

Abstract
Alcohol use disorder (AUD) persists as a devastating public health problem; widely effective pharmacological treatments are needed. Evidence from rodent models suggests that stimulating brain receptors for the neuropeptide nociceptin/orphanin FQ (NOP) can decrease ethanol drinking. We characterized the effects of the mu opioid peptide (MOP) receptor agonist buprenorphine and the buprenorphine analog (2S)-2-[(5R,6R,7R,14S)-N-cyclopropylmethyl-4,5-epoxy-6,14-ethano-3-hydroxy-6 methoxymorphinan-7-yl]-3,3-dimethylpentan-2-ol (BU08028), which stimulates MOP and NOP receptors, in a translational nonhuman primate model of AUD. Rhesus monkeys drank a 4% ethanol solution 6 h per day, 5 days per week via an operant behavioral panel in their home cages. To assess behavioral selectivity, monkeys responded via a photo-optic switch to earn food pellets. After characterizing the acute effects of BU08028 (0.001–0.01 mg/kg, i.m.) and buprenorphine (0.003–0.056 mg/kg, i.m.), the drugs were administered chronically using a model of pharmacotherapy assessment that incorporates clinical aspects of AUD and treatment. Acutely, both drugs decreased ethanol drinking at doses that did not affect food-maintained responding. During chronic treatment, effects of BU08028 and buprenorphine were maintained for several weeks without development of tolerance or emergence of adverse effects. BU08028 was ~0.5 and 1.0 log units more potent in acute and chronic studies, respectively. The selective NOP receptor agonist SCH 221510 also selectively decreased ethanol intakes when given acutely (0.03–1.0 mg/kg, i.m.), whereas the MOP antagonist naltrexone (1.7–5.6 mg/kg, i.m.) decreased both ethanol intake and food pellets delivered. These data demonstrate that bifunctional MOP/NOP agonists, which may have therapeutic advantages to MOP-selective drugs, can decrease alcohol drinking in nonhuman primates.
Subject terms: Addiction, Behavioural methods
Introduction
Alcohol use disorder (AUD) contributes to 88,000 deaths and costs the US ~$250 billion each year; fewer than 10% of individuals with AUD receive treatment [1–4]. Existing Food and Drug Administration-approved medications (disulfiram, naltrexone, and acamprosate) are not widely prescribed due to limited efficacy and adverse effects that limit compliance [5, 6]. The critical need to develop novel medications for treating AUD was emphasized in the National Institute on Alcohol Abuse and Alcoholism’s 2017–2021 strategic plan.
The neuropeptide nociceptin/orphanin FQ (NOP) receptor (formerly called the “opioid receptor-like” or “ORL1” receptor) is a G-protein-coupled receptor classified in the opioid receptor family based largely on sequence homology [7, 8]. Neither endogenous opioid peptides nor classical drugs acting at mu opioid peptide (MOP), kappa opioid (KOP), or delta opioid (DOP) receptors possess appreciable affinity for NOP receptors [9]. Over the last decade, interest in NOP receptor pharmacology has grown following the discovery that NOP receptor stimulation can produce analgesia in the absence of side effects associated with MOP receptor agonists (e.g., respiratory depression, itch, constipation, and addiction). Whereas initial work focused on the NOP peptide, non-peptidic agonists have subsequently been synthesized [10, 11]. More recently, “bifunctional” MOP/NOP receptor agonists were developed that show promise as non-addictive analgesics [12–15], including the buprenorphine analog BU08028 [12, 16]. We recently demonstrated the lack of abuse potential of bifunctional agonists, including BU08028, in nonhuman primates [12, 13].
Beyond their potential as non-addictive analgesics, recent research has supported the potential of NOP receptors as targets for medications to treat other psychiatric conditions including AUD [17–19]. For example, NOP receptor agonists can decrease neurochemical and neurophysiological effects of ethanol in rats [20, 21]. Moreover, administration of the NOP peptide or NOP receptor agonists can attenuate behavioral effects of ethanol in rodent models, including ethanol self-administration, ethanol seeking, and symptoms of ethanol withdrawal [22–30]. More recently, it was reported that antagonism or genetic deletion of NOP receptors also decreased ethanol drinking [31, 32], and that a NOP receptor antagonist reduced heavy drinking in a clinical trial [33]. These results suggest that NOP receptor internalization and/or desensitization may influence behavioral effects (see ref. [32]). The complex pharmacology of NOP receptors merits further research, particularly in the context of AUD medications.
Although important knowledge can be gained about the neuropharmacology of ethanol in rodents, the ability to generate clinically relevant phenotypes related to long-term drinking is limited. Alternately, nonhuman primates have several advantages that render them a useful translational bridge between rodents and potential clinical use. Nonhuman primates are the animals most similar to humans in terms of genetics, neuroanatomy, and pharmacokinetics, and show similar neurobiological consequences of long-term alcohol use [34–38]. Moreover, monkeys and rodents differ in the distribution and function of brain opioid receptors [39–42]. For example, MOP/NOP receptor agonists including BU08028 have been shown to have rewarding/reinforcing effects in rodents but not in monkeys [12, 13, 43, 44]. Prominent differences in NOP distribution include relatively higher NOP binding in the human and nonhuman primate basal ganglia, frontal cortex, and amygdala [45, 46] in contrast to relatively higher NOP receptor binding in the rodent midbrain [47–50]. Positron emission tomography imaging studies using NOP receptor-selective radiotracers have been consistent with autoradiographic studies in supporting a similar distribution in monkeys and humans [51–53]. Considering the importance of these brain regions in addiction, species differences provide further rationale for nonhuman primate studies to bridge the gap between rodents and humans.
The present studies characterized the effects of the MOP/NOP receptor agonist BU08028 and its parent compound buprenorphine in a nonhuman primate model of AUD. Drugs were administered to rhesus monkeys in a self-administration procedure in which ethanol was freely available in 6 h sessions 5 days per week. To assess whether alterations in consumption resulted from behavioral effects unrelated to ethanol reinforcement, monkeys also responded using a photo-optic switch to receive food pellets during drinking sessions. At MOP receptors, BU08028 and buprenorphine have similar affinities (2.14 and 1.52 nM, respectively) and efficacies (21.1% vs. 28.7% compared with [D-Ala2, N-MePhe4, Gly-ol]-enkephalin). At NOP receptors, however, BU08028 has ~10-fold higher affinity (Ki = 8.5 vs. 77.4 nM) and higher efficacy (48.0% vs. 15.5% compared with the NOP peptide [16]). Both drugs block DOP and KOP receptors with affinities similar to their MOP receptor affinity [16]. It is important to note that species differences have been documented in the effects of drugs that target MOP and NOP receptors [39–42]. For example, although the antinociceptive effects of buprenorphine are mediated in part by NOP receptors in rodents, NOP receptors do not appear to play a role in buprenorphine-induced antinociception in monkeys [39, 54]. Thus, the comparison of buprenorphine and BU08028 permits an assessment of how the effects of a MOP receptor partial agonist on ethanol consumption differ from those of a bifunctional MOP/NOP receptor agonist. In addition, we compared the acute effects of these drugs with those of the NOP receptor agonist SCH 221510, which exhibits 217-fold selectivity in binding and 58-fold selectivity in stimulating NOP vs. MOP receptors [11], and the MOP receptor antagonist naltrexone.
Although acute administration of drugs provides an initial indication of potential clinical effectiveness, assessment of the effects of chronic treatment is critical for predictive validity with respect to the results of clinical trials [55, 56]. Considerations include whether tolerance is observed to the therapeutic effects of drugs and whether any adverse drug effects emerge. Thus, we examined the effects of chronic treatment with BU08028 and buprenorphine using a translational model of pharmacotherapy evaluation that incorporates aspects of AUD and treatment [57–59]. Clinically relevant features of the model include: (1) subjects’ extensive history of ethanol use, (2) an individual-subject design wherein the dose of treatment drug is adjusted based on effect rather than a group design in which all monkeys receive identical treatment regimens, and (3) concurrent measurement of food-maintained responding to monitor potential adverse effects. This procedure has demonstrated good predictive validity with respect to the results of clinical trials for cocaine use disorder [57, 58, 60].
Materials and methods
Subjects
Five adult female rhesus monkeys (Macaca mulatta) were housed individually in 0.76 × 0.83 × 0.83 m stainless steel cages in a vivarium maintained at 24 °C and ~30% humidity with a 12:12 light/dark cycle. All monkeys had been trained to drink a solution of 4% ethanol in water using schedule-induced polydipsia [38, 61, 62] and had been consuming ethanol 5 days per week for ~1.5 years with a mean (±SD) lifetime intake of 815.5 ± 81.6 g/kg. Monkeys were fed 1 g nutritionally complete banana-flavored pellets (Bio-serv; Flemington, NJ), delivered during the behavioral session (see below), supplemented with fruit/vegetables. Monkeys received enough food to maintain healthy weights as determined by daily visual inspection and periodic veterinary examinations. All procedures were approved by the Wake Forest University Animal Care and Use Committee, and were performed in accordance with the 2011 National Research Council Guidelines for the Care and Use of Mammals in Neuroscience and Behavioral Research.
Apparatus
Each monkey’s cage contained an operant behavioral panel (“drinking panel”) that provided access to ethanol and food pellets during each session [38, 61]. Each panel contained two drinking spouts, two sets of three lights (red, amber, and green) positioned above each spout, a photo-optic switch positioned below one of the spouts, and a centrally positioned opening containing a dowel. For all experimental sessions described below, only one spout was active and provided ethanol; the monkeys had ad libitum access to water. Each panel was connected to a 0.5- gallon ethanol bottle positioned atop a balance that continuously monitored its weight. Pulling the dowel activated a solenoid that allowed ethanol to flow. The ethanol was gravity-fed through the spout when it was displaced. Application of negative pressure could increase the rate of flow through the spout, allowing the monkey to have complete control over the volume she consumed.
Ethanol and food self-administration
Sessions began at 11:00a.m. Monday through Friday with illumination of the lights above the ethanol spout. Throughout the 6- h session, ethanol was freely available when the dowel was pulled. Monkeys also self-administered food pellets under a fixed ratio 1 schedule of reinforcement using the photo-optic switch. The daily food pellet allotment was divided into three “meals,” which began 0, 120, and 240 min after the start of the session. Each meal lasted until 1/3 of the daily ration had been delivered or until the next meal period began.
Acute drug administration
When daily ethanol intakes and food pellet deliveries were stable, a single dose of BU08028 (0.001–0.01 mg/kg), buprenorphine (0.003–0.056 mg/kg), SCH 221510 (0.03–1.0 mg/kg), naltrexone (1.7–5.6 mg/kg), or vehicle was administered as an i.m. injection 30 min prior to the start of alcohol access (60 min for BU08028). Typically, drugs were administered on Tuesdays and Fridays; vehicle was administered on Thursdays. Each monkey received each dose and vehicle at least twice, with replicates averaged within a monkey for analysis.
Chronic drug administration
A similar approach to chronic drug treatment was used as was described previously [57, 59]. Briefly, once ethanol intake and food pellet deliveries were stable, treatment with BU08028 or buprenorphine commenced. The starting dose was chosen based on each monkey’s sensitivity during acute dosing. A dose of BU08028 or buprenorphine was administered 7 days per week at the same time each day (60 or 30 min prior to the start of the session, respectively). Drinking sessions were conducted 5 days per week (Monday through Friday). For BU08028, the initial dose was administered every other day for 2 weeks. This every-other-day dosing regimen was used because the analgesic effects of BU08028 have a duration of action of ~48 h [12]. After 2 weeks, effects indicated that daily dosing was necessary (see below). At the end of each week of treatment (i.e., Friday), the average ethanol intake of the last 3 days was calculated and compared with baseline using a t-test. A decision was made, based on the observed effects (or lack thereof) on ethanol intake and food-maintained responding, to either increase the dose of treatment drug or to keep it the same for the following week. When a dose was reached that resulted in persistent decreases in ethanol consumption (3-day average significantly below baseline), treatment was terminated and drinking was measured for an additional 3 weeks.
Data analysis
For acute studies, each drug was analyzed separately. Data for ethanol consumed and food pellets delivered were analyzed separately using a one-way repeated-measures analysis of variance (ANOVA) with a post-hoc Dunnett’s test to determine which doses produced effects significantly different from vehicle. For chronic studies, three time points (baseline, the week with the largest treatment drug effect, and the third week after the end of treatment) were compared with a one-way ANOVA followed by a post-hoc Tukey’s multiple comparisons test. In all cases, differences were considered significant when p < 0.05.
Determination of blood ethanol concentrations
Several blood samples were collected during the months before drug testing began. For each monkey, 20 μL samples were collected from the saphenous vein 6 h after the onset of the behavioral session. Blood samples were sealed in airtight vials containing 500 μL of ultrapure high-performance liquid chromatography water and 20 μL of 200 mg% isopropanol (internal standard) and stored at −4 °C until assayed using gas chromatography (Agilent 7890A GC system with G1888 Network Headspace Autosampler; Santa Clara, CA) supplied with a flame ionization detector and Agilent ChemStation integrator. Once all data had been collected, several more samples were collected in each monkey. The goal of these studies was to determine whether ethanol metabolism changed over time, which could occur due to chronic administration of opioid compounds, long-term ethanol consumption, passage of time, or a combination of factors.
Drugs
Ethanol (95% ethyl alcohol; The Warner-Graham Company; Cockeysville, MD) was diluted each morning in water purified by reverse osmosis. BU08028 HCl was dissolved in a solution of dimethyl sulfoxide (DMSO)/10% (mass/vol) (2-hydroxypropyl)-β-cyclodextrin in a ratio of 3:97 as previously described [63]. SCH 221510 (Tocris Bioscience; Bristol, UK) was prepared at a final concentration of 5 mg/mL in a vehicle (15% Tween80, 15% DMSO and 70% sterile water) as follows. Solid SCH 221510 was dissolved in 100% DMSO, then Tween80 was added. The mixture was heated and vortexed until clear, then water was added and the vial was heated/vortexed until the compound was dissolved. Naltrexone HCl and buprenorphine HCl (National Institute on Drug Abuse) were dissolved in sterile water.
Results
Effects of acute BU08028 or buprenorphine administration
When BU08028 was administered prior to the drinking session, there was a main effect of dose on ethanol intake (F3,12 = 13.33; Fig. 1, top left) but not food pellets delivered (Fig. 1, bottom left). Post-hoc testing revealed that ethanol intake was significantly reduced by 0.01 mg/kg BU08028. Similarly, for buprenorphine (Fig. 1, right column), there was a main effect of dose on ethanol intake (F3,12 = 23.45) but not on food pellets delivered. Ethanol consumption was significantly decreased by 0.03 and 0.056 mg/kg buprenorphine.
Fig. 1.
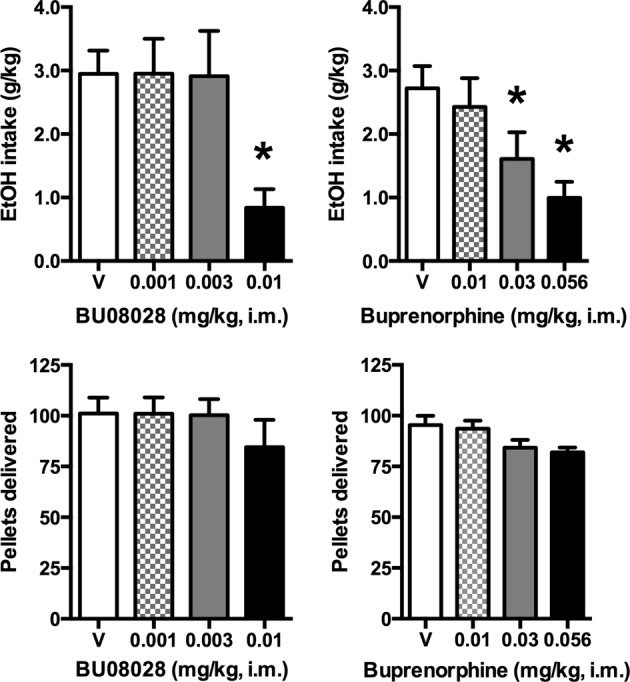
Effects of acute BU08028 and buprenorphine on ethanol drinking and food pellet deliveries. Top row, ethanol intake. Bottom row, food pellet deliveries. Bars represent mean ± SEM, n = 5; *p < 0.05 compared with vehicle (V)
Effects of acute SCH 221510 or naltrexone administration
After administration of SCH 221510 (Fig. 2, left column), there was a main effect of dose on ethanol intake (F2,8 = 8.30) but not food pellet deliveries. Ethanol intake was significantly reduced by 1.0 mg/kg SCH 221510. In contrast, after naltrexone administration (Fig. 2, right column), there was a main effect of drug on both ethanol intake (F3,12 = 10.16) and food pellet deliveries (F3,12 = 7.83). Both were decreased significantly after 3.0 and 5.6 mg/kg naltrexone. For all four drugs, no clear relationships were observed between baseline behavior (ethanol intake and food pellet deliveries) and individual differences in potency or efficacy of treatments.
Fig. 2.
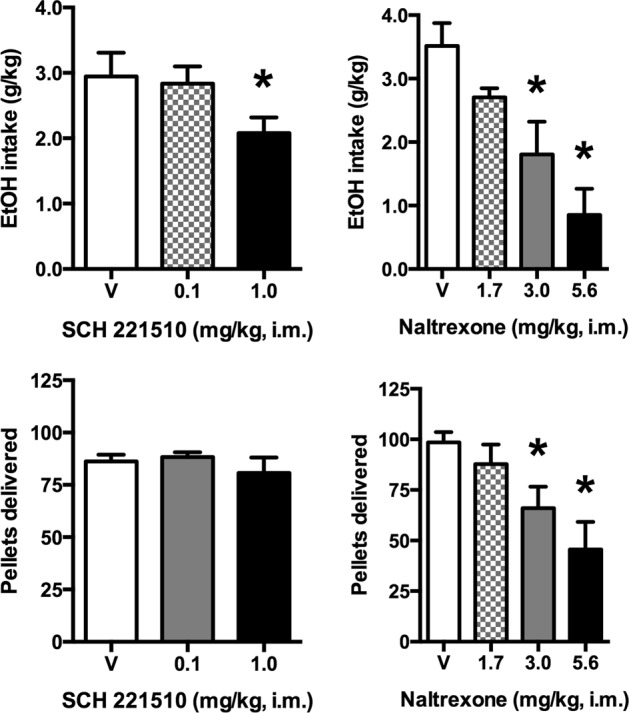
Effects of acute SCH 221510 and naltrexone on ethanol drinking and food pellet deliveries. Top row, ethanol intake. Bottom row, food pellet deliveries. Bars represent mean ± SEM, n = 5; *p < 0.05 compared with vehicle (V)
Effects of chronic BU08028 administration
Effects of chronic BU08028 treatment are depicted in Fig. 3. For each monkey, a starting dose of BU08028 was selected based on the monkey’s sensitivity to BU08028 during acute studies. At the end of each treatment week, for each subject a decision was made based on observed effects (or lack thereof) on ethanol intake and food-maintained responding to continue or increase the BU08028 dose for the next week. In four subjects, a significant decrease in drinking was observed after 1 week of every-other-day administration. However, tolerance developed to this effect in all four animals. During every-other-day dosing, it was common to see a greater effect on the day of drug administration than on the day drug was not given (e.g., R-1680). Based on these observations, BU08028 was subsequently administered daily. In R-1543, ethanol intake was decreased by 0.003 mg/kg BU08028 and further decreased when the dose was raised to 0.01 mg/kg. In R-1680, 0.01 mg/kg BU08028 produced a significant decrease in ethanol drinking during the first week, which was maintained for two additional weeks of treatment. In R-1624 and R-1544, daily treatment with 0.017 mg/kg significantly decreased ethanol intake. In these four subjects, minimal or no effects on food-maintained responding were observed during BU08028 treatment and ethanol intakes returned to baseline by week 2 after discontinuation of treatment. In contrast to these four subjects, no persistent effects of BU08028 were observed in R-1542. In this monkey, drinking tended to decrease on day 4 and 5 of treatment but returned to baseline by day 7. Interestingly, this monkey was the most sensitive to the effects of BU08028 on food-maintained responding. In all monkeys, a transient decrease in food-maintained responding was observed upon termination of treatment. When ethanol drinking data from the week of the largest BU08028 effect were averaged across monkeys and compared with baseline and the third week after termination of treatment (Fig. 3, lower right), there was a significant main effect of condition (F2,8 = 19.54); post-hoc testing indicated that the week of BU08028 treatment was significantly different from baseline.
Fig. 3.

Effects of chronic BU80828 treatment on ethanol drinking and food pellet deliveries. For first five graphs, left ordinate: ethanol (EtOH) intake (circles; filled circles represent data collected during BU08028 treatment); right ordinate: food pellets delivered (squares); abscissa: day of exposure to each condition, which is indicated across the top of each panel. Point above BL represents mean (±SD) ethanol dose consumed at baseline before BU08028 treatment. Vertical dotted lines indicate days during every-other-day (qod) treatment on which BU08028 was administered. Asterisks indicate weeks when average ethanol intake differed significantly from baseline. Lower right: mean (±SEM) ethanol intake at baseline, during the week of the maximum BU08028 effect and during the third week after BU08028 treatment ended. sid, once-per-day dosing. n = 5; *p < 0.05
Effects of chronic buprenorphine administration
Effects of chronic buprenorphine treatment are depicted in Fig. 4; the approach to testing was the same as that for BU08028. In R-1543 and R-1624, buprenorphine decreased ethanol intake during the first week of treatment. This effect was maintained or slightly increased when the dose was increased by one log unit over the subsequent 6 weeks. In R-1680, a significant decrease in ethanol intake was observed during treatment with 0.01 mg/kg buprenorphine treatment, but tolerance developed to this effect. Eventually, treatment was terminated after a significant decrease was observed after 2 weeks of treatment with 0.3 mg/kg buprenorphine. In R-1544, tolerance developed to decreases that were observed during treatment with lower buprenorphine doses. In this monkey, however, no decrease was observed after 2 weeks of treatment with 0.3 mg/kg buprenorphine. Finally, as was observed with BU08028, R-1542 was the least sensitive to buprenorphine. Ethanol intake was slightly but significantly lower than baseline during the second week of treatment with 0.3 mg/kg buprenorphine, but the difference was not significant when the dose was increased to 0.56 mg/kg. When drinking data from the week of the largest buprenorphine effect were averaged across monkeys and compared with baseline and the third week after termination of treatment (Fig. 4, lower right), there was a significant main effect of condition (F2,8 = 7.57); post-hoc testing indicated that the week of buprenorphine treatment was significantly different from baseline. Across monkeys, other similarities to BU08028 treatment included a general lack of effect on food-maintained responding during drug administration, except for R-1542, and some disruption in pellets delivered upon termination of treatment. Buprenorphine-induced decreases in ethanol drinking tended to persist longer after termination of treatment than did BU08028-induced decreases.
Fig. 4.

Effects of chronic buprenorphine treatment on ethanol drinking and food pellet deliveries. For the first five graphs, left ordinate: ethanol (EtOH) intake (circles; filled circles represent data collected during buprenorphine [BUPR] treatment); right ordinate: food pellets delivered (squares); abscissa: day of exposure to each condition, which is indicated across the top of each panel. Point above BL represents mean (±SD) ethanol dose consumed at baseline before buprenorphine treatment. Vertical dotted lines indicate days during every-other-day (qod) treatment on which buprenorphine was administered. Asterisks indicate weeks when average ethanol intake differed significantly form baseline. Lower right: mean (±SEM) ethanol intake at baseline, during the week of the maximum buprenorphine effect and during the third week after BU08028 treatment ended. sid, once-per-day dosing. n = 5; *p < 0.05
Ethanol pharmacokinetics did not change during the experiment
The relationship between ethanol consumed and resulting blood ethanol concentration (BEC) was compared between samples collected in the months prior to initiation of drug testing and samples collected after all drug testing was completed. There was no significant difference in either the slopes or the y-intercepts of the two regression lines (Fig. 5).
Fig. 5.
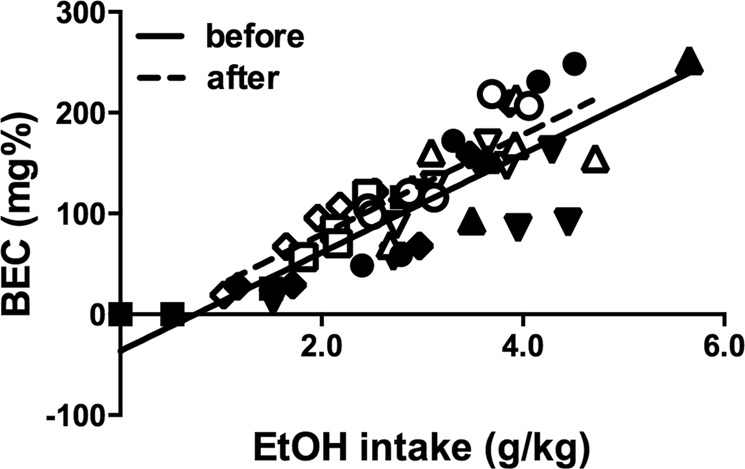
Relationship between amount of ethanol consumed and blood ethanol concentration (BEC). Points represent single determinations from individual subjects (n = 5), one shape per subject. Solid symbols and line depict samples collected before the drug treatments began; open symbols and dashed line represent data collected after the last drug treatment. Lines represent best fit results of linear regression
Discussion
Efforts to develop analgesics safer than currently used opioids have identified “bifunctional” MOP/NOP receptor agonists, including BU08028, which can provide pain relief with reduced side effects [12–16]. Beyond their role in analgesia, in part because the NOP peptide can inhibit the beta-endorphin neurons that may mediate alcohol abuse [64, 65], NOP receptors have been investigated in rodent models of AUD. These studies have consistently demonstrated that stimulating NOP receptors can decrease the abuse-related effects of ethanol [22–30]. The present studies compared the effects of the MOP receptor agonist buprenorphine and the bifunctional MOP/NOP agonist BU08028 in rhesus monkeys who had free access to a 4% ethanol solution 6 h per day, 5 days per week. Monkeys consumed ~3.0 g/kg per day, equivalent to ~12 drinks per day. Despite this relatively high ethanol intake, the lack of observed withdrawal signs on weekends indicated that monkeys were not physically dependent on ethanol. Moreover, the relationship between ethanol consumed and resulting BECs did not change over the course of these experiments, indicating that no metabolic tolerance occurred. In addition, neither the duration of monkeys’ menstrual cycles nor pattern of drinking across the cycle was altered during these experiments [62].
Acute administration of both BU08028 and buprenorphine decreased ethanol drinking, consistent with previous studies of NOP receptor agonists and buprenorphine in rodents [22–30, 66], and buprenorphine in monkeys [67]. Acutely, BU08028 was ~0.5 log units more potent than buprenorphine. Importantly, decreases in ethanol drinking were observed at doses that did not significantly alter food-maintained responding. The lack of effect of buprenorphine on food-maintained responding is consistent with most studies in nonhuman primates [e.g., [68, 69]], although effects on food-maintained responding have been observed under some conditions depending on the monkeys’ experimental history and the schedule of reinforcement used [70–72]. The behavioral selectivity observed in the present study supports the conclusion that decreases in drinking were due to reductions in the reinforcing/motivational effects of ethanol rather than to sedation, motor impairments, or other behavioral effects.
A critical aspect of testing potential medications is the assessment of effects of chronic administration [55, 56]. In the context of this study, it was important to assess whether tolerance would develop to the ability of BU08028 and buprenorphine to decrease ethanol drinking and whether any adverse effects would emerge. In four of five monkeys, BU08028 significantly decreased ethanol intake without tolerance development. Buprenorphine produced decreases in ethanol drinking in the same four subjects, although the effect was modest in one subject (R-1680), and in another subject (R-1544) tolerance developed. In chronic studies, BU08028 was more potent and somewhat more efficacious than buprenorphine in most monkeys. Termination of drug treatment resulted in temporary disruption of food-maintained responding in some animals, possibly a sign of withdrawal from chronic treatment with BU08028, as NOP receptor agonists have been shown to decrease gastric motility [73, 74]. Levels of ethanol consumption gradually returned to baseline levels; reductions of drinking persisted longer after buprenorphine treatment. Interestingly, in R-1542, no persistent decreases in ethanol consumption were produced by either drug, despite disruption of food-maintained responding. No distinguishing characteristics have been identified to indicate why results in this monkey differed from the other four subjects.
As described above, BU08028 and buprenorphine have been characterized in vitro as MOP and NOP receptor partial agonists [16]. However, the contribution of NOP receptors to the behavioral effects of buprenorphine appears to differ between rodents and monkeys. In contrast to results in rodents [54, 75], blocking NOP receptors in rhesus monkeys did not alter buprenorphine-induced antinociception, respiratory depression, or scratching [39]. Thus, although a role for NOP receptors has been demonstrated in the ability of buprenorphine to decrease ethanol drinking in rats [25], it is likely that its effects on ethanol consumption in the present and only previous nonhuman primate study [67] resulted primarily from activity at MOP receptors. The observed decreases are consistent with previous studies of the effects of buprenorphine on alcohol consumption. In a previous rodent study [25], ethanol drinking was increased by low doses of buprenorphine, whereas higher buprenorphine doses decreased ethanol intake. The authors suggested that this biphasic effect resulted from stimulation of MOP at lower doses and NOP at higher doses. That study used as subjects rats genetically bred for high ethanol intake and have upregulated NOP receptor function [76, 77]. In another study using outbred rats, low-to-intermediate buprenorphine doses increased ethanol intakes under some conditions and decreased drinking under others [66]. In addition to rat strain, there were several differences in experimental design that may have contributed to the discrepant effects including extent of daily ethanol access and time before the session that buprenorphine was administered. In contrast, in the present and previous nonhuman primate studies [67], lower buprenorphine doses did not increase ethanol intakes. Nonetheless, in all four studies as well as one other rodent study that tested only one dose [78], higher doses of buprenorphine decreased ethanol drinking.
Taken together, the present data suggest that the ability of buprenorphine to decrease ethanol drinking is shared by the bifunctional MOP/NOP receptor agonist BU08028. In acute studies, BU08028 was more potent than buprenorphine. In addition, in chronic studies, BU08028 was more potent in most monkeys and somewhat more effective. In general the findings support the view developed from studies of the analgesic effects of these drugs that combining NOP and low-efficacy MOP agonists could prove advantageous compared with MOP agonists alone [12–16]. For example, addition of NOP receptor activity could permit the use of lower doses of a MOP receptor agonist, enhance efficacy in decreasing alcohol drinking, widen the therapeutic window, reduce the rate or magnitude of tolerance development, and reduce side effects including abuse potential. For example, although remifentanil, oxycodone, and buprenorphine maintained self-administration in rhesus monkeys under a progressive-ratio schedule, BU088028 did not function as a reinforcer [12, 13].
Future studies will be necessary to characterize the relative contribution of MOP and NOP receptors to the therapeutic effects of bifunctional MOP/NOP agonists such as BU08028. A role for both receptor systems in the ability of buprenorphine to decrease alcohol self-administration would be consistent with its effects on cocaine self-administration [79]. In the present studies, the acute effects of SCH 221510, a NOP receptor agonist with 217-fold selectivity in binding and 58-fold selectivity in stimulating NOP vs. MOP receptors [11], were similar to those of buprenorphine and BU08028. This result may suggest that NOP receptor stimulation alone is sufficient to reduce alcohol drinking, in agreement with rodent studies [26, 27, 30]. However, the dose of SCH 221510 required to reduce ethanol drinking was higher than the dose needed to produce antinociception in a previous study [80]. Although the discrepancy might be explained by different efficacy requirements between the two assays, the large potency difference could also indicate that the reduction in drinking resulted from activity at other receptors. Additional study of NOP receptor-selective agonists, administered chronically, and combination studies of bifunctional MOP/NOP receptor agonists with NOP and MOP receptor antagonists will be required to fully understand the relative contributions of MOP and NOP receptors to the behavioral effects of bifunctional agonists. Moreover, it will be important to assess the involvement of KOP receptors, which can mediate and modulate the abuse-related effects of ethanol [81–83]. This is particularly important, because buprenorphine and BU08028 are KOR antagonists with relatively high affinity (2.5 ± 1.2 nM and 5.6 ± 1.3 nM, respectively [16]). Finally, it will be important to examine whether MOP/NOP and NOP receptor agonists selectively decrease alcohol intake in males; sex differences have been observed in the effects of low-efficacy MOP receptor agonists on antinociception and on rates of food-maintained responding [71, 84].
Due to the enormous need for non-abusable analgesics in response to the ongoing epidemic of opioid abuse, it is likely that clinical development of bifunctional MOP/NOP receptor agonists will be prioritized. NOP-acting drugs were included in the list of the National Institute on Drug Abuse’s medication development priorities as part of the National Institutes of Health HEAL (Helping to End Addiction Long-term) Initiative [85]. The results of the present studies extend promising results from rodent models that suggest that such drugs are candidates as novel medications for AUD as well.
Funding and disclosure
This work was supported by NIH/NIAAA grants P01 AA021099 and P50 AA026117, and NIH/NIDA grant R01 DA007315. The authors declare no conflict of interest. In the past 3 years, SMH received research funding and consulting fees from Orexigen Therapeutics, Inc. MCK received honorariums from Johns Hopkins University, Duke University, Nanjing University of Chinese Medicine, Trevena, Inc., and Grünenthal GmbH. He had research contracts with Biogen MA, Inc. and Orexigen Therapeutics, Inc.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.U.S. Department of Health and Human Services (HHS), Office of the Surgeon General, Facing Addiction in America. The surgeon general’s report on alcohol, drugs, and health. Washington, DC: HHS; 2016. [PubMed]
- 2.Sacks JJ, Gonzales KR, Bouchery EE, Tomedi LE, Brewer RD. national and state costs of excessive alcohol consumption. Am J Prev Med. 2010;2015:e73–79. doi: 10.1016/j.amepre.2015.05.031. [DOI] [PubMed] [Google Scholar]
- 3.Stahre M, Roeber J, Kanny D, Brewer RD, Zhang X. Contribution of excessive alcohol consumption to deaths and years of potential life lost in the United States. Prev Chronic Dis. 2014;11:130293.4. doi: 10.5888/pcd11.130293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmidt LA. Recent developments in alcohol services research on access to care. Alcohol Res. 2016;38:27–33. [PMC free article] [PubMed] [Google Scholar]
- 5.Miller PM, Book SW, Stewart SH. Medical treatment of alcohol dependence: a systematic review. Int J Psychiatry Med. 2011;42:227–66. doi: 10.2190/PM.42.3.b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rezvani AH, Lawrence AJ, Arolfo MP, Levin ED, Overstreet DH. Novel medication targets for the treatment of alcoholism: preclinical studies. Recent Pat CNS Drug Discov. 2012;7:151–62. doi: 10.2174/157488912800673182. [DOI] [PubMed] [Google Scholar]
- 7.Mollereau C, Parmentier M, Mailleux P, Butour JL, Moisand C, Chalon P, et al. ORL1, a novel member of the opioid receptor family. Cloning, functional expression and localization. FEBS Lett. 1994;341:33–38. doi: 10.1016/0014-5793(94)80235-1. [DOI] [PubMed] [Google Scholar]
- 8.Toll L, Bruchas MR, Calo’ G, Cox BM, Zaveri NT. Nociceptin/orphanin FQ receptor structure, signaling, ligands, functions, and interactions with opioid systems. Pharm Rev. 2016;68:419–57. doi: 10.1124/pr.114.009209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Butour JL, Moisand C, Mazarguik H, Mollereau C, Meunier JC. Recognition and activation of the opioid receptor-like ORL 1 receptor by nociceptin, nociceptin analogs and opioids. Eur J Pharmacol. 1997;321:97–103. doi: 10.1016/s0014-2999(96)00919-3. [DOI] [PubMed] [Google Scholar]
- 10.Jenck F, Wichmann J, Dautzenberg FM, Moreau JL, Ouagazzal AM, Martin JR, et al. A synthetic agonist at the orphanin FQ/nociceptin receptor ORL1: anxiolytic profile in the rat. Proc Natl Acad Sci USA. 2000;97:4938–43. doi: 10.1073/pnas.090514397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Varty GB, Lu SX, Morgan CA, Cohen-Williams ME, Hodgson RA, Smith-Tohran A, et al. The anxiolytic-like effects of novel, orally active nociceptin opioid receptor agonist 8-[bis(2-methylphenyl)methyl]-3-phenyl-8-azabicyclo[3.2.1]octan-3-ol (SCH 221510) J Pharm Exp Ther. 2008;326:672–82. doi: 10.1124/jpet.108.136937. [DOI] [PubMed] [Google Scholar]
- 12.Ding H, Czoty PW, Kiguchi N, Cami-Kobeci G, Sukhtantar DD, Nader MA, et al. A novel orvinol analog, BU08028, as a safe opioid analgesic without abuse liability in primates. Proc Natl Acad Sci USA. 2016;113:EE5511–8. doi: 10.1073/pnas.1605295113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ding H, Kiguchi N, Yasuda D, Daga PR, Polgar WE, Lu JJ, et al. A bifiunctional nociceptin and mu opioid receptor agonist is analgesic without opioid side effects in nonhuman primates. Sci Transl Med. 2018;10:eaar3483. doi: 10.1126/scitranslmed.aar3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin AP, Ko MC. The therapeutic potential of nociceptin/orphanin FQ receptor agonists as analgesics without abuse liability. ACS Chem Neurosci. 2013;4:214–24. doi: 10.1021/cn300124f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zaveri NT. Nociceptin opioid receptor (NOP) as a therapeutic target: progress in translation from preclinical research to clinical utility. J Med Chem. 2016;59:7011–28. doi: 10.1021/acs.jmedchem.5b01499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khroyan TV, Polgar WE, Cami-Kobeci G, Husbands SM, Zaveri NT, Toll L. The first universal opioid ligand, (2S)-2-[(5R,6R,7R,14S)-N-cyclopropylmethyl-4,5-epoxy-6,14-ethano-3-hydroxy-6-methoxymorphinan-7-yl]-3,3-dimethylpentan-2-ol (BU08028): characterization of the in vitro profile and in vivo behavioral effects in mouse models of acute pain and cocaine-induced reward. J Pharm Exp Ther. 2011;336:952–61. doi: 10.1124/jpet.110.175620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Witkin JM, Statnick MA, Rorick-Kehn LM, Pintar JE, Ansonoff M, Chen Y, et al. The biology of Nociceptin/Orphanin FQ (N/OFQ) related to obesity, stress, anxiety mood, and drug dependence. Pharm Ther. 2014;141:283–99. doi: 10.1016/j.pharmthera.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heilig M, Egli M. Pharmacological treatment of alcohol dependence: target symptoms and target mechanisms. Pharm Ther. 2006;111:855–76. doi: 10.1016/j.pharmthera.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 19.Aubin HJ, Daeppen JB. Emerging pharmacotherapies for alcohol dependence: a systematic review focusing on reduction in consumption. Drug Alcohol Depend. 2013;133:15–29. doi: 10.1016/j.drugalcdep.2013.04.025. [DOI] [PubMed] [Google Scholar]
- 20.Kallupi M, Oleata CS, Luu G, Teshima K, Ciccocioppo R, Roberto M. MT-7716, a novel selective nonpeptidergic NOP receptor agonist, effectively blocks ethanol-induced increase in GABAergic transmission in the rat central amygdala. Front Integr Neurosci. 2014;8:18. doi: 10.3389/fnint.2014.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kallupi M, Varodayan FP, Oleata CS, Correia D, Luu G, Roberto M. Nociceptin/orphanin FQ decreases glutamate transmission and blocks ethanol-induced effects in the central amygdala of naïve and ethanol-dependent rats. Neuropsychopharmacology. 2014;39:1081–92. doi: 10.1038/npp.2013.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aziz AM, Brothers S, Sartor G, Holm L, Heilig M, Wahlestedt C, et al. The nociceptin/orphanin FQ receptor agonist SR-8993 as a candidate therapeutic for alcohol use disorders: validation in rat models. Psychopharmacology. 2016;233:3553–63. doi: 10.1007/s00213-016-4385-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ciccocioppo R, Panocka I, Polidori C, Regoli D, Massi M. Effect of nociceptin on alcohol intake in alcohol-preferring rats. Psychopharmacology. 1999;141:220–4. doi: 10.1007/s002130050828. [DOI] [PubMed] [Google Scholar]
- 24.Ciccocioppo R, Economidou D, Fedeli A, Angeletti S, Weiss F, Heilig M, et al. Attenuation of ethanol self-administration and of conditioned reinstatement of alcohol-seeking behaviour by the antiopioid peptide nociceptin/orphanin FQ in alcohol-preferring rats. Psychopharmacology. 2004;172:170–8. doi: 10.1007/s00213-003-1645-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ciccocioppo R, Economidou D, Rimondini R, Sommer W, Massi M, Heilig M. Buprenorphine reduces alcohol drinking through activation of the nociceptin/orphanin FQ-NOP receptor system. Biol Psychiatry. 2007;61:4–12. doi: 10.1016/j.biopsych.2006.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ciccocioppo R, Stopponi S, Economidou D, Kuriyama M, Kinoshita H, Heilig M, et al. Chronic treatment with novel brain-penetrating selective NOP receptor agonist MT-7716 reduces alcohol drinking and seeking in the rat. Neuropsychopharmacology. 2014;39:2601–10. doi: 10.1038/npp.2014.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Guglielmo G, Martin-Fardon R, Teshima K, Ciccocioppo R, Weiss F. MT-7716, a potent NOP receptor agonist, preferentially reduces ethanol seeking and reinforcement in post-dependent rats. Addict Biol. 2015;20:643–51. doi: 10.1111/adb.12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Economidou D, Fedeli A, Fardon RM, Weiss F, Massi M, Ciccocioppo R. Effect of novel nociceptin/orphanin FQ–NOP receptor ligands on ethanol drinking in alcohol-preferring msP rats. Peptides. 2006;27:3299–306. doi: 10.1016/j.peptides.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Economidou D, Cippitelli A, Stopponi S, Braconi S, Clementi S, Ubaldi M, et al. Activation of brain NOP receptors attenuates acute and protracted alcohol withdrawal symptoms in the rat. Alcohol Clin Exp Res. 2011;35:747–55. doi: 10.1111/j.1530-0277.2010.01392.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuzmin A, Kreek MJ, Bakalkin G, Liljequist S. The nociceptin/orphanin FQ receptor agonist Ro 64- 6198 reduced alcohol self-administation and prevents relapse-like alcohol drinking. Neuropsychopharmacology. 2007;32:902–10. doi: 10.1038/sj.npp.1301169. [DOI] [PubMed] [Google Scholar]
- 31.Kallupi M, Scuppa G, de Guglielmo G, Calò G, Weiss F, Statnick MA, et al. Genetic deletion of the nociceptin/orphanin FQ receptor in the rat confers resilience to the development of drug addiction. Neuropsychopharmacology. 2017;42:695–706. doi: 10.1038/npp.2016.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rorick-Kehn LM, Ciccocioppo R, Wong CJ, Witkin JM, Martinez-Grau MA, Stopponi S, et al. A novel, orally bioavailable nociceptin receptor antagonist. LY2940094, reduced ethanol self-administration and ethanol seeking in animal models. Alcohol Clin Exp Res. 2016;40:945–54. doi: 10.1111/acer.13052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Post A, Smart TS, Jackson K, Mann J, Mohs R, Rorick-Kehn L, et al. Proof-of-concept study to assess the nociceptin receptor antagonist LY2940094 as a new treatment for alcohol dependence. Alcohol Clin Exp Res. 2016;40:1935–44. doi: 10.1111/acer.13147. [DOI] [PubMed] [Google Scholar]
- 34.Phillips KA, Bales KL, Capitanio JP, Conley A, Czoty PW, ‘t Hart BA, et al. Why primate models matter. Am J Prima. 2014;76:801–27. doi: 10.1002/ajp.22281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weerts EM, Fantegrossi WE, Goodwin AK. The value of nonhuman primates in drug abuse research. Exp Clin Psychopharmacol. 2007;15:309–27. doi: 10.1037/1064-1297.15.4.309. [DOI] [PubMed] [Google Scholar]
- 36.Baker EJ, Farro J, Gonzales S, Helms C, Grant KA. Chronic alcohol self-administration in monkeys shows long-term quantity/frequency categorical stability. Alcohol Clin Exp Res. 2014;38:2835–43. doi: 10.1111/acer.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grant KA, Bennett AJ. Advances in nonhuman primate alcohol abuse and alcoholism research. Pharm Ther. 2003;100:235–55. doi: 10.1016/j.pharmthera.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 38.Vivian JA, Green HL, Young JE, Majersky LS, Thomas BW, Shively CA, et al. Induction and maintenance of ethanol self-administration in cynomolgus monkeys (Macaca fascicularis): long-term characterization of sex and individual differences. Alcohol Clin Exp Res. 2001;25:1087–97. [PubMed] [Google Scholar]
- 39.Cremeans CM, Gruley E, Kyle DJ, Ko MC. Roles of μ-opioid receptors and nociceptin/orphanin FQ peptide receptors in buprenorphine-induced physiological responses in primates. J Pharm Exp Ther. 2012;343:72–81. doi: 10.1124/jpet.112.194308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bowen CA, Fischer BD, Mello NK, Negus SS. Antagonism of the antinociceptive and discriminative stimulus effects of morphine by 3-methoxynaltrexone and naltrexone in rhesus monkeys. J Pharm Exp Ther. 2002;30:264–73. doi: 10.1124/jpet.302.1.264. [DOI] [PubMed] [Google Scholar]
- 41.Ko MC, Divin MF, Woods JH, Traynor JR. Differential in vivo potencies of naltrexone and 6beta-naltrexol in the monkey. J Pharm Exp Ther. 2006;316:772–9. doi: 10.1124/jpet.105.094409. [DOI] [PubMed] [Google Scholar]
- 42.Mansour A, Khachaturian H, Lewis ME, Akil H, Watson SJ. Anatomy of CNS opioid receptors. Trends Neurosci. 1988;11:308–14. doi: 10.1016/0166-2236(88)90093-8. [DOI] [PubMed] [Google Scholar]
- 43.Khroyan TV, Zaveri NT, Polgar WE, Orduna J, Olsen C, Jiang F, et al. SR 15435[1-(1-(bicyclo[3.3.1]nonan-9-yl)piperidin-4-yl)indolin-2-one], a novel mixed nociceptin/orphanin FQ/mu-opioid receptor partial agonist: analgesic and rewarding properties in mice. Eur J Pharmacol. 2007;560:29–35. doi: 10.1124/jpet.106.111997. [DOI] [PubMed] [Google Scholar]
- 44.Toll L, Khroyan TV, Polgar WE, Jiang F, Olsen C, Zaveri NT. Comparison of the antinociceptive and antirewarding effects of novel bifunctional nociceptin receptor/mu-opioid: implications for therapeutic applications. J Pharm Exp Ther. 2009;331:946–53. doi: 10.1124/jpet.109.157446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peluso J, LaForge KS, Matthes HW, Kreek MJ, Kieffer BL, Gaveriaux-Ruff C. Distribution of nociceptin/orphanin FQ transcript in human central nervous system and immune cells. J Neuroimmunol. 1998;81:184–92. doi: 10.1016/s0165-5728(97)00178-1. [DOI] [PubMed] [Google Scholar]
- 46.Bridge KE, Wainwright A, Reilly K, Oliver KR. Autoradiographic localization of (125)i[Tyr(14)] nociceptin/orphanin FQ binding sites in macaque primate CNS. Neuroscience. 2003;118:513–23. doi: 10.1016/s0306-4522(02)00927-2. [DOI] [PubMed] [Google Scholar]
- 47.Florin S, Meunier J, Constentin J. Autoradiographic localization of [3H]nociceptin binding sites in the rat brain. Brain Res. 2000;880:11–16. doi: 10.1016/s0006-8993(00)02669-x. [DOI] [PubMed] [Google Scholar]
- 48.Letchworth SR, Mathis JP, Rossi GC, Bodnar RJ, Pasternak GW. Autoradiographic localization of 125I [Tyr14] orphanin FQ/nociceptin and 125I [Tyr10] orphanin FQ/nociceptin (1–11) binding sites in rat brain. J Comp Neurol. 2000;24:319–29. [PubMed] [Google Scholar]
- 49.Maidment NT, Chen Y, Tan AM, Murphy NP, Leslie FM. Rat ventral midbrain dopamine neurons express the orphanin FQ/nociceptin receptor ORL-1. NeuroReport. 2002;13:1137–40. doi: 10.1097/00001756-200207020-00013. [DOI] [PubMed] [Google Scholar]
- 50.Meunier JC, Mollereau C, Toll L, Suaudeau C, Moisand C, Alvinerie P, et al. Isolation and structure of the endogenous agonist of the opioid receptor-like ORL1 receptor. Nature. 1995;377:532–5. doi: 10.1038/377532a0. [DOI] [PubMed] [Google Scholar]
- 51.Kimura Y., Fujita M., Hong J., Lohith T. G., Gladding R. L., Zoghbi S. S., Tauscher J. A., Goebl N., Rash K. S., Chen Z., Pedregal C., Barth V. N., Pike V. W., Innis R. B. Brain and Whole-Body Imaging in Rhesus Monkeys of 11C-NOP-1A, a Promising PET Radioligand for Nociceptin/Orphanin FQ Peptide Receptors. Journal of Nuclear Medicine. 2011;52(10):1638–1645. doi: 10.2967/jnumed.111.091181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hostetler ED, Sanabria-Bohórquez S, Eng W, Joshi AD, Patel S, Gibson RE, et al. Evaluation of [18F]MK-0911, a positron emission tomography (PET) tracer for opioid receptor-like 1 (ORL1), in rhesus monkey and human. Neuroimage. 2013;68:1–10. doi: 10.1016/j.neuroimage.2012.11.053. [DOI] [PubMed] [Google Scholar]
- 53.Narenden R, Ciccocioppo R, Lopresti B, Paris J, Himes ML, Mason NS. Nociceptin receptors in alcohol use disorders: a positron emission tomography study using [11C]NOP-1A. Biol Psychiatry. 2017;84:708–14. doi: 10.1016/j.biopsych.2017.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khroyan TV, Polgar WE, Jiang F, Zaveri NT, Toll L, Nociceptin/orphanin FQ. receptor activation attenuates antinociception induced by mixed nociceptin/orphanin FQ/mu-opioid receptor agonists. J Pharm Exp Ther. 2009;331:946–53. doi: 10.1124/jpet.109.156711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haney M, Spealman R. Controversies in translational research: drug self-administration. Psychopharmacology. 2008;199:403–19. doi: 10.1007/s00213-008-1079-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Czoty PW, Stoops WW, Rush CR. Evaluation of the “pipeline” for development of medications for cocaine use disorder: a review of translational preclinical, human laboratory, and clinical trial research. Pharm Rev. 2016;68:533–62. doi: 10.1124/pr.115.011668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Czoty PW, Gould RW, Martelle JL, Nader MA. Prolonged attenuation of the reinforcing strength of cocaine be chronic d-amphetamine in rhesus monkeys. Neuropsychopharmacology. 2011;36:539–47. doi: 10.1038/npp.2010.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Czoty PW, Martelle SE, Gould RW, Nader MA. Effects of chronic methylphenidate on cocaine self-administration under a progressive-ratio schedule of reinforcement in rhesus monkeys. J Pharm Exp Ther. 2013;345:374–82. doi: 10.1124/jpet.113.204321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Czoty PW, Blough BE, Fennell TR, Snyder RW, Nader MA. Attenuation of cocaine self-administration by chronic oral phendimetrazine in rhesus monkeys. Neuroscience. 2016;324:367–76. doi: 10.1016/j.neuroscience.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gould RW, Czoty PC, Nader SH, Nader MA. Effects of varenicline on the reinforcing and discriminative stimulus effects of cocaine in rhesus monkeys. J Pharm Exp Ther. 2011;339:678–86. doi: 10.1124/jpet.111.185538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grant KA, Leng X, Green HL, Szeliga KT, Rogers LS, Gonzales SW. Drinking typography established by schedule induction predicts chronic heavy drinking in a monkey model of ethanol self-administration. Alcohol Clin Exp Res. 2008;32:1824–38. doi: 10.1111/j.1530-0277.2008.00765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thomas SG, Czoty PW. Effect of menstrual cycle on ethanol drinking in rhesus monkeys. Drug Alcohol Depend. 2019;194:205–9. doi: 10.1016/j.drugalcdep.2018.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cami-Kobeci G, Polgar WE, Khroyan TV, Toll L, Husbands SM. Structural determinants of opioid and NOP activity in derivatives of buprenorphine. J Med Chem. 2011;54:6531–7. doi: 10.1021/jm2003238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Herz A. Endogenous opioid systems and alcohol addiction. Psychopharmacology. 1997;129:99–111. doi: 10.1007/s002130050169. [DOI] [PubMed] [Google Scholar]
- 65.Wagner EJ, Ronnekleiv OK, Grandy DK, Kelly MJ. The peptide orphanin FQ inhibits [beta]-endorphin neurons and neurosecretory cells in the hypothalamic arcuate nucleus by activating an inwardly-rectifying K+conductance. Neuroendocrinology. 1998;67:73–82. doi: 10.1159/000054301. [DOI] [PubMed] [Google Scholar]
- 66.June HL, Cason CR, Chen SH, Lewis MJ. Buprenorphine alters ethanol self-administration in rats: dose-response and time-dependent effects. Psychopharmacology. 1998;140:29–37. doi: 10.1007/s002130050735. [DOI] [PubMed] [Google Scholar]
- 67.Carroll ME, Carmon GN, May SA, Buzalsky S, Larson C. Buprenorphine’s effects on self-administration of smoked cocaine base and orally delivered phencyclidine, ethanol and saccharin in rhesus monkeys. J Pharm Exp Ther. 1992;261:26–37. [PubMed] [Google Scholar]
- 68.Negus SS, Mello NK, Linsenmayer DC, Jones RM, Portoghese PS. Delta opioid antagonist effects of buprenorphine in rhesus monkeys. Behav Pharmacol. 2002;13:557–70. doi: 10.1097/00008877-200211000-00005. [DOI] [PubMed] [Google Scholar]
- 69.Paronis CA, Bergman J. Buprenorphine and opioid antagonism, tolerance, and naltrexone-participated withdrawal. J Pharm Exp Ther. 2011;336:488–95. doi: 10.1124/jpet.110.173823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mello NK, Bree MP, Lukas SE, Mendelson JH. Buprenorphine effects on food-maintained responding in Macaque monkeys. Pharm Biochem Behav. 1985;23:765–8. doi: 10.1016/0091-3057(85)90111-x. [DOI] [PubMed] [Google Scholar]
- 71.Schwienteck Kathryn L., Negus S. Stevens, Banks Matthew L. Sex differences in the effectiveness of buprenorphine to decrease rates of responding in rhesus monkeys. Behavioural Pharmacology. 2019;30(4):358–362. doi: 10.1097/FBP.0000000000000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Withey SL, Paronis CA, Bergman J. Concurrent assessment of the antinociceptive and behaviorally disruptive effects of opioids in squirrel monkeys. J Pain. 2018;19:728–40. doi: 10.1016/j.jpain.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sobczak M, Cami-Kobeci G, Salaga M, Husbands SM, Fichna J. Novel mixed NOP/MOP agonist BU08070 alleviates pain and inhibits gastrointestinal motility in mouse models mimicking diarrhea-predominant irritable bowel syndrome symptoms. Eur J Pharmacol. 2014;736:63–69. doi: 10.1016/j.ejphar.2014.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fichna J, Sobczak M, Mokrowiecka A, Cygankiewicz AI, Zakrzewski PK, Cenac N, et al. Activation of the endogenous nociceptin system by selective nociceptin receptor agonist SCH 221510 produces antitransti and antinociceptive effect: a novel strategy for treatment of diahrrea-prominent IBS. Neurogastroenterol Motil. 2014;26:1539–50. doi: 10.1111/nmo.12390. [DOI] [PubMed] [Google Scholar]
- 75.Lutfy K, Eitan S, Bryant CD, Yang YC, Saliminejad N, Walwyn W, et al. Buprenorphine-induced antinociception is mediated by mu-opioid receptors and compromised by concomitant activation of opioid receptor-like receptors. J Neurosci. 2003;23:10331–7. doi: 10.1523/JNEUROSCI.23-32-10331.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hansson AC, Cippitelli A, Sommer WH, Fedeli A, Bjork K, Soverchia L, et al. Variation at the rat Crhr1 locus and sensitivity to relapse into alcohol seeking induced by environmental stress. Proc Natl Acad Sci USA. 2006;103:15236–41. doi: 10.1073/pnas.0604419103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Economidou D, Hansson AC, Weiss F, Terasmaa A, Cippitelli A, Fedeli A, et al. Dysregulation of nococeptin/orphanin FQ activity in the amygdala is linked to excessive alcohol drinking in the rat. Biol Psychiatry. 2008;64:211–8. doi: 10.1016/j.biopsych.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Martin A, Pilotto R, Singer G, Oei TP. The suppression of ethanol self injection by buprenorphine. Pharm Biochem Behav. 1983;19:985–6. doi: 10.1016/0091-3057(83)90403-3. [DOI] [PubMed] [Google Scholar]
- 79.Kallupi M, Shen Q, de Guglielmo G, Yasuda D, Journigan VB, Zaveri NT, et al. Buprenorphine requires concomitant activation of NOP and MOP receptors to reduce cocaine consumption. Addict Biol. 2018;23:585–95. doi: 10.1111/adb.12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kangas BD, Bergman J. Operant nociception in nonhuman primates. Pain. 2014;155:1821–8. doi: 10.1016/j.pain.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Anderson RI, Becker HC. Role of the dynorphin/kappa opioid receptor system in the motivational effects of ethanol. Alcohol Clin Exp Res. 2017;41:1402–18. doi: 10.1111/acer.13406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Karkhanis A, Holleran KM, Jones SR. Dynorphin/kappa opioid receptor signaling in preclinical models of alcohol, drug and food addiction. Int Rev Neurobiol. 2017;136:53–88. doi: 10.1016/bs.irn.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 83.Kissler JL, Walker BM. Dissociating motivational from physiological withdrawal in alcohol dependence: role of central amygdala □-opioid receptors. Neuropsychopharmacology. 2016;41:560–7. doi: 10.1038/npp.2015.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Negus SS, Mello NK. Opioid antinociception in ovariectomized monkeys: comparison with antinociception in males and effects of estradiol replacement. J Pharm Exp Ther. 1999;290:1132–40. [PubMed] [Google Scholar]
- 85.Rasmussen K, White DA, Acri JB. NIDA’s medication development priorities in response to the opioid crisis: ten most wanted. Neuropsychopharmacology. 2019;44:657–9. doi: 10.1038/s41386-018-0292-5. [DOI] [PMC free article] [PubMed] [Google Scholar]