (n = 3 in all cases) (Fig. 4A) within 20–30 min. A similar amount of inhibition was observed with 5 μm NDGA within 40–60 min in a first set of experiments with FMRFa (data not shown).
Fig. 4.
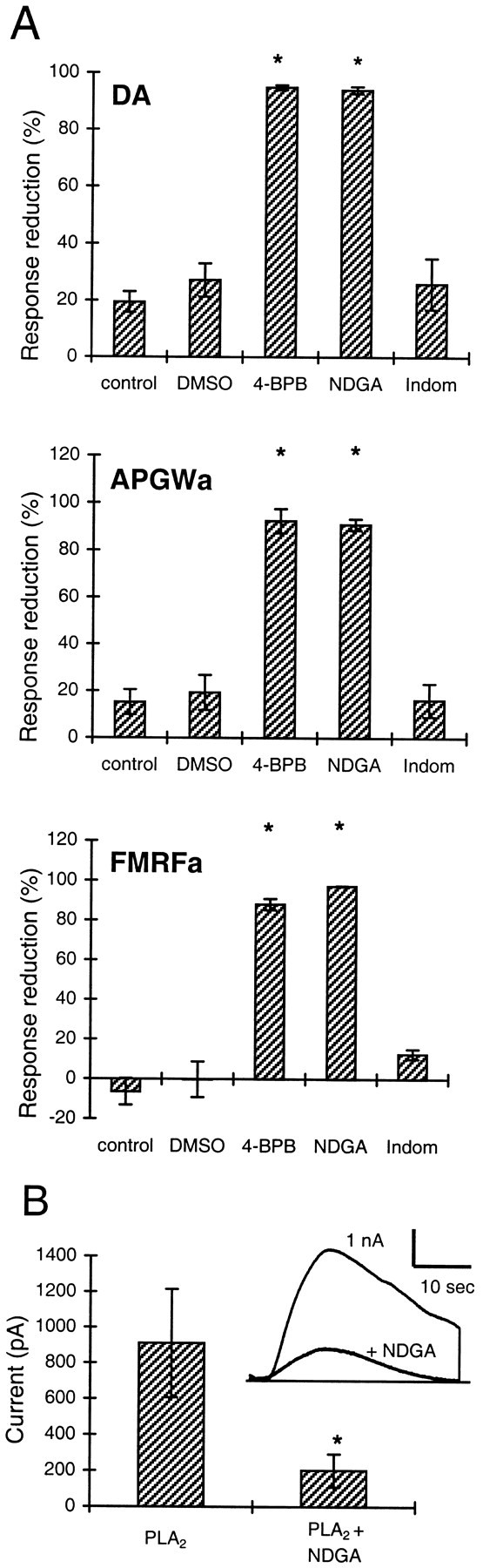
Inhibitors of phospholipase and lipoxygenase reduce the responses to dopamine, APGWa, and FMRFa. A, Percentage reduction of the potassium current elicited by dopamine, APGWa, and FMRFa (top, middle, andbottom, respectively) after perfusion of the cells with standard medium (control), 0.01% DMSO-containing medium (DMSO), the inhib itor of AA-release 4-BPB (10 μm), the lipoxygenase inhibitor NDGA (15 μm), and the cyclooxygenase inhibitor indomethacin (Indom; 5 μm). Asterisks indicate significant difference with respect to the DMSO control (unpaired ttest; p < 0.01). Apart from controls in whichn = 6, experimental groups weren = 3 for each agonist. B, NDGA also blocks the current induced by bee venom-derived PLA2, indicating that exogenous PLA2acts specifically to produce AA. Shown are the average response amplitudes to applications of PLA2 (50 U/ml) in the absence and presence of 15 μm NDGA (n = 11). NDGA was applied for 10 min before PLA2 was tested.Inset, Superimposed traces showing a control response to PLA2 and the strong reduction of this response by NDGA (responses from the same cell). *p < 0.01; paired t test.
Indomethacin is an inhibitor of the cyclooxygenase pathway. InAplysia ganglia, indomethacin inhibits the formation of prostaglandines, with an IC50 of 0.5 μm. The lipoxygenase pathway is only affected at higher concentrations (IC50 of >10 μm) (Piomelli et al., 1987a). Perfusion of the LGCs during ∼25 min with 5 μmindomethacin did not affect the current responses to dopamine, APGWa, and FMRFa compared with the control situation in which the cells were perfused with standard medium containing 0.01% DMSO (Fig.4A). At this concentration, DMSO did not affect the responses. As a final additional test of the involvement of the lipoxygenase pathway, we used the lipoxygenase blocker AA-861 (Yamada et al., 1994) on dopamine and APGWa responses, both of which were significantly reduced (dopamine, 64.8 ± 4.1% block with 4 μm AA-861; n = 3; APGWa, 61.6 ± 2.1 and 84.1 ± 0.1% block with 4 and 10 μm AA-861, respectively; n = 3 for both concentrations; unpairedt test; p ≤ 0.01, tested against 0.05% ethanol controls; n = 3).
To demonstrate that the exogenous bee venom-derived PLA2that we used specifically forms AA and starts the same cascade as the agonists, we tested whether NDGA blocks the PLA2-induced response. Figure 4B shows that NDGA (15 μm) blocks the response to PLA2 (50 U/ml) by 80 ± 4% (n = 11). In three of these experiments, responses to PLA2 were evoked repeatedly before NDGA was applied, which confirmed that control responses to PLA2were stable.
We conclude from these data that (1) AA metabolites of the lipoxygenase pathway, formed after activation of the receptors for dopamine, APGWa, and FMRFa, are responsible for generation of the potassium current, and (2) convergence onto the potassium channel occurs at an early step in the signal transduction path, i.e., at or even upstream of the level of AA.
Involvement of G-protein βγ subunits
We next asked which G-protein subunit mediates the responses. In several cases, βγ subunits of heterotrimeric G-proteins have been implicated in the activation of PLA2 (Jelsema and Axelrod, 1987; Murayama et al., 1990). Because the responses to dopamine, APGWa, and FMRFa are all G-protein-dependent (van Tol-Steye et al., 1997) and mediated by AA, as demonstrated above, our strategy was to confine the action of the βγ subunits. To this end, we investigated the effect of a synthetic peptide, peptide G, which was demonstrated previously to interfere with βγ-mediated signaling (Koch et al., 1993; Macrez et al., 1997). Peptide G consists of 28 amino acids and is derived from the βγ-binding sequence of βARK1 (Koch et al., 1993). After three control dopamine applications, peptide G was injected into the cell (final concentration estimated to be ≤50 μm), after which dopamine was applied again. Injection of peptide G caused an irreversible reduction of the dopamine response by 79 ± 4% (n = 5), whereas control injections with carrier had hardly any effect (8 ± 1% reduction of the dopamine response;n = 3) (Fig.5A).
Fig. 5.

βγ-interacting peptide (peptide G) inhibits the potassium current response. A, Reduction of the response to dopamine after injection of peptide G (79 ± 4%;n = 5) or injection of carrier (8 ± 1%;n = 3). *p < 0.001; unpairedt test. B, Induction of the sustained K+ current as a result of irreversible activation of the G-protein. Repeated application of agonist with 200 μm GTP-γ-S in the pipette induces a sustained current (b), which is the irreversible part of the response. In the right panel, it is also seen as the increased holding current before the response. Shown are the first and last response to the agonist, in this example FMRFa;arrows indicate application of agonist during the step to −40 mV. The sustained current is only detected at −40 mV, when the driving force for potassium is large. Notice that the holding current at −80 mV (a) remains constant.C1, Effect of peptide G on the sustained potassium current, induced as in B, using dopamine as agonist. Sustained current amplitude was measured once every 2 min. Injection of peptide G (indicated by arrow) is done after the last dopamine application. (Concentration of peptide G in injection electrode was 5 mm, and final concentration is estimated to be ≤50 μm.) C2, Example trace of the immediate effect of peptide G on the sustained current at −40 mV.Arrow marks injection of peptide G. C3, Like B2 but with injection of carrier instead of peptide G. Obviously, the carrier has no effect.
The βγ-interacting peptide in principle might, apart from competing with effector molecules for binding free βγ, also compete with Gα for binding to βγ and thus reduce the amount of functional heterotrimeric G-protein available for activation by receptors (but seeKoch et al., 1994; Macrez et al., 1997). To rule out this option, we induced sustained and irreversible activation of the G-protein by repeated applications (approximately six) of agonist, either dopamine or FMRFa, with the nonhydrolyzable GTP-analog GTP-γ-S (200 μm) in the pipette medium (Fig. 5B). This procedure results in the development of a sustained outward current that only decays over a period of several minutes after washout of the agonist (cf. van Tol-Steye et al., 1997). Figure 5, C1 andC2, illustrates the effect of peptide G on this sustained current. Injection of the peptide caused a rapid reduction of the current (>50% within 1 min). Injection of carrier solution, however, hardly affected the sustained current (Fig. 5C3). The results obtained for dopamine and FMRFa were highly similar (bothn = 4). Thus, also when the G-protein is irreversibly activated, the peptide has an inhibitory effect on the potassium current re- sponse. This suggests that this inhibitory effect is a result of interference with βγ-induced activation of effector (PLA2) and not of reduction of the amount of functional heterotrimeric G-protein.
Effects of cAMP on agonist-induced responses
Previous work in our laboratory demonstrated that the dopamine-induced hyperpolarization of the LGCs is inhibited by the cAMP-analog 8-cpt-cAMP and by forskolin (which activates adenylyl cyclase) and IBMX (an inhibitor of phosphodiesterase) (De Vlieger et al., 1986). To test whether the outward current responses induced by dopamine, APGWa, and FMRFa are all inhibited by cAMP, we used caged cAMP. NPE-caged cAMP (209 μm) was included in the pipette medium and allowed to diffuse into the cell after establishing the whole-cell configuration. Caged cAMP was photolyzed by a flash of UV light (360 nm), as was confirmed in similar experiments on voltage-gated calcium currents known to be increased by cAMP-analogs in other cells of Lymnaea (Dreijer and Kits, 1995). When a flash was given just before application of agonist (at a membrane potential of −40 mV), the potassium current evoked by the agonist was greatly reduced [83.8 ± 1.4% (n = 2), 87.5 ± 1.9% (n = 2), and 66.5 ± 4.0% (n = 5) for dopamine, APGWa, and FMRFa, respectively] (Figure 6A). The flash itself had virtually no effect (data not shown). If the flash was given when the agonist-induced current was at its maximum, the outward current declined rapidly (within 5 sec), as shown in Figure6B (dopamine, n = 3; APGWa,n = 2; FMRFa, n = 6). Thus, cAMP not only prevents the response but also inhibits an ongoing response. In the continuous presence of agonist, the current recovered within 30–50 sec to the level before the flash. Probably the recovery reflects phosphodiesterase activity in the cell rather than desensitization of the cAMP effect because the current was reinhibited after a second flash (n = 8) (Fig. 6C, APGWa). The experiments presented here indicate that the responses induced by dopamine, APGWa, and FMRFa in the LGCs are all inhibited by cAMP. This conclusion was further substantiated in experiments using either extracellularly applied 8-cpt-cAMP, a membrane-permeable nonhydrolyzable cAMP-analog, or intracellularly applied cAMP (Fig.6D, dopamine; other data not shown).
Fig. 6.

cAMP-dependent phosphorylation inhibits the responses to dopamine, APGWa, and FMRFa. A1, Peak current amplitudes as a function of pulse number. a–cdenote traces depicted in corresponding panels in A.A2, Superimposed K+ current responses to dopamine, APGWa, or FMRFa as indicated, before (a) and directly after (b) flash-photolysis of NPE-caged cAMP and after ≥60 sec recovery (c). Arrows indicate timing of the flashes 2–3 sec before agonist application. NPE-caged cAMP was added to the pipette solution at 209 μm. All responses were evoked at a potential of −40 mV. Uncaged cAMP strongly reduces the responses, although it has no effect on the holding current at −40 mV.B, Superimposed traces of a response interrupted by flash-photolysis of caged cAMP at the peak of the potassium current and the response after ≥60 sec recovery. Inhibition of current responses by cAMP is fast and reversible. Shown is an example with dopamine.C, Effect of repeated flashes on the current response in the continuous presence of agonist. After the first decrease attributable to uncaging of cAMP, the response recovers to a level similar to that before the flash, whereas a second flash induces a similar inhibition of the response. Shown is an example with APGWa.D, Percentage reduction of the dopamine response after 3 min perfusion with 1 mm 8-cpt-cAMP, with and without the PKA inhibitor wiptide (100 μm) in the pipette medium. The inhibitory effect of 8-cpt-cAMP is strongly reduced by wiptide.n = 3 for both groups. *p < 0.005; unpaired t test.
To study whether cAMP acts by stimulation of PKA, we tested the effect of 8-cpt-cAMP on the dopamine response in the presence of the PKA inhibitor wiptide. Figure 6D shows that, when wiptide (100 μm) was included in the electrode, the inhibitory effect of 8-cpt-cAMP (1 mm) on the dopamine response was significantly smaller than in the absence of wiptide (31 ± 5% vs 89 ± 2%; n = 3 for both treatments; unpaired t test; p < 0.005). Wiptide had no effect on the dopamine response. We conclude that the inhibition of the agonist-induced potassium current by cAMP and analogs is mediated by PKA-dependent phosphorylation.
Effects of phorbol ester on agonist-induced responses
To investigate whether PKC modulates the responses to the three agonists under study also, we tested the effects of TPA, a phorbol ester activator of PKC. Bath perfusion of TPA (50 nm) caused a significant reduction of the current induced by all three agonists (Fig. 7A). After ∼16 min, the currents induced by dopamine, APGWa, and FMRFa were reduced by 71.3 ± 4.0% (n = 5), 65.9 ± 5.7% (n = 3), and 88.5 ± 1.3% (n = 3), respectively. The PKC inhibitor staurosporin (1 μm) significantly reduced the inhibition by TPA of the response to FMRFa to 16.2 ± 3.1 (unpaired t test;p < 0.001; n = 3) (Fig.7B). Staurosporin did not affect the response to FMRFa. These data suggest that the responses induced by dopamine, APGWa, and FMRFa are all inhibited by PKC.
Fig. 7.
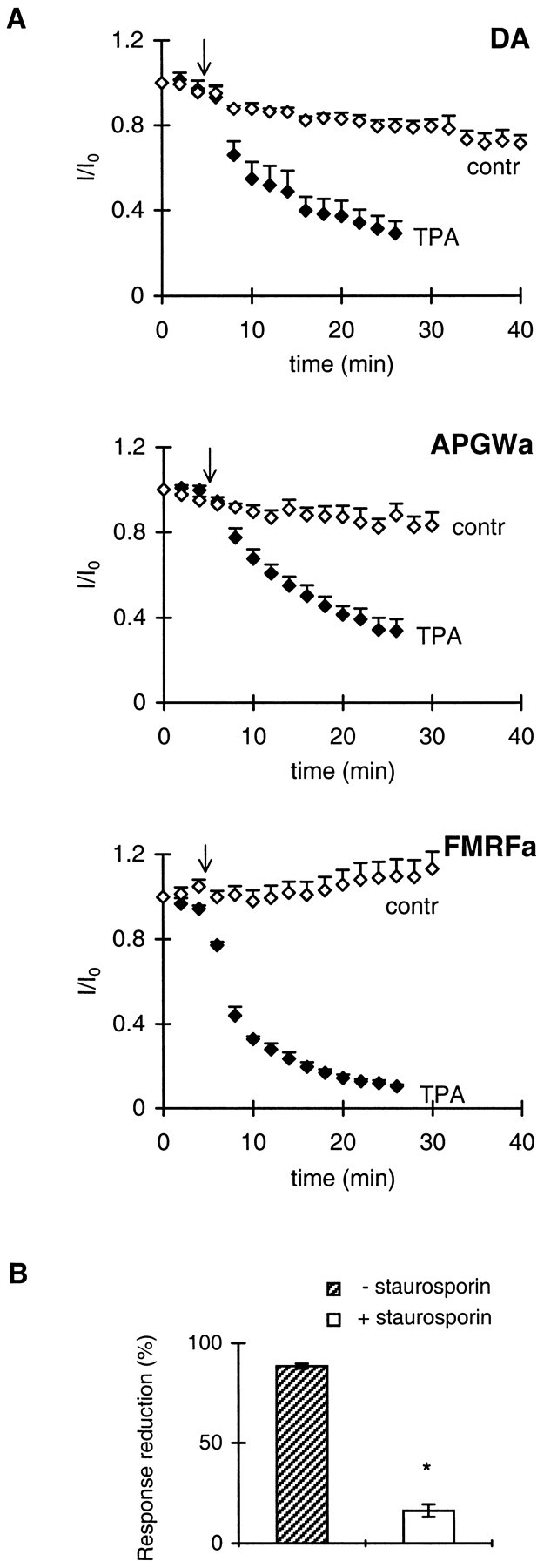
Phosphorylation by protein kinase C inhibits the responses to dopamine, APGWa, and FMRFa. A, Repetitively induced responses to dopamine, APGWa, and FMRFa as indicated, in the absence and presence of TPA. Current amplitudes (I) are normalized to the current response at t = 0 (I0).Arrows indicate the start of bath perfusion with 50 nm TPA (black diamonds;n = 3–5 for each agonist). Control current responses (contr, open diamonds;n = 5–6 for each agonist) were obtained from experiments performed without change of medium. SEMs are indi cated only by upward error bars. TPA strongly reduces the responses to the agonists. B, Percentage reduction of the FMRFa response after ∼16 min perfusion with 50 nm TPA or with a combination of TPA and the PKC inhibitor staurosporin (1 μm). Staurosporin strongly reduces the inhibitory effect of TPA. *p < 0.005; unpaired ttest; n = 3 for both groups.
Effects of phosphatase inhibitor
Because the K+ current responses are inhibited by PKA- and PKC-dependent phosphorylation, it is to be expected that inhibition of phosphatase activity will have similar effects. To test this, we studied the effect of the phosphatase inhibitor okadaic acid (Cohen et al., 1990) on the dopamine-induced response. When okadaic acid was included in the patch electrode (1–2 μm), the dopamine-induced response decreased within ∼30 min to 48.2 ± 6.7% of the initial value, measured directly after rupture of the seal (n = 5; data not shown). Controls with 1% DMSO in the patch electrode did not show a decrease of the dopamine-induced current within the same period of time (n = 2; data not shown).
The results with cAMP, TPA, and okadaic acid suggest that phosphorylation of the channel or a component in the AA pathway inhibits the responses, or alternatively, that the AA pathway involves a dephosphorylation step.
The AA pathway and the phosphorylation–dephosphorylation cycle
To test at what site PKA and PKC interact with the signaling cascades activated by dopamine, APGWa, and FMRFa, we examined the effect of PKA and PKC stimulation on the signal transduction pathway downstream of AA formation. Thus, the potassium current response to application of exogenous PLA2 (50 U/ml) was measured before and after 10 min perfusion of 8-cpt-cAMP (100 μm;n = 6), TPA (50 nm; n = 3), the inactive phorbol ester 4α-PMA (50 nm;n = 3), and DMSO-containing medium (0.05%;n = 4) (Fig. 8). After perfusion with 8-cpt-cAMP, the response to PLA2 was reduced by 88 ± 5%, which was significantly different (unpairedt test; p < 0.01) from the effect of perfusion with DMSO-containing medium (reduction of 11 ± 15%). TPA, however, had no effect (Fig. 8). This suggests that PKA inhibits the agonist-induced potassium current by phosphorylation of a site downstream of phospholipase activation, e.g., the potassium channel. In contrast, PKC phosphorylation targets a site upstream of AA, e.g., the receptors, the G-protein, or the endogenous phospholipase. This last option cannot be excluded in view of possible differences between the bee venom PLA2 used in our experiments and the endogenous phospholipase in LGCs.
Fig. 8.

Potassium current response induced by PLA2 is inhibited by 8-cpt-cAMP but not by TPA.A, Effect of 10 min perfusion with 0.1 mm8-cpt-cAMP (n = 6), 50 nm TPA (n = 3), and the corresponding controls 0.05% DMSO (n = 4) and 50 nm 4α-PMA (n = 3) on the potassium current response to PLA2 (50 U/ml). Responses after perfusion (I) are normalized to responses before perfusion (I0). *p < 0.01, significant difference from control; unpaired t test.B, Current responses to PLA2 before and after perfusion with 8-cpt-cAMP and before and after perfusion with TPA, as indicated. Differences in response kinetics are caused by differences in duration of PLA2 application and perfusion rate. 8-cpt-cAMP, TPA, and corresponding control solutions were applied by Y-tube.
To further specify the site of action of PKC, we induced sustained and irreversible activation of the G-protein by repeated applications of FMRFa by means of the nonhydrolyzable GTP-analog GTP-γ-S (200 μm) as described above (Fig. 5B). This procedure results in the development of a sustained outward current, which only slowly runs down after washout of the agonist (van Tol-Steye et al., 1997). TPA caused a rapid reduction of this sustained current, as shown in Figure 9A. Within 4 min of perfusion, TPA (50 nm) reduced the sustained current by 56 ± 8% (n = 5), which was significantly different from the reduction of 33 ± 7% under control conditions (perfusion with 4α-PMA or DMSO; n= 8; p < 0.05; unpaired t test) (Fig.9B). These results and those from Figure 8 imply that PKC must act at a site downstream of the receptor but upstream of AA and may either target the active G-protein subunit mediating the response or the endogenous phospholipase.
Fig. 9.

TPA inhibits the potassium current induced by irreversible activation of the G-protein. A, The sustained current (measured as Ihold at −40 mV; see legend of Fig. 5B) as a function of time after perfusion with TPA (50 nm) or with the inactive phorbol ester 4α-PMA (50 nm). Perfusion of TPA or 4α-PMA (indicated by bars) is started directly after repetitive FMRFa applications. TPA (left) causes a rapid decrease of the sustained current compared with the decrease under control conditions, i.e., with 4α-PMA (right).B, Average reduction of the sustained current after 4 min of perfusion of TPA (56 ± 8%; n = 5) or control medium with inactive phorbol ester 4α-PMA or DMSO (33 ± 7%; n = 8). *p < 0.05; unpaired t test.
DISCUSSION
Three main conclusions can be drawn from the present study. First, the activation of S-like potassium channels in LGCs by dopamine, APGWa, and FMRFa involves AA and its metabolism by lipoxygenases. Thus, convergence of dopamine, FMRFa, and APGWa onto the potassium conductance resides at or upstream of AA. Second, AA release is probably mediated by G-protein βγ subunits, which activate the phospholipase. Third, the responses to the agonists are all affected by PKA and PKC, of which PKC may target a site upstream of the point of convergence and PKA downstream of this point. A schematic representation of the most likely signal transduction pathway used by dopamine, APGWa, and FMRFa to activate S-like K+channels is given in Figure 10.
Fig. 10.

Minimal hypothetical scheme of the signal transduction routes involved in regulation of the receptor-driven K+ channels. Boxes indicate that different isoforms may be used. Dashed arrows indicate equally probable options. PL, Phospholipase;LO, lipoxygenase.
Dopamine, APGWa, and FMRFa activate a single type of K+ channel via lipoxygenase metabolites of arachidonic acid
Several lines of evidence suggest that activation of the common K+ conductance in LGCs by dopamine, APGWa, and FMRFa be accomplished by the release of AA and lipoxygenase metabolites. First, inhibitors of AA formation block the responses to the three agonists. Second, the responses are mimicked by application of AA and PLA2, which generates AA from membrane lipids, and there is occlusion of the responses to PLA2 and FMRFa. Third, the responses are blocked by inhibitors of lipoxygenases but not of cyclooxygenases. Apparently, lipoxygenase metabolites cause, directly or not, opening of the K+ channel. Because the above results hold for all three agonists, convergence may occur either at the level of the G-protein, the phospholipase, or AA.
4-BPB not only inhibits AA formation by PLA2 but also by the combined action of phospholipase C and diacylglycerol lipase (Blackwell and Flower, 1983; Piomelli et al., 1987a). Our results with 4-BPB, therefore, do not specify the type of phospholipase activated by the three agonists.
The presently described responses are closely related to other receptor-driven K+ currents that are all activated by FMRFa via formation of lipoxygenase metabolites, i.e., S and S-like currents in Aplysia sensory neurons (Piomelli et al., 1987a;Belardetti et al., 1989; Buttner et al., 1989), in identified neurons of Helisoma (Bahls et al., 1992), and in the caudodorsal cells of Lymnaea (Kits et al., 1997). Our data imply that, apart from FMRFa, dopamine and the neuropeptide APGWa also couple to the AA pathway. Dopamine-induced activation of PLA2 was shown to occur after D2 receptor expression in Chinese hamster ovary cells (Vial and Piomelli, 1995) and is suspected to occur in striatal cells (Schinelli et al., 1994).
Molluscan S-like currents were recently suggested to derive from a novel class of voltage-independent K+ channels cloned from mammals and characterized by the occurrence of four transmembrane segments and two pore-forming P domains per subunit (Fink et al., 1998; Patel et al., 1998). Overall, these channels (notably TREK-1) show a strong similarity to S channels. They are activated by AA and inhibited by PKA-dependent phosphorylation when expressed in COS cells. However, they do not respond to lipoxygenase products of AA. Also, the S-like current in Lymnaea differs from these channels because it does not follow Goldman–Hodgkin–Katz rectification at potentials negative to −90 mV and shows a much stronger block by TEA and Ba2+ (Kits et al., 1997;van Tol-Steye et al., 1997). The limited amount of homology between the novel channels (∼30%) suggests a large potential variability that may account for these differences.
Effects of βγ-interacting peptide suggest a role for the Gβγ subunit
We tested the effect of peptide G, a βγ-interacting peptide derived from the βγ-binding sequence of βARK1, on the dopamine-induced response under standard conditions and on the prolonged dopamine- and FMRFa-induced responses obtained with GTP-γ-S in the electrode. In both cases, peptide G inhibited the K+ current, suggesting that βγ subunits are the active G-protein subunits mediating the signal from receptor to effector. The controls with GTP-γ-S showed that the inhibitory effect of peptide G is not caused by a reduction of the amount of functional heterotrimeric G-protein, resulting from competition of the peptide with Gα for binding βγ.
The simplest hypothesis incorporating both the G-protein βγ subunit and AA in the signal transduction pathway is to assume that, after G-protein dissociation, βγ causes activation of a phospholipase. As discussed above, AA might be released from membrane phospholipids by PLA2 or by the combined action of PLC and diacylglycerol lipases. PLC can be directly activated by βγ subunits, and PLA2 also has been suggested to be activated, whether or not directly, by βγ subunits (Jelsema and Axelrod, 1987; Murayama et al., 1990; Exton, 1997). Alternatively, AA might activate a G-protein, after which the K+ channels are directly activated by Gβγ. Such a scheme was proposed to explain AA modulation of GIRKs in atrial cells (Kurachi et al., 1989). It is, however, very unlikely that this hypothesis explains our results. The result that TPA inhibited the K+ current induced by GTP-γ-S, but not the PLA2-induced current, suggests that there is no G-protein acting downstream of PLA2. Moreover, preliminary experiments failed to show an effect of peptide G on the PLA2-induced current response (K. Kits and J. C. Lodder, unpublished observations).
The concentration of peptide G we used to inject was 5 mm. We estimate that, at maximum, ∼1% of the cell volume was injected, implying that the final concentration of the peptide was always <50 μm. At this concentration, the K+currents were inhibited by 50% or more. A similar IC50value (76 μm) was reported for inhibition of βγ-stimulated βARK1 activity by peptide G in rod outer segment membranes (Koch et al., 1993), suggesting that the affinity of βγ for the peptide is similar in both cases, despite possible differences between molluscan and mammalian βγ. A much lower IC50value (65 nm), however, was found for inhibition by peptide G of angiotensin II-mediated stimulation of L-type calcium channels in rat myocytes (Marcez et al., 1997).
All responses are modulated by phosphorylation at two different sites in the AA pathway
All three responses are inhibited by (analogs of) cAMP and by PKC-activating phorbol ester. These effects were relieved in the presence of PKA and PKC inhibitors, respectively, indicating that phosphorylation by PKA and PKC mediates the inhibition. Moreover, the dopamine response was reduced by the phosphatase inhibitor okadaic acid. Apparently, phosphorylation counteracts the signaling induced by dopamine, APGWa, and FMRFa.
The observation that 8-cpt-cAMP also inhibited the outward current induced by exogenous PLA2 suggests that PKA-dependent phosphorylation occurs at a site downstream of AA release, e.g., the channel. It cannot be excluded that inhibition of PKA by lipoxygenase metabolites of AA (or possibly activation of a phosphatase) is the actual trigger for opening of the K+ channel. However, if dopamine would inhibit PKA activity, it does not achieve this via inhibition of adenylyl cyclase, because dopamine does not inhibit adenylyl-cyclase in LGCs (Werkman et al., 1990). An alternative and more simple explanation is that PKA-dependent phosphorylation forms a modulatory pathway. Either way, the fact that not only cAMP but also okadaic acid affects the responses suggests that a basal level of phosphorylation controls the activity of the K+channels. The presumed mammalian S-like channel, TREK-1, has a PKA consensus site in its cytoplasmic C-terminal region, which is critical for 8-cpt-cAMP mediated downmodulation of the channel (Patel et al., 1998).
Interestingly, a similar relationship as found here between AA-dependent activation of K+ channels and (de)phosphorylation has been reported in sensory neurons ofAplysia. The S channels in these cells are activated by FMRFa via 12-lipoxygenase metabolites of AA, and they are inhibited by serotonin via PKA-dependent phosphorylation of the channel or an associated protein (Shuster et al., 1985; Belardetti and Siegelbaum, 1988). In addition to the AA metabolite-dependent effect on the S channel, which is thought to be independent of (de)phosphorylation (Belardetti et al., 1989; Buttner et al., 1989), FMRFa also reverses PKA-dependent phosphorylation in the sensory neurons (Belardetti and Siegelbaum, 1988; Sweatt et al., 1989; Shi and Belardetti, 1991). There are indications that dephosphorylation is secondary to AA formation and not dependent on inhibition of adenylyl-cyclase or stimulation of phosphodiesterase activity (Volterra and Siegelbaum, 1988; Sweatt et al., 1989; Shi and Belardetti, 1991). A similar mechanism might be active in the LGCs.
Although both PKA and PKC suppress the responses to dopamine, APGWa, and FMRFa, there is a clear differential effect of the two kinases. Unlike 8-cpt-cAMP, the phorbol ester TPA failed to inhibit PLA2-induced K+ currents. TPA, however, inhibited the GTP-γ-S-activated sustained current. Therefore, PKC most likely targets a site upstream of AA, presumably the Gβγ subunit or the phospholipase. Interestingly, LymnaeaG-protein β subunits contain six consensus sites for PKC phosphorylation (Knol et al., 1994; H. van Heerikhuizen, unpublished observations).
Thus, the responses to all three agonists are affected by PKA and PKC, of which PKC may act at a site upstream of the putative point of convergence (AA) and PKA downstream of AA. This suggests a modulatory role for receptors that stimulate PKA- or PKC-activity in LGCs. Candidate agonists for such receptors are acetylcholine and conopressin that induce depolarizations in LGCs, an effect that is also produced by injections of cAMP (Stoof et al., 1984). The ability of conopressin to activate PKC in Lymnaea anterior lobe neurons was recently demonstrated in our lab (P. F. van Soest, unpublished observations).
Footnotes
We thank Dr. Arjen B. Brussaard and Dr. Paul F. van Soest for comments on this manuscript.
Correspondence should be addressed to Dr. Karel S. Kits, Department of Neurophysiology, Research Institute Neurosciences, Vrije Universiteit, De Boelelaan 1087, 1081 HV Amsterdam, The Netherlands.
REFERENCES
- 1.Bahls FH, Richmond JE, Smith WL, Haydon PG. A lipoxygenase pathway of arachidonic acid metabolism mediates FMRFamide activation of a potassium current in an identified neuron of Helisoma. Neurosci Lett. 1992;138:165–168. doi: 10.1016/0304-3940(92)90497-u. [DOI] [PubMed] [Google Scholar]
- 2.Belardetti F, Siegelbaum SA. Up- and down-modulation of single K+ channel function by distinct second messengers. Trends Neurosci. 1988;11:233–238. doi: 10.1016/0166-2236(88)90132-4. [DOI] [PubMed] [Google Scholar]
- 3.Belardetti F, Campbell WB, Falck JR, Demontis G, Rosolowsky M. Products of heme-catalyzed transformation of the arachidonate derivative 12-HPETE open S-type K+ channels in Aplysia. Neuron. 1989;3:497–505. doi: 10.1016/0896-6273(89)90208-0. [DOI] [PubMed] [Google Scholar]
- 4.Blackwell GJ, Flower RJ. Inhibition of phospholipase. Br Med Bull. 1983;3:260–264. doi: 10.1093/oxfordjournals.bmb.a071830. [DOI] [PubMed] [Google Scholar]
- 5.Bolshakov VY, Gapon SA, Katchman AN, Magazanik LG. Activation of a common potassium channel in molluscan neurones by glutamate, dopamine and muscarinic agonist. J Physiol (Lond) 1993;468:11–33. doi: 10.1113/jphysiol.1993.sp019757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buttner N, Siegelbaum SA, Volterra A. Direct modulation of Aplysia S-K+ channels by a 12-lipoxygenase metabolite of arachidonic acid. Nature. 1989;342:553–555. doi: 10.1038/342553a0. [DOI] [PubMed] [Google Scholar]
- 7.Clapham DE, Neer EJ. G protein βγ subunits. Annu Rev Pharmacol Toxicol. 1997;37:167–203. doi: 10.1146/annurev.pharmtox.37.1.167. [DOI] [PubMed] [Google Scholar]
- 8.Cohen P, Holmes CFB, Tsukitani Y. Okadaic acid: a new probe for the study of cellular regulation. Trends Biochem Sci. 1990;15:98–102. doi: 10.1016/0968-0004(90)90192-e. [DOI] [PubMed] [Google Scholar]
- 9.Critz SD, Baxter DA, Byrne JH. Modulatory effects of serotonin, FMRFa, and myomodulin on the duration of action potentials, excitability, and membrane currents in tail sensory neurons of Aplysia. J Neurophysiol. 1991;66:1912–1926. doi: 10.1152/jn.1991.66.6.1912. [DOI] [PubMed] [Google Scholar]
- 10.De Vlieger TA, Lodder JC, Stoof JC, Werkman TR. Dopamine receptor stimulation induces a potassium dependent hyperpolarization response in growth hormone producing neuroendocrine cells of the gastropod mollusc Lymnaea stagnalis. Comp Biochem Physiol. 1986;83C:429–433. doi: 10.1016/0742-8413(86)90148-9. [DOI] [PubMed] [Google Scholar]
- 11.Dreijer AMC, Kits KS. Multiple second messenger routes enhance two high-voltage-activated calcium currents in molluscan neuroendocrine cells. Neuroscience. 1995;64:787–800. doi: 10.1016/0306-4522(94)00446-c. [DOI] [PubMed] [Google Scholar]
- 12.Exton JH. Cell signalling through guanine-nucleotide-binding regulatory proteins (G proteins) and phospholipases. Eur J Biochem. 1997;243:10–20. doi: 10.1111/j.1432-1033.1997.t01-1-00010.x. [DOI] [PubMed] [Google Scholar]
- 13.Fink M, Lesage F, Duprat F, Heurteaux C, Reyes R, Fosset M, Lazdunski M. A neuronal two P domain K+ channel stimulated by arachidonic acid and polyunsaturated fatty acids. EMBO J. 1998;17:3297–3308. doi: 10.1093/emboj/17.12.3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geraerts WPM. Control of growth by the neurosecretory hormone of the light green cells in the freshwater snail Lymnaea stagnalis. Gen Comp Endocrinol. 1976;29:61–67. doi: 10.1016/0016-6480(76)90007-1. [DOI] [PubMed] [Google Scholar]
- 15.Geraerts WPM, Smit AB, Li KW, Vreugdenhil E, van Heerikhuizen . Neuropeptide gene families that control reproductive behaviour and growth in molluscs. In: Osborne NN, editor. Current aspects of the neurosciences, Vol 3. MacMillan; London: 1991. pp. 255–304. [Google Scholar]
- 16.Isomoto S, Kondo C, Kurachi Y. Inwardly rectifying potassium channels: their molecular heterogeneity and function. Jpn J Physiol. 1997;47:11–39. doi: 10.2170/jjphysiol.47.11. [DOI] [PubMed] [Google Scholar]
- 17.Jelsema CL, Axelrod J. Stimulation of phospholipase A2 activity in bovine rod outer segments by the βγ subunits of transducin and its inhibition by the α subunit. Proc Natl Acad Sci USA. 1987;84:3623–3627. doi: 10.1073/pnas.84.11.3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kits KS, Lodder JC, Veerman MJ. Phe-Met-Arg-Phe-amide activates a novel voltage-dependent K+ current through a lipoxygenase pathway in molluscan neurones. J Gen Physiol. 1997;110:611–628. doi: 10.1085/jgp.110.5.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knol JC, Roovers E, Van Kesteren ER, Planta RJ, Vreugdenhil E, Van Heerikhuizen H. A G-protein β subunit that is expressed in the central nervous system of the mollusc Lymnaea stagnalis identified through cDNA cloning. Biochim Biophys Acta. 1994;1222:129–133. doi: 10.1016/0167-4889(94)90035-3. [DOI] [PubMed] [Google Scholar]
- 20.Koch WJ, Inglese J, Stone WC, Lefkowitz RJ. The binding site for the βγ subunits of heterotrimeric G proteins on the β-adrenergic receptor kinase. J Biol Chem. 1993;268:8256–8260. [PubMed] [Google Scholar]
- 21.Koch WJ, Hawes BE, Inglese J, Luttrell LM, Lefkowitz RJ. Cellular expression of the carboxyl terminus of a G protein-coupled receptor kinase attenuates Gβγ-mediated signalling. J Biol Chem. 1994;269:6193–6197. [PubMed] [Google Scholar]
- 22.Kurachi Y, Ito H, Sugimoto T, Shimizu T, Miki I, Ui M (1989) Arachidonic acid metabolites as intracellular modulators of the G protein-gated cardiac K+channel. Nature [Erratum (1989) 338: 360] 337:555–557. [DOI] [PubMed]
- 23.Macrez N, Morel J-L, Kalkbrenner F, Viard P, Schultz G, Mironneau J. A βγ dimer derived from G13 transduces the angiotensin AT1 receptor signal to stimulation of Ca2+ channels in rat portal vein myocytes. J Biol Chem. 1997;272:23180–23185. doi: 10.1074/jbc.272.37.23180. [DOI] [PubMed] [Google Scholar]
- 24.Murayama T, Kajiyama Y, Nomura Y. Histamine-stimulated and GTP-binding proteins-mediated phospholipase A2 activation in rabbit platelets. J Biol Chem. 1990;265:4290–4295. [PubMed] [Google Scholar]
- 25.Needleman P, Turk J, Jakschik BA, Morrison AR, Lefkowith JB. Arachidonic acid metabolism. Annu Rev Biochem. 1986;55:69–102. doi: 10.1146/annurev.bi.55.070186.000441. [DOI] [PubMed] [Google Scholar]
- 26.Patel AJ, Honoré E, Maingret F, Lesage F, Fink M, Duprat F, Lazdunski M. A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J. 1998;17:4283–4290. doi: 10.1093/emboj/17.15.4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Piomelli D, Volterra A, Dale N, Siegelbaum SA, Kandel ER, Schwartz JH, Belardetti F. Lipoxygenase metabolites of arachidonic acid as second messengers for presynaptic inhibition of Aplysia sensory cells. Nature. 1987a;328:38–43. doi: 10.1038/328038a0. [DOI] [PubMed] [Google Scholar]
- 28.Piomelli D, Shapiro E, Feinmark SJ, Schwartz JH. Metabolites of arachidonic acid in the nervous system of Aplysia: possible mediators of synaptic modulation. J Neurosci. 1987b;7:3675–3686. doi: 10.1523/JNEUROSCI.07-11-03675.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sasaki K, Sato M. A single GTP-binding protein regulates K+-channels coupled with dopamine, histamine and acetylcholine receptors. Nature. 1987;325:259–262. doi: 10.1038/325259a0. [DOI] [PubMed] [Google Scholar]
- 30.Schinelli S, Paolillo M, Corona GL. Opposing actions of D1- and D2-dopamine receptors on arachidonic acid release and cyclic AMP production in striatal neurons. J Neurochem. 1994;62:944–949. doi: 10.1046/j.1471-4159.1994.62030944.x. [DOI] [PubMed] [Google Scholar]
- 31.Shi RY, Belardetti F. Serotonin inhibits the peptide FMRFa response through a cyclic AMP-independent pathway in Aplysia. J Neurophysiol. 1991;66:1847–1857. doi: 10.1152/jn.1991.66.6.1847. [DOI] [PubMed] [Google Scholar]
- 32.Shuster MJ, Camardo JS, Siegelbaum SA, Kandel ER. Cyclic AMP-dependent protein kinase closes the serotonin-sensitive K+ channels of Aplysia sensory neurones in cell-free membrane patches. Nature. 1985;313:392–395. doi: 10.1038/313392a0. [DOI] [PubMed] [Google Scholar]
- 33.Smit AB, Vreugdenhil E, Ebberink RHM, Geraerts WPM, Klootwijk J, Joosse J. Growth-controlling molluscan neurons produce the precursor of an insulin-related peptide. Nature. 1988;331:535–538. doi: 10.1038/331535a0. [DOI] [PubMed] [Google Scholar]
- 34.Stoof JC, De Vlieger TA, Lodder JC. Opposing roles for D-1 and D-2 dopamine receptors in regulating the excitability of growth hormone-producing cells in the snail Lymnaea stagnalis. Eur J Pharmacol. 1984;106:431–435. doi: 10.1016/0014-2999(84)90735-0. [DOI] [PubMed] [Google Scholar]
- 35.Sweatt JD, Volterra A, Edmonds B, Karl KA, Siegelbaum SA, Kandel ER. FMRFa reverses protein phosphorylation produced by 5-HT and cAMP in Aplysia sensory neurons. Nature. 1989;342:275–278. doi: 10.1038/342275a0. [DOI] [PubMed] [Google Scholar]
- 36.Van der Steen WJ, Van der Hoven NP, Jager JC. A method for breeding and studying freshwater snails under continuous water change with some remarks on growth and reproduction in Lymnaea stagnalis. Neth J Zool. 1969;19:131–139. [Google Scholar]
- 37.Van Tol-Steye H, Lodder JC, Planta RJ, van Heerikhuizen H, Kits KS. Convergence of multiple G-protein-coupled receptors onto a single type of potassium channel. Brain Res. 1997;777:119–130. doi: 10.1016/s0006-8993(97)01096-2. [DOI] [PubMed] [Google Scholar]
- 38.Vial D, Piomelli D. Dopamine D2 receptors potentiate arachidonate release via activation of cytosolic, arachidonate-specific phospholipase A2. J Neurochem. 1995;64:2765–2772. doi: 10.1046/j.1471-4159.1995.64062765.x. [DOI] [PubMed] [Google Scholar]
- 39.Volterra A, Siegelbaum SA. Role of two different guanine nucleotide-binding proteins in the antagonistic modulation of the S-type K+ channel by cAMP and arachidonic acid metabolites in Aplysia sensory neurons. Proc Natl Acad Sci USA. 1988;85:7810–7814. doi: 10.1073/pnas.85.20.7810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Werkman TR, Schepens E, De Vlieger TA, Stoof JC. Cyclic AMP production in the central nervous system of the snail Lymnaea stagnalis is stimulated by forskolin and 5-hydroxytryptamine but is not affected by dopamine. Comp Biochem Physiol. 1990;95C:163–168. [Google Scholar]
- 41.Wickman K, Clapham DE. Ion channel regulation by G proteins. Physiol Rev. 1995;75:865–885. doi: 10.1152/physrev.1995.75.4.865. [DOI] [PubMed] [Google Scholar]
- 42.Yamada M, Terzic A, Kurachi Y. Regulation of potassium channels by G-protein subunits and arachidonic acid metabolites. Methods Enzymol. 1994;238:394–422. doi: 10.1016/0076-6879(94)38036-8. [DOI] [PubMed] [Google Scholar]