

Abstract
Major depressive disorder (MDD) is the most prevalent mental disorder that affects more than 200 million people worldwide. Recent large-scale genome-wide association studies (GWAS) have identified multiple risk variants that show robust association with MDD. Nevertheless, how the identified risk variants confer risk of MDD remains largely unknown. To identify risk variants that are associated with gene expression in human brain and to identify genes whose expression change may contribute to the susceptibility of MDD, we systematically integrated the genetic associations from a large-scale MDD GWAS (N = 480,359) and brain expression quantitative trait loci (eQTL) data (N = 494) using a Bayesian statistical framework (Sherlock). Sherlock integrative analysis showed that FLOT1 was significantly associated with MDD (P = 6.02 × 10−6), suggesting that risk variants may contribute to MDD susceptibility through affecting FLOT1 expression. We further examined the expression level of FLOT1 in MDD cases and controls and found that FLOT1 was significantly upregulated in brains and peripheral blood of MDD cases compared with controls (European sample). Interestingly, we found that FLOT1 expression was also significantly upregulated in peripheral blood of first-episode drug-naive MDD cases compared with controls (P = 1.01 × 10−7, Chinese sample). Our study identified FLOT1 as a novel MDD risk gene whose expression level may play a role in MDD. In addition, our findings also suggest that risk variants may confer risk of MDD through affecting expression of FLOT1. Further functional investigation of FLOT1 may provide new insights for MDD pathogenesis.
Subject terms: Genetics, Depression
Introduction
MDD is a common mental disorder with complex etiology. The core symptoms of MDD include depressed mood, loss of interests, hopelessness, sleep disturbance and anxiety [1]. As the most prevalent mental disorder (the life time prevalence is about 15% [2, 3]), MDD affects over 200 million people worldwide [4]. In fact, MDD is the leading cause of disability worldwide and is a major contributor to the global burden of disease [5–7]. Though MDD becomes a major threat to global health and imposes great economic burden on society [8], the etiology of MDD remains largely unknown.
Accumulating evidence indicates that MDD is caused by a combination of genetic and environmental factors. Several environmental factors, including major life changes, stress, traumatic events, childhood abuse, and substance abuse were reported to have a role in MDD [9]. In addition to environmental factors, genetic factors also have pivotal role in MDD. The heritability of MDD is estimated around 40% [10], indicating the important role of genetic factors in MDD. To identify the genetic risk variants for MDD, numerous genetic studies were carried out in world populations [11–14]. Though several promising candidate genes (e.g., SIRT1 and LHPP) have been reported [15], identification of MDD risk variants and genes remains a major challenge in psychiatric genetics. For example, though genetic variants near SIRT1 and LHPP were reported to be significantly associated with MDD in Chinese population [15], recent large-scale GWAS did not found significant association between these risk variants and MDD in European populations [16]. The frequency of the risk alleles of the identified risk variants (near SIRT1 and LHPP) shows dramatic difference in Chinese and European populations (i.e., the risk alleles are common in Chinese population. However, the frequency of the risk alleles are very low in European population), suggesting the population heterogeneity of MDD risk variants.
In addition to population heterogeneity, the relative low heritability of MDD also impedes the identification of risk variants and genes. In 2012, the Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium carried out a large-scale GWAS in European population [13]. Though more than 75,000 subjects were included in their final analysis, no genome-wide significant variants were identified. In 2016, Hyde et al. performed a larger GWAS (a total of 459,472 individuals were used) and successfully identified 15 risk loci for MDD [16]. In 2018, Wray et al. conducted a large-scale genome-wide association meta-analysis (135,458 cases and 344,901 controls were included) and identified 44 risk variants for MDD [17]. Recently, Howard et al. carried out the largest MDD GWAS meta-analysis (246,363 cases and 561,190 controls were included) and reported 102 independent risk variants [18]. Though genetic study of MDD have made great achievements in recent years and over 100 genome-wide significant risk variants have been identified, how the reported risk variants affect MDD pathogenesis remains elusive. Considering the fact that most of identified MDD risk variants are located in non-coding region, it is likely that these risk variants exert their effects on MDD through affecting gene expression. To identify MDD risk variants that were associated with gene expression in human brain and to identify genes whose expression change may have a role in MDD, we carried out a Bayesian integrative analysis (Sherlock) through combining genetic associations from a large-scale GWAS of MDD and brain eQTL data. Sherlock integrative analysis identified 18 genes whose expression level may have a role in MDD. We further explored the expression level of these 18 genes and found that FLOT1 was consistently upregulated in brain and peripheral blood of MDD cases compared with controls (European populations). Interestingly, we found that FLOT1 was also significantly upregulated in peripheral blood of first-episode drug-naive MDD cases compared with controls (Chinese populations). Our study identifies FLOT1 as a novel risk gene for MDD and suggests that risk variants may contribute to MDD susceptibility through affecting FLOT1 expression.
Materials and methods
MDD GWAS used in this study
The genetic associations from a large-scale MDD GWAS [17] were used in this study. Wray et al. performed a genome-wide association meta-analysis recently and identified 44 genome-wide significant risk variants for MDD [17]. Briefly, seven MDD cohorts were included (including PGC29 [13], deCODE, Generation Scotland [19, 20], GERA [21], iPSYCH [22], UK Biobank [23], and 23andme [16]) in study of Wray et al. and a total of 135,458 MDD cases and 344,901 controls were used in the final meta-analysis. Sample recruitment, diagnosis of MDD, genotyping, and statistical analyses can be found in individual cohort. We first downloaded the genome-wide associations from the PGC website (which includes GWAS summary statistics from a combined meta-analysis of PGC29 and five cohorts (deCODE, GenScotland, GERA, iPSYCH, and UK Biobank). We then conducted a meta-analysis though combining the samples from above six cohorts and 23andme [14, 16]. Meta-analysis was performed as previously described [14] using PLINK [24] (the fixed-effect model was used).
Brain eQTL data
Previous studies have reported that the dorsolateral prefrontal cortex (DLPFC) may have pivotal roles in MDD [25, 26], suggesting that the DLPFC may represent a proper brain region to study the effects of MDD risk variants. We thus used the DLPFC to identify genes whose expression level were affected by the reported MDD risk variants. Brain expression quantitative trait loci (eQTL) from Ng et al. [27] were used in this study. Ng et al. created a multi-omic resource through using brain tissues from the DLPFC. In brief, 2093 individuals of European ancestry were collected by Ng et al. and genotyping was performed with the Illumina OmniQuad Express platform and the Affymetrix Genome-Wide HumanSNP Array6.0. They then created an xQTL resources through integrating genotypes with omic data, including gene expression (measured with RNA sequencing, RNA Seq), DNA methylation, and histone acetylation. Expression (quantified with RNA Sequencing) and genotype data of 494 individuals were used to perform eQTL analysis using Spearman’s rank correlation approach. More detailed information about brain tissue collection, genotyping, RNA quantification, quality control, and statistical analyses can be found in the original study [27].
Integration of MDD GWAS and brain eQTL using Sherlock integrative analysis
Most of the genome-wide significant MDD risk variants are located in non-coding region [17], implying that these variants may confer MDD risk through regulating gene expression. If expression level of a specific gene has a role in MDD, genetic variants that affect the expression of this gene are also likely to confer the risk of MDD. That is, risk variants contribute to MDD susceptibility through influencing gene expression. Thus, eQTL data may have important roles in dissecting genetic basis of MDD. To identify genes whose expression change may confer MDD risk, we integrated the single nucleotide polymorphism (SNP) associations from a large-scale MDD GWAS [17] and brain eQTL data from Ng et al. [27] using the Sherlock integrative analysis approach [28]. Briefly, Sherlock utilizes a Bayesian statistical framework to infer genes whose expression alternations influence the pathogenesis of complex diseases by using both cis (proximal to the gene) and trans (distal to the gene or on different chromosomes) genetic variations. For a given gene (whose expression level may have a role in MDD), usually there are several SNPs influence the expression level of this gene in a disease-related tissue (considering MDD is a mental disorder, brain may represent the most relevant tissue for exploring MDD pathogenesis). These expression-associated SNPs (hereafter referred to as eSNPs) may locate in different regions (e.g., promoter and enhancer regions) of the human genome and they may act synergistically to modulate the expression level of the given gene. Allelic differences (or genotype variation) at any of these eSNPs will affect gene expression level, and change of gene expression level eventually alter the disease susceptibility. Therefore, the eQTL of the given gene should overlap significantly with the risk loci of the disease.
Through integrating the GWAS associations and the eQTL data from a related tissue, Sherlock can identify genes whose expression change may have a role in disease risk. For a given gene, Sherlock calculates the Bayes factor for each eSNP individually and the sum of logarithm of Bayes factor (LBF) of each eSNP constitutes the total LBF score. The LBF score of a gene reflects the association strength between this gene and the disease. For example, a LBF of 5.0 means that the gene is more likely to be associated with the disease (exp(5.0) = ~148 times) than no association. For each SNP, there are three scenarios: (1) If this SNP is significantly associated with gene expression and disease simultaneously, a positive LBF score will be given. (2) If this SNP is only associated with gene expression (but is not associated with disease), a negative LBF score will be assigned. (3) Disease-only associations (i.e., this SNP is only associated with disease, not associated with gene expression) have no effect to the score. The P thresholds for cis and trans association were set to 1.0 × 10−3 and 5.0 × 10−5, respectively (as recommended by the default settings of Sherlock). And the P threshold for MDD GWAS associations was set to 1.0 × 10−3. More detailed information about the principle of Sherlock, statistical model, LBF calculation can be found in the original paper [28].
Expression analysis of FLOT1 in brains of MDD cases and controls
Sherlock identify MDD-associated genes under the assumption that gene expression change may have a role in the pathogenesis of MDD. To further explore if the genes identified by Sherlock integrative analysis were dysregulated in MDD cases compared with controls, we compared the expression level of the genes identified by Sherlock in brains of MDD cases and controls using the expression data from the study of Labonte et al. [29]. Briefly, Labonte et al. conducted a transcriptome analysis using brain tissues of 26 MDD cases (13 males and 13 females) and 22 controls (13 males and 9 females). Through dividing the individuals into females and males, Labonte et al. identified transcriptional change in male and female MDD cases compared with control male and female subjects. Tissues from six brain regions (including the dorsolateral prefrontal cortex (DLPFC), ventromedial prefrontal cortex (VMPFC), orbitofrontal cortex (OFC), ventral subiculum (VSUB), anterior insula (AINS), and nucleus accumbens (NAC)) were included in the study of Labonte et al. and transcriptome was obtained using RNA sequencing. More detail information can be found in the original publication [29].
In addition to the DLPFC, previous studies also indicated the pivotal role of hippocampus in MDD [30–32]. Many studies repeatedly reported that MDD cases have smaller hippocampal volumes than healthy individuals [30, 31, 33], suggesting the involvement of hippocampus in MDD. Therefore, we explored the expression of genes identified by Sherlock integrative analysis in hippocampal tissues of MDD cases and controls using expression data from Duric et al. [34]. In brief, Duric et al. collected post-mortem hippocampal tissues (two subfields were collected, including the CA1 pyramidal cell layer and the dentate gyrus granule cell layer) from 21 MDD cases and 18 healthy controls. Genome-wide gene expression analysis was performed with the human whole-genome expression MI Ready microarrays (Microarray, Inc.). More detailed information about sample collection, tissue isolation, RNA extraction, and expression quantification can be found in the original paper.
Expression analysis of FLOT1 in blood of MDD cases and controls (European sample)
In addition to genetic study and expression analysis in brains of MDD cases, gene expression analysis in blood is also a complementary approach to explore the potential role of a specific gene in disease [35, 36]. Furthermore, gene expression analysis in blood also provides a possibility to explore if FLOT1 can be used as a potential biomarker for MDD diagnosis in future. We thus examined FLOT1 expression in blood of MDD cases and controls using the expression data from Jansen et al. [37]. Briefly, Jansen et al. measured gene expression in peripheral blood of MDD cases and controls using a large-scale sample from the Netherlands Study of Depression and Anxiety. Three groups were used in the study of Jansen et al., including current MDD group (N = 882), remitted MDD group (N = 635), and control group N = 331). The Composite Interview Diagnostic Instrument (version 2.1) [38] was used for MDD diagnosis and blood was drown in the morning (8:30–9:30). RNA was extracted with Qiagen kit and gene expression was quantified the Affymetrix U219 arrays. More detailed information about MDD diagnosis, blood collection, RNA extraction, gene measurement, quality control, and statistical analyses can be found in the study of Jansen et al. [37].
Expression analysis of FLOT1 in blood of first-episode drug-naive MDD cases and controls (Chinese sample)
To further investigate if FLOT1 was dysregulated in MDD cases, we quantified FLOT1 expression in first-episode drug-naive MDD cases and controls using real-time quantitative PCR (qPCR). Briefly, 50 first-episode drug-naive MDD cases (22 males and 28 females, 35.22 ± 13.47 years) and 46 controls (23 males and 23 females, 39.59 ± 10.24 years) were recruited from the First people’s hospital of Yunnan province. MDD cases were diagnosed by experienced psychiatrists using the DSM-5 criteria. The MDD patient must experience at least five following symptoms in two weeks: (1) Mood depression; (2) Loss of interest or pleasure; (3) Significant weight changes; (4) Insomnia or hypersomnia; (5) Psychomotor agitation or retardation; (6) Low energy or fatigue; (7) Feeling of worthless; (8) Difficulty of thinking and concentration; and (9) Thought of death repeatedly. Cases with substance abuse or other psychiatric disorders were excluded from this study. The 17-item Hamilton Rating Scale for Depression (HAMD17) was used to assess the level of depression of MDD cases. The controls were also recruited from the First people’s hospital of Yunnan province and subjects with physical diseases and mental disorders were excluded. Informed consent was provided by all of the participants and this study was approved by Institutional Review Board (IRB) of Kunming institute of zoology, Chinese Academy of Sciences.
RNA was extracted from the peripheral blood using Trizol Reagent (Invitrogen, 10296028). The PrimeScript™ RT reagent Kit (Takara, RR047A) was used for reverse transcription and a total of 1 µg RNA was reverse transcribed according to the manufacturer’s instruction. TB Green™ Premix Ex Taq™ II (Tli RNaseH Plus) (Takara, RR820A) was used to quantify the FLOT1 expression level. The QuantStudio™12 K Flex (Applied Biosystems) instrument was used to conduct the real-time quantitative PCR (qPCR). The qPCR primers for FLOT1 gene are as follows: Forward primer: 5’-CTCGAACAGCTCAAGTCCAAAAAG-3′; Reverse primer: 5′-GCCATCTCGATCTCACTCAGGTACT-3′. ACTB was used as the internal control and the qPCR primers of ACTB are as follows: Forward primer: 5′-CATGTACGTTGCTATCCAGGC-3′; Reverse primer: 5′-CTCCTTAATGTCACGCACGAT-3′. The expression of FLOT1 in each individual was normalized to the expression of ACTB. The analysis of qPCR data was based on the Delta Ct (ΔCt) values and fold change (FC) was determined using 2−ΔΔCt approach [39]. All reactions were run in triplicate and Student’s t-test was used to test if FLOT1 expression in MDD cases was significantly different from controls.
Expression analysis of FLOT1 in different human tissues
To explore the expression profiling of FLOT1 gene in different human tissues, we utilized the gene expression data from the GTEx (Genotype-Tissue Expression) project (v6p release) [40]. We downloaded the gene expression data (i.e., median gene expression values) of 53 human tissues [40]. The expression level of genes were quantified by RNA sequencing (read per kilobase per million (RPKM) value was used to reflect the expression level of each gene). More detailed information about tissue collection, RNA extraction, expression quantification and data processing can be found on the GTEx website (http://gtexportal.org/) and related publications (https://gtexportal.org/home/publicationPage).
Expression analysis of FLOT1 in different cell types of the central nervous system
To explore the potential function of FLOT1 in brain, we explored FLOT1 expression in different cell types of the central nervous system using the data from Zhang et al. [41]. Zhang et al. isolated different cell types (including microglia/macrophage, astrocytes, oligodendrocytes, endothelial cells, and neurons) from the human brain and performed transcriptome analysis (using RNA sequencing method). We extracted the expression values (fragments per kilobase of transcript per million mapped reads (FPKM)) of FLOT1 in different cell types and compared the expression level of FLOT1 in different cell types. To compare if FLOT1 has similar expression pattern in different cell types of human and mouse brains, we also explored the expression of FLOT1 in different cell types of mouse brain using data from Cahoy et al. [42].
Temporal expression pattern of FLOT1 in developing and adult human brains
To explore the potential role of FLOT1 in the central nervous system, we examined the temporal expression pattern of FLOT1 in developing and adult human brain. Two independent expression data sets were used in this study. The first expression dataset was from the BrainSpan (Atlas of the Developing Human Brain (http://www.brainspan.org/) [43]. Gene expression values (based on RNA sequencing) of FLOT1 in the prefrontal cortex (PFC) (N = 42) was downloaded and transformed as previously described [44]. The transformed expression level was used to plot the temporal expression pattern of FLOT1 in developing and adult human brain. The second expression dataset was from the study of Colantuoni et al. [45]. Colantuoni et al. explored the temporal dynamics of gene expression in human prefrontal cortex using serial post-mortem brains from fetal to aging stages. The post-mortem brain tissues (PFC) of 267 normal (without neuropathological or neuropsychiatric diagnosis) subjects were included and transcriptome were measured with microarrays. More detail information about can be found in the original paper [45].
Association between rs2523593 and neuroticism
Neuroticism is an important risk factor for MDD [46, 47] and previous studies have showed significant genetic correlation between MDD and neuroticism [48, 49]. Our Sherlock integrative analysis showed that SNP rs2523593 may confer risk of MDD through affecting FLOT1 expression. To test if rs2523593 is also associated with neuroticism, we examined the association between rs2523593 and neuroticism using the data from the study of Nagel et al. [50]. Nagel et al. carried out a large GWAS meta-analysis (N = 449,484) on neuroticism and identified over 100 independent significant risk loci (P < 5 × 10−8) for neuroticism. We extracted the association significance between rs2523593 and neuroticism using a subset sample (N = 390,278) from Nagel et al. [50].
Results
Sherlock integrative analysis identifies 18 genes that showed significant association with MDD
To infer genes whose expression perturbations may confer risk of MDD, we used the Sherlock statistical inference method to integrate the SNP associations from a large-scale MDD GWAS [17] and brain eQTL [27]. Sherlock integrative analysis identified 18 genes whose expression change may contribute to MDD susceptibility (FDR < 0.01) (Table 1). These genes include BTN3A3, CCHCR1, PPP1R18, GPANK1, CLIC1, ZNF193, NOTCH4, BTN3A2, AGER, VARS2, DDAH2, LY6G5B, C4B, HLA-C, HIST1H4C, FLOT1, MICA, and C4A. For each gene, at least one SNP showed significant association with MDD and the expression of this gene simultaneously, suggesting that these genetic variants may confer MDD risk through affecting the expression of these genes. Of note, many of the risk genes identified by Sherlock are located in the MHC region, suggesting that genes in MHC region may have pivotal roles in the pathogenesis of MDD. The MDD risk SNPs that showed the most significant association with gene expression are rs13195402 (associated with expression of BTN3A2, eQTL P = 5.01 × 10−29) and rs2523593 (associated with expression of HLA-C, C4A, and FLOT1, eQTL P = 7.25 × 10−25, 4.98 × 10−19 and 1.37 × 10−15, respectively).
Table 1.
Top MDD-associated genes predicted by Sherlock integrative analysis
| Gene | Gene positiona | MHC | LBFb | P valuec | Supporting SNP | Cis or Trans | GWAS P valued | eQTL P valuee | FDRf |
|---|---|---|---|---|---|---|---|---|---|
| BTN3A3 | chr6:26440700–26453643 | No | 7.86 | 5.02 × 10–7 | rs35627490 | Cis | 4.43 × 10−7 | 2.65 × 10−4 | <0.01 |
| rs34150729 | Cis | 4.38 × 10−8 | 2.58 × 10−4 | ||||||
| CCHCR1 | chr6:31110216–31126015 | Yes | 5.50 | 5.02 × 10−7 | rs2523593 | Cis | 4.67 × 10−7 | 3.02 × 10−12 | <0.01 |
| PPP1R18 | chr6:30644166–30655672 | Yes | 5.42 | 5.02 × 10−7 | rs2523593 | Cis | 4.67 × 10−7 | 1.63 × 10−5 | <0.01 |
| GPANK1 | chr6:31629006-31634060 | Yes | 5.16 | 5.02 × 10−7 | rs2523593 | Cis | 4.67 × 10−7 | 2.93 × 10−7 | <0.01 |
| CLIC1 | chr6:31698358–31707540 | Yes | 5.15 | 5.02 × 10−7 | rs2523593 | Cis | 4.67 × 10−7 | 4.05 × 10−7 | <0.01 |
| ZNF193 | chr6:28192664–28201260 | No | 4.70 | 5.02 × 10−7 | rs13207689 | Cis | 4.74 × 10−8 | 2.50 × 10−6 | <0.01 |
| NOTCH4 | chr6:32162620–32191844 | Yes | 4.60 | 5.02 × 10−7 | rs2523593 | Cis | 4.67 × 10−7 | 4.13 × 10−7 | <0.01 |
| BTN3A2 | chr6:26365387–26378546 | No | 4.24 | 5.02 × 10−7 | rs13195402 | Cis | 4.79 × 10−9 | 5.01 × 10−29 | <0.01 |
| AGER | chr6:32148745–32152101 | Yes | 3.99 | 5.02 × 10−7 | rs2523593 | Cis | 4.67 × 10−7 | 4.00 × 10−4 | <0.01 |
| VARS2 | chr6:30876019–30894236 | Yes | 3.78 | 5.02 × 10−7 | rs2523593 | Cis | 4.67 × 10−7 | 1.16 × 10−11 | <0.01 |
| DDAH2 | chr6:31694815–31698394 | Yes | 3.77 | 5.02 × 10−7 | rs2523593 | Cis | 4.67 × 10−7 | 3.67 × 10−4 | <0.01 |
| LY6G5B | chr6:31637944–31641553 | Yes | 3.73 | 5.02 × 10−7 | rs2523593 | Cis | 4.67 × 10−7 | 2.06 × 10−5 | <0.01 |
| C4B | chr6:31982539–32003195 | Yes | 3.66 | 5.02 × 10−7 | rs2523593 | Cis | 4.67 × 10−7 | 2.49 × 10−4 | <0.01 |
| HLA-C | chr6:31236526–31239907 | Yes | 3.58 | 5.02 × 10−6 | rs2523593 | Cis | 4.67 × 10−7 | 7.25 × 10−25 | <0.01 |
| HIST1H4C | chr6:26104104–26104518 | No | 3.52 | 5.02 × 10−6 | rs34546498 | Cis | 4.05 × 10−7 | 1.58 × 10−7 | <0.01 |
| FLOT1 | chr6:30695486–30710510 | Yes | 3.33 | 6.02 × 10−6 | rs28849176 | Cis | 3.25 × 10−5 | 1.72 × 10−2 | <0.01 |
| rs2523593 | Cis | 4.67 × 10−7 | 1.37 × 10−15 | ||||||
| MICA | chr6:31371356–31383092 | Yes | 3.33 | 6.02 × 10−6 | rs2523593 | Cis | 4.67 × 10−7 | 6.66 × 10−12 | <0.01 |
| C4A | chr6:31949801–31970458 | Yes | 3.21 | 6.02 × 10−6 | rs2523593 | Cis | 4.67 × 10−7 | 4.98 × 10−19 | <0.01 |
FLOT1 was shown in bold
agene locations were based on hg19
bLBF, logarithm of Bayes factor. The LBF score of a gene reflects the association strength between this gene and MDD. For example, a LBF of 5.0 means that the gene is more likely to be associated with the disease (exp(5.0) = ~148 times) than no association
cP value from Sherlock integrative analysis. Larger LBF corresponds to smaller P value
dGWAS P value indicates the association significance between this SNP and MDD
eeQTL P value indicates the association significance between this SNP and gene expression in brain
fFDR was corrected by Benjamini–Hochberg procedure
Dysregulation of FLOT1 in brains of MDD cases compared with controls
Sherlock integrative analysis identified 18 risk genes that showed significant association (FDR < 0.01) with MDD (Table 1). Of note, Sherlock identifies MDD risk genes under the assumption that expression change of the predicted genes (from Sherlock analysis) may have a role in MDD. We thus compared the expression level of the significant genes identified by Sherlock in brains of MDD cases and controls. As previous studies showed that the dorsolateral prefrontal cortex (DLPFC) and anterior insula (AINS) may have pivotal roles in MDD [51–53], we compared the expression level of the significant genes (i.e., genes listed in Table 1) in the DLPFC and AINS of MDD cases and controls using the expression data from Labonte et al. [29]. We first examined expression level of significant genes identified by Sherlock analysis in the DLPFC. Among the genes listed in Table 1, only two genes (FLOT1 and HLA-C) showed significant difference (P < 0.05, uncorrected) in the DLPFC of MDD cases compared with controls (Fig. 1a, b). Of note, both FLOT1 and HLA-C were significantly upregulated in the DLPFC of MDD cases compared with controls (P = 0.040 and 0.039, respectively). To further validate the dysregulation of FLOT1 and HLA-C in brains of MDD cases, we compared the expression of FLOT1 and HLA-C in the AINS. Again, we found that both FLOT1 and HLA-C were significantly upregulated in the AINS of MDD cases compared with controls (P = 0.046 and 0.027, respectively) (Fig. 1c, d). These expression results suggest that dysregulation of FLOT1 and HLA-C may have a role in MDD.
Fig. 1.

Significant upregulation of FLOT1 in brains of MDD cases compared with controls. a, c Compared with controls, FLOT1 was significantly upregulated in the dorsolateral prefrontal cortex (DLPFC) and anterior insula (AINS) of MDD cases. b, d HLA-C was significantly upregulated in the dorsolateral prefrontal cortex (DLPFC) and anterior insula (AINS) of MDD cases. One-tailed Student’s t-test was used to compare if the different was significant (P < 0.05)
Our above expression analysis suggested that dysregulation of FLOT1 and HLA-C may have a role in MDD. To further verify the dysregulation of FLOT1 and HLA-C in brains of MDD cases, we examined FLOT1 and HLA-C expression using an independent expression dataset (i.e., expression data from Duric et al. [34]). We found that HLA-C expression was not significantly changed in brains of MDD cases compared with controls in the study of Duric et al. [34]. Intriguingly, we found that FLOT1 showed a trend of significant upregulation in brains of MDD cases compared with controls (1.53 fold upregulation in MDD cases, P = 0.07) in the study of Duric et al. It should be noted that the P values of FLOT1 and HLA-C were not corrected for multiple testing. After correcting for multiple testing (Bonferroni correction), these two genes did not show significant expression difference (corrected P > 0.05) in MDD cases and controls. As FLOT1 showed consistent upregulation in studies of Labonte et al. [29] and Duric et al. [34], we used single-tailed Student’s t-test to explore if FLOT1 was significantly upregulated in MDD cases compared with controls. More work is need to elucidate if FLOT1 and HLA-C were significantly dysregulated in MDD cases. Taken together, our Sherlock integrative analysis suggests that MDD-associated genetic variants may confer risk of MDD through affecting FLOT1 expression. Consistent with this hypothesis, we found that FLOT1 showed a trend of significant upregulation in MDD cases compared with controls, suggesting FLOT1 dysregulation may have a pivotal role in MDD.
Dysregulation of FLOT1 in peripheral blood of MDD cases compared with controls (European sample)
Expression of FLOT1 was significantly upregulated in brain of MDD cases in study of Labonte et al. In addition, FLOT1 also showed a trend of significant upregulation in brain of MDD cases in study of Duric et al. These expression data suggest that dysregulation of FLOT1 may have a role in MDD. To further verify the dysregulation of FLOT1 in MDD, we examined FLOT1 expression in peripheral blood of MDD cases and controls using data from Jansen et al. [37]. Consistent with the significant upregulation in brains of MDD cases, we found that FLOT1 was also significantly upregulated in peripheral blood of current MDD cases compared with controls (P = 0.031, 882 cases and 331 controls). Interestingly, we noticed that FLOT1 was also significantly upregulated in peripheral blood of remitted MDD cases compared with controls (P = 0.035, 635 remitted cases and 331 controls). When current MDD cases and remitted cases were combined, FLTO1 showed significant upregulation in current and remitted MDD cases compared controls (P = 0.020, 1,517 cases and 331 controls). Taken together, the significant upregulation of FLOT1 in both brains and bloods of MDD cases suggests that dysregulation of FLOT1 may have a role in MDD.
Dysregulation of FLOT1 in first-episode drug-naive MDD cases compared with controls (Chinese sample)
FLOT1 showed a consistent significant upregulation in brains and peripheral blood of MDD cases compared with controls, implying that dysregulation of FLOT1 may have a role in MDD. However, considering that the MDD cases recruited in study of Labonte et al. [29] and Jasen et al. [37] were not first-episode drug-naive patients, these studies could not exclude the potential effects of depression medications (e.g., using of antidepressants) on gene expression. We thus investigated FLOT1 expression in first-episode drug-naive MDD cases and controls using qPCR. Consistent with the significant upregulation observed in brains and peripheral blood of MDD cases in European sample, we found that FLOT1 expression was significantly elevated in first-episode drug-naive MDD cases (Chinese sample) compared with controls in Chinese sample (P = 1.01 × 10−7, two-tailed Student’s t-test) (Fig. 2). The significance was strengthened after three outlying MDD cases were removed (P = 1.01 × 10−10). Significant upregulation of FLOT1 in peripheral blood of first-episode drug-naive MDD cases provide further evidence that support the involvement of FLOT1 in MDD. Collectively, the consistent upregulation of FLOT1 observed in independent MDD cases supports that dysregulation of FLOT1 may have a pivotal role in MDD. Sherlock integrative analysis and expression results suggest that genetic variants may confer risk of MDD through affecting FLOT1 expression.
Fig. 2.
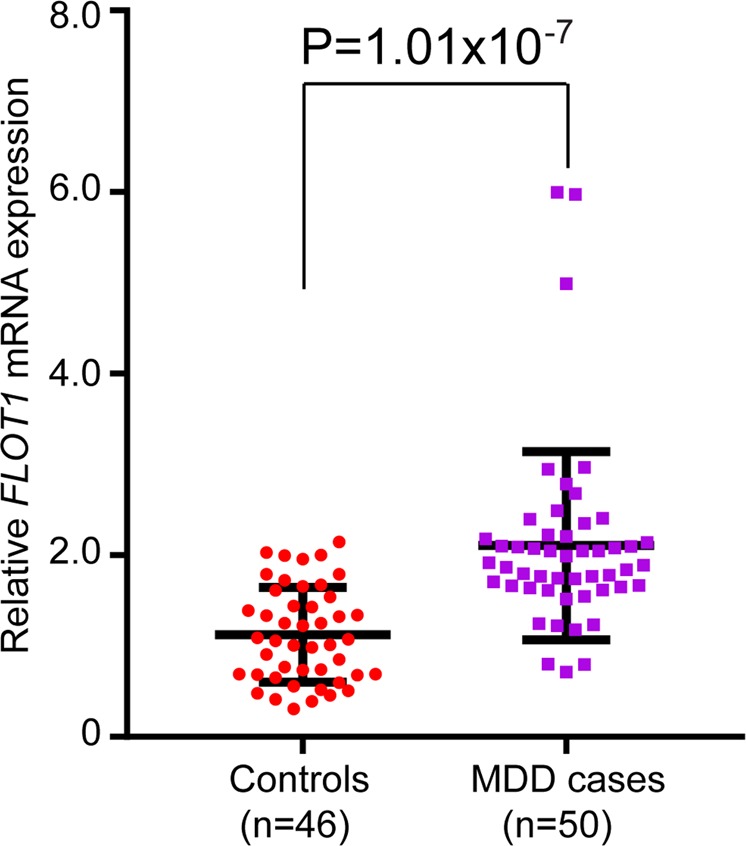
Significant upregulation of FLOT1 in peripheral blood of first-episode drug-naive MDD cases compared with controls. Quantitative PCR showed that FLOT1 was significantly upregulated (P = 1.01 × 10−7) in peripheral blood of first-episode drug-naive MDD cases compared with controls. Expression of FLOT1 was quantified in 46 controls and 50 MDD cases. Two-tailed Student’s t-test was used to compare if the different was significant (P < 0.05)
FLOT1 is widely expressed in diverse human tissues
Our Sherlock integrative analysis suggests that FLOT1 is a MDD risk gene whose expression level may have a role in MDD. Consistent with this, we found that FLOT1 was dysregulated in MDD cases compared controls. To further explore the potential function of FLOT1, we examined FLOT1 expression in diverse human tissues using data from the GTEx. We found that FLOT1 was widely expressed in different human tissues, with the highest expression level in whole blood (Fig. 3). This expression data implies that FLOT1 may have a role in different human tissues.
Fig. 3.

FLOT1 is widely expressed in diverse human tissues. RNA sequencing-based expression data from GTEx was used to explore FLOT1 expression. FLOT1 was abundantly expressed in different human tissues, with the highest expression level in blood
FLOT1 is widely expressed in different cell types of the central nervous system
We further explored the expression of FLOT1 in different cell types (including astrocytes, neurons, oligodendrocytes, and microglia cells) of the central nervous system. We found that FLOT1 was widely expressed in different cell types of the human brain (Fig. 4a). To compare if FLOT1 has similar expression patterns in different cell types of the human and mouse brains, we examined FLOT1 expression in different cell types of the mouse brain. Compared with the relatively low expression level observed in different cell types of the human brain, we found that the expression level of Flot1 was high in different cell types of the mouse brain (Fig. 4b). Of note, we found that Flot1 has the highest expression level in neurons and microglia cells of mouse brain (Fig. 4b). These expression data imply that FLOT1 may have a role in the central nervous system.
Fig. 4.

Expression of FLOT1 in different cell types of human and mouse brain. a FLOT1 was expressed in different cell types (including neuron, microglia, oligodendrocytes, and astrocytes) of human brain. b FLOT1 was abundantly expressed in different cell types of mouse brain, with the relatively high expression level in neurons and microglia cells. c, d Expression of FLOT1 was relatively low at early developmental stage. With the progress of development, FLOT1 expression was gradually increased, and peaked at adult stage. c Expression data from the BrainSpan was used for plotting. d Expression data from the BrainCloud was used for plotting
Temporal expression pattern of FLOT1 in developing and adult human brains
We explored the expression pattern of FLOT1 in developing and adult human brain using the expression data from the BrainSpan [43] and Colantuoni et al. [45]. We found that the expression of FLOT1 was relatively low at early developmental stage in both data sets (Fig. 4c, d). However, FLOT1 expression was gradually increased with the progress of development, and peaked at adult stage (Fig. 4c, d). These temporal expression pattern suggests that FLOT1 may have different roles during brain development.
FLOT1 expression-associated SNP (rs2523593) showed significant association with neuroticism
Previous studies have revealed that MDD showed significant genetic correlation with neuroticism [17, 50], an important risk factor for MDD [46] and anxiety [54]. Our Sherlock integrative analysis indicated that rs2523593 was significantly associated with MDD and FLOT1 expression simultaneously (Table 1), suggesting that SNP rs2523593 may confer risk of MDD through affecting FLOT1 expression. Considering the significant genetic correlation between MDD and neuroticism, we hypothesized that rs2523593 may also be associated with neuroticism. We thus explored the association between rs2523593 and neuroticism using the data from the study of Nagel et al. [50]. We found that rs2523593 was also significantly associated with neuroticism (P = 2.47 × 10−5). Intriguingly, we found that the MDD risk-associated allele (i.e., T allele) also increases the risk of neuroticism. The association between rs2523593 and neuroticism provided additional evidence that supports rs2523593 may represent a promising risk SNP for MDD and neuroticism.
Discussion
Recent large-scale GWAS have identified multiple risk variants that show strong association with MDD. Nevertheless, how the identified risk variants confer MDD risk remains largely unknown. Considering the fact that most of the identified MDD risk variants were located in non-coding region, it is possible that these MDD-associated variants confer risk of MDD through regulating gene expression. To identify MDD risk genetic variants that were associated with gene expression in human brains and to prioritize genes whose expression change may have a role in MDD, we systematically integrated the genetic associations from the a large-scale MDD GWAS and brain eQTL using Sherlock integrative analysis. We identified 18 genes whose perturbations may have a role in MDD. Further expression analysis showed that FLOT1 was consistently upregulated in brains and peripheral blood of MDD cases compared with controls (European sample). Interestingly, we found that FLOT1 was significantly upregulated in first-episode drug-naive MDD cases (Chinese sample). Our integrative analysis and gene expression results provide convergent lines of evidence that supports the potential involvement of FLOT1 in MDD.
FLOT1 encodes Flotillin-1, a caveolae-associated integral membrane protein. The function of FLOT1 has not been fully determined. However, previous studies have suggested that Flotillin-1 may have pivotal roles in the central nervous system [55, 56]. The protein sequence of FLOT1 is highly conserved from Drosophila to humans [57], suggesting the important role of FLOT1. In addition, FLOT1 was showed to be abundantly expressed in Drosophila brain [57], implying that FLOT1 may have a role in the central nervous system. Consistently, it has been reported that FLOT1 is involved in neuronal differentiation, axon outgrowth, and regeneration [55]. These lines of evidence demonstrated the important role of FLOT1 in the central nervous system. Interestingly, Cremona et al. [56] showed that Flotillin-1 played an important role in the internalization of dopamine transporter (DAT) and the glial glutamate transporter EAAT2, members of two different neurotransmitter transporters families. In addition to dopamine transporter and the glial glutamate transporter, a recent study of Reisinger et al. revealed that Flotillin-1 modulates serotonergic neurotransmission through interacting with the serotonin transporter [58]. Considering the important role of serotonin transporter, dopamine transporter and glutamate transporter in MDD [59–63], it is possible that FLOT1 may be involved in MDD through modulating the internalization of dopamine transporter and the glial glutamate transporter EAAT2. However, more work is needed to investigate the exact role of FLOT1 in the pathogenesis of MDD.
In addition to playing important roles in the central nervous system, recent studies also revealed that FLOT1 has important roles in cancer. Dysregulation of FLOT1 expression was reported in different cancers, including epithelial ovarian tumors [64], transitional cell carcinoma [65], nasopharyngeal carcinoma [66], and renal cell carcinoma [67]. Moreover, studies also showed that FLOT1 regulates migration, invasion, and proliferation of tumor cells [68, 69]. These studies demonstrated the important role of FLOT1 in cancer. Intriguingly, it has been reported that depression is a risk factor for cancer prognosis [70]. Besides, previous studies also showed that prevalence of MDD in cancer patients was higher compared with the general population [71, 72].
Compared with the high expression level in mouse brain, we found that FLOT1 expression was relatively low in different cell types of the human brain. These results suggest that FLOT1 may have different roles in the human and mouse brain. In addition, these observations also implicate that we need to consider the difference of FLOT1 expression pattern when we use transgenic (or knockout) mouse model to study the function of Flot1 in the central nervous system (i.e., mouse model may not fully mimic (or reflect) the function of FLOT1 in humans as this gene has different expression level in the human and mouse brain).
We found that the MDD risk SNP rs2523593 was also associated with neuroticism. Previous studies indicated that MDD has a high genetic correlation with neuroticism [48, 49] and individuals with higher score in neuroticism tend to be more susceptible to MDD [46, 73], suggesting that exploring the genetic basis of neuroticism may provide insights into etiology of MDD. Of note, we found that the association significance between rs2523593 and neuroticism was less significant than MDD. This could be due to the smaller sample size of neuroticism GWAS (N = 390,278) compared with MDD GWAS (N = 480,359). More work is needed to explore if rs2523593 is a risk variant for neuroticism.
Sherlock integrative analysis identified 18 MDD risk genes (Table 1) whose expression change may have a role in MDD. In addition to FLOT1, following genes (including NOTCH4, C4A and C4B) may also be relevant to MDD from the aspect of biological function. Previous studies have indicated the important role of Notch signaling pathway in neuronal development and differentiation [74, 75]. Interestingly, it has been reported that NOTCH4 is associated with schizophrenia [76], a severe psychiatric disorder. C4A and C4B encode complement C4-A and C4-B, respectively. A recent study revealed that C4 protein played an important role in synapse development and C4 genes (C4A and C4B) were associated with schizophrenia [77], suggesting the pivotal roles of C4A and C4B in the central nervous system. Collectively, these lines of evidence suggest that NOTCH4, C4A and C4B may also have a role in MDD. Nevertheless, more work is needed to explore the role of these genes in MDD.
There are several limitations of this study. First, though our Sherlock integrative analysis showed that genetic variants may confer risk of MDD through regulating FLOT1 expression. Due to the complexity of linkage disequilibrium and gene regulation, the causal (or functional) variants that regulate the expression of FLOT1 and the exact regulatory mechanism remain elusive. Second, it is possible that FLOT1 have a role in neurotransmission as previous studies have shown that Flotillin-1 played an important role in the internalization of dopamine transporter (DAT) and the glial glutamate transporter EAAT2. However, currently we still do not know the role of FLOT1 dysregulation in MDD. Third, we examined the association between FLOT1 and MDD in Chinese population by using the CONVERGE data (including 5303 MDD cases and 5337 controls) [15]. We found that rs2523593 was not significantly associated with MDD in Chinese population (P = 0.70). However, A SNP (rs115494986, which showed low level LD with rs2523593 (r2 = 0.20)) near FLOT1 is marginally associated with MDD in CONVERGE (P = 0.029). Further validation of the association between FLOT1 and MDD in independent samples is needed. Fourth, our Sherlock integrative analysis identified 18 genes whose expression perturbation may have a role in MDD (Table 1). Nevertheless, we only focused on FLOT1 in this study as expression of FLOT1 was consistently upregulated in MDD cases compared with controls. Further work is needed to investigate if other genes predicted by Sherlock integrative analysis have a role in MDD.
In summary, our study provides convergent lines of evidence that support FLOT1 is a new MDD risk gene. Our results suggested that MDD-associated variants may confer MDD risk through affecting FLOT1 expression. The significant upregulation of FLOT1 in MDD also suggests that FLOT1 may be targeted in future as a potential marker for future therapeutics and diagnostics.
Funding and disclosure
This study was equally supported by the National Key Research and Development Program of China (Stem Cell and Translational Research) (2016YFA0100900) and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB13000000) (to X.-J.L). Also was supported by the Key Research Project of Yunnan Province (2017FA008 to X.-J.L), and the Applied basic research project of Yunnan province (2017FE468–129).
Acknowledgements
We would like to thank the research participants and employees of 23andMe for making this work possible. We thank the following members of the 23andMe Research Team: Michelle Agee, Babak Alipanahi, Adam Auton, Robert K. Bell, Katarzyna Bryc, Sarah L. Elson, Pierre Fontanillas, Nicholas A. Furlotte, David A. Hinds, Karen E. Huber, Aaron Kleinman, Nadia K. Litterman, Jennifer C. McCreight, Matthew H. McIntyre, Joanna L. Mountain, Elizabeth S. Noblin, Carrie A.M. Northover, Steven J. Pitts, J. Fah Sathirapongsasuti, Olga V. Sazonova, Janie F. Shelton, Suyash Shringarpure, Chao Tian, Joyce Y. Tung, Vladimir Vacic, and Catherine H. Wilson, who generated and made the summary statistics available for us, which made this work possible.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Jingmei Zhong, Shiwu Li, Wanli Zeng and Xiaoyan Li
References
- 1.Kennedy SH. Core symptoms of major depressive disorder: relevance to diagnosis and treatment. Dialog- Clin Neurosci. 2008;10:271–7. doi: 10.31887/DCNS.2008.10.3/shkennedy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hasin DS, Goodwin RD, Stinson FS, Grant BF. Epidemiology of major depressive disorder: results from the National Epidemiologic Survey on Alcoholism and Related Conditions. Arch Gen Psychiatry. 2005;62:1097–106. doi: 10.1001/archpsyc.62.10.1097. [DOI] [PubMed] [Google Scholar]
- 3.Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R) JAMA. 2003;289:3095–105. doi: 10.1001/jama.289.23.3095. [DOI] [PubMed] [Google Scholar]
- 4.GBD 2015 Disease and Injury Incidence and Prevalence Collaborators.. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388:1545–602. doi: 10.1016/S0140-6736(16)31678-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJ. Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet. 2006;367:1747–57. doi: 10.1016/S0140-6736(06)68770-9. [DOI] [PubMed] [Google Scholar]
- 6.Wittchen HU, Jacobi F, Rehm J, Gustavsson A, Svensson M, Jonsson B, et al. The size and burden of mental disorders and other disorders of the brain in Europe 2010. Eur Neuropsychopharmacol. 2011;21:655–79. doi: 10.1016/j.euroneuro.2011.07.018. [DOI] [PubMed] [Google Scholar]
- 7.Ferrari AJ, Charlson FJ, Norman RE, Patten SB, Freedman G, Murray CJ, et al. Burden of depressive disorders by country, sex, age, and year: findings from the global burden of disease study 2010. PLoS Med. 2013;10:e1001547. doi: 10.1371/journal.pmed.1001547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greenberg PE, Fournier AA, Sisitsky T, Pike CT, Kessler RC. The economic burden of adults with major depressive disorder in the United States (2005 and 2010) J Clin Psychiatry. 2015;76:155–62. doi: 10.4088/JCP.14m09298. [DOI] [PubMed] [Google Scholar]
- 9.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (5th ed), Arlington: American Psychiatric Publishing. ISBN 978-0-89042-555-8. 2013.
- 10.Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry. 2000;157:1552–62. doi: 10.1176/appi.ajp.157.10.1552. [DOI] [PubMed] [Google Scholar]
- 11.Lewis CM, Ng MY, Butler AW, Cohen-Woods S, Uher R, Pirlo K, et al. Genome-wide association study of major recurrent depression in the U.K. population. Am J Psychiatry. 2010;167:949–57. doi: 10.1176/appi.ajp.2010.09091380. [DOI] [PubMed] [Google Scholar]
- 12.Rietschel M, Mattheisen M, Frank J, Treutlein J, Degenhardt F, Breuer R, et al. Genome-wide association-, replication-, and neuroimaging study implicates HOMER1 in the etiology of major depression. Biol Psychiatry. 2010;68:578–85. doi: 10.1016/j.biopsych.2010.05.038. [DOI] [PubMed] [Google Scholar]
- 13.Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM, Breen G, et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry. 2012;18:497–511. doi: 10.1038/mp.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li X, Luo Z, Gu C, Hall LS, McIntosh AM, Zeng Y, et al. Common variants on 6q16.2, 12q24.31 and 16p13.3 are associated with major depressive disorder. Neuropsychopharmacology. 2018;43:2146–53. doi: 10.1038/s41386-018-0078-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.CONVERGE consortium*.. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature. 2015;523:588–91. doi: 10.1038/nature14659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hyde CL, Nagle MW, Tian C, Chen X, Paciga SA, Wendland JR, et al. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat Genet. 2016;48:1031–6. doi: 10.1038/ng.3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet. 2018;50:668–81. doi: 10.1038/s41588-018-0090-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Howard DM, Adams MJ, Clarke TK, Hafferty JD, Gibson J, Shirali M et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat Neurosci. 2019;10.1038/s41593-41018-40326-41597. Electronic version published on Feb 4, 2019. [DOI] [PMC free article] [PubMed]
- 19.Smith BH, Campbell A, Linksted P, Fitzpatrick B, Jackson C, Kerr SM, et al. Cohort Profile: Generation Scotland: Scottish Family Health Study (GS:SFHS). The study, its participants and their potential for genetic research on health and illness. Int J Epidemiol. 2013;42:689–700. doi: 10.1093/ije/dys084. [DOI] [PubMed] [Google Scholar]
- 20.Fernandez-Pujals AM, Adams MJ, Thomson P, McKechanie AG, Blackwood DH, Smith BH, et al. Epidemiology and heritability of major depressive disorder, stratified by age of onset, sex, and illness course in generation Scotland: Scottish Family Health Study (GS:SFHS) PLoS ONE. 2015;10:e0142197. doi: 10.1371/journal.pone.0142197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Banda Y, Kvale MN, Hoffmann TJ, Hesselson SE, Ranatunga D, Tang H, et al. Characterizing Race/Ethnicity and Genetic Ancestry for 100,000 Subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) Cohort. Genetics. 2015;200:1285–95. doi: 10.1534/genetics.115.178616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pedersen CB, Bybjerg-Grauholm J, Pedersen MG, Grove J, Agerbo E, Baekvad-Hansen M, et al. The iPSYCH2012 case-cohort sample: new directions for unravelling genetic and environmental architectures of severe mental disorders. Mol Psychiatry. 2018;23:6–14. doi: 10.1038/mp.2017.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Allen NE, Sudlow C, Peakman T, Collins R. UK biobank data: come and get it. Sci Transl Med. 2014;6:224ed224. doi: 10.1126/scitranslmed.3008601. [DOI] [PubMed] [Google Scholar]
- 24.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koenigs M, Huey ED, Calamia M, Raymont V, Tranel D, Grafman J. Distinct regions of prefrontal cortex mediate resistance and vulnerability to depression. J Neurosci. 2008;28:12341–8. doi: 10.1523/JNEUROSCI.2324-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koenigs M, Grafman J. The functional neuroanatomy of depression: distinct roles for ventromedial and dorsolateral prefrontal cortex. Behav Brain Res. 2009;201:239–43. doi: 10.1016/j.bbr.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ng B, White CC, Klein HU, Sieberts SK, McCabe C, Patrick E, et al. An xQTL map integrates the genetic architecture of the human brain’s transcriptome and epigenome. Nat Neurosci. 2017;20:1418–26. doi: 10.1038/nn.4632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He X, Fuller CK, Song Y, Meng Q, Zhang B, Yang X, et al. Sherlock: detecting gene-disease associations by matching patterns of expression QTL and GWAS. Am J Hum Genet. 2013;92:667–80. doi: 10.1016/j.ajhg.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Labonte B, Engmann O, Purushothaman I, Menard C, Wang J, Tan C, et al. Sex-specific transcriptional signatures in human depression. Nat Med. 2017;23:1102–11. doi: 10.1038/nm.4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.den Heijer T, Tiemeier H, Luijendijk HJ, van der Lijn F, Koudstaal PJ, Hofman A, et al. A study of the bidirectional association between hippocampal volume on magnetic resonance imaging and depression in the elderly. Biol Psychiatry. 2011;70:191–7. doi: 10.1016/j.biopsych.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 31.Schmaal L, Veltman DJ, van Erp TG, Samann PG, Frodl T, Jahanshad N, et al. Subcortical brain alterations in major depressive disorder: findings from the ENIGMA Major Depressive Disorder working group. Mol Psychiatry. 2016;21:806–12. doi: 10.1038/mp.2015.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Otte C, Gold SM, Penninx BW, Pariante CM, Etkin A, Fava M, et al. Major depressive disorder. Nat Rev Dis Prim. 2016;2:16065. doi: 10.1038/nrdp.2016.65. [DOI] [PubMed] [Google Scholar]
- 33.Geerlings MI, Gerritsen L. Late-life depression, hippocampal volumes, and hypothalamic-pituitary-adrenal axis regulation: a systematic review and meta-analysis. Biol Psychiatry. 2017;82:339–50. doi: 10.1016/j.biopsych.2016.12.032. [DOI] [PubMed] [Google Scholar]
- 34.Duric V, Banasr M, Licznerski P, Schmidt HD, Stockmeier CA, Simen AA, et al. A negative regulator of MAP kinase causes depressive behavior. Nat Med. 2010;16:1328–32. doi: 10.1038/nm.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ayalew M, Le-Niculescu H, Levey DF, Jain N, Changala B, Patel SD, et al. Convergent functional genomics of schizophrenia: from comprehensive understanding to genetic risk prediction. Mol Psychiatry. 2012;17:887–905. doi: 10.1038/mp.2012.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wingo AP, Velasco ER, Florido A, Lori A, Choi DC, Jovanovic T, et al. Expression of the PPM1F gene is regulated by stress and associated with anxiety and depression. Biol Psychiatry. 2018;83:284–95. doi: 10.1016/j.biopsych.2017.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jansen R, Penninx BW, Madar V, Xia K, Milaneschi Y, Hottenga JJ, et al. Gene expression in major depressive disorder. Mol Psychiatry. 2016;21:444. doi: 10.1038/mp.2015.94. [DOI] [PubMed] [Google Scholar]
- 38.Wittchen HU. Reliability and validity studies of the WHO—Composite International Diagnostic Interview (CIDI): a critical review. J Psychiatr Res. 1994;28:57–84. doi: 10.1016/0022-3956(94)90036-1. [DOI] [PubMed] [Google Scholar]
- 39.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 40.GTEx Consortium*.. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–13. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron. 2016;89:37–53. doi: 10.1016/j.neuron.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–78. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–9. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang CP, Li X, Wu Y, Shen Q, Zeng Y, Xiong Q, et al. Comprehensive integrative analyses identify GLT8D1 and CSNK2B as schizophrenia risk genes. Nat Commun. 2018;9:838. doi: 10.1038/s41467-018-03247-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, Leek JT, et al. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature. 2011;478:519–23. doi: 10.1038/nature10524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kendler KS, Myers J. The genetic and environmental relationship between major depression and the five-factor model of personality. Psychol Med. 2010;40:801–6. doi: 10.1017/S0033291709991140. [DOI] [PubMed] [Google Scholar]
- 47.Kendler KS, Neale MC, Kessler RC, Heath AC, Eaves LJ. A longitudinal twin study of personality and major depression in women. Arch Gen Psychiatry. 1993;50:853–62. doi: 10.1001/archpsyc.1993.01820230023002. [DOI] [PubMed] [Google Scholar]
- 48.Smith DJ, Escott-Price V, Davies G, Bailey ME, Colodro-Conde L, Ward J, et al. Genome-wide analysis of over 106 000 individuals identifies 9 neuroticism-associated loci. Mol Psychiatry. 2016;21:1644. doi: 10.1038/mp.2016.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luciano M, Hagenaars SP, Davies G, Hill WD, Clarke TK, Shirali M, et al. Association analysis in over 329,000 individuals identifies 116 independent variants influencing neuroticism. Nat Genet. 2018;50:6–11. doi: 10.1038/s41588-017-0013-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nagel M, Jansen PR, Stringer S, Watanabe K, de Leeuw CA, Bryois J, et al. Meta-analysis of genome-wide association studies for neuroticism in 449,484 individuals identifies novel genetic loci and pathways. Nat Genet. 2018;50:920–7. doi: 10.1038/s41588-018-0151-7. [DOI] [PubMed] [Google Scholar]
- 51.Kupfer DJ, Frank E, Phillips ML. Major depressive disorder: new clinical, neurobiological, and treatment perspectives. Lancet. 2012;379:1045–55. doi: 10.1016/S0140-6736(11)60602-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grimm S, Beck J, Schuepbach D, Hell D, Boesiger P, Bermpohl F, et al. Imbalance between left and right dorsolateral prefrontal cortex in major depression is linked to negative emotional judgment: an fMRI study in severe major depressive disorder. Biol Psychiatry. 2008;63:369–76. doi: 10.1016/j.biopsych.2007.05.033. [DOI] [PubMed] [Google Scholar]
- 53.Belden AC, Barch DM, Oakberg TJ, April LM, Harms MP, Botteron KN, et al. Anterior insula volume and guilt: neurobehavioral markers of recurrence after early childhood major depressive disorder. JAMA Psychiatry. 2015;72:40–48. doi: 10.1001/jamapsychiatry.2014.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hettema JM, Neale MC, Myers JM, Prescott CA, Kendler KS. A population-based twin study of the relationship between neuroticism and internalizing disorders. Am J Psychiatry. 2006;163:857–64. doi: 10.1176/ajp.2006.163.5.857. [DOI] [PubMed] [Google Scholar]
- 55.Stuermer CA. Reggie/flotillin and the targeted delivery of cargo. J Neurochem. 2011;116:708–13. doi: 10.1111/j.1471-4159.2010.07007.x. [DOI] [PubMed] [Google Scholar]
- 56.Cremona ML, Matthies HJ, Pau K, Bowton E, Speed N, Lute BJ, et al. Flotillin-1 is essential for PKC-triggered endocytosis and membrane microdomain localization of DAT. Nat Neurosci. 2011;14:469–77. doi: 10.1038/nn.2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Galbiati F, Volonte D, Goltz JS, Steele Z, Sen J, Jurcsak J, et al. Identification, sequence and developmental expression of invertebrate flotillins from Drosophila melanogaster. Gene. 1998;210:229–37. doi: 10.1016/S0378-1119(98)00064-X. [DOI] [PubMed] [Google Scholar]
- 58.Reisinger SN, Kong E, Molz B, Humberg T, Sideromenos S, Cicvaric A, et al. Flotillin-1 interacts with the serotonin transporter and modulates chronic corticosterone response. Genes Brain Behav. 2019;18:e12482. doi: 10.1111/gbb.12482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Amsterdam JD, Newberg AB, Soeller I, Shults J. Greater striatal dopamine transporter density may be associated with major depressive episode. J Affect Disord. 2012;141:425–31. doi: 10.1016/j.jad.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hsiao MC, Lin KJ, Liu CY, Schatz DB. The interaction between dopamine transporter function, gender differences, and possible laterality in depression. Psychiatry Res. 2013;211:72–77. doi: 10.1016/j.pscychresns.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 61.Huang CC, Lu RB, Shih MC, Yen CH, Huang SY. The dopamine transporter gene possibly affects personality traits in patients with early-onset major depressive disorder. Acta Neuropsychiatr. 2013;25:227–34. doi: 10.1111/acn.12019. [DOI] [PubMed] [Google Scholar]
- 62.Zink M, Vollmayr B, Gebicke-Haerter PJ, Henn FA. Reduced expression of glutamate transporters vGluT1, EAAT2 and EAAT4 in learned helpless rats, an animal model of depression. Neuropharmacology. 2010;58:465–73. doi: 10.1016/j.neuropharm.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 63.McCullumsmith RE, Meador-Woodruff JH. Striatal excitatory amino acid transporter transcript expression in schizophrenia, bipolar disorder, and major depressive disorder. Neuropsychopharmacology. 2002;26:368–75. doi: 10.1016/S0893-133X(01)00370-0. [DOI] [PubMed] [Google Scholar]
- 64.Li J, Zuo X, Shi J, Zhang J, Duan X, Xu G. Flotillin 1 is differentially expressed in human epithelial ovarian tumors. Neoplasma. 2018;65:561–71. doi: 10.4149/neo_2018_170714N483. [DOI] [PubMed] [Google Scholar]
- 65.Guan Y, Song H, Zhang G, Ai X. Overexpression of flotillin-1 is involved in proliferation and recurrence of bladder transitional cell carcinoma. Oncol Rep. 2014;32:748–54. doi: 10.3892/or.2014.3221. [DOI] [PubMed] [Google Scholar]
- 66.Cao S, Cui Y, Xiao H, Mai M, Wang C, Xie S, et al. Upregulation of flotillin-1 promotes invasion and metastasis by activating TGF-beta signaling in nasopharyngeal carcinoma. Oncotarget. 2016;7:4252–64. doi: 10.18632/oncotarget.6483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Y, Li J, Song Y, Chen F, Pei Y, Yao F. Flotillin-1 expression in human clear-cell renal cell carcinoma is associated with cancer progression and poor patient survival. Mol Med Rep. 2014;10:860–6. doi: 10.3892/mmr.2014.2310. [DOI] [PubMed] [Google Scholar]
- 68.Butz H, Szabo PM, Khella HW, Nofech-Mozes R, Patocs A, Yousef GM. miRNA-target network reveals miR-124as a key miRNA contributing to clear cell renal cell carcinoma aggressive behaviour by targeting CAV1 and FLOT1. Oncotarget. 2015;6:12543–57. doi: 10.18632/oncotarget.3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang FQ, Zhang HM, Chen SJ, Yan Y, Zheng JH. MiR-506 is downregulated in clear cell renal cell carcinoma and inhibits cell growth and metastasis via targeting FLOT1. PLoS ONE. 2015;10:e0120258. doi: 10.1371/journal.pone.0120258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pasquini M, Biondi M. Depression in cancer patients: a critical review. Clin Pract Epidemiol Ment Health. 2007;3:2. doi: 10.1186/1745-0179-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maneeton B, Maneeton N, Reungyos J, Intaprasert S, Leelarphat S, Thongprasert S. Prevalence and relationship between major depressive disorder and lung cancer: a cross-sectional study. Onco Targets Ther. 2014;7:815–21. doi: 10.2147/OTT.S60000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hu LY, Liu CJ, Yeh CM, Lu T, Hu YW, Chen TJ, et al. Depressive disorders among patients with gastric cancer in Taiwan: a nationwide population-based study. BMC Psychiatry. 2018;18:272. doi: 10.1186/s12888-018-1859-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zinbarg RE, Mineka S, Bobova L, Craske MG, Vrshek-Schallhorn S, Griffith JW, et al. Testing a hierarchical model of neuroticism and its cognitive facets: Latent structure and prospective prediction of first onsets of anxiety and unipolar mood disorders during 3 years in late adolescence. Clin Psychol Sci. 2016;4:805–24. doi: 10.1177/2167702615618162. [DOI] [Google Scholar]
- 74.Redmond L, Oh SR, Hicks C, Weinmaster G, Ghosh A. Nuclear Notch1 signaling and the regulation of dendritic development. Nat Neurosci. 2000;3:30–40. doi: 10.1038/71104. [DOI] [PubMed] [Google Scholar]
- 75.Scheer N, Groth A, Hans S, Campos-Ortega JA. An instructive function for Notch in promoting gliogenesis in the zebrafish retina. Development. 2001;128:1099–107. doi: 10.1242/dev.128.7.1099. [DOI] [PubMed] [Google Scholar]
- 76.Ikeda M, Aleksic B, Yamada K, Iwayama-Shigeno Y, Matsuo K, Numata S, et al. Genetic evidence for association between NOTCH4 and schizophrenia supported by a GWAS follow-up study in a Japanese population. Mol Psychiatry. 2013;18:636–8. doi: 10.1038/mp.2012.74. [DOI] [PubMed] [Google Scholar]
- 77.Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530:177–83. doi: 10.1038/nature16549. [DOI] [PMC free article] [PubMed] [Google Scholar]