

Abstract
The basolateral amygdala (BLA) is implicated in responding to affective stimuli. Dopamine (DA) is released in the BLA during numerous conditions; however, the neurophysiological effects of DA in the BLA have not been examined in depth. In this study, the effects of DA receptor manipulation on spontaneous and afferent-driven neuronal firing were examined using in vivo extracellular single-unit recordings in parallel with systemic and iontophoretic drug application, and stimulation of the substantia nigra/ventral tegmental area in the rat. The effects of DA receptor activation in the BLA were found to depend on the characteristics of the BLA neuron examined, causing an increase in the firing rate of putative interneurons and a decrease in the firing of identified projection neurons. Additionally, DA receptor activation attenuated short-latency spikes evoked by electrical stimulation of prefrontal cortical and mediodorsal thalamic inputs to the BLA while potentiating the responses evoked by electrical stimulation of sensory association cortex.
DA receptor activation can thus attenuate BLA projection neuron firing via two mechanisms: (1) by a direct inhibition, and (2) by indirect actions mediated via activation of BLA interneurons. This is hypothesized to lead to a global filtration of weaker inputs. Moreover, DA potentiates sensory inputs and attenuates medial prefrontal cortex inputs to the BLA. Conditions in which DA is released in the BLA, such as during the presentation of an affective stimulus, will lead to a potentiation of the strongest sensory input and a dampening of cortical inhibition over the BLA, thus augmenting the response to affective sensory stimuli.
Keywords: dopamine, basolateral amygdala, projection neuron, interneuron, afferent, electrophysiology, iontophoresis, substantia nigra, ventral tegmental area, modulation
The importance of the amygdala in autonomic, endocrine, and motor responses to affective stimuli and affective conditioning has long been recognized (for review, seeAggleton, 1992; Rolls, 1992; Davis et al., 1994). The amygdala may be divided into several nuclei based on cytoarchitectural, histochemical, connectional, and functional criteria (for review, see Price et al., 1987; Swanson and Petrovich, 1998). Amygdalar areas that are key to affective conditioning and responding include the basolateral amygdala (BLA) nuclei (lateral, basolateral, and basomedial nucleus) and the central nuclei (Roozendaal et al., 1991; Falls and Davis, 1995; Maren et al., 1996; Killcross et al., 1997; Sajdyk and Shekhar, 1997; Soltis et al., 1997). The BLA receives projections from areas including the medial prefrontal cortex (mPFC; Otterson, 1989; McDonald et al., 1996), sensory association cortex (McDonald and Mascagni, 1996; Shi and Cassell, 1997), and thalamus (Van Vulpen and Verwer, 1989), whose integrity are necessary to permit affective conditioning (Gaffan et al., 1988, 1993; Gaffan and Murray, 1990; Campeau and Davis, 1995;Poremba and Gabriel, 1997). It is suggested that the primary flow of information through the amygdala during affective responding involves sensory input to the BLA, which projects to the central nucleus, and onto autonomic and neuroendocrine centers (Pitkanen et al., 1997) to produce autonomic and neuroendocrine responses (LeDoux et al., 1988;Hitchcock and Davis, 1991; Yeomans and Pollard, 1993). Moreover, the BLA projections to the nucleus accumbens (NAc; McDonald, 1991), a limbic region involved in the production of affective motor behavior (Mogenson et al., 1980; Le Moal and Simon, 1991), may be involved in the motor response to affective stimuli (Cador et al., 1989).
Systemic and direct alterations of dopamine (DA) transmission within the BLA are known to produce significant effects on affective conditioning and responding (Weldon et al., 1991; Hitchcott et al., 1997; Munro and Kokkinidis, 1997; Lamont and Kokkinidis, 1998; Nader and LeDoux, 1999). Furthermore, DA has been implicated in either the etiology or treatment of certain symptoms of schizophrenia, depression, and anxiety (Reynolds, 1983; Seeman, 1990; Grace, 1991, 1992; Deutch, 1992; Pitchot et al., 1992; Brown and Gershon, 1993; Wingerson et al., 1996). Each of these disorders is associated with symptoms that are characteristic of amygdala dysfunction, and furthermore, these patients are reported to exhibit anatomical abnormalities in the amygdala and in areas synaptically connected with the BLA (Berman et al., 1986; Arnold et al., 1991; Drevets et al., 1992, 1997; Pakkenberg, 1992; Raine et al., 1992; Bogerts et al., 1993).
There is converging evidence that DA plays a significant role in amygdala function. Thus, the elements necessary for functional DA transmission have been shown to be present in the BLA (Swanson, 1982;Loughlin and Fallon, 1983; Scibilia et al., 1992; Revay et al., 1996;Asan, 1997). Furthermore, studies have shown that DA levels are increased in the BLA during learning (Hori et al., 1993) and in response to stressful or predictive stimuli (Herman et al., 1982; Coco et al., 1992; Harmer and Phillips, 1999; Inglis and Moghaddam, 1999). Nonetheless, the neurophysiology of DA in the BLA has not been examined in detail. Previous studies examining this aspect of DA action (Wepsic and Austin, 1971; Ben-Ari and Kelly, 1976; Bashore et al., 1978;Spehlmann and Norcross, 1984; Wang and Rebec, 1996) have reported heterogenous results. Moreover, even though the BLA consists of two basic neuronal subtypes [i.e., the pyramidal-like projection neuron and the inhibitory interneuron (McDonald, 1985, 1992)], previous studies have not segregated the effects of DA based on the neuron subtype examined. Additionally, there has been virtually no examination of the effects of DA on the neurophysiology of afferent inputs to this region.
In this study, we examine the electrophysiological effects of DA receptor activation on afferents to the BLA, such as mediodorsal thalamus (MD), mPFC, and sensory association cortex (Te3). Additionally, the effects of DA receptor activation on the two basic neuronal subtypes of the BLA are examined.
Parts of this paper have been presented in abstract form (Rosenkranz and Grace, 1998).
MATERIALS AND METHODS
Materials
DA, the nonselective DA agonist apomorphine, the DA D1 agonist SKF-38393, the D1 antagonist SCH-23390, the D2 agonist quinpirole, and the D2 antagonist raclopride, were purchased from Research Biochemicals (Natick, MA). The nonselective DA antagonist haloperidol was a generous gift from McNeil Laboratories.l-glutamic acid (glutamate) was purchased from Sigma (St. Louis, MO).
Electrophysiological recordings
In vivo extracellular single-unit or population field potential electrophysiological recordings were performed in anesthetized male Sprague Dawley rats (250–400 gm). All procedures followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and were approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh. Animals were housed in pairs, supplied with food and water ad libitum, and maintained on a 12 hr light/dark cycle. Rats were anesthetized with 8% chloral hydrate (400 mg/kg, i.p). Additional supplemental doses of chloral hydrate were administered intraperitoneally when necessary. Temperature was monitored with a rectal temperature probe (model 4600; Precision Thermometer, Yellow Springs, OH), and maintained at 37–38°C using a heat control unit and heating pad (Fintronics, Orange, CT). The rat was mounted in a stereotaxic device (Narishige, Tokyo, Japan), incisions were made in the scalp to expose the skull, burr holes were drilled into the skull, and the dura was removed in an area overlying the BLA [−5.3 lateral (L), −3.3 caudal (C) from bregma]. Depending on the experiment, an additional hole for the stimulating electrode was drilled in the skull overlying one of the following: the mPFC [infralimbic/prelimbic cortex, +2.7 rostral (R), −0.7 L, −4.3 ventral (V)], mediodorsal thalamus [MD; −2.1 C, −0.5 L, −5.3 V), secondary sensory cortex (Te3; −6.5 L, −5.0 C, −5.2 V), substantia nigra/ventral tegmental area (SN/VTA; −5.6 C, −0.8 L, −8.0 V) or NAc (+2.2 R, −1.4 L, −7.2 V). Structures were localized using a stereotaxic atlas (Paxinos and Watson, 1997). Single-barrel electrodes were constructed using a vertical microelectrode puller (PE-2; Narishige), and the recording barrel filled with 2% Pontamine sky blue in 2 m NaCl (impedance measured in vivo ranged between 10 and 20 MΩ for single-unit recording and 4–8 MΩ for population field potential recordings, both measured at 1 kHz). Recording electrodes were slowly lowered into the amygdala via a micromanipulator (MO-8; Narishige). Bipolar concentric stimulating electrodes were lowered into one of the other remaining structures, and stimulation was delivered using a Grass S88 stimulator (Quincy, MA), with the intensity ranging between 75 and 900 μA with a duration of 0.2–0.3 msec. Stimulation pulses were photoelectrically isolated (PSIU6G; Grass). At the completion of each experiment, recording sites were marked by ejection of Pontamine sky blue to mark the recording site.
Data collection
Signals from the recording electrode were amplified by a headstage connected to the preamplifier before being fed into a window discriminator (Fintronics discriminator/amplifier) and displayed on an oscilloscope (V-134; Hitachi, Tokyo, Japan) and an audio monitor (AM5; Grass). The data were also stored on video tapes after being digitized (DR-390, Neurocorder; NeuroData, New York, NY). Data were simultaneously collected using a Microstar board for data acquisition and online data monitoring using software developed in this laboratory (Neuroscope), and stored on a personal computer (Gateway 2000, model P5–100XL) for subsequent off-line analysis.
Iontophoretic application of drug
Multibarrel microelectrodes (five barrels; Activational Systems, Warren, MI) were constructed using a vertical microelectrode puller (PE-2; Narishige), and the tip was broken back under microscopic control. The central barrel of the microelectrode was filled with 2% Pontamine sky blue in 2 m NaCl for electrophysiological recordings. One of the outer barrels was filled with 4 mNaCl for automatic current balancing, and various drug solutions were used in the remaining barrels. Drug barrels of the multibarrel pipette were filled with 50 or 100 mm DA, pH 4.5, 20–100 mm glutamate, pH 8.0, 20–40 mm NMDA, pH 8.0, 10–30 mm SKF 38393, pH 4.5, 10 mm quinpirole, pH 4.5, or 100–200 mm GABA, pH 4.0. All drugs were dissolved in 10 mm NaCl. Drugs dissolved in solutions of acidic pH were ejected with (+) iontophoretic current, and drugs dissolved in solutions of basic pH were ejected with (−) iontophoretic current (E104B; Fintronics). Retaining currents of the opposite polarity ranged between 8 and 10 nA. Iontophoretic drug currents ranged between 2 and 32 nA, although currents of up to 110 nA were tested in a few cases to ensure consistency of effects at high ejection currents. The ejection of glutamate or NMDA was often done using timed, repetitive pulses with a duration of 15 or 30 sec, with a 30 or 45 sec delay between pulses.
Systemic drug administration
Drugs were dissolved in distilled water, or in the case of haloperidol, dilute lactic acid, to a final concentration of 0.5 mg/ml. Drugs were administered via a lateral tail vein (or in a few cases intraperitoneally) in volumes of 0.05–0.4 ml in ascending doses in a dose–response fashion. Saline was administered as a control.
Data analysis
The particulars of the data analysis depended on the type of neuronal activity monitored:
Spontaneous spike discharge. Single units were isolated (with a signal-to-noise ratio of ≥3:1, and a minimal duration of 1.0 msec was set to exclude spikes that were not of somatodendritic origin; Humphrey, 1979), and stable baseline firing rates were obtained for a minimum of 1 min (typically 5 min) before drug administration. After stable baseline data were collected, systemic or iontophoretic drug administration was performed. After each dose of systemically administered drug, neuronal activity was recorded for a minimum of 4 min before a subsequent administration occurred. These predrug and postdrug administration epochs were compared using paired ttests (α = 0.05 for significance). If a DA agonist was administered first, antagonists were administered 10–30 min after the final agonist administration, while continuously recording neuronal activity.
Additionally, the duration of the action potentials recorded from BLA units was quantified as the time from the initial change from baseline to the return to baseline. The distribution of firing rates was also examined and fit to population curves (Jandel Table Curve). Furthermore, the distribution of firing rates was examined as a function of action potential duration. Using firing rate population distributions and firing rate distributions as a function of action potential duration, a cutoff of 0.5 Hz was used to segregate fast- and slow-firing neurons.
Electrically evoked responses. Electrical stimulus pulses were often delivered during electrode penetration to search for units that exhibited evoked responses (0.6 Hz, 0.2–0.7 mA, 0.2 msec duration). Evoked responses consisted of single-unit responses or evoked field potentials. Single units were operationally defined as monosynaptic if their latency was <25 msec; they showed very little shift in latency when increasing the stimulus intensity, yet they showed some range (1–2 msec) in latency distribution (“jitter”), ruling out antidromic activation. Stimulus intensities were varied to determine an evoked spike response probability of ∼50% in the case of single units, or half-maximal amplitude in the case of evoked field potentials. The magnitude of the evoked field potential was quantified as the absolute voltage change from baseline to the peak of the positive deflections, or the trough of the negative deflections. After stable baselines were recorded, drugs were administered systemically as above, and drug-induced changes in the evoked spike probability and field potential amplitude were measured. A minimum of 150 sweeps was obtained before and after drug administration at various time points. If the neuron was spontaneously spiking, a minimum of 1–2 min was given after electrical stimulation before basal firing rate was recorded.
Substantia nigra/VTA stimulation. After stable baselines were obtained from spontaneously spiking neurons or neurons activated by iontophoretic application of glutamate, the substantia nigra/ventral tegmental area was electrically stimulated to induce dopamine release in the amygdala. The stimulus parameters used consisted of pulse trains of 10–20 Hz, 0.2 msec duration, 0.5–0.6 mA, for a period of 1–2 sec. Prestimulus firing rate values were compared to firing rate values of a 15 sec epoch that immediately followed the SN/VTA stimulation. In many cases, this process was repeated for the same neuron after several minutes had elapsed, and firing had returned to baseline levels. In some of these repeated stimulation trials, haloperidol was administered systemically in an attempt to block the effects of later SN/VTA stimulation trains as a way of confirming dopaminergic mediation of the response.
Antidromically evoked activity. Electrical stimulations were performed as above. An antidromic response was defined as the ability of evoked spikes to follow stimulation frequencies of > 250 Hz, displayed constant response latency, display collision with spontaneously occurring spikes when possible, and be evoked from an area receiving BLA projections. In the majority of cases, a test for collision of evoked and spontaneous spikes was not feasible because of the lack of spontaneous spiking in many antidromically activated units.
Glutamate-evoked activity. Responses to glutamate were tested in one of two ways: (1) constant glutamate ejection (+10 to +40 nA) was used to maintain a stable baseline of neuronal activity, or (2) glutamate was ejected in pulses, as described above. After obtaining a stable firing rate, or a stable response to pulsed glutamate, DA or DA agonists were co-iontophoresed (as described above), and changes in firing rate that were induced by the iontophoretic application of DA or DA agonists was examined. During constant glutamate iontophoresis, the glutamate-induced spike discharge (minimum 60 sec) was compared to the firing rate during DA co-iontophoresis. Alternatively, when glutamate was applied using 15 or 30 sec duration pulses, several consecutive pulses were averaged, and the responses compared to the effects obtained to pulsed glutamate recorded during DA co-iontophoresis.
To examine the statistical significance of the drug effects, Student'st tests were used. A group t test was applied, and if the results were not statistically significant, individualt tests were used on the results from single neurons to determine how many neurons of the particular treatment group displayed a significant change concomitant with the treatment. Post hoc layered Bonferroni was used to maintain multiple comparisons at α = 0.05. Where violations of assumptions of normality and homogeneity of variance were present, appropriate nonparametric tests were used.
Histology
Verification of recording and stimulating electrode sites was obtained histologically. Rats were killed by overdose with anesthetic, decapitated, and the brains were removed and fixed in 10% formalin for a minimum of 24 hr. Brains were cryoprotected with 30% sucrose in 0.1m phosphate buffer and were then frozen and sliced with a cryostat or with a sliding microtome into 40 μm coronal sections. Mounted sections were then stained with cresyl violet. Recording sites were identified by the blue spot caused by ejection of Pontamine sky blue (see Electrophysiological recordings). Stimulating sites were identified as the end of the stimulating electrode track.
RESULTS
Evidence for two subtypes of BLA neurons
As seen in previous reports, the basal firing rate of most spontaneously spiking neurons in the amygdala was very low (mean ± SEM, 0.91 ± 2.47 Hz; n = 179; range, 0–22.3 Hz), with evidence of a large population of neurons that did not fire spontaneously during the recording period. After closer examination, neurons could be placed into one of two normal distributions (r2 > 0.98) with significantly different mean firing rates (p < 0.05; groupt test; Fig. 1): fast-firing (mean, 2.81 ± 0.52 Hz; n = 55; range, 0.51–22.3 Hz) and slow-firing (mean, 0.071 ± 0.013 Hz; n = 124; range, 0–0.50 Hz) neurons. Based on the distribution of neuronal firing rates, a cutoff of 0.5 Hz was used to segregate the two populations. Hence “slow-firing” will be used to indicate neurons that fire at <0.5 Hz, whereas “fast-firing” will be used to denote neurons that fire at >0.5 Hz. This segregation of neuronal types also corresponded to differences in action potential waveform (Fig.2). The spike durations of a 90 neuron sample were examined (34 fast-spiking neurons, mean, 2.56 ± 0.380 Hz; 56 slow-spiking neurons, mean, 0.18 ± 0.02 Hz; Fig. 2). Slowly firing neurons displayed longer duration action potential waveforms (mean, 2.71 ± 0.10 msec) than fast-firing neurons (mean, 1.64 ± 0.095 msec; p < 0.001; group t test).
Fig. 1.

Neuronal firing rate distribution supports the presence of two populations of BLA neurons. A, Firing rate distribution of neurons recorded in the BLA. There is a bimodal distribution that may be separated into two normal populations (p < 0.05; n = 179 neurons) with 0.5 Hz as a cutoff. Representative firing rate histograms of two spontaneously firing neurons, a fast-firing (B) and a slow-firing (C) neuron.
Fig. 2.

Parcellation of fast- and slow-firing neurons of the BLA by spike duration. A, Example trace of a fast-spiking neuron that displays a short spike duration (1.2 msec).B, Example trace of a slow-firing neuron that displays a long spike duration (4.0 msec). C, A plot of spike duration by firing rate for a sample of 90 BLA neurons demonstrates that fast-firing neurons (>0.5 Hz) tend to display short spike durations, whereas slow-firing neurons (<0.5 Hz) tend to display longer duration spikes (p < 0.01).
To further validate the parcellation of neuronal subtypes, projection neurons were identified by antidromic activation from either the mPFC or NAc (Fig. 3). Antidromically activated BLA neurons, as defined by a fixed response latency, collision with spontaneously occurring spikes when possible, and the ability to follow high-frequency stimulation, fired spontaneously at slow firing rates (n = 5; mean, 0.263 ± 0.128 Hz) or not at all (n = 16). A sample of antidromically activated neurons also displayed significantly longer spike durations than fast-firing neurons [n = 12; mean, 2.49 ± 0.47 msec;p < 0.001; group t test; there were no differences noted between the spike duration, response latency (mean latency, 12.1 msec; range, 7–20 msec), or projection target of slow-firing and nonspontaneously firing neurons antidromically driven from the mPFC or NAc]. By contrast, none of the fast-firing neurons could be activated antidromically.
Fig. 3.
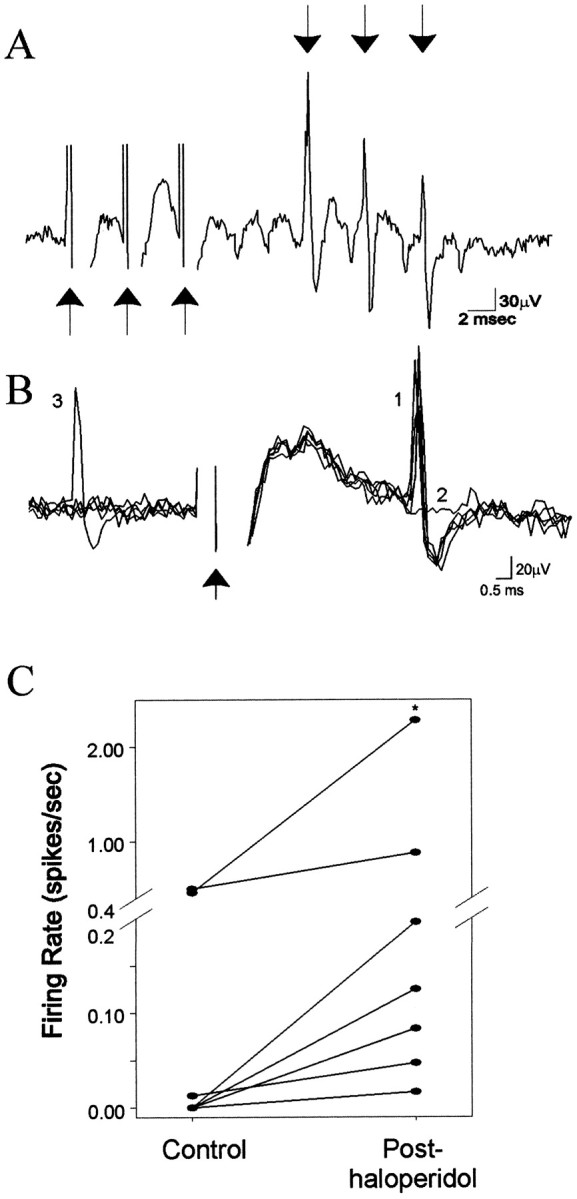
Antidromically activated, slow-firing BLA neurons display an increase in firing rate after DA receptor blockade.A, An example trace of a BLA neuron that follows high-frequency (300 Hz) electrical stimulation of the NAc.Arrows pointing up indicate stimulation, and arrows pointing down indicate spikes, respectively. B, Traces of antidromically evoked spikes in a slow-firing neuron in response to single NAc stimulation. The spikes display constant response latency (1) and collision (2) with a spontaneously occurring spike (3). Arrow indicates stimulus artifact. C, BLA neurons that could be antidromically activated from NAc stimulation display an increase in firing rate after systemic administration of the DA antagonist haloperidol (n = 7; *p < 0.05; Wilcoxon; circles represent neuronal firing rates prehaloperidol and posthaloperidol administration).
DAergic effects on spontaneous or microiontophoretic glutamate-evoked firing
Systemic administration of saline had no substantial effect on neuronal firing rate (two of two neurons). After intravenous drug administration, when significant effects occurred, changes of firing rate could be observed within 2 min. Systemic administration of the dopaminergic agonists apomorphine (n = 10), SKF-38393 (n = 6), or quinpirole (n = 4) had variable effects on the firing rate of spontaneously spiking neurons (10 of 20 neurons increased firing rate, 7 of 20 neurons decreased firing rate). However, the response observed could be differentiated based on the neuronal firing rate. Because of the similarity in responses, all DA agonists were grouped together. Fast-firing (>0.5 Hz) neurons displayed an increase in firing rate after systemic DA agonist administration (Fig. 4; 10 of 11 neurons; 10 rats; p = 0.016; Wilcoxon; mean firing rate ± SEM; pre-DA agonist, 2.57 ± 0.749 Hz; post-DA agonist, 5.44 ± 1.898 Hz; maximum change, >3000% of baseline; mean change, 256% of baseline), whereas the firing rate of slowly firing neurons was attenuated (Fig. 4; seven of nine neurons; eight rats; p = 0.0156; Wilcoxon; pre-DA agonist, 0.371 ± 0.199 Hz; post-DA agonist, 0.191 ± 0.092 Hz; maximum change, 0% of baseline; mean change, 33% of baseline) by similar doses of the same drugs [doses as follows: (1) DA agonists (initial doses of 0.05–0.15 mg): apomorphine, mean initial dose, 0.14 ± 0.016 mg/kg, ranging from 0.09 to 0.31 mg/kg, and a final dose as high as 0.61 mg/kg; SKF-38393, initial dose, 0.05–0.15 mg, corresponding to a mean initial dose 0.12 ± 0.002 mg/kg, ranging from 0.11 to 0.13 mg/kg and a final dose as high as 0.50 mg/kg; quinpirole, initial dose, 0.16 ± 0.03 mg/kg, ranging from 0.13 to 0.290 mg/kg, and a final dose as high as 0.58 mg/kg; (2) DA antagonists (initial doses of 0.1–0.25 mg): haloperidol, initial dose, 0.30 ± 0.050 mg/kg, ranging from 0.13 to 0.65 mg/kg, and a final dose as high as 0.65 mg/kg; raclopride, initial dose, 0.26 ± 0.086 mg/kg, ranging from 0.13 to 0.49 mg/kg, with a final dose as high as 0.58 mg/kg; SCH 23390, initial dose, 0.21 ± 0.017 mg/kg, ranging from 0.13–0.23 mg/kg, with a final dose as high as 0.75 mg/kg]. In several cases, the initial dose was repeated with subsequent injections given at double the previous dose, in a dose–response manner. In the instances in which more than one dose of DA agonist was administered, supplementary doses potentiated the effects of the initial dose or produced an effect when the initial dose was subthreshold for producing a result. In most cases, the effects were reversible after systemic administration of DA receptor antagonists (raclopride, three of four neurons; haloperidol, five of seven neurons; and SCH-23390, three of three neurons). It was also observed in 4 of 11 cases that administration of a DA antagonist after the DA agonist not only reversed the effects of the agonist, but further altered the firing rate beyond baseline levels. This may be attributable to a combination of two factors: (1) removal of DA agonist-induced actions and (2) attenuation of baseline DAergic tone on the neuron. Furthermore, systemic administration of dopamine antagonists alone decreased the firing rate of fast firing neurons (Fig. 5; five of six neurons; six rats; pre-DA antagonist, 2.83 ± 0.92 Hz, post-DA antagonist, 0.93 ± 0.46 Hz; p = 0.040; group t test; maximum change, 2% of baseline; mean change, 18% of baseline) and increased the firing rate of slowly firing neurons (Fig. 4; eight of nine neurons; eight rats; pre-DA antagonist, 0.110 ± 0.077 Hz; post-DA antagonist, 1.21 ± 0.965 Hz; p = 0.0195; Wilcoxon; maximum change, >1000% of baseline; mean change, >1000% of baseline; haloperidol, mean initial dose, 0.10 ± 0.025 mg/kg, ranging from 0.03 to 0.16 mg/kg, with a final dose as high as 0.32 mg/kg).
Fig. 4.
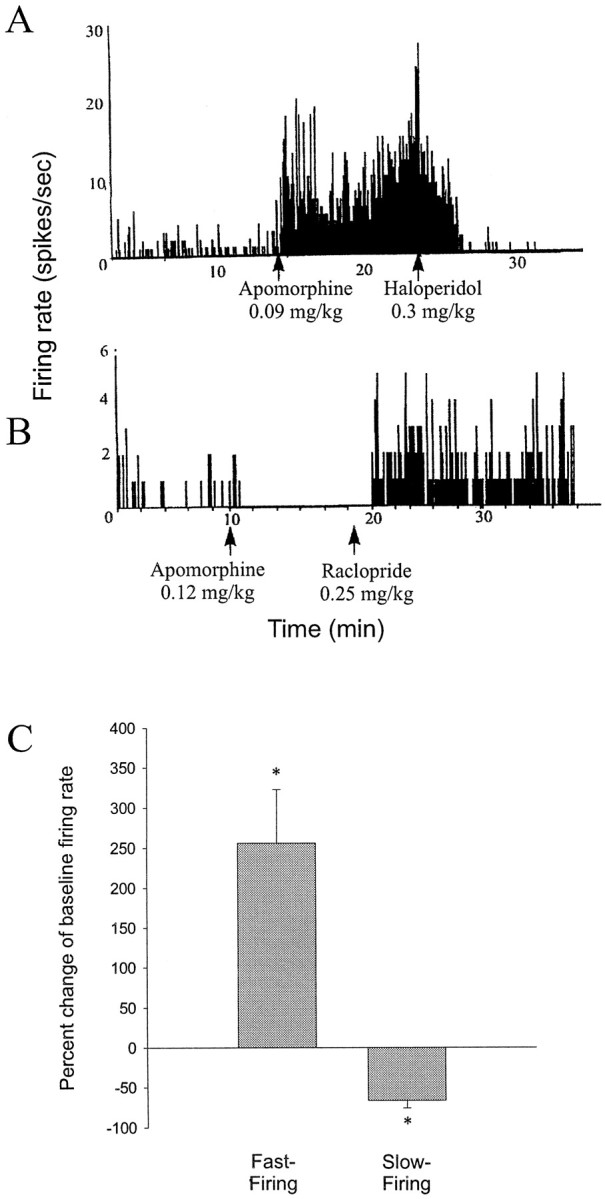
Opposite effects of DA receptor activation on fast-firing and slow-firing BLA neurons. A, Firing rate histogram of a fast-firing neuron that displays an increase in firing rate after systemic administration of the DA agonist apomorphine (0.09 mg/kg, i.v.) that is reversed after systemic administration of the DA antagonist haloperidol (0.30 mg/kg, i.v.). B, Firing rate histogram of a slow-firing neuron that displays a decrease in firing rate after systemic administration of apomorphine (0.12 mg/kg, i.v.) that is reversed after systemic administration of the DA antagonist raclopride (0.25 mg/kg, i.v.). Arrowsindicate the time of drug administration. C, DA agonist administration has opposite effects on fast-firing (n = 10 of 11 neurons) and slow-firing (n = 7 of 9 neurons) neurons of the BLA (*p < 0.05; error bars indicate mean ± SEM; see Results for dose ranges).
Fig. 5.
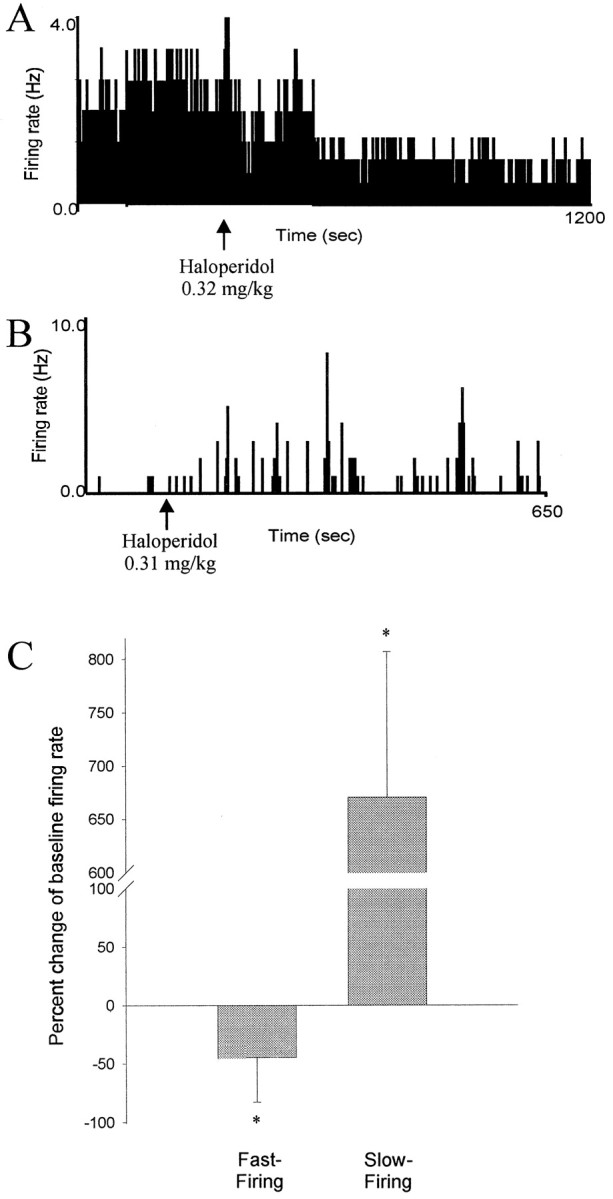
Opposite effects of DA receptor blockade on fast-firing and slow-firing BLA neurons. A, Firing rate histogram of a fast-firing neuron that displays a decrease in firing rate after systemic administration of the DA antagonist haloperidol (0.32 mg/kg, i.v.). B, Firing rate histogram of a slow-firing neuron that displays an increase in firing rate after systemic administration of haloperidol (0.31 mg/kg, i.v.).Arrows indicate time of drug administration.C, Opposite effects of systemic administration of DA antagonists on fast-firing (5 of 6 neurons) and slow-firing (8 of 9 neurons) neurons of the BLA (*p < 0.05; error bars indicate mean ± SEM; see Results for dose ranges).
Consistent with this, the response of neurons that exhibited antidromic activation from electrical stimulation of the NAc was also qualitatively different from that of the fast-firing neurons. Thus, the firing rates of these neurons increased after systemic administration of haloperidol alone (Fig. 3; seven of seven neurons; six rats; prehaloperidol, 0.27 ± 0.18 Hz; posthaloperidol, 0.52 ± 0.32 Hz; p = 0.016;Wilcoxon; each neuron displayed an increase in firing rate p < 0.05, individualt tests with Bonferroni corrections; haloperidol, mean initial dose, 0.61 ± 0.057 mg/kg, ranging from 0.40–0.85 mg/kg, with a maximal dose of 0.85 mg/kg).
To further demonstrate that the response was dopamine-mediated in the BLA, two strategies were taken: microiontophoresis of DA in the BLA and electrical stimulation of the SN/VTA, which is the locus of the DA cell afferents to the BLA. Electrical stimulation of SN/VTA in three rats yielded results similar to the effects observed with systemic administration of DA agonists. Thus, fast-firing neurons showed an increase in firing after SN/VTA stimulation (Fig.6; 9 of 10 neurons; pre-SN stimulation, 2.87 ± 0.828 Hz; post-SN stimulation, 4.51 ± 1.242 Hz;p = 0.0091; group t test), whereas slow-firing neurons showed a decrease in firing rate [Fig. 6; four of four neurons; pre-SN stimulation, 0.34 ± 0.097 Hz; post-SN stimulation, 0.11 ± 0.046 Hz; p = 0.066; groupt test; however, each individual neuron showed a significant change in firing rate; p < 0.05; individualt tests (with Bonferroni correction)]. The effects of SN/VTA stimulation were attenuated by systemic haloperidol administration (Fig. 6; 0.55–0.68 mg/kg), independent of the effects of haloperidol on basal firing rate. The effects began within seconds of, or during, SN/VTA stimulation and exhibited durations of up to 1 min. Additionally, iontophoretic glutamate-evoked excitation of nonspontaneously spiking neurons was depressed by SN stimulation [Fig.6; four of four neurons; pre-SN stimulation, 16.1 ± 8.9 Hz; post-SN stimulation, 8.1 ± 4.2 Hz; mean change, 53.6 ± 3.8% of baseline; p > 0.05; group t test; however, each individual neuron displayed a significant (p < 0.05, individual t tests with Bonferroni correction) decrease in firing rate after SN/VTA stimulation].
Fig. 6.
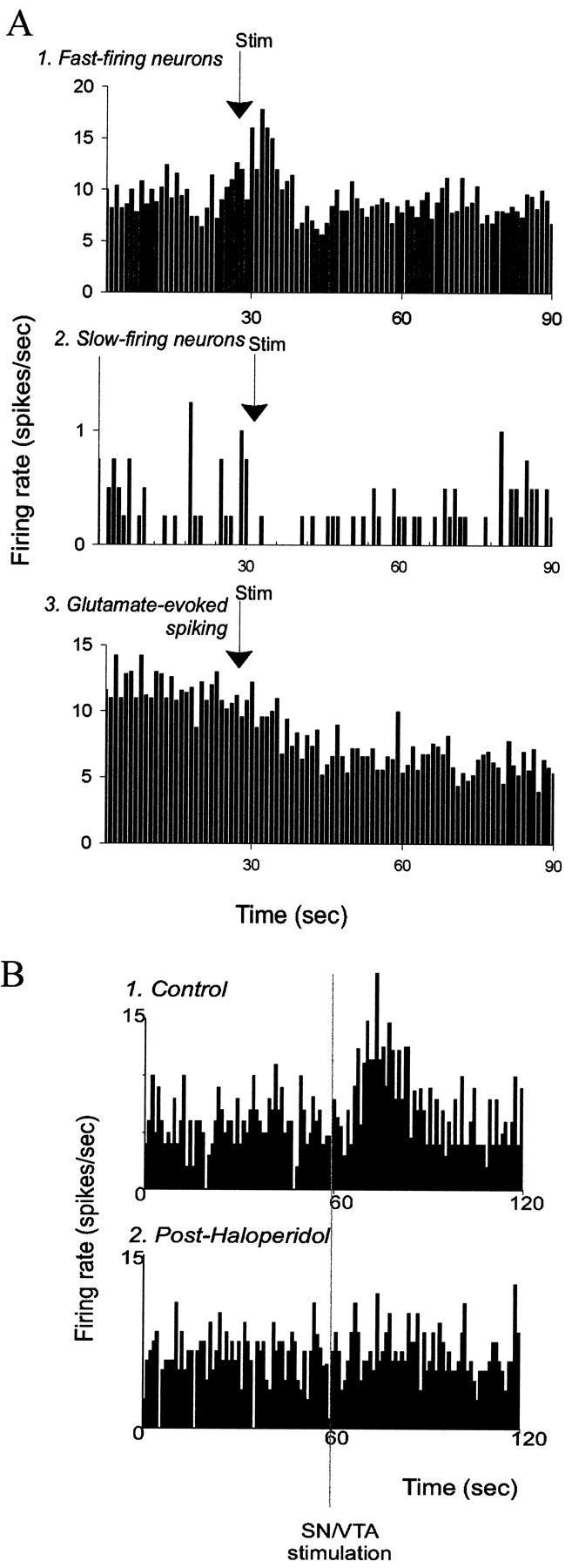
Electrical stimulation of the SN/VTA exerts opposite effects on fast-firing and slow-firing neurons.A1, Averaged composites of neuronal responses to SN/VTA stimulations (stimulus time histograms for several neurons were added together and divided by the number of stimulus presentations). SN/VTA stimulation increases the firing rate of fast-firing neurons of the BLA (n = 5; p < 0.05), decreases the firing rate of slow-firing neurons (n = 4; p< 0.05) (2), and attenuates the glutamate-evoked excitation of neurons that did not display spontaneous spike discharge (n = 4; p < 0.05) (3). Arrows indicate onset of electrical stimulation (1–2 sec; 10–20 Hz; 0.5–0.6 mA).B, Example firing rate histogram of the response of a fast-firing neuron to SN/VTA stimulation before (1) and after (2) systemic administration of the DA antagonist haloperidol (0.67 mg/kg, i.v.).Vertical line indicates beginning of 1 sec SN/VTA stimulation at 10 Hz, 0.2 msec durations, 0.6 mA.
Co-microiontophoresis of DA and glutamate produced consistent results. In our iontophoresis recordings, most of the neurons exhibited spontaneous spike discharge only during iontophoresis of glutamate. All but one of the neurons excited by iontophoretic glutamate displayed an attenuation in firing rates during co-iontophoresis of DA (Fig.7; 20 of 21 neurons; seven rats;p < 0.01; group t test). In the neurons tested with variable iontophoretic currents of DA, the firing rate of the neurons was attenuated in a current-dependent manner (Fig. 7). The effects of iontophoretic DA were attenuated by systemic administration of haloperidol (n = 2 of 2). Iontophoresis of the specific DA agonists SKF-38393 (n = 4 of 5;p = 0.013; group t test) and quinpirole [n = 3 of 4; p = 0.11; groupt test; however, three of four neurons displayed significant changes with individual t tests; p < 0.05 (with Bonferroni correction)] also attenuated the firing of BLA neurons induced by glutamate iontophoresis. In no case in which DA or DA agonist was administered alone did a nonfiring neuron begin firing spontaneously. Additionally, a set of neurons antidromically activated from NAc stimulation were induced to fire with iontophoretic glutamate application, and the firing was attenuated by co-iontophoresis of DA (Fig. 7B; n = 3; the firing rate of each neuron was significantly attenuated, p < 0.05; individual t tests with Bonferroni correction).
Fig. 7.
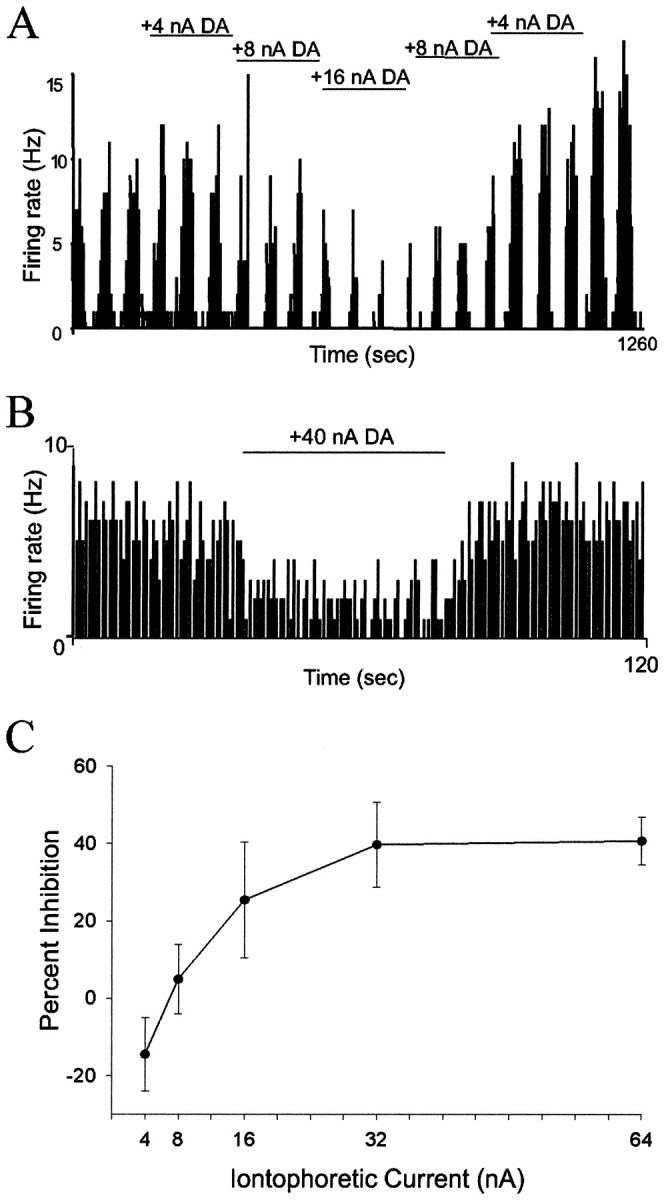
Microiontophoretic application of DA attenuates BLA neuronal firing induced by co-microiontophoresis of glutamate.A, Firing rate histogram showing that DA attenuates the neuronal firing induced by iontophoretically applied pulses of glutamate (−20 nA) in a current-dependent manner. B,Firing rate histogram showing that DA attenuates the neuronal firing induced by continuous iontophoretic application of glutamate (−20 nA) in an antidromically activated neuron. C, Plot of the current dependency of the attenuation of neuronal firing by iontophoretic application of DA (n = 20 neurons).
DAergic effects on afferent-evoked responses
The BLA receives afferent input from a number of cortical structures, most of which are known to use glutamate as a neurotransmitter (for review, see Somogyi et al., 1998). Several of these cortical areas are believed to have a potent effect on BLA activity (Pare and Gaudreau, 1996) via glutamatergic projections. To examine the effects of DA on afferent-driven activity, responses evoked by electrical stimulation of several regions that send projections to the BLA were examined. In all cases, only the short-latency responses were analyzed. After stimulation of the MD, short-latency, putative monosynaptic (Fig. 8; n = 15; mean latency, 16.9 msec; range, 5–26 msec), and occasionally multisynaptic responses were evoked. Very few neurons that displayed both antidromic and orthodromic responses were encountered, and these were not included in this study. Except where noted, the responses evoked from electrical stimulation of MD, mPFC, and Te3 were recorded from neurons that were not spontaneously spiking or spiked spontaneously at such low firing rates that quantification of spontaneous firing rates was not appropriate. The action potential duration of a sample of units that displayed spikes evoked from MD, mPFC, and Te3 stimulation was 2.8 ± 0.10 msec; n= 15. Given the low spontaneous firing rates and the long-duration action potentials, the majority of the evoked responses were probably recorded from projection neurons. The probability of an evoked response after MD stimulation was significantly attenuated by systemic administration of apomorphine (Fig. 8; six of seven neurons; seven rats; preapomorphine response probability, 0.49 ± 0.04; postapomorphine response probability, 0.26 ± 0.10;p < 0.05; group t test, 51 ± 16% of baseline; apomorphine mean initial dose, 0.11 ± 0.010 mg/kg, ranging from 0.08 to 0.16 mg/kg, and subsequent injections repeated or doubled the dose, until, maximally, a dose of 0.60 mg/kg was achieved). This attenuation was typically observed within 3 min of drug administration and was reversed after administration of a DA antagonist (raclopride, n = 2 of 2 neurons; mean initial dose, 0.22 ± 0.085 mg/kg, ranging from 0.13 to 0.30 mg/kg, with a maximal dose of 0.30 mg/kg, or haloperidol, n = 4 of 5 neurons; mean dose, 0.27 ± 0.054 mg/kg, ranging from 0.17 to 0.35 mg/kg, with a maximal dose of 0.67 mg/kg). These, and effects of DA agonists observed on other afferents, were dose-dependent in the ranges tested, such that lower doses produced little or no effects, and higher doses produced subsequently greater effects on the same neuron or when compared to another neuronal response to an afferent input. Furthermore, extended periods of stimulation alone had no effect on the evoked-response probability. In two cases, a short-latency MD-evoked response was observed in spontaneously spiking neurons (Fig.9). Systemic administration of apomorphine attenuated the short-latency MD input, but at the same time moderately increased the spontaneous firing rate of these neurons (Fig.9).
Fig. 8.
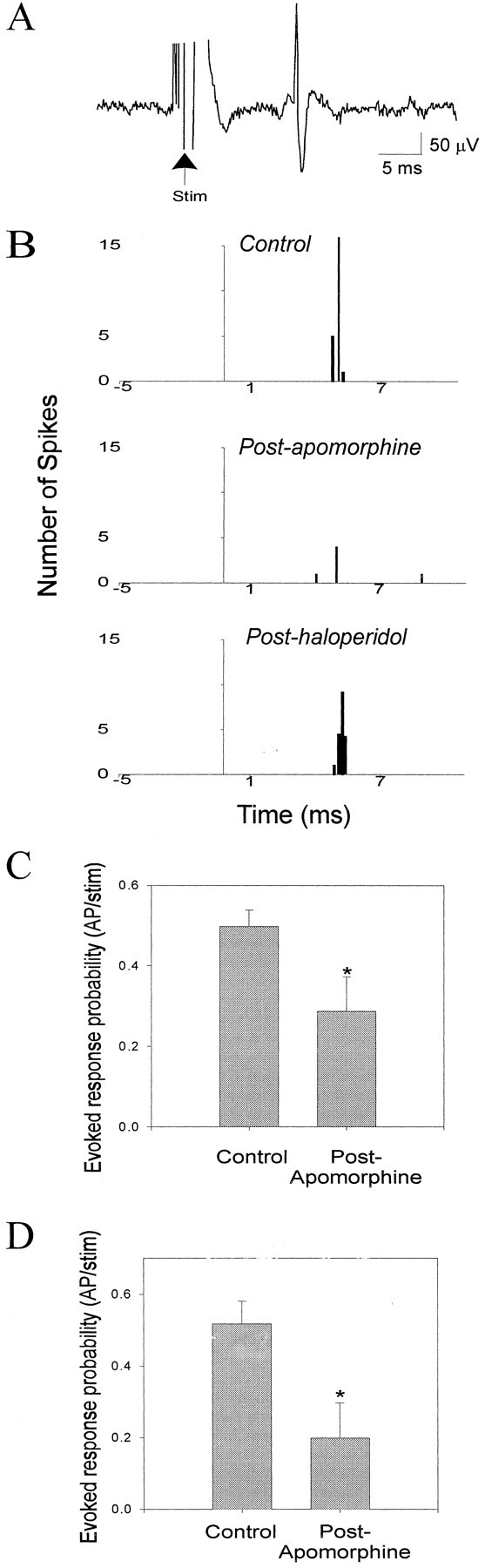
DA receptor activation attenuates mPFC- and MD-evoked short-latency responses in BLA neurons. A, An example trace of an MD-evoked short-latency response in a BLA neuron that did not fire spontaneously. Arrow indicates electrical stimulation artifact. B, Repre- sentative PSTHs of the attenuation of MD-evoked responses in a nonspontaneously firing BLA neuron by systemic administration of apomorphine (0.10 mg/kg) and its reversal by systemic administration of haloperidol (0.30 mg/kg). C, MD-evoked short-latency responses are attenuated after systemic administration of DA agonists (6 of 7 neurons; *p < 0.05; error bars indicate mean ± SEM). D, mPFC-evoked short-latency responses are attenuated after systemic administration of DA agonists (5 of 5 neurons; *p < 0.05; error bars indicate mean ± SEM, see Results for dose ranges).
Fig. 9.
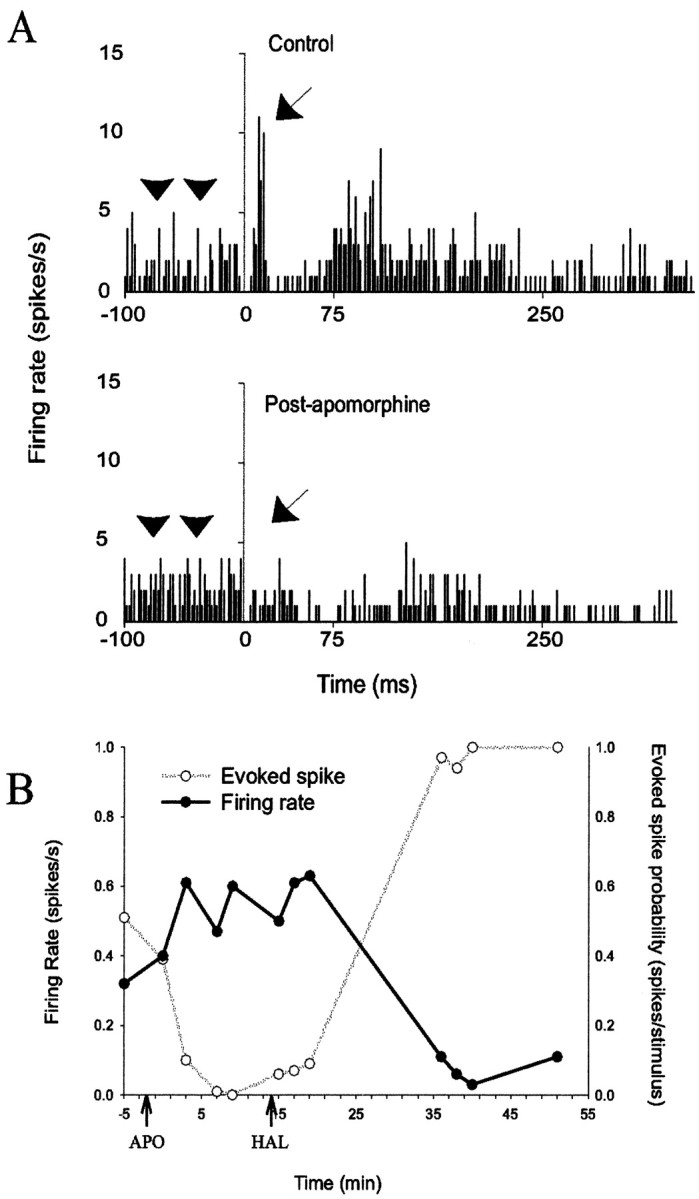
DA agonist administration has opposite effects on MD-evoked and spontaneous firing recorded from the same neuron.A, PSTH indicating the presence of a short-latency evoked response (arrow) in a spontaneously firing neuron (top). The short-latency evoked response is attenuated, yet the spontaneous firing rate (arrowheads, the prestimulus firing rate) is increased (p < 0.05; bottom) after systemic administration of the DA agonist apomorphine (0.10 mg/kg). B, Graphic representation of the opposite changes in firing rate and MD-evoked short-latency responses recorded in the same neuron displayed inA. Additionally, systemic administration of haloperidol (0.65 mg/kg) reversed the effects of apomorphine and caused a further suppression of neuronal firing rates and potentiation of evoked spike probability to levels that surpassed predrug baseline.
If the activity of these neurons is in fact driven by glutamatergic inputs, one interpretation of these results is a differential modulation of glutamatergic inputs by DA and specifically implies the potentiation of another glutamatergic input, leading to an increased basal firing rate. In light of this, two other inputs were examined; the input from mPFC (infralimbic and prelimbic) and the input from sensory association cortex (Te3). Both send substantial projections to the lateral nucleus of the BLA and represent two classes of inputs to the amygdala: sensory and limbic. Short-latency, presumed monosynaptic evoked responses from mPFC stimulation (mean latency, 17 msec; range, 15–20 msec) were attenuated after systemic apomorphine administration (Fig. 9; 5 of 5 neurons; five rats; pre-DA agonist response probability, 0.52 ± 0.06; post-DA agonist response probability, 0.20 ± 0.10; p = 0.012; group t test; 35 ± 13% of baseline; apomorphine mean initial dose, 0.46 ± 0.052 mg/kg, ranging from 0.19 to 0.61 mg/kg, with a maximal dose of 0.78 mg/kg). By contrast, short-latency evoked responses from Te3 (Fig.10; mean latency, 11.5 msec; range, 7–13 msec) were potentiated after systemic apomorphine (9 of 10 neurons; nine rats; pre-DA agonist response probability, 0.51 ± 0.07; post-DA agonist response probability, 0.79 ± 0.09;p = 0.001; group t test, 162 ± 12.7% of baseline; apomorphine mean initial dose, 0.28 ± 0.057 mg/kg, ranging from 0.13 to 0.37, with a maximal dose of 0.74 mg/kg). These distinct effects on mPFC and Te3 inputs to the BLA were induced by similar doses of drug and were reversed after systemic administration of haloperidol (mPFC, n = 3 of 4; mean initial dose, 0.55 ± 0.02 mg/kg, ranging from 0.39 to 0.61 mg/kg; Te3,n = 2 of 3; mean initial dose, 0.53 ± 0.035 mg/kg, ranging from 0.49 to 0.60 mg/kg).
Fig. 10.
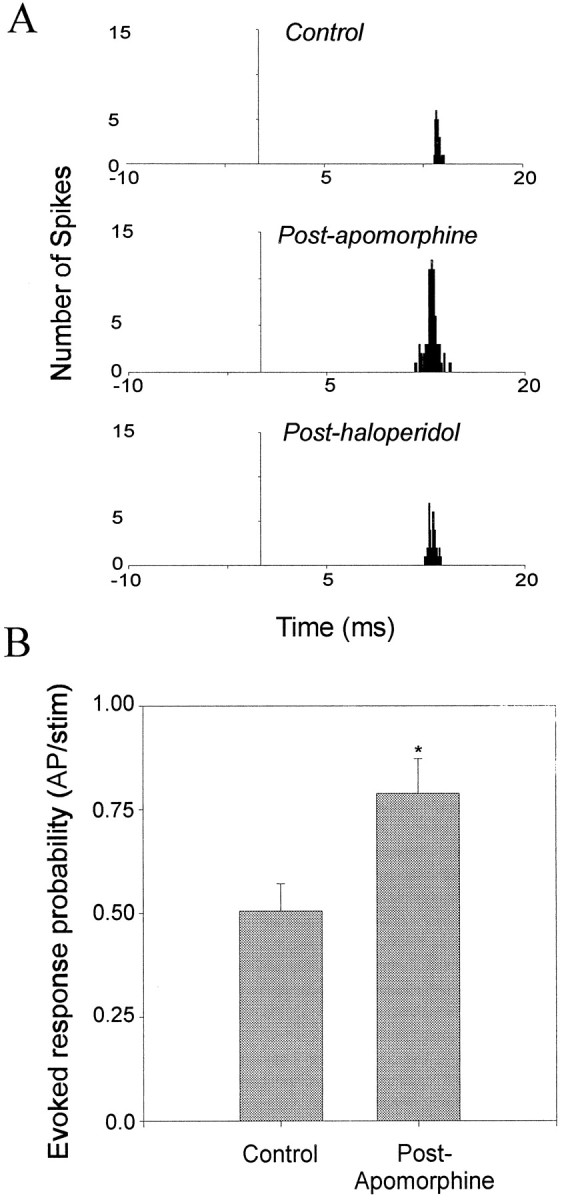
DA receptor activation potentiates Te3-evoked short-latency responses in BLA neurons. A,Representative PSTHs from a nonspontaneously firing BLA neuron that displays potentiation of the Te3-evoked short-latency response after systemic administration of apomorphine (0.37 mg/kg). This effect is reversed after systemic administration of haloperidol (0.49 mg/kg).B, Systemic administration of apomorphine potentiates Te3-evoked short-latency responses in BLA neurons (9 of 10 neurons; *p < 0.05; t test; error bars indicate mean ± SEM; see Results for dose ranges).
Similarly, MD- and mPFC-evoked field potentials (n = 3 of 4, each) were attenuated after systemic DA agonist administration (Fig. 11; MD: apomorphine mean dose, 0.73 ± 0.150 mg/kg; range, 0.58–0.88 mg/kg; mPFC: apomorphine mean dose, 0.72 ± 0.094 mg/kg; range, 0.51–1.0 mg/kg). The field potentials evoked by either MD or mPFC stimulation could be divided into four primary components; two positive and two negative components, labeled P1, P2, N1, and N2, respectively (Fig. 11). The amplitude of MD- and mPFC-evoked responses was dependent on the stimulus intensity. The average ratio of preapomorphine and postapomorphine administration for mPFC-evoked field potentials was: P1, 0.65 ± 0.14,p < 0.05 group t test; P2, 0.62 ± 0.14, p < 0.05 group t test; N1, 0.66 ± 0.20, p > 0.05 group t test; and N2, 0.536 ± 0.18, p < 0.05 group t test. The average ratios of preapomorphine and postapomorphine administration for MD-evoked field potentials were: P1, 0.88 ± 0.12; P2, 0.73 ± 0.15; N1, 0.86 ± 0.21; and N2, 1.04 ± 0.41 (none were significant with group t test, presumably because of the large variance; however, in three of four cases, the MD-evoked field potential displayed a prominent attenuation after apomorphine administration).
Fig. 11.
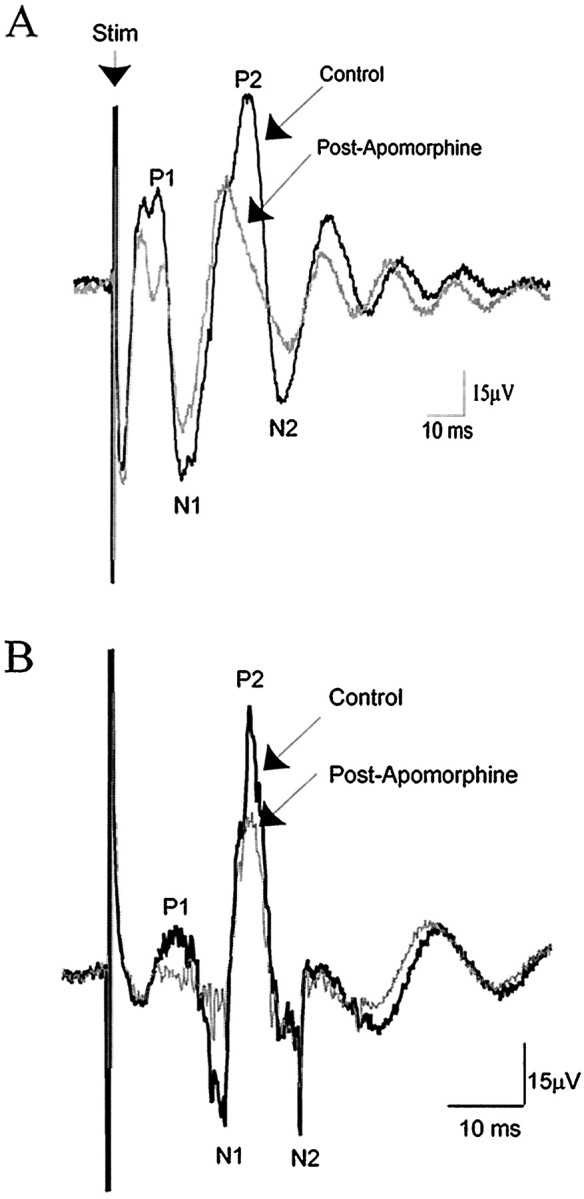
DA receptor activation attenuates MD- and mPFC-evoked population field potentials recorded in the BLA. Traces of mPFC-evoked (A) and MD-evoked population field potentials before (black) and after (gray) systemic administration of apomorphine (mPFC, 0.88 mg/kg; MD, 0.58 mg/kg). Each trace represents 30 averaged recorded field potentials. Arrow (Stim) marks stimulus artifact.
DISCUSSION
This study examined the effects of DA receptor activation on spontaneous activity and afferent drive of BLA neurons. The results indicate that DA receptor activation decreases the spontaneous firing rate of slowly firing neurons but increases the firing rate of fast-firing BLA neurons. Moreover, the effects of DA receptor activation on afferent drive depended on the afferent examined.
BLA neuron subtypes
BLA neurons were parcellated into two types based on their action potential duration, antidromic activation, and their firing rates, with neurons firing at <0.5 Hz comprising one population, and neurons firing at >0.5 Hz belonging to a separate population. This division is consistent with evidence presented here and in other studies showing distinct physiological properties of identified BLA neuronal subtypes (Washburn and Moises, 1992; Rainnie et al., 1993; Sugita et al., 1993) and indicates that slow-firing BLA neurons with long-duration spikes are typically projection neurons, whereas the fast-firing, short-spike duration neurons are almost exclusively inhibitory interneurons (Pare and Gaudreau, 1996; Lang and Pare, 1998). Thus, antidromically activated neurons displayed very low firing rates and long spike durations, and none of the fast firing neurons with short-spike durations could be antidromically activated. It is not clear whether these criteria would be valid in all other preparations, because the firing rates of neurons in the BLA may differ depending on the anesthetic and stage of the sleep–wake cycle (Ben-Ari et al., 1974).
Effects of DA on spontaneous firing of BLA neuron subtypes
Slow-firing, presumed projection neurons exhibited a decrease in firing rate after systemic administration of DA agonists, which could be reversed after systemic administration of DA antagonists. Furthermore, systemic administration of DA antagonists alone increased the firing of slow-firing and nonfiring neurons, some of which were identified as projection neurons using antidromic activation. Taken together, these results imply that the spontaneous firing of projection neurons is attenuated by DA receptor activation and that there may be a tonic DAergic inhibition on BLA projection neurons.
When using systemic drug administration, it is unclear whether the effects of DA receptor activation are localized to the BLA. Thus, we undertook co-iontophoresis of DA and glutamate in the BLA. When performing glutamate iontophoresis, the probability of recording from projection neurons is increased. These neurons comprise ∼80% of BLA neurons (McDonald, 1985, 1992) and tend to have a larger soma size, yielding more distributed field potentials (Humphrey and Schmidt, 1990), and are thus much easier to record with the larger five-barrel microiontophoresis pipettes than are the smaller interneurons (for a detailed discussion, see Stone, 1985). Consistent with this observation, and similar to earlier studies (Ben-Ari and Kelly, 1976;Spehlmann and Norcross, 1984), DA attenuated the firing of BLA neurons induced to fire with co-iontophoresis of glutamate. All of the presumed projection neurons exposed to glutamate iontophoresis in this study were not spiking spontaneously when the glutamate current was retained, and additionally, several were antidromically activated from the NAc. Thus, DA receptor activation within the BLA is sufficient to attenuate BLA projection neuron firing and is likely to occur via a direct postsynaptic effect of DA on projection neurons.
Fast-firing, presumed interneurons display an increase in firing rate after systemic administration of DA agonists that is reversed by DA antagonist administration. Similarly, systemic administration of DA antagonists alone decreases the firing rate of the presumed interneurons. This may provide a mechanism by which DA has indirect inhibitory actions on projection neurons of the BLA. These interneurons display extensive axonal arborization, with many terminals onto the somata of BLA pyramidal neurons (McDonald, 1985; Carlsen, 1988; Smith et al., 1998), and can thus exert powerful modulation over pyramidal neuron firing (Lang and Pare, 1997). Earlier studies examining spontaneous firing rates and using the indirect DA agonist amphetamine, and DA antagonists, yielded heterogeneous results (Wepsic and Austin, 1971; Bashore et al., 1978; Wang and Rebec, 1996). A parsimonious reconciliation with our data are that these earlier studies did not parcellate responses based on neuronal subtype. Additionally, some of these studies were performed in awake rats, which may represent a substantially different situation.
To further test whether this response was a DA-mediated effect, the SN/VTA was electrically stimulated at parameters similar to those shown to evoke DA release in the BLA (Garris and Wightman, 1994). SN/VTA stimulation increased the firing rate of fast-spiking neurons while decreasing the firing rate of slowly firing neurons. Additionally, SN/VTA stimulation decreased the iontophoretic glutamate-evoked excitation of nonfiring neurons in the BLA. These effects were attenuated after systemic administration of the DA antagonist haloperidol. The stimulus parameters used were not substantially different from the spontaneous discharge characteristics exhibited by DA neurons when they are in a burst firing mode (Grace and Bunney, 1983; Freeman et al., 1985), demonstrating the potential physiological relevance of modulation of BLA neuronal activity by endogenous DA.
This study indicates that the direction of the effects of DA on neuronal firing rates is neuron subtype-specific, and not firing rate-dependent. Thus, nonfiring neurons, as well as some antidromically activated neurons, could be induced to fire by iontophoretic application of glutamate to rates that in some cases exceeded 10 Hz; nevertheless, DA receptor activation still decreased the firing rate of these neurons.
One puzzling result is that both the D1 and D2 DA receptor agonists produced similar effects on neuronal firing. Further studies are ongoing to determine whether this is the case under other conditions. However, it is conceivable that activation of the two classes of DA receptors may have similar effects but via different substrates (Hoffman and Johnston, 1998, in hippocampus), receptor localization on neuronal subtypes (Gaspar et al., 1995, DA receptors in cortex; Mahanty and Sah, 1998, glutamate receptors in BLA), or glutamate–receptor interaction (as seen in striatum, Cepeda et al., 1993).
One broad interpretation of results pertaining to DA receptor activation and firing rate is that DA nonspecifically decreases the firing of projection neurons, and by doing so decreases the general excitatory output of the BLA. By modulating the diffuse BLA projection systems, DA may exert broad effects on motoric (NAc) and autonomic (central nucleus of the amygdala and hypothalamus) function, as well as alterations of attention (MD/PFC) and arousal (indirect projections to nucleus basalis, Kretteck and Price, 1977, 1978; McDonald, 1987, 1991;Savander et al., 1997; Pitkanen and Amaral 1998). Additionally, this combined DA and GABA inhibition may function as a noise filter, wherein only the strongest inputs into the BLA can drive the amygdalar contribution to behavior.
DA modulation of afferent inputs to the BLA
DA receptor activation appears to exert opposite effects on neuronal responses evoked from Te3 and mPFC (and MD), areas that are representative of sensory and limbic inputs, respectively. Specifically, the sensory input was potentiated by DA, whereas the limbic inputs were attenuated. However, the mechanism of these actions is unclear. For example, these opposite effects may be attributable to presynaptic downmodulation (Maura et al., 1988; Pennartz et al., 1992;O'Donnell and Grace, 1994; Delle Donne et al., 1996; Flores-Hernandez et al., 1997; Nicola and Malenka, 1997) or postsynaptic potentiation of glutamate afferent systems, as described in other preparations. Thus, in the striatum, DA receptor activation is proposed to have opposing modulatory effects on NMDA and AMPA/kainate receptors (Cepeda et al., 1993), and in the BLA there is evidence for different ratios of NMDA and AMPA receptors at certain synapses (Mahanty and Sah, 1999) (but seeWeisskopf and LeDoux, 1999).
Tentative criteria were established to operationally define monosynaptic, polysynaptic, and antidromic responses. However, the possibility of excitation via a collateral of antidromically activated neighboring neurons could not be excluded because of a continuum of antidromic and orthodromic response latencies. Although stimulation of fibers of passage could also not be excluded, it is unlikely to have been the primary source of excitation from cortical areas that send dense projections to the BLA.
Integrated function of DA in the BLA
DA in the BLA appears to exert a dual filtering role: (1) a nonspecific filtering accomplished via GABAergic interneurons that is hypothesized to attenuate the weaker inputs, and (2) a selectivity filter accomplished by modulation of sensory and limbic inputs in opposite directions. Thus, DA may provide a mechanism by which sensory inputs become more capable of driving projection neuron firing than the limbic inputs examined. It is interesting to note that sensory inputs (albeit only sensory thalamus inputs have been examined in depth to date) are potentiated during associative conditioning (McKernan and Shinnick-Gallagher, 1997; Rogan et al., 1997). The potentiated inputs may be representative of a particular sensory stimulus that has acquired affective valence and has become more adept at driving the affective responses of the organism (Campeau and Davis, 1995; Rogan and LeDoux, 1995). This behavioral tendency is potentiated by DA in the BLA (Gasbarri et al., 1993; Lamont and Kokkinidis, 1998; but see also Nader and LeDoux, 1999), perhaps by a combined augmentation of weaker inputs and a potentiation of already potentiated sensory inputs. The mPFC input has been hypothesized to suppress amygdala-mediated responses (Al Maskati and Zbrozyna, 1989; Wan and Swerdlow, 1997), perhaps via feedforward inhibition (J. A. Rosenkranz and A. A. Grace, unpublished observations; similarly, with regards to perirhinal/entorhinal cortical inputs, Lang and Pare, 1998). However, during situations in which a sensory-driven affective response is appropriate, such as behavioral responding during presentation of a stressful sensory stimulus, DA is released in the amygdala (Coco et al., 1992; Inglis and Moghaddam, 1999). In this situation, we hypothesize that the mPFC afferent inhibitory influence over the BLA output is reduced by DA receptor activation, allowing sensory-driven affective responses to predominate.
Our results indicate that DA plays a powerful modulatory role in the BLA, a site involved in affective learning and affective responding. Thus, DA receptor activation, via cooperative modulation of interneurons, projection neurons, and excitatory inputs, filters particular afferent inputs based on their strength and their source. This study provides the first description of differential effects of DA receptor activation on the projection neurons and presumptive interneurons, as well as on sensory and limbic afferents into the BLA. This provides a neurophysiological substrate for behavioral changes noted under conditions of artificially manipulated DA transmission in the BLA, and more importantly, under conditions in which DA is released in the BLA, such as during the presentation of an aversive sensory stimulus and during some forms of learning.
Footnotes
This work was supported by National Institutes of Health Grants MH57440, MH45156 (A.A.G.), and NS 07433 (J.A.R.). We acknowledge the excellent technical assistance of Nicole MacMurdo and Brian Lowry.
Correspondence should be addressed to J. Amiel Rosenkranz, Department of Neuroscience, 446 Crawford Hall, University of Pittsburgh, Pittsburgh, PA 15260. E-mail: rosenk@bns.pitt.edu.
REFERENCES
- 1.Aggleton JP. The functional effects of amygdala lesions in humans: a comparison with findings from monkeys. In: Aggleton JP, editor. The amygdala: neurobiological aspects of emotions, memory, and mental dysfunction. Wiley-Liss; New York: 1992. pp. 485–503. [Google Scholar]
- 2.Al Maskati HF, Zbrozyna AW. Stimulation in prefrontal cortex inhibits cardiovascular and motor components of the defense reaction in rats. J Auton Nerv Syst. 1989;28:117–126. doi: 10.1016/0165-1838(89)90084-2. [DOI] [PubMed] [Google Scholar]
- 3.Arnold SA, Hyman BT, Van Hoesen GW, Damasio AR. Some cytoarchitectural abnormalities of the entorhinal cortex in schizophrenia. Arch Gen Psychiatry. 1991;48:625–632. doi: 10.1001/archpsyc.1991.01810310043008. [DOI] [PubMed] [Google Scholar]
- 4.Asan E. Ultrastructural features of tyrosine-hydroxylase-immunoreactive afferents and their targets in the rat amygdala. Cell Tissue Res. 1997;288:449–469. doi: 10.1007/s004410050832. [DOI] [PubMed] [Google Scholar]
- 5.Bashore TR, Rebec GV, Groves PM. Alterations of spontaneous neuronal activity in the caudate-putamen, nucleus accumbens and amygdaloid complex of rats produced by d-amphetamine. Pharmacol Biochem Behav. 1978;8:467–474. doi: 10.1016/0091-3057(78)90086-2. [DOI] [PubMed] [Google Scholar]
- 6.Ben-Ari Y, Kelly JS. Dopamine evoked inhibition of single cells of the feline putamen and basolateral amygdala. J Physiol (Lond) 1976;256:1–21. doi: 10.1113/jphysiol.1976.sp011308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ben-Ari Y, Le Gal La Salle G, Champagnat J-C. Lateral amygdala unit activity: I. Relationship between spontaneous and evoked activity. Electroencephalogr Clin Neurophysiol. 1974;37:449–461. doi: 10.1016/0013-4694(74)90086-8. [DOI] [PubMed] [Google Scholar]
- 8.Berman KF, Zec RF, Weinberger DR. Physiologic dysfunction of dorsolateral prefrontal cortex in schizophrenia. II. Role of neuroleptic treatment, attention, and mental effort. Arch Gen Psychiatry. 1986;43:126–135. doi: 10.1001/archpsyc.1986.01800020032005. [DOI] [PubMed] [Google Scholar]
- 9.Bogerts B, Lieberman JA, Ashtari M, Bilder RM, Degreef G, Lerner G, Johns C, Masiar S. Hippocampus-amygdala volumes and psychopathology in chronic schizophrenia. Biol Psychiatry. 1993;33:236–246. doi: 10.1016/0006-3223(93)90289-p. [DOI] [PubMed] [Google Scholar]
- 10.Brown AS, Gershon S. Dopamine and depression. J Neural Transm. 1993;91:75–109. doi: 10.1007/BF01245227. [DOI] [PubMed] [Google Scholar]
- 11.Cador M, Robbins TW, Everitt BJ. Involvement of the amygdala in stimulus-reward association: interaction with the ventral striatum. Neuroscience. 1989;30:77–86. doi: 10.1016/0306-4522(89)90354-0. [DOI] [PubMed] [Google Scholar]
- 12.Campeau S, Davis M. Involvement of subcortical and cortical afferents to the lateral nucleus of the amygdala in fear conditioning measured with fear-potentiated startle in rats trained concurrently with auditory and visual conditioned stimuli. J Neurosci. 1995;15:2312–2327. doi: 10.1523/JNEUROSCI.15-03-02312.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carlsen J. Immunocytochemical localization of glutamate decarboxylase in the rat basolateral amygdaloid nucleus, with special reference to GABAergic innervation of amygdalostriatal projection neuron. J Comp Neurol. 1988;273:513–526. doi: 10.1002/cne.902730407. [DOI] [PubMed] [Google Scholar]
- 14.Cepeda C, Buchwald NA, Levine MS. Neuromodulatory actions of DA in the neostriatum are dependent upon the excitatory amino acid receptor subtypes activated. Proc Natl Acad Sci USA. 1993;90:9576–9580. doi: 10.1073/pnas.90.20.9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coco ML, Kuhn CM, Ely TD, Kilts CD. Selective activation of mesoamygdaloid dopamine neurons by conditioned stress: attenuation by diazepam. Brain Res. 1992;590:39–47. doi: 10.1016/0006-8993(92)91079-t. [DOI] [PubMed] [Google Scholar]
- 16.Davis M, Rainnie D, Cassell M. Neurotransmission in the amygdala related to fear and anxiety. Trends Neurosci. 1994;17:208–214. doi: 10.1016/0166-2236(94)90106-6. [DOI] [PubMed] [Google Scholar]
- 17.Delle Donne KT, Sesack SR, Pickel VM. Ultrastructural immunocytochemical localization of neurotensin and the dopamine D2 receptor in the rat nucleus accumbens. J Comp Neurol. 1996;371:552–566. doi: 10.1002/(SICI)1096-9861(19960805)371:4<552::AID-CNE5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 18.Deutch AY. The regulation of subcortical dopamine systems by the prefrontal cortex: interactions of central dopamine systems and the pathogenesis of schizophrenia. J Neural Transm [Suppl] 1992;36:61–89. doi: 10.1007/978-3-7091-9211-5_5. [DOI] [PubMed] [Google Scholar]
- 19.Drevets WC, Videen TO, Price JL, Preskorn SH, Carmichael ST, Raichle ME. A functional anatomical study of unipolar depression. J Neurosci. 1992;12:3628–3641. doi: 10.1523/JNEUROSCI.12-09-03628.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drevets WC, Price JL, Simpson JR, Jr, Todd RD, Reich T, Vannier M, Raichle ME. Subgenual prefrontal cortex abnormalities in mood disorders. Nature. 1997;386:824–827. doi: 10.1038/386824a0. [DOI] [PubMed] [Google Scholar]
- 21.Falls WA, Davis M. Lesions of the central nucleus of the amygdala block conditioned excitation, but not conditioned inhibition of fear as measured with the fear-potentiated startle effect. Behav Neurosci. 1995;109:379–387. doi: 10.1037//0735-7044.109.3.379. [DOI] [PubMed] [Google Scholar]
- 22.Flores-Hernandez J, Galarraga E, Bargas J. Dopamine selects glutamatergic inputs to neostriatal neurons. Synapse. 1997;25:185–195. doi: 10.1002/(SICI)1098-2396(199702)25:2<185::AID-SYN9>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 23.Freeman AS, Meltzer LT, Bunney BS. Firing properties of substantia nigra dopaminergic neurons in freely moving rats. Life Sci. 1985;36:1983–1994. doi: 10.1016/0024-3205(85)90448-5. [DOI] [PubMed] [Google Scholar]
- 24.Gaffan D, Murray EA. Amygdalar interaction with the mediodorsal nucleus of the thalamus and the ventromedial prefrontal cortex in stimulus-reward learning in the monkey. J Neurosci. 1990;10:3479–3493. doi: 10.1523/JNEUROSCI.10-11-03479.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaffan EA, Gaffan D, Harrison S. Disconnection of the amygdala from visual association cortex impairs visual reward-association learning in monkeys. J Neurosci. 1988;8:3144–3150. doi: 10.1523/JNEUROSCI.08-09-03144.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gaffan D, Murray EA, Fabre-Thorpe M. Interaction of the amygdala with the frontal lobe in reward memory. Eur J Neurosci. 1993;5:968–975. doi: 10.1111/j.1460-9568.1993.tb00948.x. [DOI] [PubMed] [Google Scholar]
- 27.Garris PA, Wightman RM. In vivo voltammetric measurement of evoked extracellular dopamine in the rat basolateral amygdaloid nucleus. J Neurophysiol. 1994;478:239–249. doi: 10.1113/jphysiol.1994.sp020246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gasbarri A, Introini-Cillison IB, Packard MG, Pacitti C, McGaugh JL. Interaction of cholinergic-dopaminergic systems in the regulation of memory storage in aversively motivated learning tasks. Brain Res. 1993;627:72–78. doi: 10.1016/0006-8993(93)90750-h. [DOI] [PubMed] [Google Scholar]
- 29.Gaspar P, Bloch B, Le Moine C. D1 and D2 receptor gene expression in the rat frontal cortex: cellular localization in different classes of efferent neurons. Eur J Neurosci. 1995;7:1050–1063. doi: 10.1111/j.1460-9568.1995.tb01092.x. [DOI] [PubMed] [Google Scholar]
- 30.Grace AA. Phasic versus tonic dopamine release and the modulation of dopamine system responsitivity: a hypothesis for the etiology of schizophrenia. Neuroscience. 1991;41:1–23. doi: 10.1016/0306-4522(91)90196-u. [DOI] [PubMed] [Google Scholar]
- 31.Grace AA. The depolarization block hypothesis of neuroleptic action: Implications for the etiology and treatment of schizophrenia. J Neural Transm. 1992;36:91–131. doi: 10.1007/978-3-7091-9211-5_6. [DOI] [PubMed] [Google Scholar]
- 32.Grace AA, Bunney BS. Intracellular and extracellular electrophysiology of nigral dopaminergic neurons. 1. Identification and characterization. Neuroscience. 1983;10:301–315. doi: 10.1016/0306-4522(83)90135-5. [DOI] [PubMed] [Google Scholar]
- 33.Harmer CJ, Phillips GD. Enhanced dopamine efflux in the amygdala by a predictive, but not a non-predictive, stimulus: facilitation by prior repeated d-amphetamine. Neuroscience. 1999;90:119–130. doi: 10.1016/s0306-4522(98)00464-3. [DOI] [PubMed] [Google Scholar]
- 34.Herman JP, Guillonneau D, Dantzer R, Scatton B, Semerdjian-Rouquier L, Le Moal M. Differential effects of inescapable footshocks and of stimuli previously paired with inescapable footshocks on dopamine turnover in cortical and limbic areas of the rat brain. Life Sci. 1982;30:2207–2214. doi: 10.1016/0024-3205(82)90295-8. [DOI] [PubMed] [Google Scholar]
- 35.Hitchcock JM, Davis M. Efferent pathway of the amygdala involved in conditioned fear as measured with the fear-potentiated startle paradigm. Behav Neurosci. 1991;105:826–842. doi: 10.1037//0735-7044.105.6.826. [DOI] [PubMed] [Google Scholar]
- 36.Hitchcott PK, Bonardi CMT, Phillips GD. Enhanced stimulus-reward learning by intra-amygdala administration of a D3 dopamine receptor agonist. Psychopharmacology. 1997;133:240–248. doi: 10.1007/s002130050397. [DOI] [PubMed] [Google Scholar]
- 37.Hoffman DA, Johnston D. Downregulation of transient K+ channels in dendrites of hippocampal CA1 pyramidal neurons by activation of PKA and PKC. J Neurosci. 1998;18:3521–3528. doi: 10.1523/JNEUROSCI.18-10-03521.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hori K, Tanaka J, Nomura M. Effects of discrimination learning on the rat dopamine release: a microdialysis study. Brain Res. 1993;621:296–300. doi: 10.1016/0006-8993(93)90119-8. [DOI] [PubMed] [Google Scholar]
- 39.Humphrey DR. Extracellular single unit recording methods. In: Humphrey DR, editor. Electrophysiological techniques. Society for Neuroscience; Bethesda, MD: 1979. pp. 199–259. [Google Scholar]
- 40.Humphrey DR, Schmidt EM. Extracellular single-unit recording methods. In: Boulton AA, Baker GB, Vanderwolf CH, editors. Neurophysiological techniques, neuromethods, Vol 15. Humana; Totowa, NJ: 1990. [Google Scholar]
- 41.Inglis FM, Moghaddam B. Dopaminergic innervation of the amygdala is highly responsive to stress. J Neurochem. 1999;72:1088–1094. doi: 10.1046/j.1471-4159.1999.0721088.x. [DOI] [PubMed] [Google Scholar]
- 42.Killcross S, Robbins TW, Everitt BJ. Different types of fear-conditioned behaviour mediated by separate nuclei within amygdala. Nature. 1997;388:377–380. doi: 10.1038/41097. [DOI] [PubMed] [Google Scholar]
- 43.Kretteck JE, Price JL. Projections from the amygdaloid complex to the cerebral cortex and thalamus in the rat and cat. J Comp Neurol. 1977;172:687–722. doi: 10.1002/cne.901720408. [DOI] [PubMed] [Google Scholar]
- 44.Kretteck JE, Price JL. Amygdaloid projections to subcortical structures within the basal forebrain and brainstem in the rat and cat. J Comp Neurol. 1978;178:225–254. doi: 10.1002/cne.901780204. [DOI] [PubMed] [Google Scholar]
- 45.Lamont EW, Kokkinidis L. Infusion of the dopamine D1 receptor antagonist SCH 23390 into the amygdala blocks fear expression in a potentiated startle paradigm. Brain Res. 1998;795:128–136. doi: 10.1016/s0006-8993(98)00281-9. [DOI] [PubMed] [Google Scholar]
- 46.Lang EJ, Pare D. Similar inhibitory processes dominate the response of cat lateral amygdaloid projection neurons to their various afferents. J Neurophysiol. 1997;77:341–352. doi: 10.1152/jn.1997.77.1.341. [DOI] [PubMed] [Google Scholar]
- 47.Lang EJ, Pare D. Synaptic responsiveness of interneurons of the cat lateral amygdaloid nucleus. Neuroscience. 1998;83:877–889. doi: 10.1016/s0306-4522(97)00420-x. [DOI] [PubMed] [Google Scholar]
- 48.Le Moal M, Simon H. Mesocorticolimbic dopaminergic network: functional and regulatory roles. Physiol Rev. 1991;71:155–234. doi: 10.1152/physrev.1991.71.1.155. [DOI] [PubMed] [Google Scholar]
- 49.LeDoux JE, Iwata J, Cicchetti P, Reis DJ. Different projections of the central amygdaloid nucleus mediate autonomic and behavioral correlates of conditioned fear. J Neurosci. 1988;8:2517–2529. doi: 10.1523/JNEUROSCI.08-07-02517.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loughlin SE, Fallon JH. Dopaminergic and non-dopaminergic projections to amygdala from substantia nigra and ventral tegmental area. Brain Res. 1983;262:334–338. doi: 10.1016/0006-8993(83)91029-6. [DOI] [PubMed] [Google Scholar]
- 51.Mahanty NK, Sah P. Calcium-permeable AMPA receptors mediate long-term potentiation in interneurons in the amygdala. Nature. 1998;394:683–687. doi: 10.1038/29312. [DOI] [PubMed] [Google Scholar]
- 52.Mahanty NK, Sah P. Excitatory synaptic inputs to pyramidal neurons of the lateral amygdala. Eur J Neurosci. 1999;11:1217–1222. doi: 10.1046/j.1460-9568.1999.00528.x. [DOI] [PubMed] [Google Scholar]
- 53.Maren S, Aharonov G, Stote DL, Faneslow MS. N-methyl-D-aspartate receptors in the basolateral amygdala are required for both acquisition and expression of conditioned fear in rats. Behav Neurosci. 1996;110:1365–1374. doi: 10.1037//0735-7044.110.6.1365. [DOI] [PubMed] [Google Scholar]
- 54.Maura G, Giardi A, Raiter M. Release-regulating D-2 dopamine receptors are located on striatal glutamatergic nerve terminals. J Pharmacol Exp Ther. 1988;247:680–684. [PubMed] [Google Scholar]
- 55.McDonald AJ. Immunohistochemical identification of γ-aminobutyric acid-containing neurons in the rat basolateral amygdala. Neurosci Lett. 1985;53:203–207. doi: 10.1016/0304-3940(85)90186-7. [DOI] [PubMed] [Google Scholar]
- 56.McDonald AJ. Organization of amygdaloid projections to the mediodorsal thalamus and prefrontal cortex: a fluorescence retrograde transport study in the rat. J Comp Neurol. 1987;262:46–58. doi: 10.1002/cne.902620105. [DOI] [PubMed] [Google Scholar]
- 57.McDonald AJ. Organization of amygdaloid projections to the prefrontal cortex and associated striatum in the rat. Neuroscience. 1991;44:1–14. doi: 10.1016/0306-4522(91)90247-l. [DOI] [PubMed] [Google Scholar]
- 58.McDonald AJ. Projection neurons of the basolateral amygdala: a correlative Golgi and retrograde tract tracing study. Brain Res Bull. 1992;28:179–185. doi: 10.1016/0361-9230(92)90177-y. [DOI] [PubMed] [Google Scholar]
- 59.McDonald AJ, Mascagni F. Cortico-cortical and cortico-amygdaloid projections of the rat occipital cortex: a phaseolus vulgaris study. Neuroscience. 1996;71:37–54. doi: 10.1016/0306-4522(95)00416-5. [DOI] [PubMed] [Google Scholar]
- 60.McDonald AJ, Mascagni F, Guo L. Projections of the medial and lateral prefrontal cortices to the amygdala: a phaseolus vulgaris leucoagglutinin study in the rat. Neuroscience. 1996;71:55–75. doi: 10.1016/0306-4522(95)00417-3. [DOI] [PubMed] [Google Scholar]
- 61.McKernan MG, Shinnick-Gallagher P. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature. 1997;390:607–611. doi: 10.1038/37605. [DOI] [PubMed] [Google Scholar]
- 62.Mogenson GJ, Jones DL, Yim CY. From motivation to action: functional interface between the limbic system and the motor system. Prog Neurobiol. 1980;14:69–97. doi: 10.1016/0301-0082(80)90018-0. [DOI] [PubMed] [Google Scholar]
- 63.Munro LJ, Kokkinidis L. Infusion of quinporole and muscimol into the ventral tegmental area inhibits fear-potentiated startle: implications for the role of dopamine in fear expression. Brain Res. 1997;746:231–238. doi: 10.1016/s0006-8993(96)01225-5. [DOI] [PubMed] [Google Scholar]
- 64.Nader K, LeDoux J. The dopaminergic modulation of fear: quinporole impairs the recall of emotional memories in rats. Behav Neurosci. 1999;113:152–165. doi: 10.1037//0735-7044.113.1.152. [DOI] [PubMed] [Google Scholar]
- 65.Nicola SM, Malenka RC. Dopamine depresses excitatory and inhibitory synaptic transmission by distinct mechanisms in the nucleus accumbens. J Neurosci. 1997;17:5697–5710. doi: 10.1523/JNEUROSCI.17-15-05697.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.O'Donnell P, Grace AA. Tonic D2-mediated attenuation of cortical excitation in nucleus accumbens neurons recorded in vitro. Brain Res. 1994;634:105–112. doi: 10.1016/0006-8993(94)90263-1. [DOI] [PubMed] [Google Scholar]
- 67.Otterson OP. Connections of the amygdala of the rat. IV: Corticoamygdaloid and intraamygdaloid connections as studies with axonal transport of horseradish peroxidase. J Comp Neurol. 1989;205:30–48. doi: 10.1002/cne.902050104. [DOI] [PubMed] [Google Scholar]
- 68.Pakkenberg B. The volume of the mediodorsal thalamic nucleus in treated and untreated schizophrenics. Schizophrenia Res. 1992;7:95–100. doi: 10.1016/0920-9964(92)90038-7. [DOI] [PubMed] [Google Scholar]
- 69.Pare D, Gaudreau H. Projection neurons and interneurons of the lateral and basolateral amygdala: distinct firing patterns and differential relation to theta and delta rhythms in conscious cats. J Neurosci. 1996;16:3334–3350. doi: 10.1523/JNEUROSCI.16-10-03334.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Paxinos G, Watson C. The rat brain in stereotaxic coordinates, Ed 3. Academic; San Diego: 1997. [DOI] [PubMed] [Google Scholar]
- 71.Pennartz CMA, Dolleman-Van der Weel MJ, Kitai ST, Lopes da Silva FH. Presynaptic dopamine D1 receptors attenuate excitatory and inhibitory limbic inputs to the shell region of the rat nucleus accumbens studies in vitro. J Neurophysiol. 1992;67:1325–1334. doi: 10.1152/jn.1992.67.5.1325. [DOI] [PubMed] [Google Scholar]
- 72.Pitchot W, Ansseau M, Gonzalez Moreno A, Hansenne M, von Frenckell R. Dopaminergic function in panic disorder: comparison with major and minor depression. Biol Psychiatry. 1992;32:1004–1011. doi: 10.1016/0006-3223(92)90061-4. [DOI] [PubMed] [Google Scholar]
- 73.Pitkanen A, Amaral DG. Organization of the intrinsic connections of the monkey amygdaloid complex: projections originating in the lateral nucleus. J Comp Neurol. 1998;398:431–458. doi: 10.1002/(sici)1096-9861(19980831)398:3<431::aid-cne9>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 74.Pitkanen A, Savander V, LeDoux JE. Organization of intra-amygdaloid circuitries in the rat: an emerging framework for understanding functions of the amygdala. Trends Neurosci. 1997;20:517–523. doi: 10.1016/s0166-2236(97)01125-9. [DOI] [PubMed] [Google Scholar]
- 75.Poremba A, Gabriel M. Amygdalar lesions block discriminative avoidance learning and cingulothalamic training-induced neuronal plasticity in rabbits. J Neurosci. 1997;17:5237–5244. doi: 10.1523/JNEUROSCI.17-13-05237.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Price JL, Russchen FT, Amaral DG. The limbic region. II: The amygdaloid complex. In: Bjorklund A, Swanson LW, editors. Handbook of chemical neuroanatomy, Vol 5, Integrated systems of the CNS, Part I. Elsevier Science; Amsterdam: 1987. [Google Scholar]
- 77.Raine A, Lencz T, Reynolds GP, Harrison G, Sheard C, Medley I, Reynolds LM, Cooper JE. An evaluation of structural and functional prefrontal deficits in schizophrenia: MRI and neuropsychological measures. Psychiatry Res. 1992;45:123–137. doi: 10.1016/0925-4927(92)90006-p. [DOI] [PubMed] [Google Scholar]
- 78.Rainnie DG, Asprodini EK, Shinnick-Gallagher P. Intracellular recordings from morphologically identified neurons of the basolateral amygdala. J Neurophysiol. 1993;69:1350–1362. doi: 10.1152/jn.1993.69.4.1350. [DOI] [PubMed] [Google Scholar]
- 79.Revay R, Vaughan R, Grant S, Kuhar MJ. Dopamine transporter immunohistochemistry in median eminence, amygdala, and other areas of the rat brain. Synapse. 1996;22:93–99. doi: 10.1002/(SICI)1098-2396(199602)22:2<93::AID-SYN1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 80.Reynolds GP. Increased concentrations and lateral asymmetry of amygdala dopamine in schizophrenia. Nature. 1983;305:527–529. doi: 10.1038/305527a0. [DOI] [PubMed] [Google Scholar]
- 81.Rogan MT, LeDoux JE. LTP is accompanied by commensurate enhancement of auditory-evoked responses in a fear conditioning circuit. Neuron. 1995;15:127–136. doi: 10.1016/0896-6273(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 82.Rogan MT, Staubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390:604–607. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- 83.Rolls ET. The amygdala: neurobiological aspects of emotions, memory, and mental dysfunction. Wiley-Liss; New York: 1992. Neurophysiology and functions of the primate amygdala. pp. 143–165. [Google Scholar]
- 84.Roozendaal B, Koolhaus JM, Bohus B. Attenuated cardiovascular, neuroendocrine, and behavioral responses after a single footshock in central amygdala lesioned male rats. Physiol Behav. 1991;50:771–775. doi: 10.1016/0031-9384(91)90016-h. [DOI] [PubMed] [Google Scholar]
- 85.Rosenkranz JA, Grace AA. Afferent inputs into the basolateral amygdala from the mediodorsal thalamus are modulated by dopamine. Soc Neurosci Abstr. 1998;24:690. [Google Scholar]
- 86.Sajdyk TJ, Shekhar A. Excitatory amino acid receptor antagonists block the cardiovascular and anxiety responses elicited by gamma-aminobutyric acid A receptor blockade in the basolateral amygdala of rats. J Pharmacol Exp Ther. 1997;283:969–977. [PubMed] [Google Scholar]
- 87.Savander V, LeDoux JE, Pitkanen A. Interamygdaloid projections of the basal and accessory basal nuclei of the rat amygdaloid complex. Neuroscience. 1997;76:725–735. doi: 10.1016/s0306-4522(96)00371-5. [DOI] [PubMed] [Google Scholar]
- 88.Scibilia RJ, Lachowicz JE, Kilts CD. Topographic nonoverlapping distribution of D1 and D2 dopamine receptors in the amygdaloid nuclear complex of the rat brain. Synapse. 1992;11:146–154. doi: 10.1002/syn.890110208. [DOI] [PubMed] [Google Scholar]
- 89.Seeman P. Atypical neuroleptics: role of multiple receptors, endogenous dopamine, and receptor linkage. Acta Psychiatry Scand. 1990;82:14. doi: 10.1111/j.1600-0447.1990.tb05280.x. [DOI] [PubMed] [Google Scholar]
- 90.Shi C-J, Cassell MD. Cortical, thalamic, and amygdaloid projections of the rat temporal cortex. J Comp Neurol. 1997;382:153–175. [PubMed] [Google Scholar]
- 91.Smith Y, Pare JF, Pare D. Cat intraamygdaloid inhibitory network: ultrastructural organization of parvalbumin-immunoreactive elements. J Comp Neurol. 1998;391:164–179. doi: 10.1002/(sici)1096-9861(19980209)391:2<164::aid-cne2>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 92.Soltis RP, Cook JC, Gregg AE, Sanders BJ. Interaction of GABA and excitatory amino acids in the basolateral amygdala: role in cardiovascular regulation. J Neurosci. 1997;17:9367–9374. doi: 10.1523/JNEUROSCI.17-23-09367.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Somogyi P, Tamas G, Lujan R, Buhl EH. Salient features of synaptic organisation in the cerebral cortex. Brain Res Rev. 1998;26:113–135. doi: 10.1016/s0165-0173(97)00061-1. [DOI] [PubMed] [Google Scholar]
- 94.Spehlmann R, Norcross K. Decreased sensitivity of neurons in the basolateral amygdala to dopamine and noradrenaline iontophoresis after a kindling stimulus. Exp Neurol. 1984;83:204–210. doi: 10.1016/0014-4886(84)90059-1. [DOI] [PubMed] [Google Scholar]
- 95.Stone TW. Microiontophoresis and pressure injection. IBRO handbook series: methods in neuroscience. Wiley-Interscience; New York: 1985. [Google Scholar]
- 96.Sugita S, Tanaka E, North RA. Membrane properties and synaptic potentials of three types of neurone in rat lateral amygdala. J Physiol (Lond) 1993;460:705–718. doi: 10.1113/jphysiol.1993.sp019495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Swanson LW. The projections of the ventral tegmental area and adjacent regions: a combined fluorescent retrograde tracer and immunofluorescence study in the rat. Brain Res Bull. 1982;9:321–353. doi: 10.1016/0361-9230(82)90145-9. [DOI] [PubMed] [Google Scholar]
- 98.Swanson LW, Petrovich GD. What is the amygdala? Trends Neurosci. 1998;21:323–331. doi: 10.1016/s0166-2236(98)01265-x. [DOI] [PubMed] [Google Scholar]
- 99.Van Vulpen EHS, Verwer RWH. Organization of projections from the mediodorsal nucleus of the thalamus to the basolateral complex of the amygdala in the rat. Brain Res. 1989;500:389–394. doi: 10.1016/0006-8993(89)90337-5. [DOI] [PubMed] [Google Scholar]
- 100.Wan F-J, Swerdlow NR. The basolateral amygdala regulates sensorimotor gating of acoustic startle in the rat. Neuroscience. 1997;76:715–724. doi: 10.1016/s0306-4522(96)00218-7. [DOI] [PubMed] [Google Scholar]
- 101.Wang Z, Rebec GV. Amygdaloid neurons respond to clozapine rather than haloperidol in behaving rats pretreated with intra-amygdaloid amphetamine. Brain Res. 1996;711:64–72. doi: 10.1016/0006-8993(95)01401-2. [DOI] [PubMed] [Google Scholar]
- 102.Washburn MS, Moises HC. Electrophysiological and morphological properties of rat basolateral amygdaloid neurons in vitro. J Neurosci. 1992;12:4066–4079. doi: 10.1523/JNEUROSCI.12-10-04066.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Weisskopf MG, LeDoux JE. Distinct populations of NMDA receptors at subcortical and cortical inputs to principal cells of the lateral amygdala. J Neurophysiol. 1999;81:930–934. doi: 10.1152/jn.1999.81.2.930. [DOI] [PubMed] [Google Scholar]
- 104.Weldon DA, Travis ML, Kennedy DA. Posttraining D1 receptor blockade impairs odor conditioning in neonatal rats. Behav Neurosci. 1991;105:450–458. doi: 10.1037//0735-7044.105.3.450. [DOI] [PubMed] [Google Scholar]
- 105.Wepsic JG, Austin GM. The neurophysiological effects of amphetamine upon the cat amygdala. In: Eleftheriou BE, editor. Neurobiology of the amygdala. Plenum; New York: 1971. pp. 623–640. [Google Scholar]
- 106.Wingerson DK, Cowley DS, Kramer GL, Petty F, Roy-Byrne PP. Effect of benzodiazepines on plasma levels of homovanillic acid in anxious patients and control subjects. Psychiatry Res. 1996;65:53–59. doi: 10.1016/0165-1781(96)02886-7. [DOI] [PubMed] [Google Scholar]
- 107.Yeomans JS, Pollard BA. Amygdala efferents mediating electrically evoked startle-like responses and fear potentiation of acoustic startle. Behav Neurosci. 1993;107:596–610. doi: 10.1037//0735-7044.107.4.596. [DOI] [PubMed] [Google Scholar]