

Abstract
The Drosophila memory gene amnesiac(amn) has been proposed to encode a neuropeptide protein, which includes regions homologous to vertebrate pituitary adenylyl cyclase-activating peptide (PACAP; Feany and Quinn, 1995). Definitive experiments to link this gene to memory formation, however, have not yet been accomplished (Kandel and Abel, 1995). The experiments described here demonstrate that the putative amntranscript is involved in adult memory formation. With the use of aUAS–amn+ transgene, we show complete rescue of memory defects in amn28A, a mutant allele caused by the insertion of a GAL4 enhancer trap transposon (Moore et al., 1998). Study of theamn28A reporter reveals widespread expression in the adult brain but also enriched expression in the embryonic and larval nervous systems. To begin addressing the temporal requirement of amn in memory, we asked whether the memory defects could be rescued by restricting transgenic expression to the adult stage. A heat-shock regimen shown previously to rescue fully the amn ethanol sensitivity defect (Moore et al., 1998) failed to rescue the memory defect. These results, coupled with previous genetic and anatomical studies, suggest that adult memory formation and ethanol sensitivity have different temporal and spatial requirements for amn.
Keywords: Drosophila, neuropeptide, Pavlovian learning, neurogenetics, mutants, behavioral rescue, olfactory memory, associative learning, neurodevelopment, cAMP signaling, ethanol sensitivity
Efforts to understand the genetic basis of learning and memory are being advanced in fruit flies via a combination of forward and reverse genetic strategies (for review, seeDubnau and Tully, 1998). Initially, behavioral screens for learning/memory mutants identified two genes, dunce andrutabaga, both of which encode enzymatic components of the cAMP second messenger pathway. Subsequently, reverse genetic disruptions of other enzymatic steps of this pathway reinforced the notion that cAMP signaling is important for olfactory associative learning in Drosophila.
More recently, amnesiac (amn), another gene identified from the behavioral mutant screen (Quinn et al., 1979), has been linked to cAMP signaling (Feany and Quinn, 1995; Moore et al., 1998). Feany and Quinn (1995) identified a new mutation,amnP19A, based on its ability to suppress the female sterility of dnc mutants. This mutation was produced by the insertion of a P-element transposon, which permitted cloning of the genomic region. Focusing on a nearby transcription unit, the authors reported that amn encoded a neuropeptide homologous to vertebrate pituitary adenylyl cyclase-activating peptide (PACAP). Feany and Quinn did not providein vivo evidence that the putative AMN protein or proteins were expressed, were defective in mutant flies, or functioned during memory formation. Instead, the conclusion that amnencoded this transcript was based solely on their claim of a single nucleotide difference between wild-type andamn1 DNA sequences.
This key evidence subsequently was shown to be erroneous byMoore et al. (1998), who were addressing the role of amn in a different Drosophila behavior, ethanol sensitivity. With this assay they identified two additional amn mutations:amnchpd andamnX8. In contrast to Feany and Quinn (1995), Moore et al. (1998) determined by genomic and cDNA sequencing that the amn1 and wild-type open reading frames (ORFs), in fact, were identical. Consequently, they performed transgenic rescue experiments to establish a link between the amn transcript and ethanol sensitivity. Induced expression of the putativeamn+ transcript in adults was sufficient to rescue the ethanol sensitivity defect of amnmutants.
The findings of Moore et al. (1998) left unresolved a role for this putative transcription unit in adult memory formation. With the use of two independent transgenic approaches, we have resolved this issue. Our results show that the amn adult memory and ethanol sensitivity defects are caused by a disruption of the same gene. Our findings also indicate, however, that the spatial and temporal requirements for amn in these two behavioral processes are distinct.
MATERIALS AND METHODS
Fly stocks
amn28A carries a mini-P[w+] element at cytological location 19A (Ferveur et al., 1995) and has been shown to be an allele of amn with regard to ethanol sensitivity (Moore et al., 1998). amnX8 was isolated from excisions of the P[w+] insertion inamn28A and deletes theamn ORF (see Fig. 1A). BothUAS–amn+ andhs-amn+ transgenes carry the same DNA sequence from the amn genomic region: a 744 bp fragment extending from 164 bp upstream of the start codon to 40 bp past the end of the ORF (see Fig. 1A). The generation and characterization of hs-amn transgenic lines have been described previously (Moore et al., 1998). Of the five independent transformant lines, hs-amn+-7 was shown to rescue fully the ethanol sensitivity defect ofamnX8 in the adult after three daily heat-shocks at 37°C. For this reason, this study focuses only on that transgenic hs-amn+ line. TheUAS–amn+ line was made by inserting the 744 bp fragment into pUAST (Brand and Perrimon, 1993), followed by standard microinjection techniques. One transgenic line was generated, carrying the UAS–amn+transgene on the third chromosome. Southern blot determinations of theUAS–amn+ andhs-amn+-7 insertion sites are available on request.
Fig. 1.
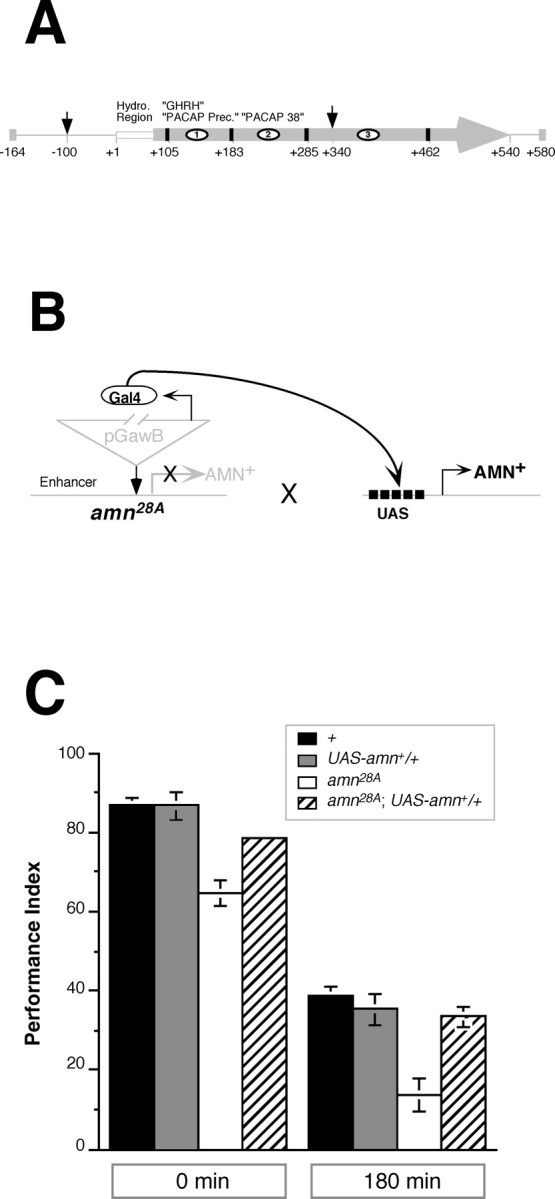
Transgenic expression ofamn+ rescues the memory defect ofamn28A mutants. A, Structure of the amnesiac locus. This schematic diagram details molecular aspects of theamnesiac gene as per Feany and Quinn (1995) with corrections from Moore et al. (1998). The 744 bp rescuing fragment described in this paper extends from 164 bp upstream of the start codon (−164) to 40 bp past the end of the open reading frame (+580). The primary transcription unit encodes a putative 180 amino acid protein with sequence features characteristic of neuropeptide precursors. These include a 25 amino acid hydrophobic domain at the N terminus, which may function as a signal sequence, and four pairs of basic residues (black boxes), which may function as protease cleavage sites. The three putative processed peptides consist of (1) a 24 amino acid N-terminal fragment with homology to the growth-hormone releasing hormone (GHRH) and to the mammalian pituitary adenylyl cyclase-activating peptide precursor (PACAP), (2) a 32 amino acid fragment with homology to mature PACAP-38 neuropeptide, and (3) a distal 57 amino acid fragment. The amn28A mutation was produced by the insertion of a pGawB P-element 100 bp upstream of the translation start site and a second 1.4 kb insertion at position +340 (arrows). TheamnX8 mutation is a deletion of the genomic region extending from −100 to somewhere between +451 and +587 bp. B, Genetic strategy to express GAL4-driven transgenes. Endogenous enhancer elements near the amngene presumably drive expression of theamn28A GAL4 protein, encoded within the pGawB P-element. When expressed alone, this GAL4 protein has no effect. When expressed in flies that also carry aUAS–amn+ transgene, however, GAL4 binds to its UAS recognition element and drives the expression of AMN+. In the absence of GAL4 theUAS–amn+ transgene is not expressed.C, Rescue of adult olfactory memory.Memory retention was quantified in wild-type (+), mutant (amn28A), and transgenic (UAS–amn+/+ andamn28A; UAS–amn+/+) males immediately (0 min) or 3 hr (180 min) after training. At both time points the memory retention in transgenic males with no GAL4 expression (UAS–amn+/+) is not significantly different from that in wild-type flies (p = 0.96 and p = 0.44) and is significantly higher than that in mutant amn28A males (p < 0.001 and p < 0.001). In contrast, memory retention inamn28A mutants expressing anamn+ transgene (amn28A;UAS–amn+/+) is significantly higher than that in mutantamn28A males (p < 0.001 and p = 0.001) and is not significantly different from that inUAS–amn+/+ (control) flies (p = 0.06 and p = 0.69). These data indicate that GAL4-induced expression of theUAS–amn+ transgene is sufficient to rescue the memory defect of amn28Amutants. The mean ± SEM PI is plotted for males of each genotype;n = 8 PIs per group. The p values were derived from planned pairwise comparisons (α′ = 0.013) after one-way ANOVAs were done separately at each time point.
General behavioral methods
Olfactory conditioning. The 2-to-3-d-old adult flies were subjected to the olfactory conditioning procedure of Tully and Quinn (1985), with minor modifications (see Tully et al., 1994). Briefly, groups of ∼100 flies received one training session, during which they were exposed sequentially to one odor (CS+) paired with footshock and then a second odor (CS−) without footshock. Conditioned odor avoidance was tested either immediately (0 min) or 180 min after the training session. During the test trial the flies were exposed simultaneously to the CS+ and CS− in a T-maze. After 2 min the flies were trapped in either T-maze arm, anesthetized, and counted. From this distribution, a performance index (PI) was calculated so that a 50:50 distribution (no memory) yielded a PI of zero and a 0:100 distribution away from the CS+ yielded a PI of 100.
Shock reactivity. To assess the flies’ ability to sense footshock and to escape from it, we attached “grid tubes” to each T-maze arm. DC current (60 V) was applied to one arm of the T-maze but not to the other. Naive (untrained) flies were lowered to the center of the T-maze, and their electroshock avoidance was quantified as above (see Luo et al., 1992).
Olfactory acuity. To assess the flies’ ability to sense odors, we passed two odors through the T-maze arms as in the conditioning experiments. Naive flies were lowered to the center of the T-maze, and their odor avoidance was quantified as above (see Boynton and Tully, 1992).
Statistics
Because of the nature of its mathematical derivation, PIs are distributed normally. Hence, these data from behavioral experiments were evaluated via one- or two-way ANOVAs. Subsequent planned pairwise comparisons were adjusted for experiment-wise error (α′), keeping the overall α = 0.05.
PCR analysis
Three sets of primers were used to verify the mutant and wild-type genomic loci as well as the presence of theUAS–amn+ transgene. For detection of 789, ∼2200, and 1135 bp products specific to the wild-type locus, amn28A locus, and UAS–amn+ transgene, respectively, the primers used were amn–WT-1 (5′-GAATGTGCGCTGGTATTGGCG-3′) and D–amn-1 (Moore et al., 1998) (5′-CGGATTATACGGCGTATGTGCAAGCC-3′); amn–28A-1 (5′-GCGAAAGCTAAGCAAATAAACAAGC-3′) and amn–WT-2 (5′-TTGGCTTCTGGATTGTTCTAGTG-3′); and amn–UAS-1 (5′-TGCCTGCAGGTCGGAGTACTG-3′) and amn–WT-2. The PCR was performed with KlenTaq DNA polymerase as specified by the manufacturer (Clontech, Palo Alto, CA). The cycling parameters were the same for all primer sets: 1 cycle for 3 min at 94°C; 35 cycles for 30 sec at 94°C, 30 sec at 58°C, and 2.5 min at 68°C. Genomic DNA was isolated fromamn28A oramn28A;UAS–amn+/+ orUAS–amn+/+ flies, and wild-type males and then subjected to PCR amplification. Reaction products were resolved by agarose gel electrophoresis. Only the 789 or 2200 bp products were detected in wild-type oramn28A males, respectively. Only the 789 and 1135 bp products were detected inUAS–amn+ males, and only the 2200 and 1135 bp products were detected inamn28A;UAS–amn+males. These PCR data confirmed the genotypes of the populations of flies bred for the behavioral experiments.
GFP and lacZ reporter studies
Adult and larva. Adult brains were dissected and processed as previously described (Connolly et al., 1996). Third instar larval brains were dissected in PBS plus 4% fresh paraformaldehyde, washed extensively in PBS, cleared in 80% glycerol, and mounted in 100% glycerol.
Embryos. For GFP detection, stage 16 embryos were collected on egg plates, dechorionated in 100% bleach for 3 min, rinsed in water for 1 min, and visualized in PBS under a glass coverslip. For detection of the lacZ reporter gene, antibody staining was performed by standard procedures. The primary antibody was rabbit anti-lacZ (1:1000; Sigma, St. Louis, MO); the secondary antibody was peroxidase-conjugated goat anti-rabbit IgG (1:500; Jackson ImmunoResearch, West Grove, PA). The antibody complexes were visualized by staining in DAB peroxidase (Sigma). Stained embryos were cleared in 70% glycerol and mounted in 100% glycerol. All images were captured digitally with the Spot CCD camera (Diagnostic Instruments, Sterling Heights, MI) under bright-field or Nomarski optics on a Zeiss Axiophot microscope (Carl Zeiss, Thornwood, NY).
Northern analysis
Total RNA from whole adult flies was isolated with the TriZOL reagent (BRL, Bethesda, MD). The poly(A+) fraction was purified with magnetized oligo-dT beads (Dynal, Great Neck, NY). The purified poly(A+) RNA was fractionated by formaldehyde–agarose gel electrophoresis and transferred to a ZetaProbe nylon membrane (Bio-Rad, Richmond, CA) in 10× SSC. The RNA on the dried membrane was fixed by UV-cross-linking at 2500 μJ (Stratalinker, Stratagene, La Jolla, CA). The membrane was hybridized with a 744 bp fragment encoding the entireamnesiac ORF in high-stringency Church and Gilbert Buffer, washed extensively, and exposed to Kodak BioMax film (Rochester, NY).
RESULTS
Genetic complementation of mutant amn alleles for olfactory associative memory
As a necessary genetic foundation for this work, we first assessed the genetic complementation among three mutant alleles (amn1,amn28A, andamnX8) with respect to memory formation. This was critical, because theamn1 lesion is unknown (see above) and the amn28A andamnX8 mutations are large molecular perturbations that potentially could affect multiple transcripts. All three mutants failed to complement each other for memory retention either immediately (initial learning) or 180 min (3 hr memory) after training (Table 1), indicating thatamn28A andamnX8 are bona fide mutations of theamn gene along with amn1. In contrast to the recessivity ofamn1 andamnX8, theamn28A mutation appears semidominant—but significantly so only for initial learning. More pertinently for the subsequent rescue experiments, all homozygous mutants scored significantly lower than wild-type flies.
Table 1.
Genetic complementation of mutant and wild-typeamn alleles for olfactory memory
| Genotype | Initial learning1-a | 3 hr memory1-b | ||
|---|---|---|---|---|
| Mean ± SEM | p | Mean ± SEM | p | |
| +/+1-c | 87 ± 2 | 37 ± 3 | ||
| amn1/+ | 81 ± 21-d | 0.294 | 36 ± 71-d | 0.844 |
| amn28A/+ | 71 ± 41-d | 0.004 | 28 ± 51-d | 0.138 |
| amnX8/+ | 80 ± 31-d | 0.228 | 39 ± 41-d | 0.730 |
| amn1/amn1 | 59 ± 41-d | <0.001 | 16 ± 21-d | <0.001 |
| amn28A/amn28A | 55 ± 31-d | <0.001 | 20 ± 41-d | 0.003 |
| amnX8/amnX8 | 48 ± 41-d | <0.001 | 17 ± 41-d | <0.001 |
| amn1/amn28A | 67 ± 41-e | 0.126 | 21 ± 41-e | 0.443 |
| amn1/amnX8 | 62 ± 41-e | 0.608 | 20 ± 41-e | 0.496 |
| amn28A/amnX8 | 53 ± 41-e | 0.218 | 18 ± 51-e | 0.688 |
n = 6 PIs per group.
n = 8 PIs per group.
This amn+ allele, which was carried in a w (CS10) stock, is aw1118 allele that was out-crossed for 10 generations to a wild-type Can-S stock and shows normal olfactory learning/memory, olfactory acuity, and shock reactivity (Dura et al., 1993).
Planned pairwise comparison (α′ = 0.006) to +/+ after a one-way ANOVA, with genotype as a main effect. Data from initial learning and memory were analyzed separately.
Planned pairwise comparison (α′ = 0.006) toamn1/amn1after a one-way ANOVA, with genotype as a main effect. Data from initial learning and memory were analyzed separately.
In behavioral control experiments we assayed “task-relevant” sensorimotor responses for each of the new homozygous mutants. No significant differences were detected between wild-type and mutantamn flies for olfactory acuity or shock reactivity (Table2). These data suggest that the reduced performance of mutant flies resulted specifically from defects in the associative process.
Table 2.
Olfactory acuity and shock reactivity of mutants
| Strain | Olfactory acuity2-a | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OCT dilution | MCH dilution | Shock reactivity2-a | ||||||||||
| 100 | 10−2 | 100 | 10−2 | 60 V | 20 V | |||||||
| Mean ± SEM | p | Mean ± SEM | p | Mean ± SEM | p | Mean ± SEM | p | Mean ± SEM | p | Mean ± SEM | p | |
| w(CS10) | 57 ± 6 | 27 ± 6 | 68 ± 4 | 27 ± 6 | 83 ± 3 | 39 ± 5 | ||||||
| amnX8 | 53 ± 42-b | 0.729 | 20 ± 62-b | 0.396 | 58 ± 52-b | 0.246 | 19 ± 62-b | 0.376 | 81 ± 42-c | 0.728 | 37 ± 72-c | 0.760 |
| amn28A | 60 ± 62-b | 0.674 | 31 ± 62-b | 0.667 | 62 ± 92-b | 0.515 | 27 ± 92-b | 0.988 | 79 ± 32-c | 0.536 | 39 ± 32-c | 0.917 |
n = 8 PIs per group.
Planned pairwise comparison (α′ = 0.006) tow(CS10) after a two-way ANOVA, with strain and odor/dilution as main effects and a strain × odor/dilution interaction term.
Planned pairwise comparison (α′ = 0.006) tow(CS10) after a two-way ANOVA, with strain and voltage as main effects and a strain × voltage interaction term.
Enhancer trap-driven expression of amn+rescues the mutant memory defect
We capitalized on the fact that theamn28A mutation results from the insertion of a GAL4-containing P-element (pGawB) in the amntranscription unit (Fig.1A,B) (Moore et al., 1998). In many such cases, endogenous enhancers of the target gene drive the expression of GAL4 in a spatiotemporal pattern similar to that of the wild-type (but now disrupted) transcript (Han et al., 1996). Thus, it seemed possible to rescue the memory defect associated with the amn28A mutation by using its inherent GAL4 expression, in turn, to drive the expression of aUAS–amn+ transgene. To accomplish this, we inserted a 744 bp genomic fragment ofamn+ into pUAST (Brand and Perrimon, 1993), a P-transformation cassette containing five GAL4-binding sites (Fig. 1A,B). [Thisamn+ genomic fragment was identical to the one used by Moore et al. (1998) to rescue the amnethanol sensitivity defect.] P-element-mediated transformation with this construct yielded a single transgenic line,UAS–amn+.
For the rescue experiments we crossedamn28A females toUAS–amn+ males and tested the male progeny (amn28A/Y;UAS–amn+/+)for memory retention immediately (0 min) or 180 min after training. We found that expression of UAS–amn+rescued the memory deficit of amn28Amutant males (Fig. 1C). In addition, we also observed rescue of the partial performance defect of heterozygousamn28A/+ females (Table 1; results not shown).
Four additional observations indicated that this rescue was authentic and specific. First, PCR analysis confirmed the genotypes of the wild-type, mutant, and transgenic populations used for behavioral experiments (see Materials and Methods). Second, sensorimotor controls for olfactory avoidance and shock reactivity were normal in the mutants (Table 2) (cf. Bolwig et al., 1995). Third, memory was still mutant inamn28A males with the same genetic background as the UAS–amn+ strain (Fig. 1C). Fourth, memory was not greater than normal whenUAS–amn+ was overexpressed in a wild-type background, using several additional enhancer trap lines with GAL4 expression patterns in various regions of the CNS (data not shown). Together, these experiments establish conclusively that the minimal ORF (180 amino acids) ofamn+ is sufficient to rescue fully the amn mutant memory defect.
The amn28Aenhancer is expressed in the nervous system throughout development
We visualized the expression pattern of theamn28A P-GAL4 element, using a UAS-driven green fluorescent protein (GFP). Widespread expression was observed throughout the adult central brain with no apparent preferential expression (Fig.2A) (cf. Han et al., 1996). More importantly for the results summarized below, expression of the amn28A GAL4 protein was not confined to adulthood. In third instar larvae, GFP was expressed in a punctate pattern throughout the central brain and ventral ganglion (Fig. 2B). In late embryos, GFP expression showed a highly restricted pattern that appeared to be mainly neuronal (Fig.2C). In this case we obtained a higher resolution view by immunohistochemical detection of a UAS-driven β-galactosidase. With this reporter gene we found that theamn28A GAL4 protein was expressed clearly in a subset of neurons along the ventral nerve cord in the CNS (Fig. 2D,E) and in segmented clusters of neurons with clearly discernible neuropilar projections (arrowheads) in the peripheral nervous system (Fig.2D,F). Such widespread spatiotemporal expression left open the question of the temporal requirement foramn during adult memory formation.
Fig. 2.
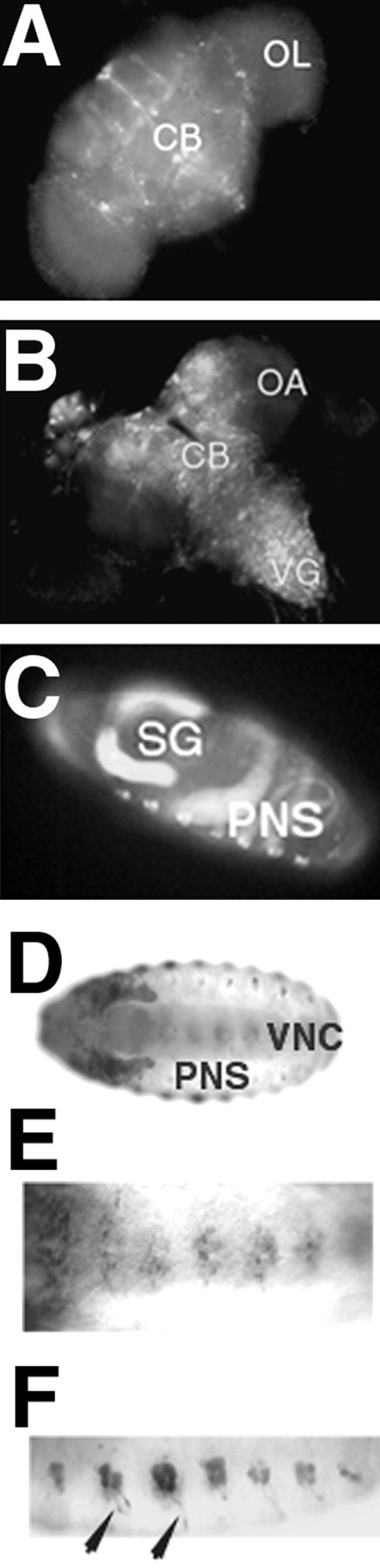
Enhancer trap expression of theamn28A is widespread in the developing nervous system. Expression of the GAL4 yeast protein contained in the P-element insertion ofamn28A mutants was visualized by GAL4-driven expression of UAS–GFP (green fluorescent protein [S65T]) in amn28A;UAS–GFP/+ males oramn28A/+;UAS–GFP/+ females. In addition, expression was visualized by GAL4-driven expression of UAS–lacZ inamn28A;UAS–lacZ/+males oramn28A/+;UAS–lacZ/+ females. A, GFP expression in adults was widespread throughout the central brain (CB) and, to a lesser extent, in the optic lobes (OL). B, GFP expression in the CNS of third instar larvae was widespread in the central brain (CB), ventral ganglion (VG), and, to a lesser extent, in the optic lobe anlagen (OA). C, GFP expression in the embryo was most apparent in non-neuronal tissue, such as the salivary glands (SG) and gut (which is common for many PGAL4 lines) and peripheral cells that appear to be neuronal. D–F, Whole-mount views of lacZimmunoreactivity in the embryo revealed expression in the ventral nerve cord (VNC; D, E), and PNS (D, F).
Induced expression of an hs-amn+transgene in adults fails to rescue the mutant memory deficit
To begin addressing the temporal requirements of theamn gene for the rescue of memory, we capitalized on previous studies of amn in ethanol sensitivity (Moore et al., 1998). Using theamnX8;hs-amn+-7transgenic line, Moore et al. (1998) showed that a rigorous heat-shock regimen in adults—one 60 min heat-shock per day for 3 d with testing 24 hr after the last heat shock— was necessary and sufficient to rescue fully the ethanol sensitivity defect of amn mutants. We used the same heat-shock regimen on the same transgenic flies (amnX8;hs-amn+-7) but failed to observe any rescue of the memory defects ofamn (Fig. 3A,B). Despite these negative results for the transgenic rescue ofamn mutant memory, we observed robust induction of thehs-amn+-7 transgene 3 and 24 hr after the heat-shock protocol (Fig. 3D).
Fig. 3.
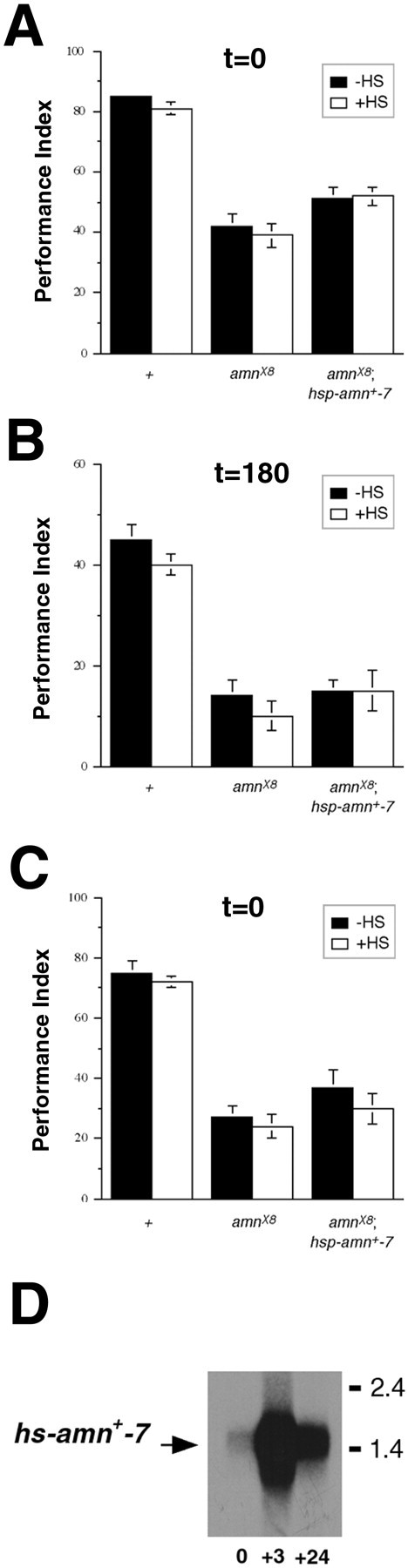
Induced expression ofhs-amn+ in adult flies does not rescue the memory defect of amnX8mutants. As in Moore et al. (1998), wild-type (+), mutant (amnX8), and transgenic (amnX8;hs-amn+-7) adults were grown at 25°C and then were subjected as adults to three 60 min heat-shocks (to 37°C) (+HS) or not (−HS) 24 hr before receiving one training session (see Fig. 1 for methods). Immediate (O min; A) and 180 min memory (B) was quantified thereafter. This heat-shock regimen produced no significant effects on memory retention for transgenic flies at either time point (p = 0.85 and p = 0.92 for t = 0 andt = 180, respectively); n = 6 PIs per group. C, The same transgenic flies (and control flies) were raised at 18°C, rather than 25°C, and then were subjected as adults to one 60 min heat-shock per day (to 35°C) for 3 d in the period 24 hr before testing. Here again, heat-shock produced no effect (p = 0.30). ForA–C, planned pairwise comparisons (α′ = 0.05) were done after a one-way ANOVA with strain/hs as a main effect;n = 4 PIs per group. D, TransgenicamnX8,hs-amn+-7 flies were raised at 25°C and then were subjected to the heat-shock protocol described byMoore et al. (1998), which was used for the behavioral experiments summarized in A. Poly(A+) RNA was isolated before and 3 or 24 hr after the heat-shock treatment. In contrast to the minimal levels of expression before heat-shock, this heat-shock regimen induced high levels ofhs-amn+ expression, which lasted for >24 hr.
In fact, induction of thehs-amn+-7 transgene by using the heat-shock protocol of Moore et al. (1998) was so great that we considered whether such expression might be deleterious for memory formation. Accordingly, we attempted two different heat-shock regimens. First, we applied the same conditions as Moore at al. (1998) but reduced the heat-shock time from 60 to 45 min. This failed to yield rescue inamnX8;hs-amn+-7transgenic flies (data not shown). Second, we raised flies at 18°C, rather than 25°C, and gave adults one 60 min heat-shock per day at 35°C for 3 d with testing 24 hr after the last heat shock. This heat-shock protocol also failed to produce any rescue of memory (Fig.3C). Although these results cannot exclude an adult role foramn in memory, they begin to argue that amnexpression during development may be critical to rescue the memory defects of mutant adults (see below).
DISCUSSION
Transgenic expression of amn+rescues the memory defect in amn adults
Rescue of the amn mutant memory defect with the GAL4-driven expression of aUAS–amn+ transgene establishes the molecular identity of amn (see Kandel and Abel, 1995). Although Feany and Quinn (1995) first suggested that this transcription unit corresponded to the amn gene, their only evidence in support of this notion subsequently was shown to be insufficient (Moore et al., 1998) (see above). Moore et al. (1998) discovered, however, that mutations in the amn gene produced defects in adult ethanol sensitivity and that inducible expression of anhs-amn+ transgene could rescue this mutant defect. Our results clearly establish a role for amnin both ethanol sensitivity and olfactory memory.
Olfactory memory and ethanol sensitivity may reflect different spatial and temporal requirements for amn
The observation that induced expression ofhs-amn+ in the adult rescues ethanol sensitivity, but not olfactory memory, suggests different roles foramn in the two processes. In fact, the neuroanatomical requirements for olfactory memory and ethanol sensitivity appear to be different. Targeted disruption, or chemical ablation, of mushroom bodies abolishes olfactory associative learning (de Belle and Heisenberg, 1994; Connolly et al., 1996) but does not appear to affect ethanol sensitivity (Moore et al., 1998). Thus, different spatial requirements for amn expression likely underlie these two behavioral effects.
Adult memory formation and ethanol sensitivity also may have different temporal requirements for amn. Expression of anamn+ transgene in adults is sufficient to rescue mutant ethanol sensitivity, but not mutant memory. Although this latter outcome may derive from ectopic (misexpression) ofamn+ in the CNS, this scenario appears unlikely. Widespread overexpression of theUAS–amn+ transgene in the adult CNS, driven by a variety of enhancer trap lines, does not disrupt olfactory memory (data not shown). Conversely, induced ectopic expression of the hs-amn+ transgene does not worsen olfactory memory inamnX8 mutants (Fig.3A–C). More generally, similar inducible transgenic approaches for the linotte and Volado memory mutants have yielded full rescue (Bolwig et al., 1995; Grotewiel et al., 1998).
Instead, our results and those from other laboratories point to a developmental role for amn. Mutant alleles of amncan suppress the female sterility of dunce mutants (Feany and Quinn, 1995), suggesting an early biological role for this gene. In studies of amn1 mutants, Hitier et al. (1998) have identified two developmental defects in the calyces of mushroom bodies, neural centers required for adult olfactory learning:amn1 calyces have a greater-than-normal volume and do not show normal developmental plasticity. Consistent with these functional observations, the expression of amn28A is widespread in the embryonic and larval nervous systems. Collectively, these results suggest that optimal memory formation in the adult depends on proper amn function in the mushroom bodies during development.
Complexities of amn neuropeptide function
A definitive description of the amn gene product(s) will facilitate greatly the functional studies of amn. As first described by Feany and Quinn (1995), analysis of the deduced amino acid sequence of the amn ORF suggests that it encodes a secreted protein with consensus cleavage sites that might give rise to three peptides (see Fig. 1A). Two of these peptides show weak homologies to mammalian PACAP and growth hormone-releasing hormone (GHRH), and the third appears novel. The homology to vertebrate PACAP, in fact, linked amn to the cAMP signaling pathway and, accordingly, supported the authors’ claim to have identified the correct transcript. This link also directedMoore et al. (1998) to assess other genetic and pharmacological aspects of cAMP signaling for ethanol sensitivity. To that end, cAMP signaling clearly is involved in adult ethanol sensitivity.
Such a clear-cut conclusion is not yet possible for the role ofamn in memory formation. Our data suggest that one or more of these putative neuropeptides are involved in the development of brain structures that normally subserve adult olfactory memory (cf.deBelle and Heisenberg, 1994; Connolly et al., 1996). Perhaps the PACAP-like neuropeptide is responsible for this neurodevelopment. Mammalian PACAP activates the cAMP pathway in Drosophilaneurons (Zhong, 1995). Moreover, other “learning/memory” genes inDrosophila encode enzymatic components involved in cAMP-signaling, and mutations in some of these other genes also yield developmental abnormalities (Dubnau and Tully, 1998), including synaptic defects at the larval neuromuscular junction (Hannan and Zhong, 1998).
Taken together, these observations underscore the need for further experiments that use inducible transgenes to discern whether the memory defects of the cAMP mutants derive from maldevelopment or a more acute defect in cAMP signaling in adults. To date, such experiments have been accomplished only for dunce, revealing a combination of developmental and acute etiologies (Dauwalder and Davis, 1995).
By the way, an amn peptide with homology to mammalian GHRH also may support such a developmental role. Critically, none of the putative AMN peptides has been detected yet in situ or evaluated for its effects on neuronal function. Future experiments that use transgenes expressing only one processed peptide or that identify the corresponding neuropeptide receptor(s) promise to shed light on the pleiotropic functions of amn.
Continued studies of amn also promise to illuminate the mechanisms of peptide signaling in neuronal development and function. Our work has established that animals carrying a complete knock-out ofamn are healthy and viable nevertheless, and the behavioral defects of mutants can be modulated with small transgenes of limited complexity. Studies of the role of amn in plasticity even may bear on human cognitive function. Behavioral properties of associative memory have been shown to be similar between fruit flies and mammals, thereby suggesting similar underlying molecular mechanisms (DeZazzo and Tully, 1995; Dubnau and Tully, 1998). In accordance, homologs of several Drosophila genes have been implicated in synaptic and behavioral plasticity in other invertebrates and in vertebrates (Bailey et al., 1996). Such “functional homology” predicts that the biological etiologies of particular gene mutations will be similar. With this perspective we anticipate that the study ofamn neuropeptide function may help to inform the genetic basis of developmental learning disability in humans.
Footnotes
This work was supported by National Institutes of Health Grant NS32480 and the John A. Hartford Foundation (to T.T.) and by National Research Service Award NS09763 (to J.D.). We thank John Connolly, Josh Dubnau, Scott Gossweiler, Heidi Hammer, and Kate Pedatella for contributions to this work and the Bloomington Stock Center for fly stocks.
J.D. and S.X. contributed equally to this study.
Correspondence should be addressed to Dr. Tim Tully at the above address.
REFERENCES
- 1.Bailey CH, Bartsch D, Kandel ER. Toward a molecular definition of long-term memory storage. Proc Natl Acad Sci USA. 1996;93:13445–13452. doi: 10.1073/pnas.93.24.13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bolwig GM, Del Vecchio M, Hannon G, Tully T. Molecular cloning of linotte in Drosophila: a novel gene that functions in adults during associative learning. Neuron. 1995;15:829–842. doi: 10.1016/0896-6273(95)90174-4. [DOI] [PubMed] [Google Scholar]
- 3.Boynton S, Tully T. latheo, a new gene involved in associative learning and memory in Drosophila melanogaster, identified from P-element mutagenesis. Genetics. 1992;131:655–672. doi: 10.1093/genetics/131.3.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 5.Connolly JB, Roberts IRH, Armstrong JD, Kaiser K, Forte M, Tully T, O’Kane CH. Associative learning disrupted by impaired Gs signaling in Drosophila mushroom bodies. Science. 1996;274:2104–2106. doi: 10.1126/science.274.5295.2104. [DOI] [PubMed] [Google Scholar]
- 6.Dauwalder B, Davis RL. Conditional rescue of the dunce learning/memory and female fertility defects with Drosophila or rat transgenes. J Neurosci. 1995;15:3490–3499. doi: 10.1523/JNEUROSCI.15-05-03490.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Belle JS, Heisenberg M. Associative odor learning in Drosophila abolished by chemical ablation of mushroom bodies. Science. 1994;263:692–695. doi: 10.1126/science.8303280. [DOI] [PubMed] [Google Scholar]
- 8.DeZazzo J, Tully T. Dissection of memory formation: from behavioral pharmacology to molecular genetics. Trends Neurosci. 1995;18:212–217. doi: 10.1016/0166-2236(95)93905-d. [DOI] [PubMed] [Google Scholar]
- 9.Dubnau J, Tully T. Gene discovery in Drosophila: new insights for learning and memory. Annu Rev Neurosci. 1998;21:407–444. doi: 10.1146/annurev.neuro.21.1.407. [DOI] [PubMed] [Google Scholar]
- 10.Dura J-M, Preat T, Tully T. Identification of linotte, a new gene affecting learning and memory in Drosophila melanogaster. J Neurogenet. 1993;9:1–14. doi: 10.3109/01677069309167272. [DOI] [PubMed] [Google Scholar]
- 11.Feany MB, Quinn WG. A neuropeptide gene defined by the Drosophila memory mutant amnesiac. Science. 1995;268:869–873. doi: 10.1126/science.7754370. [DOI] [PubMed] [Google Scholar]
- 12.Ferveur JF, Stoertkuhl KF, Stocker RF, Greenspan RJ. Genetic feminization of brain structures and changed sexual orientation in male Drosophila. Science. 1995;267:902–905. doi: 10.1126/science.7846534. [DOI] [PubMed] [Google Scholar]
- 13.Grotewiel MS, Beck CD, Wu KH, Zhu XR, Davis RL. Integrin-mediated short-term memory in Drosophila. Nature. 1998;391:455–460. doi: 10.1038/35079. [DOI] [PubMed] [Google Scholar]
- 14.Han P-L, Meller V, Davis RL. The Drosophila brain revisited by enhancer detection. J Neurobiol. 1996;31:88–102. doi: 10.1002/(SICI)1097-4695(199609)31:1<88::AID-NEU8>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 15.Hannan F, Zhong Y. Second messenger systems underlying plasticity at the neuromuscular junction. In: Budnick V, Gramates L, editors. International review of neurobiology. Academic; New York: 1998. pp. 119–138. [DOI] [PubMed] [Google Scholar]
- 16.Hitier R, Heisenberg M, Preat T. Abnormal mushroom body plasticity in the Drosophila memory mutant amnesiac. NeuroReport. 1998;9:2717–2719. doi: 10.1097/00001756-199808240-00006. [DOI] [PubMed] [Google Scholar]
- 17.Kandel ER, Abel T. Neuropeptides, adenylyl cyclase, and memory storage [comment]. Science. 1995;268:825–826. doi: 10.1126/science.7754367. [DOI] [PubMed] [Google Scholar]
- 18.Luo L, Tully T, White K. Human amyloid precursor protein ameliorates behavioral deficit of flies deleted for Appl gene. Neuron. 1992;9:595–605. doi: 10.1016/0896-6273(92)90024-8. [DOI] [PubMed] [Google Scholar]
- 19.Moore MS, DeZazzo J, Luk AY, Tully T, Singh CM, Heberlein U. Ethanol intoxication in Drosophila: genetic and pharmacological evidence for regulation by the cAMP pathway. Cell. 1998;93:997–1007. doi: 10.1016/s0092-8674(00)81205-2. [DOI] [PubMed] [Google Scholar]
- 20.Quinn W, Sziber PP, Booker R. The Drosophila memory mutant amnesiac. Nature. 1979;277:212–214. doi: 10.1038/277212a0. [DOI] [PubMed] [Google Scholar]
- 21.Tully T, Quinn WG. Classical conditioning and retention in normal and mutant Drosophila melanogaster. J Comp Physiol [A] 1985;157:263–277. doi: 10.1007/BF01350033. [DOI] [PubMed] [Google Scholar]
- 22.Tully T, Preat T, Boynton SC, Del Vecchio M. Genetic dissection of consolidated memory in Drosophila. Cell. 1994;79:35–47. doi: 10.1016/0092-8674(94)90398-0. [DOI] [PubMed] [Google Scholar]
- 23.Zhong Y. Mediation of PACAP-like neuropeptide transmission by coactivation of Ras/Raf and cAMP signal transduction pathways in Drosophila. Nature. 1995;375:588–592. doi: 10.1038/375588a0. [DOI] [PubMed] [Google Scholar]