

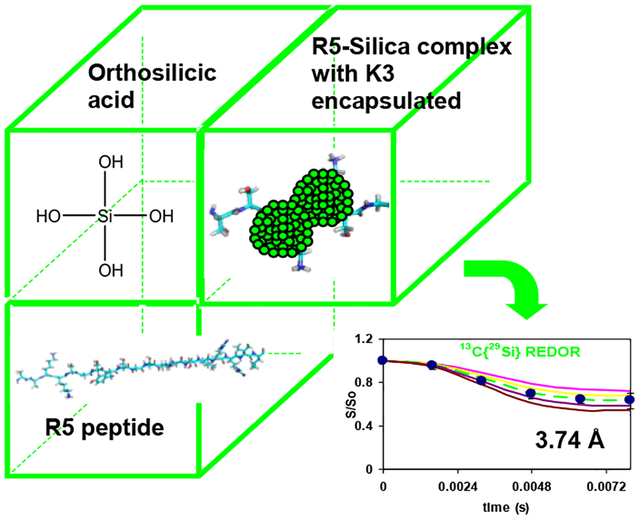
Abstract
Diatoms are unicellular algae that construct cell walls called frustules by the precipitation of silica, using special proteins that order the silica into a wide variety of nanostructures. The diatom species Cylindrotheca fusiformis contains proteins called silaffins within its frustules, which are believed to assemble into supramolecular matrices that serve as both accelerators and templates for silica deposition. Studying the properties of these biosilicification proteins has allowed the design of new protein and peptide systems that generate customizable silica nanostructures, with potential generalization to other mineral systems. It is essential to understand the mechanisms of aggregation of the protein and its co-precipitation with silica. We continue previous investigations into the peptide R5, derived from silaffin protein sil1p, shown to independently catalyze the precipitation of silica nanospheres in vitro. We used the solid-state NMR techniques 13C{29Si} and 15N{29Si}REDOR to investigate the structure and interactions of R5 in complex with co-precipitated silica. These experiments are sensitive to the strength of magnetic dipole-dipole interactions between the 13C nuclei in R5 and the 29Si nuclei in the silica, and thus yield distance between parts of R5 and 29Si in silica. Our data show strong interactions and short internuclear distances of 3.74 ± 0.20 Å between 13C=O Lys3 and silica. The Cα and Cβ nuclei, on the other hand show little or no interaction with 29Si. This selective proximity between the K3 C=O and the silica supports a previously proposed mechanism of rapid silicification of the antimicrobial peptide KSL (KKVVFKVKFK) through an imidate intermediate. This study reports for the first time a direct interaction between the N-terminus of R5 and silica, leading us to believe that the N-terminus of R5 is a key component in the molecular recognition process and a major factor in silica morphogenesis.
Keywords: solid-state NMR, biomineralization, biosilification, silica, R5
Graphical Abstract
Introduction:
Biomineralization is the process by which living organisms produce and pattern minerals to serve a variety of functions, including the hardening or stiffening of existing tissues and the formation of protective layers1–4. Some examples of bio-patterned minerals include silicates in algae, carbonates and silica in marine protists (diatoms) and invertebrates, and calcium phosphate and carbonates in vertebrates. Interestingly, biomineral crystals often have unique morphological features created by the deployment, in both invertebrates and vertebrates, of special proteins acting as either catalytic or collaborative agents. For example, an intrinsically disordered mollusk shell prismatic protein was found to modulate calcium carbonate crystal growth5, and ferritin protein cages are involved in iron oxide biomineralization in organisms ranging in complexity from archaea to humans6. It is therefore of great interest to elucidate the mechanisms by which these proteins carry out their biomineralization functions, the comprehension of which could lead to the engineering of novel molecular systems and the customized creation of nanostructured materials. Accordingly, much recent work has been done studying the methods by which silica nanostructures are generated in biological contexts, and how the associated proteins and mimics thereof are able to co-precipitate with silica in vitro7–10.
Diatoms, single-celled eukaryotic algae found both in marine and freshwater habitats, are the primary generators of biogenic silica. Indeed, diatoms help to regulate the ocean’s biogeochemical cycle by polymerizing the silicic acid to biogenic silica used to construct their cell walls, known as frustules11, 12. A prevailing hypothesis of protein-induced silica formation in the diatom species Cylindrotheca fusiformis holds that within the silica deposition vesicle basic proteins called silaffins assemble into supramolecular matrices that serve to accelerate and template silica deposition1, 13–16. In vitro, silaffins induce formation of silica nanospheres from silicic acid solutions. In attempting to study the properties of these silaffins in C. fusiformis, Sumper and Kröger identified the polypeptide sil1p encoded by the sil1 gene and found that this polypeptide caused the precipitation of silica nanospheres from silica17. The sil1p polypeptide, in turn, consists of a series of 7 units, R1–7 (Figure 1), post-translationally modified to include polyamine chains and phosphorylated serines. Research has also shown that the non-post-translationally modified R5 peptide (SSKKSGSYSGSKGSKRRIL) is independently capable of mimicking the parent polypeptide by co-precipitating with silica out of a silicic acid solution so as to form nanospheres18, 19. Thus, it is possible to focus on the properties and functional behavior of R5 to distil out the mechanisms of silica patterning and the corresponding characteristics of the peptide.
Figure 1:

Primary sequence of Silaffin protein sil 1p showing R1-R7 repeats.
It is well known that proteins with charged side chains (i.e., acidic or basic) interact strongly with mineral surfaces20–22, and long-chain polyamines have been implicated in the precipitation of silica9, 16, 23. It was therefore believed that the positively charged (at the experimental pH of 7.0) amine groups in the basic residues of R5 were involved in the condensation of silica nanoparticles9, 10, 24–26, which bear negative surface charges due to the exposed hydroxyl groups. In fact, chemical shift analyses of R5 had shown that the chemical shifts of the C=O, Cα and Cβ carbons of lysine 3 (K3) experienced significant changes upon silica co-precipitation19. Therefore, to test the supposition that the chemical shift changes were indeed due to the proximity of silica, we conducted solid-state NMR studies of uniformly 13C, 15N -labeled Lys3 (K3) in the N-terminus of the R5 primary sequence co-precipitated with silica. Specifically, 13C CPMAS, 15N CPMAS, 15N{29Si} REDOR, and 13C{29Si} REDOR27 experiments were carried out. One of the central result is the observation that the carbonyl of K3 is 3.74 ± 0.20 Å from the closest silicon atom in the R5-silica complex. The implications of this finding and others are discussed in the following sections.
Materials and Methods:
Sample Preparation.
Fmoc-Ser(t-butyl)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Gly-OH, Fmoc-Arg(pbf)-OH, and Fmoc-Ile-OH were purchased from Novabiochem, Fmoc-Tyr(t-butyl)-OH from AAPPTec. Uniformly labeled 13C 99%, 15N 98% Fmoc-Lys were purchased from Cambridge Isotope Laboratory.
Peptide Synthesis.
R5 (SSKKSGSYSGSKGSKRRIL) was synthesized using uniformly labeled Lys at position 3. N-Fmoc-L-Lys u-13C 99%, 15N, 98% were used at position 3, of R5 primary sequence and unlabeled amino acids for the rest of the pepetide. R5 synthesis was performed on an automated microwave peptide synthesizer.
After synthesis and work ups, the peptide was lyophilized, and then analyzed using electrospray mass spectroscopy. There were four major fragments observed at 1010.5 (+2), 675.1 (+3), 509.3 (+4), 407.9 (+5) (see supporting document Figure S1). rK3 has a molecular weight of 2021 Da.
Biosilicification.
R5 peptide was co-precipitated with silica out of silicic acid as follow. 50 mg of R5 was dissolved in 10 mL of a 100 mM phosphate-citrate buffer with pH adjusted to 7.0. One milliliter of a 1M silicic acid solution was prepared by adding 0.15 mL of tetrametyl orthosilicate (TMOS), the precursor of silicification, to 0.85 mL of a 1 mM HCl solution. The latter solution was added to the peptide/buffer solution. The R5-silica complex precipitated out in 1 to 5 minutes. The solution was centrifuged, and the solid phase collected, washed, and dried overnight under vacuum. The R5-silica complex was later packed in a 4 mm rotor for ssNMR spectroscopy. We hereafter refer to the synthesized R5-silica complex in this study as “rK3sil”.
NMR Experiments.
Solid-state NMR experiments were performed on a home-built spectrometer attached to a 11.74 T superconducting magnet and operating at Larmor frequencies of 500.29 MHz for 1H, 125.76 MHz for 13C, 99.325 MHz for 29Si, and 50.696 MHz for 15N. Pulse lengths were 4.0 μs for 1H 90, 7.0 μs for the 13C 180°, 19 μs for the 29Si 180°, and 11 μs 15N 180°. Chemical shifts of 29Si, 13C, and 15N are reported relative to TMS, 13C-glycine, and 15N-ammonium sulfate, respectively.
29Si spectrum of rK3sil was acquired in a single channel direct detect without decoupling, transients of 80,000 and relax delay of 4 seconds (Figure S2). The 13C NMR signal was enhanced using ramped cross-polarization with a 1H-13C contact time of 1 ms. The cross polarization and magic angle spinning (CPMAS) pulse sequence is given in Figure 2a. Figure 3 shows a 13C CPMAS spectrum of rK3sil. Similarly, the 15N NMR signal was enhanced using ramped cross-polarization with a 1H-15N contact time of 1 ms. The cross polarization and magic angle spinning (CPMAS) pulse sequence is given in Figure 2d. Figure 4 shows a 15N CPMAS spectrum of rK3sil. CPMAS and REDOR experiments were performed using a 4 mm HXY triple-resonance Varian T3 magic-angle spinning probe. All experiments were done at 25°C.
Figure 2:

(a) 13C CPMAS pulse sequence. (b) XY8 phase cycling 13C{29Si} REDOR pulse sequence with alternating π pulses on both channels. (c) 29Si spin topology used in simulation of 13C{29Si} REDOR data showing nearly 180° angles between 13C=O – 29Si and 13C=O – 13Cα dipolar vectors. Arrow in the figure indicates the dipolar couplings that were varied in simulations. (d) 15N CPMAS pulse sequence. (e) XY8 phase cycling 15N{29Si} REDOR pulse sequence with alternating π pulses on both channels.
Figure 3:

1H – 13C CPMAS spectrum of rK3sil acquired at 25°C and a spinning rate of 5 kHz.
Figure 4:

1H – 15N CPMAS spectrum of rK3sil acquired at 25°C and a spinning rate of 5 kHz.
The 13C{29Si} REDOR pulse sequence is given in Figure 2b. An XY8-phase cycling 13C{29Si} REDOR sequence with alternating π-pulses in both the 13C-observed and 29Si-dephasing channel was utilized. Similarly, the 15N{29Si} REDOR pulse sequence is given in Figure 2e. An XY8-phase cycling 15N{29Si} REDOR sequence with alternating π-pulses in both the 15N-observed and 29Si-dephasing channel was utilized. REDOR experiments measure internuclear dipolar constants:
| (1) |
where bij is the dipolar coupling constant between atoms at site i and j and is in unit of rad.s−1; μ0 is the permeability of vacuum and is in unit of H.m−1; γi and γj are the gyromagnetic ratios of elements i and j, respectively and in rad·s−1·T−1; ℏ is the reduced Planck constant (in unit of J·s); and rij is the internuclear distance between nuclei i and j.
To determine the dipolar constants, two experiments were conducted for each data point. A reference S0, where the rf pulses are turned off in the 29Si channel and an experiment S, where the rf pulses are turned on in the 29Si channel. The REDOR curve is obtained by plotting vs. the number of rotor period, NTR. REDOR dephasing times were within 8.0 ms. The typical transients were 172,800 with relax delay of 1.5 s for the longest recoupling period of 40 TR in the 13C{29Si} REDOR experiment. Similarly, typical transients were 345,600 with relax delay of 1.5 s for the longest recoupling period of 32 TR. Representative 13C{29Si} REDOR spectra and 15N{29Si} REDOR spectra are illustrated in Figures 5 and Figure 6, respectively.
Figure 5:

Overlay 13C{29Si} REDOR spectra of rK3sil showing So and S acquired at 25°C and a spinning rate of 5 kHz.
Figure 6:

Overlay 15N{29Si} REDOR spectra of rK3sil showing So and S acquired at 25°C and a spinning rate of 5 kHz.
Simulations and Data Fitting.
Spin dynamics simulations for the 13C{29Si} XY8-REDOR experiments were carried out using the SIMPSON 4.1.1 package28. Powder averages were calculated using the direct algorithm without relaxation and using 232 {α, β} Euler angle sets based on the Zaremba-Conroy-Wolfsberg (ZCW) scheme28, 29 and 10 γ angles. Calculated dephasing of the carbonyl magnetization employed three 13C spins representing the C=O, Cα and Cβ carbons in the labeled lysine amino acid and one 29Si spin representing a Si atom from the silica. All 13C-13C dipolar couplings were taken into account in the simulations and a 13C – 29Si dipolar coupling with the C=O 13C spin.
Results and Discussion:
13C CPMAS, 15N CPMAS, 13C{29Si} REDOR, and 15N{29Si} REDOR of rK3sil.
The 29Si spectrum (Figure S2) showed the presence of multiple species of 29Si characterized by the degree of replacement of the −OH groups in Si(OH)4 by −OSi groups: Qn represents the species with n = { 0, 1, 2, 3, 4} such replacements30, 31. The spectrum in Figure S2 indicates a distribution between Q2 and Q3 states, which favors Q2. The Q2 species is slightly dominant in rK3sil and this can be useful in understanding the mechanism by which the peptide induced silica formation as it gives information to the number of siloxane bonds relative to the number of silanol bonds.
Extensive effort was spent optimizing the 13C CPMAS and 15N CPMAS. A representative 13C CPMAS spectrum can be found in Figure 3. The carbonyl 13C is at 176ppm and the aliphatic carbons are Cα (58 ppm), Cε (45 ppm), Cβ (38.6 ppm), Cδ (32 ppm), Cγ (28.8 ppm). Figure 4 shows a 15N CPMAS spectrum with the amine at 33 ppm and the amide at 127 ppm.
Finally, to better understand the molecular interactions of the R5 induced silica nanospheres, cross polarization (CP) 13C{29Si} REDOR, and cross polarization (CP) 15N{29Si} REDOR were performed on rK3sil with 13C, 15N-enriched K3 (Figures 5 and 6, respectively).
The NMR SIMPSON package was used to simulate the 13C{29Si}REDOR curve (Figure 7)28. Simulated REDOR dephasing of the carbonyl magnetization employed a 4-spin model comprising three 13C spins and one 29Si spin. The 13C spins represent the C=O carbon and its closest Cα, Cβ neighbors in Lys3 of R5, rK3sil. The single 29Si spin represents the Si atom in the closest shell to the amino acid, based on the encapsulation of the peptide within the silica material19. The natural abundance of 29Si is 4.7%, therefore the probability for two adjacent active spins is 0.047 times lower (equal to ~ 1 pair of 453 silicon atoms). This practically limits the number of possible active silicon spins that interact with each lysine molecule to about one. To justify the model which assumes interaction of each and every 13C spin on C=O with a 29Si spin, it is sufficient to have the closest shell comprising 20 Si atoms. In that case, a single 29Si will be found in close proximity to the labeled K3 residue in each of the R5 molecules resulting in detectable 13C – 29Si dipolar coupling. Furthermore the 13C dephasing will not be limited to 0.953 of the initial magnetization set by the natural abundance of 29Si spins. The coupling of Cα and Cβ nuclei to the 29Si nucleus depend on the orientation of K3 chain relative to the heteronucleus. The simulated dephasing fitted best the experimental data when the additional 13C – 29Si couplings for the Cα and the Cβ nuclei could be neglected, implying that an orientation in which both Cα and Cβ are beyond the maximum detectable 13C – 29Si internuclear distance of about 6Å. This is most suitable to explain the REDOR dephasing by orienting the 13C=O – Cα dipolar vector at flat or nearly 180° with respect to the 13C=O – Si dipolar vector (Figure 2c). Figure 8 shows that the aliphatic carbons (i.e., Cα, Cβ, etc…) are overlapped and together did not dephase. Due to the overlap in the aliphatic region, we could not derive direct dipolar couplings of Cα, Cβ, etc… We cannot say beyond the fact that the aliphatic carbons are more than 6Å from the silica, which imply that they are beyond the range of detectable 13C – 29Si dipolar interactions. The closest internuclear distance measured between the C=O and the single Si nucleus is 3.74 ± 0.20 Å, which corresponds to a dipolar coupling constant of 115 Hz. The reduced χ2 (with 5 data points and one degree of freedom) for the fit is 0.16.
Figure 7:

13C{29Si} REDOR decay curve of rK3sil with SIMPSON simulations. The S/So signal in the 13C{29Si} XY8-REDOR experiment (filled circles) with calculated curves using 13C=O – 29Si dipolar couplings of 95, 105, 115, 125 and 135 Hz. Best fit curve is observed for a coupling of 115 Hz corresponding to a distance of 3.74 Å. Between 28,800 and 172,800 transients were acquired for both S and So spectra of each data point with a repetition rate of 1.5s.
Figure 8:

rK3sil 13C{29Si} REDOR curve of aliphatic carbons.
This 29Si spin is assumed in the model to be interacting with the 13C=O carbon of K3. There are at least three possible mechanisms for this:
There are several reports in the literature of molecules interacting with silica through amine or amide groups (backbone or sidechain)9, 10, 23–26. It is possible that the amine group exerts electrostatic attraction towards the surface silanol groups of the nanoparticles and subsequently acts as a proton donor to catalyze silicification. The subsequent proximity of the 13C=O group to 29Si nuclei could merely be the result of a serendipitous positioning of the side chain, backbone and silica.
Eby and co-workers32 suggested an alternative based on their studies of the antimicrobial peptide KSL (KKVVFKVKFK) that mediates the rapid condensation of tetramethyl orthosilicate (TMOS) to form silica nanoparticles. The absence of a side-chain amide 15N{29Si} REDOR dephasing signal together with X-ray photoelectron spectroscopy (XPS) evidence of the formation of a backbone imidate bond suggested the possibility that the backbone was central to the condensation of siloxane bonds in the KSL-silica complex. Phosphate groups may induce tautomerization of the peptide backbone amide to an imide group. In the imidic form of the peptide, the neighboring carbonyl oxygen becomes a nucleophile that forms a pentacoordinate complex with silicic acid. This intermediate further reacts with another silicic acid monomer to form a siloxane bond, coinciding with the release of the carbonyl oxygen from the complex (implying a catalytic function of the peptide backbone). This mechanism would clearly result in a large interaction signal between 29Si and the 13C=O group.
The third possibility is that some combination of the previous two mechanisms is at play. For example, the amine group could indeed exert a long-range electrostatic force, while the condensation of silica occurs through the intervention of the pentacoordinate complex with the carbonyl oxygen.
One means of clarifying the method of silica attraction and condensation would be to carry out 15N{29Si} REDOR on the K3 side-chain amine, as in the work of Eby et al.,32, and check to see if dephasing occurs. The 15N{29Si} REDOR experiments showed no dephasing from the amine nitrogen (see Figure 9). On the other hand we saw some dephasing for the amide nitrogen (see Figure 9). This is interesting in the sense that although amine moiety might be involved electrostatically in R5 silification, the lack of dephasing from the amine nitrogen could be that the dipolar couplings between 15N spins and 29Si spins are too weak even though the side chain might interact with silica. It seems like backbone Si interactions, rather than side chain Si interactions are more important in silification.
Figure 9:

rK3sil 15N{29Si} REDOR curves of amides nitrogen (blue curve) and amine nitrogen (pink curve).
In a recent paper by Schmidt and co-workers33, it was shown that alanine interacted with the surface of SBA-15 silica through its amine −NH3+ moiety. Our REDOR study showed only strong dephasing through the carbonyl 13C and nothing from the aliphatic carbons (i.e., Cα, Cβ, etc…). Both Cα and Cβ are overlapping with the other aliphatic carbons and their individual dipolar couplings vis a vis silica could not be estimated but the entire aliphatic carbons do not dephase at all. They are likely beyond the detectable 13C – 29Si internuclear distance (i.e., > 6Å, Figure 8). Interestingly, the best-fit C=O – 29Si distance of 3.74 ± 0.20 Å is similar to that of the alanine amine 15N and silicon atoms on the surface of SBA-15 silica33. In a recent article, Brunner et al.,23 used a model substance tetraphenoxysilane labeled with 13C and 15N to carry out triple resonance solid state NMR experiments. It is worth noting that we used natural abundance silica in this article. To the best of our knowledge, this is the first time natural abundance silica (4.7%) were used successfully, in a 1H-13C-29Si solid state NMR experiment.
The results reported in this paper provide an atomic-level insight into the relative position of the peptide and the proximal silica. On account of the process of co-precipitation, the situation differs from a simple protein/peptide-surface interaction, and several possible aggregation models may be consistent with the data. However, we can make a few observations here:
The N-terminus of R5 seems to be closely oriented toward the silica matrix and as indicated by the significant chemical shift changes for several spins within K3 upon silica precipitation and by significant 13C{29Si} REDOR dephasing at the carbonyl position and 15N{29Si} dephasing at the amide position. CPMAS spectra for 13C and 15N enriched K3 indicate chemical shift changes at the 13CO site from 169.1 ppm in the neat protein to 172.4 ppm for the same site in the silica composite19. A similar trend was observed by Fernandez et al.,34 for the carbonyl site in poly-lysine upon adsorption onto silica and hydroxyapatite. Although the 13Cα and 13Cβ spins in K3 show much smaller chemical shift changes upon precipitation with silica, the amide 15NH chemical shift changes from 123 ppm in the neat peptide to 127.5 ppm in the silica composite. Therefore the large chemical shift changes observed for the 13CO and 15NH spins in K3 correspond to strong REDOR dephasing in both cases and supports the view that this portion of K3 is oriented close to the silica matrix.
The observed REDOR dephasing of the carbonyl carbon is an average over the entire sample, and thus if only a small fraction of the R5 molecules were exposed to silica, it is unlikely that any significant dephasing would have been observed. Given the substantial dephasing signal, we believe it likely that the sample contains R5 and silica intermixed with each other, rather than a “pre-aggregate” of R5 coated on the surface with silica (as proposed by Sumper and Kröger17). More experiments would need to be done to confirm this proposition.
The absence of REDOR dephasing at Cα and Cβ indicates any chemical shift changes occurring for these spins upon precipitation are due to changes in conformation. Chemical shift changes at Cγ and Cδ are minimal but upon precipitation with silica the chemical shift of Cε changes from 31.8ppm to 29.5 ppm. Interestingly, a similar trend was also observed for the Cε spins in poly-lysine adsorbed onto silica and hydroxyapatite34. The fact that no REDOR dephasing accompanies this chemical shift change may be due to the fact that a small REDOR dephasing at the Cε spin may not be observable when plotted with the signals from the other side chain aliphatic carbons, see Figure 8.
Upon precipitation with silica the side chain Nζ amine chemical shift changes from 34.5 ppm in the neat peptide to 35.4 ppm in silica, but no 15N{29Si} REDOR dephasing was observed. This is a relatively small chemical shift change compared to the >4 ppm change observed at the amide spin where REDOR dephasing was observed. The small chemical shift changes at the K3 amine and the absence of REDOR dephasing probably reflects a more distant location of the amine from the silica than occurs at the amide. If the 15Nζ spin were located at 6 Å from a 29Si spin we would expect a dipolar coupling around 13–14 Hz. Such small dipolar couplings would give rise to weak REDOR dephasing that would not be observed under our experimental conditions.
Conclusion:
A number of models have been proposed to explain how biosilicification proteins and peptides derived from these proteins induce silica precipitation and impact silica morphology. A widely-held hypothesis is that proteins and peptides aggregate in silicic acid solutions and in doing so, serve as templates and catalysts for silica formation. Kroger and colleagues assert that micelle assembly of native, zwitterionic silaffins is driven by electrostatic forces over the length of the peptide.13,17 A model where the C-terminal RRIL moiety is involved in the stabilization of R5 micelles in solution has been proposed by Knecht & Wright35 but recently disputed by Senior et al36.
To further explore these issues, a derivative of the silaffin sil1p protein, the R5 peptide 13C and 15N enriched at the K3 amino acid, was studied in complex with silica. It is shown that the K3 residue in the N-terminus of R5 does interact with silica, as evidenced by our 13C{29Si} REDOR study. The carbonyl 13C of K3 of the primary sequence of R5 is positioned at a distance of 3.74 ± 0.20 Å relative to the closest silica in the complex.
We also observed 15N{29Si}dephasing at the amide of K3. The peptide backbone in the vicinity of K3 therefore seems to be situated closer to silica. These REDOR results and the chemical shifts results obtained in earlier studies supports the hypothesis that the C=O carbon may have a role in the silification mechanism, which was proposed in a previous publication by Eby et al.,31. On the other hand, the lack of detectable REDOR dephasing in the amine moiety and the smaller chemical shift change indicates that this moiety is farther removed from the silica than is the case for the carbonyl and amide spins. Our present study also reveals that backbone Si interactions, rather than side chain Si interactions seem more important in silification.
Although the REDOR we have obtained for a single residue on R5 is insufficient to determine whether or not R5 forms aggregates in silica, further REDOR studies of peptide-silica and peptide-peptide interactions together with chemical shift data that we have already published will contribute toward obtaining a clearer picture of the structure and interactions of R5 in silica.
Supplementary Material
Acknowledgments:
This research was supported by National Science Foundation Grant CHE 1219509 and National Institute of Health Grant NIH (R01-GM 109417).
References:
- (1).Baeuerlein E; Behrens P; Epple M Handbook of Biomineralization. WILEY-VCH; 2007, ISBN 978352731641–0. [Google Scholar]
- (2).Sigel A; Sigel H; Sigel RKO Biomineralization: From Nature to Application. Metal Ions in Life Sciences 4 Wiley; 2008, ISBN 978047003525–2. [Google Scholar]
- (3).Weiner S; Lowenstam HA On biomineralization. Oxford University Press; 1989. ISBN 019504977–2. [Google Scholar]
- (4).Cuif JP; Dauphin Y; Sorauf JE Biominerals and fossils through time. Cambridge: 2011, ISBN 978052187473–1. [Google Scholar]
- (5).Ndao M; Keene E; Amos F; Rewari G; Ponce C; Estroff L; Evans JS Intrinsically disordered mollusk shell prismatic protein that modulates calcium carbonate crystal growth. Biomacromolecules 2010, 11, 2539–2544. [DOI] [PubMed] [Google Scholar]
- (6).Theil EC Ferritin: The Protein Nanocage and Iron Biomineral in Health and in Disease. Inorg. Chem 2013, 52(21), 12223–12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zane AC; Michelet C; Roehrich A; Emani PS; Drobny GP Silica Morphogenesis by Lysine-Leucine Peptides with Hydrophobic Periodicity. Langmuir 2014, 30, 7152–7161. [DOI] [PubMed] [Google Scholar]
- (8).Baio JE; Zane A; Jaeger V; Roehrich AM; Lutz H; Pfaendtner J; Drobny GP; Weidner T Diatom Mimics: Directing the Formation of Biosilica Nanoparticles by Controlled Folding of Lysine-Leucine Peptides. J. Am. Chem. Soc, 2014, 136 (43), 15134–15137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Belton DJ; Patwardhan SV; Annenkov VV; Danilovtseva EN; Perry CC From biosilicification to tailored materials: Optimizing hydrophobic domains and resistance to protonation of polyamines. Proc. Natl. Acad. Sci. U. S. A, 2008, 105(16), 5963–5968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Liang M-K; Patwardhan SV; Danilovtseva EN; Annenkov VV; Perry CC Imidazole catalyzed silica synthesis: Progress toward understanding the role of histidine in (bio)silicification. J. Mater. Res, 2009, 24(5), 1700–1708. [Google Scholar]
- (11).Siever R Silica in the oceans: Biological-geochemical interplay In “Scientists on Gaia”, MIT Press; 1991, 287–295. [Google Scholar]
- (12).Treguer P; Nelson DM; Van Bennekom AJ; DeMaster DJ; Leynaert A; Queguiner B The silica balance in the world ocean: a reestimate. Science 1995, 268, 375–379. [DOI] [PubMed] [Google Scholar]
- (13).Kröger N; Lorenz S; Brunner E; Sumper M Self-Assembly of Highly Phosphorylated Silaffins and Their Function in Biosilica Morphogenesis. Science 2002, 298, 584–586. [DOI] [PubMed] [Google Scholar]
- (14).Poulsen N; Kröger N Silica Morphogenesis by Alternative Processing of Silaffins in the Diatom Thalassiosira pseudonana. J. Biol. Chem 2004, 279, 42993–42999. [DOI] [PubMed] [Google Scholar]
- (15).Poulsen N; Sumper M; Kröger N Biosilica formation in diatoms: Characterization of native silaffin-2 and its role in silica morphogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 12075–12080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kröger N; Deutzmann R; Bergsdorf C; Sumper M Species-specific polyamines from diatoms control silica morphology. Proc. Natl. Acad. Sci. USA 2000, 97, 14133–14138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sumper M; Kröger N Silica formation in diatoms: the function of long-chain polyamines and silaffins. J. Mater. Chem 2004, 14, 2059–2065. [Google Scholar]
- (18).Kröger N; Deutzmann R; Sumper M Polycationic Peptides from Diatom Biosilica That Direct Silica Nanosphere Formation. Science 1999, 286, 1129–1132. [DOI] [PubMed] [Google Scholar]
- (19).Roehrich A; Ash J; Zane A; Masica D; Gray J; Goobes G; Drobny G Proteins at Interfaces III State of the Art. ACS SYMPOSIUM SERIES 1120 2012, 77–96. [Google Scholar]
- (20).Ndao M; Ash JA; Breen NF; Goobes G; Stayton PS; Drobny GPA 13C{31P} REDOR NMR investigation of the role of glutamic acid residues in Statherin-hydroxyapatite recognition. Langmuir 2009, 25(20), 12136–12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ndao M; Ash JA; Stayton PS; Drobny GP The role of basic amino acids in the molecular recognition of hydroxyapatite by Statherin using solid state NMR. Surface Science 2010, 604, L39–L42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Masica D; Ash JA; Ndao M; Drobny GP; Gray JJ Toward a structure determination method for biomineral-associated protein using combined solid state NMR and computational structure prediction. Structure 2010, 18, 1678–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wisser D; Brückner SI; Wisser FM; Althoff-Ospelt G; Getzschmann J; Kaskel S; Brunner E 1H–13C–29Si triple resonance and REDOR solid-state NMR—A tool to study interactions between biosilica and organic molecules in diatom cell walls. Solid State Nucl. Magn. Reson 2015, 66/67, 33–39. [DOI] [PubMed] [Google Scholar]
- (24).Coradin T; Lopez P; Biogenic Silica Patterning: Simple Chemistry or Subtle Biology? ChemBioChem, 2003, 4, 251–259. [DOI] [PubMed] [Google Scholar]
- (25).Mizutani T; Nagase H; Fujiwara N; Ogoshi H Silicic Acid Polymerization Catalyzed by Amines and Polyamines. Bull. Chem. Soc. Jpn 1998, 71, 2017–2022. [Google Scholar]
- (26).Patwardhan SV; Clarson SJ; Perry CC On the Role(s) of Additives in Bioinspired Silicification. Chem. Commun 2005, 9, 1113–1121. [DOI] [PubMed] [Google Scholar]
- (27).Gullion T; Schaefer J Rotational-Echo Double Resonance NMR. J. Magn. Reson 1989, 81, 196–200. [DOI] [PubMed] [Google Scholar]
- (28).Bak M; Rasmussen JT; Nielsen NC SIMPSON: a general simulation program for solid-state NMR spectroscopy. J. Magn. Reson 2000, 147, 296–330. [DOI] [PubMed] [Google Scholar]
- (29).Conroy H Molecular Schrödinger Equation VIII. A New Method for the Evaluation of Multidimensional Integrals. J. Chem. Phys, 1967, 47, 5307. [Google Scholar]
- (30).Maciel GE; Sintorf DW Silicon-29 Nuclear Magnetic Resonance Study of the Surface of Silica Gel by Cross Polarization and Magic-Angle Spinning. J. Am. Chem. Soc 1980, 102, 7606–7607. [Google Scholar]
- (31).Engelhardt G Multinuclear solid-state NMR in silicate and zeolite chemistry. Trends Anal. Chem 1989, 8(9), 343–347. [Google Scholar]
- (32).Eby DM; Artyuskova K; Paravastu AK; Johnson GR Probing the molecular structure of antimicrobial peptide-mediated silica condensation using X-ray photoelectron spectroscopy. J. Mater. Chem, 2012, 22, 9875–9883. [Google Scholar]
- (33).Ben Shir I; Kababya S; Amitay-Rosen T; Balazs SY; Schmidt A Molecular Level Characterization of the Inorganic−Bioorganic Interface by Solid State NMR: Alanine on a Silica Surface, a Case Study. J. Phys. Chem. B 2010, 114, 5989–5996. [DOI] [PubMed] [Google Scholar]
- (34).Femandez VL; Reimer JA; Denn MM Magnetic Resonance Studies of Polypeptides Adsorbed on Silica and Hydroxyapatite Surfaces. J. Amer. Chem Soc 1992, 114, 9634–9642 [Google Scholar]
- (35).Knecht MR; Wright DW Functional analysis of the biomimetic silica precipitating activity of the R5 peptide from Cylindrotheca fusiformis. Royal Soc. Chemistry: Chem. Commun, 2003, 3038–3039. [DOI] [PubMed] [Google Scholar]
- (36).Senior L; Crump MP; Williams C; Booth PJ; Mann S; Perrimand AW; Curnow P Structure and function of the silicifying peptide R5” J. Mat. Chem. B 2015, 3, 2607-. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.