

LPA, a blood-borne signaling lipid, induces neonatal hydrocephalus by damaging ependymal cells through LPA1/3 overactivation.
Abstract
Posthemorrhagic hydrocephalus (PHH) in premature infants is a common neurological disorder treated with invasive neurosurgical interventions. Patients with PHH lack effective therapeutic interventions and suffer chronic comorbidities. Here, we report a murine lysophosphatidic acid (LPA)–induced postnatal PHH model that maps neurodevelopmentally to premature infants, a clinically accessible high-risk population, and demonstrates ventriculomegaly with increased intracranial pressure. Administration of LPA, a blood-borne signaling lipid, acutely disrupted the ependymal cells that generate CSF flow, which was followed by cell death, phagocytosis, and ventricular surface denudation. This mechanism is distinct from a previously reported fetal model that induces PHH through developmental alterations. Analyses of LPA receptor–null mice identified LPA1 and LPA3 as key mediators of PHH. Pharmacological blockade of LPA1 prevented PHH in LPA-injected animals, supporting the medical tractability of LPA receptor antagonists in preventing PHH and negative CNS sequelae in premature infants.
INTRODUCTION
Infantile hydrocephalus is a common neurological condition that affects approximately 1 in 1000 live births (1). It is characterized by an accumulation of cerebrospinal fluid (CSF), enlargement of the fluid-filled ventricles [termed “ventriculomegaly” (VM)], increased intracranial pressure (ICP), cortical thinning, and a range of comorbid neuroanatomical changes and functional deficits. Hydrocephalus can have several causes—including neurodevelopmental malformations, infection, or intraventricular hemorrhage (IVH)—and is particularly common in premature infants with fragile cerebral vasculature (1, 2). These infants, born at gestational ages (GAs) less than 37 weeks, are at risk for severe IVH and subsequent posthemorrhagic hydrocephalus (PHH) (2). Approximately 15 to 20% of low birth weight premature infants suffer from IVH, and up to 25% of these patients develop progressive PHH (2, 3), which translates to approximately 5% of premature infants or 4 infants per 10,000 births (4). Despite the high incidence of PHH, preventive therapies are currently lacking, a situation compounded by a paucity of adequate animal models that recapitulate human hydrocephalus at medically relevant developmental stages.
One possible mediator of PHH is lysophosphatidic acid (LPA), a blood-borne signaling lipid with six known heterotrimeric GTP-binding protein–coupled receptors (GPCRs), designated LPA1-LPA6 (5). LPA is produced from lysophosphatidylcholine by the enzyme autotaxin and circulates in the blood within platelets or bound to carrier proteins such as albumin (6). Of particular relevance, blood-borne LPA levels are elevated during hemorrhage or trauma (6). Therefore, severe hemorrhage would introduce large concentrations of LPA into the brain over a prolonged period. LPA receptors (LPARs) are expressed in several central nervous system (CNS) cell types, and LPAR signaling has been reported to mediate multiple neurological and inflammatory disorders (7). Therefore, it is reasonable to predict that elevated LPA concentrations following IVH may play a role in the initiation of PHH.
The first LPAR to be identified (LPA1) (8) is expressed within the neuroproliferative ventricular zone, where it can influence neuroprogenitor cells. A previous report showed that neuroprogenitor cells were disrupted by intraventricular delivery of LPA in a fetal PHH mouse model (9). Although this model recapitulates many human comorbidities, it parallels an early fetal human neurodevelopmental age preceding even the youngest premature infants. A model that could faithfully produce hydrocephalus at ages equivalent to those seen in preterm humans at GA 22 to 34 weeks would be both mechanistically enlightening and vital to the development of therapeutic strategies for preventing or attenuating neonatal PHH. Therefore, we translated this LPA-induced model of PHH to postnatal mice.
Here, we report a new mouse model of PHH produced by intraventricular LPA delivery during postnatal life at ages analogous to those of premature infants at risk for IVH. This postnatal model recapitulates several clinical characteristics of human PHH (Table 1) and involves a specific cellular population, the ciliated ependymal monolayer that lines the ventricular system and expresses LPARs during neonatal ages. Ependymal cilia beat in a synchronized manner to distribute CSF from the ventricles (10), where it is created by the choroid plexus (11), throughout the CNS. Disrupting the development or function of these cells generates spontaneous hydrocephalus (12–14), and compromised ependymal layers have been observed in human patients with hydrocephalus (15, 16). This new model provides evidence that LPAR-mediated ependymal disruption may play a key role during the initiation of neonatal PHH.
Table 1. Clinical PHH phenotypes recapitulated by this neonatal LPA-induced mouse model.
Sequelae reported in clinical studies following cohorts of hydrocephalic patients compared to phenotypes observed in this neonatal model of PHH. IVH was mimicked by injecting LPA directly into the lateral ventricle of postnatal day 8 (P8) mice.
|
Hydrocephalus characteristics |
Clinical studies | Neonatal model |
| Bleeding/hemorrhage | Yes (1–3) | Mimicked |
| Ventricular dilation | Yes (1–3) | Yes |
| Elevated CSF pressure | Yes (1–3) | Yes |
| Early lethality | 75% untreated (41) and 20% treated (3) |
80% by 12 weeks |
| Loss of ependymal layer |
Yes (15, 16) | Yes |
| Ciliary defects | Yes (42) | Yes |
| Aqueductal occlusion | Sometimes (43) | No |
| Monocyte infiltration | Implicated in shunt failure (44) and systemic inflammation (45) |
Yes |
RESULTS
Intraventricular LPA exposure induces high-pressure hydrocephalus in neonatal mice
In a model of fetal PHH, LPA induced PHH when injected into the ventricles of embryonic day 13.5 (E13.5) mice (9), which correlates with the late first trimester in humans (Fig. 1A) (17, 18). The resultant disruptions involve effects on mitotic or newly postmitotic neuroprogenitor cells. However, this prenatal mouse model accesses a fetal neurodevelopmental stage that has limited diagnostic and therapeutic options. Therefore, we translated this model to recapitulate PHH in premature infants, who represent a clinically accessible and high-need population. Premature infants experiencing IVH, but are able to survive long enough to develop PHH, are typically GA 22 to 34 weeks, corresponding to neonatal mice at postnatal days 4 to 8 (P4 to P8) (17, 18).
Fig. 1. Intracerebral LPA exposure induces hydrocephalus in neonatal mice.
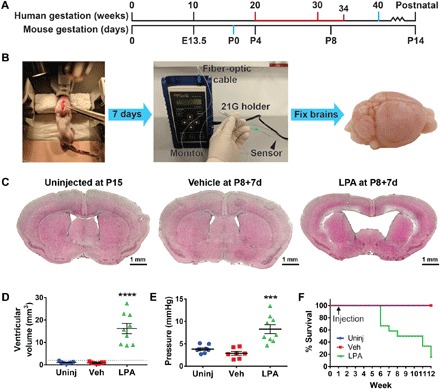
(A) Approximate correlation of human and mouse brain development, highlighting full-term birth (40 weeks versus P0; blue) and ages at high risk for PHH (20 to 34 weeks versus P4 to P8+; red). (B) Experimental approach for this neonatal model of LPA-induced hydrocephalus. Mice received stereotactic intracranial injection at P8 (left; forceps point to approximate location of injection), followed by terminal ICP measurement (center) and brain harvest 7 days later (“P8+7d”; right). 21G, 21-gauge. (C) Intracerebral injection of LPA produces VM. Representative brain sections from uninjected (Uninj), vehicle-injected (Veh), and LPA-injected P8+7d mice quantified in (D). (D) Quantification of increased lateral ventricle volume (n = 10 per experimental group). The dotted line indicates 2 SDs above the vehicle mean. (E) Increased ICP in the brains of P8+7d mice injected with LPA (n = 9) compared to brains from uninjected (n = 8) or vehicle-injected (n = 7) mice. (D and E) Symbols indicate values from individual mice. ****P < 0.0001 and ***P < 0.0005 compared to vehicle controls. (F) Kaplan-Meier survival curve over a 12-week period for uninjected mice and mice injected with vehicle or LPA at P8 (n = 7 uninjected, n = 9 vehicle, and n = 11 LPA).
We observed PHH-relevant phenotypes at each postnatal day following LPA injection, particularly at P8, which corresponds to an approximate neonatal human age when infants have higher posthemorrhage survival in addition to elevated PHH risk (Fig. 1A) (2). P8 mice also exhibited lower postoperative lethality; therefore, this age was selected for further characterization. We stereotactically injected mouse littermates into one lateral ventricle with either vehicle (control) or 112 μM LPA, to a final concentration of approximately 18 μM in CSF (Fig. 1B; for details on the concentration of LPA, see Materials and Methods and fig. S1). Animals were euthanized 7 days after injection (“P8+7d”). Brains were then harvested and analyzed for PHH end points, including VM and histological abnormalities. Most notably, LPA-injected brains demonstrated a ventricular volume that was more than 10 times larger than vehicle-injected or uninjected lateral ventricles (Fig. 1, C and D). We observed this “severe” VM in 100% of LPA-injected animals, accompanied by cortical thinning and destruction of the corpus callosum. By P8, the postnatal mouse skull is fused; therefore, VM was not accompanied by cranial expansion at P8+7d. Notably, postnatal LPA-induced PHH was not associated with overgrowth and occlusion of the rostral aqueduct, which is observed in fetal LPA-induced hydrocephalus (9), implicating distinct cellular mechanisms in a nonobstructive, or communicating, form of PHH.
PHH has two major clinical hallmarks: VM and increased ICP. While VM is commonly reported in animal models of PHH, increased ICP has not been reported in mice, reflecting the technical difficulties of assessing this benchmark in small animals. We therefore developed a protocol for measuring ICP in mouse pups (Fig. 1B) by inserting a fiber-optic pressure monitor into the lateral ventricle of sedated mice. Fiber-optic pressure measurements from mice injected with LPA demonstrated significantly increased ICP compared to uninjected and vehicle-injected cohorts (Fig. 1E).
After assessing the acute morphological effects of LPA-induced hydrocephalus, we then assayed long-term survival. In animals allowed to survive with hydrocephalus, more than 80% died within 12 weeks of age (11 weeks after injection) (Fig. 1F). These mice were typically underweight and exhibited hunched postures and unkempt coats. Together, these data demonstrate that LPA-induced PHH is chronic, progressive, and fatal without external intervention.
LPA exposure induces acute ependymal dysfunction
To observe the effects of LPA injection on various brain regions and cell types as a function of time, we harvested LPA-injected brains at 3, 6, and 24 hours after surgery. Sections were stained with H&E (hematoxylin and eosin) for histological analysis (Fig. 2, A and B). The initial insult appeared to primarily affect the ependyma, a monolayer of ciliated cells that line the ventricular system of the brain. These cells serve as a barrier between the CSF and parenchyma, and their cilia beat to generate CSF flow around the brain (10, 11).
Fig. 2. LPA affects ependymal integrity.
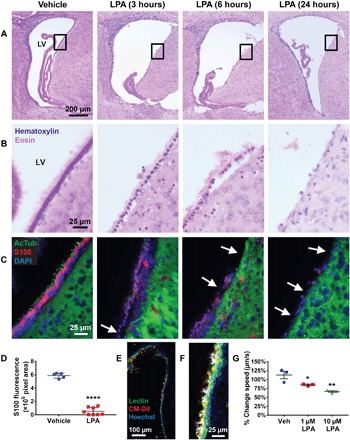
(A) Representative images of H&E-stained lateral ventricles at 3, 6, and 24 hours after LPA exposure (magnification, ×10) (n = 5). LV, lateral ventricle. (B) Magnified regions (×63) of the boxed areas shown in (A). (C) Cilia and cell bodies of lateral ventricle ependymal cells immunostained with acetylated tubulin (AcTub) (green, cilia) and S100 (red, cell body) with 4′,6-diamidino-2-phenylindole (DAPI) nuclear counterstain (blue). The arrows point to denuded sections of the ventricular wall. (D) Quantification of ependymal cell loss in P8 mice 24 hours following injection with LPA (n = 8) or vehicle (n = 5). The area of S100 immunostaining surrounding the lateral ventricles was added over five serial sections, covering 1 mm of lateral ventricle, using ImageJ. Each symbol represents total ventricular S100 fluorescence from an individual brain. ****P < 0.0001 compared to vehicle controls. (E) Single-frame image (×20) of a lateral ventricle stained with Hoechst (blue, nuclei), Lectin DyLight 488 (green, ependymal membrane), and CM-DiI (red, cilia) taken from live ciliary imaging shown in fig. S2 and movie S1. (F) Example of tracking analysis on a single frame, with colored dots overlaying beating cilia and white tracks tracing ciliary motility patterns over 10 s, taken from movie S2. (G) Quantification of the change in average ciliary movement speed from 0 to 3 hours in vehicle- and LPA-treated wells (n = 3). Symbols represent values from brain slices of the same three mice treated with vehicle, 1 μM LPA, or 10 μM LPA. (D and G) *P < 0.05 and **P < 0.005 compared to vehicle controls, as determined by analysis of variance (ANOVA) with Tukey’s post hoc test.
Ependymal cell membrane changes and nuclear rounding were evident by 3 hours after LPA injection and were accompanied by loss of basement membrane adhesion 6 hours after LPA injection, followed by substantial depletion of the ependymal monolayer at 24 hours after LPA injection (Fig. 2B). We confirmed these initial observations immunohistologically by staining for acetylated tubulin (AcTub) and S100, markers that identify ependymal cilia and cell bodies, respectively (Fig. 2C). Ciliary health declined in tandem with changes in cellular morphology, as evidenced by a loss of ciliary AcTub immunoreactivity, which declined and disappeared from 3 to 6 hours, while ependymal cell bodies (S100) were lost by 24 hours after LPA exposure (Fig. 2C). Quantitation of ependymal cell loss at 24 hours, determined by measuring the area of S100 fluorescence surrounding the lateral ventricles, demonstrated significant LPA-induced ependymal depletion (Fig. 2D).
The rapidity of ependymal cilia changes at 3 hours implicated even earlier acute events. To explore this possibility, we examined the effects of LPA on ependymal function in real time using a brain slice culture system paired with high-speed visualization of ciliary motility. Living brain slices prepared by vibratome sectioning were labeled with nontoxic membrane-permeable dyes to permit fluorescence visualization of ependymal membranes and beating cilia (Fig. 2E and fig. S2). Slices were positioned within a tissue culture plate such that one lateral ventricle wall could be imaged for 10 s at 112.3 frames/s (fps) during 30-min intervals over a 6-hour period (movie S1). After acquiring the baseline motility of each well at t = 0, sections were exposed to vehicle solution or brought to final concentrations of 1 or 10 μM LPA and then visualized with time-lapse imaging. We processed image files in Imaris to track ciliary movement (Fig. 2F and movie S2). Quantification revealed LPA dose-dependent reductions in ciliary movement speed 3 hours after LPA exposure compared to the basal ciliary motility at t = 0 (Fig. 2G). These data support LPA-dependent perturbations of ciliary motility occurring rapidly following LPA exposure.
Ependymal cilia degenerate and are phagocytosed
The above data demonstrate LPA-mediated deterioration of ependymal health and function, including the loss of AcTub and reduced ciliary motility, suggesting that structural changes in ependymal cilia may correlate with these functional perturbations. It is known that α-tubulin subunits are acetylated (AcTub) to build the microtubule axoneme, where two central microtubule singlets are coupled to nine outer microtubule doublets tightly wrapped in cellular membrane (19). Motor activity between these structures serves to drive ciliary beating; therefore, axoneme structure is integral to ciliary function. To assess whether changes in ciliary structure correlate to observed functional perturbations, we processed samples at several time points following LPA or vehicle injection, performing scanning electron microscopy (SEM) and transmission electron microscopy (TEM) on the ciliated ependymal surface. TEM revealed LPA-induced disruptions of ciliary morphology, including frequent loss of membrane integrity and occasional distortion of the 9 + 2 microtubule axoneme 6 hours after injection (Fig. 3, A to D). SEM of the apical ependymal surface also revealed ciliary tufts, which appeared kinked and disorganized, accompanied by progressive ciliary loss that resulted in an uneven ventricular surface (Fig. 3, E and F). The ventricular surface was almost fully depleted of cilia 24 hours after LPA exposure (fig. S3).
Fig. 3. Ependymal disruption precedes immune infiltration and cell death.
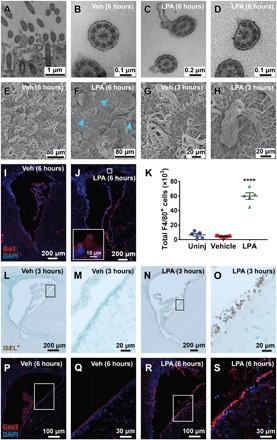
(A to D) TEM images of ependymal cilia cross sections. (A) Ciliary tuft with several distinct cilia adjacent to ependymal membrane. (B) Ciliary membrane and axoneme structure from a wild-type (WT) vehicle-injected mouse. (C) Disrupted ciliary membrane and (D) axoneme observed 6 hours after LPA exposure. (E to H) SEM images of the ventricular surface. (E) Intact ciliated ependymal surfaces were observed in vehicle-exposed brains. (F) LPA-exposed brain with ciliary loss and presence of cellular debris. The blue arrowheads point to remaining ciliary tufts 6 hours after LPA injection. (G) Vehicle-exposed cilia 3 hours after injection. (H) Possible phagocyte observed 3 hours after LPA exposure attached to shriveled and disorganized ependymal cilia. (I and J) Immunofluorescence images of Iba1+ innate immune cells in the lateral ventricle 6 hours following (I) vehicle or (J) LPA injection (×10 with ×63 inset). (K) Total number of F4/80+ CD11b+ Ly6G− macrophages and microglia isolated from brains of uninjected, vehicle-injected, or LPA-injected mice 24 hours after injection, as determined by flow cytometry. ****P < 0.0001 compared to vehicle controls. n = 5 individual brains combined from two independent experiments. (L to O) In situ end-labeling plus (ISEL+) assay indicating DNA damage 3 hours after injection (brown, fragmented DNA; light green, nuclei). (L and M) Brains exposed to vehicle were unaffected, whereas (N and O) LPA exposure caused selective ependymal DNA damage, with (L and N) ×10 magnification and (M and O) boxed areas enlarged to ×63. (P to S) Cleaved caspase 3 (Cas3) (red, with blue nuclear counterstain) immunolabeling in (P and Q) vehicle-injected and in (R and S) LPA-injected brains, which show selective ependymal apoptosis 6 hours following LPA exposure. (P and R) Images at x20 magnification with (Q and S) boxed areas enlarged to 63×.
SEM analyses at 3 and 6 hours after LPA exposure also captured a distinct population of cells on the ventricular surface with an atypical morphology (Fig. 3, G and H), possibly indicating recruitment of phagocytic immune cells. To determine whether immune cell localization was altered after LPA injection, immunofluorescence was performed 6 hours after injection. Immunostaining of ionized calcium binding adapter molecule 1 (Iba1), a macrophage and microglia marker, detected resident Iba1+ cells in the choroid plexus of vehicle-injected animals (Fig. 3I). LPA exposure resulted in a robust increase in the number of Iba1+ cells present on the ventricular surface (Fig. 3J), suggesting a migration of choroid plexus–resident or blood-derived macrophages to the damaged ependymal layer. Flow cytometric analyses confirmed that the total number of F4/80+ macrophages and microglia present in the whole brain 24 hours after injection was significantly increased in animals exposed to LPA compared to controls (Fig. 3K). These data indicate that cells of the innate immune system are recruited in response to ependymal cell damage (20) and may contribute to the development of hydrocephalus.
Ependymal cells undergo apoptotic cell death within 24 hours following LPA exposure
PHH initiation progressed from acute ciliary disruption to loss of cells along the ventricular surface, indicating that cell death pathways may be activated in ependymal cells as a response to LPA exposure. We first addressed this possibility using in situ end-labeling (ISEL+) (21), a more sensitive version of the terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) assay. This stain identified DNA fragmentation 3 hours after LPA exposure (Fig. 3, L to O). To determine whether this death was initiated by cellular apoptosis pathways, we assessed caspase activation by immunofluorescence of cleaved caspase 3 (Cas3) and detected it in ependymal cells 6 hours after LPA exposure (Fig. 3, P to S). Combined, these data support the probability that LPA induces ependymal damage, leading to apoptotic cell death, immune cell recruitment, and subsequent PHH (22, 23).
Ependymal cells express genes for multiple LPAR subtypes
To determine which LPAR subtypes might be involved in LPA-initiated ependymal changes, we used RNAscope technology to detect all six LPAR genes, Lpar1-6, in P8 tissue sections. We observed prominent Lpar1 expression and specific Lpar3 expression within the ependymal monolayer (Fig. 4, A and B), whereas Lpar2, Lpar4, Lpar5, and Lpar6 had low or nonspecific ependymal expression (fig. S4). These expression patterns are distinct from those previously reported for fetal and adult brains (24, 25), indicating that LPAR-dependent mechanisms for PHH may involve different receptor subtypes than those implicated in earlier models.
Fig. 4. LPA1 and LPA3 are involved in hydrocephalus development.
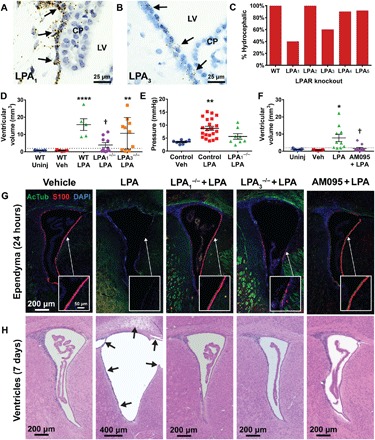
Specific LPAR expression in the ependyma was demonstrated by RNAscope for (A) Lpar1 and (B) Lpar3, (brown RNA probes with blue nuclear counterstain). CP, choroid plexus. (C) LPA was injected into specific LPAR knockout mice, and the incidence of hydrocephalus was reported as determined by VM analysis (n = 10 for LPA1-LPA4 and n = 11 for LPA5). (D) Quantification of VM in LPA1 and LPA3 knockout mice 7 days following vehicle or LPA exposure. WT, n = 5; null, n = 10. **P < 0.01 and ****P < 0.0001 versus WT vehicle-injected animals. †P < 0.001 versus WT LPA-injected. (E) ICP measurements of LPA1-null mice compared to littermate and vehicle controls 7 days after injection. n = 7 vehicle-injected pups, n = 21 LPA-injected littermates, and n = 5 LPA-injected LPA1−/− pups. **P < 0.005 compared to vehicle controls. (F) VM quantified in mice treated with AM095 or vehicle 15 min before injection of 500 μM LPA in 0.01% bovine serum albumin (BSA). n = 9 uninjected, n = 5 vehicle, n = 10 LPA, and n = 10 AM095 + LPA. *P < 0.05 compared to vehicle control and †P < 0.05 compared to LPA-injected WT. (D to F) Symbols indicate values from individual mice. Dotted lines indicate 2 SDs above the vehicle mean. (G) AcTub (green) and S100 (red) ependymal staining 24 hours after injection (×10 magnification with ×63 insets). (H) H&E-stained lateral ventricle sections 7 days after injection. Arrows indicate areas of disrupted histology.
These age-dependent LPAR subtype expression patterns suggest that susceptibility to LPA-induced PHH, along with the cell types involved in PHH initiation and progression, may vary throughout life. This hypothesis is supported by the varying effects of LPA injection at different developmental ages. We assessed VM 7 days after injection at several ages. Severe VM was observed in 100% of animals injected at P4 or P8 but in only 50% of mice injected at P14, and no significant (more than 2 SDs above the control) VM was detectable in LPA-injected adult mice (fig. S5). These results are consistent with the developmental patterns of LPAR expression and correlate with reduced LPAR subtype expression in the ventricular ependyma toward the end of neurodevelopment (24, 25).
LPA1 and LPA3 are key mediators of PHH induction
LPA exposure selectively damaged ependymal cells before the development of hydrocephalus in the early postnatal brain. Combined with LPAR in situ hybridization studies, these data implicated selective receptor overactivation. To determine the functional involvement of LPARs in PHH initiation, we attempted to induce PHH in LPA1-LPA5 single knockout mice and quantified VM at P8+7d. While PHH developed in almost all LPA2, LPA4, and LPA5 knockouts, only 40% of LPA1-null mice and 60% of LPA3-null mice developed significant VM (Fig. 4, C and D). Affected LPA1-null animals also demonstrated less severe VM compared to wild-type (WT) LPA-injected animals, and the LPA-injected LPA1−/− mean was not significantly different from controls (Fig. 4D). In other words, not only did fewer LPA1-null animals exhibit VM, but those that did were affected to a lesser degree. This LPA1 knockout–mediated protection from PHH development was not reliant on sexual dimorphism (fig. S5). LPA1 knockouts that did not develop any VM had ICP levels comparable to those of WT and heterozygous control animals (Fig. 4E and fig. S6). These results implicate LPA1 and LPA3 signaling as mediators of LPA-induced VM and PHH.
As the genetic removal of LPA1 or LPA3 reduced VM incidence and severity, we hypothesized that pharmacological interventions might also prevent LPA-induced PHH. Two LPAR antagonists were assayed to determine the effects of LPAR blockade on PHH development: AM095, an LPA1-selective antagonist (26), and Ki16425, a dual LPA1 and LPA3 antagonist (27). The amount of injected LPA was reduced to a submaximal hydrocephalus-inducing concentration to better detect the effects of AM095 and Ki16425. Intraventricular injections of antagonist [AM095 or Ki16425 (1 mg/kg)] were followed 15 min later by LPA delivered to the same location. After 7 days, brains were harvested, and VM was quantified (Fig. 4F). Attempts to use Ki16425 resulted in 44% of animals exhibiting no VM, but this reduction was not statistically significant from controls (fig. S7). These differences may reflect a pharmacokinetic or degradative issue with Ki16425. We therefore shifted our focus to AM095.
Seventy percent of vehicle-pretreated, LPA-injected animals showed significant VM. By contrast, only 20% of AM095-pretreated, LPA-injected animals exhibited VM (Fig. 4F). These results are consistent with the reduced VM severity demonstrated by LPA1-null mice (Fig. 4D). Blockade of LPA1 signaling by AM095 pretreatment or utilization of LPA1-null mice prevented LPA-induced ependymal damage at 24 hours, as demonstrated immunohistochemically by AcTub and S100 staining (Fig. 4G). Seven days following LPA exposure, LPA1-null, LPA3-null, and AM095-treated animals showed a significant increase in the number of animals with normal ventricular and cortical histology (Fig. 4, D, F, and H). These data support the use of LPA1 antagonists in prevention paradigms for LPA-induced PHH (Fig. 5).
Fig. 5. Proposed mechanism for LPAR signaling in postnatal hydrocephalus.
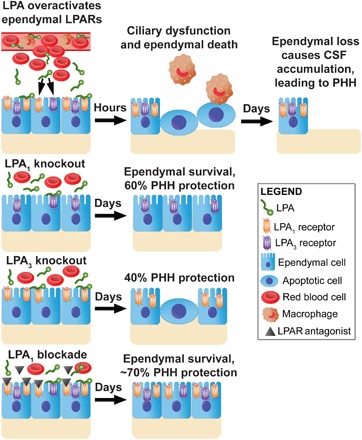
Following a severe hemorrhagic event, LPA is released into the CSF. LPA binds to LPA1 and LPA3 receptors on the ependymal cells that line the ventricles, causing GPCR overstimulation. After 6 hours, these signaling events cause the ependymal cells to become apoptotic, resulting in ciliary dysfunction and phagocyte recruitment. LPA exposure produces significant ependymal cell loss after 24 hours. With the lack of cilia-driven flow, CSF builds up in the ventricular space, leading to VM and chronic hydrocephalus. These events can be prevented with the genetic deletion of LPA1 or LPA3 or through pharmacological inhibition of LPA1 with the selective antagonist AM095, demonstrating that this effect occurs through activation of specific LPARs.
DISCUSSION
This report describes the characterization of a new mouse model for PHH generated via intraventricular LPA exposure during early postnatal life. It uses a medically targetable mechanism, LPAR signaling, at a clinically relevant age: P8 mice, which approximate to human premature infants in neonatal intensive care units who are at high risk for IVH and subsequent PHH. This mouse model replicated multiple aspects of PHH reported in humans, including elevated ICP and VM (Table 1 and Fig. 1). LPA exposure within the cerebral ventricles, which likely occurs during human intracranial hemorrhage, overactivated ependymal cell LPA1 and, to a lesser extent, LPA3, resulting in ciliary dysfunction and reduced motility along with ependymal cell damage, death, and removal. This is consistent with clinical and genetic models of PHH, which demonstrate that the reduction of cilia-driven CSF flow or removal of the CSF-parenchyma barrier is sufficient to cause CSF accumulation (12–14), leading to VM and chronic hydrocephalus (10, 11). Our postnatal model recapitulated these phenotypes both in vivo, producing chronic LPA-induced PHH, and ex vivo, demonstrating an LPA-mediated reduction in ciliary motility (Fig. 2). This ependymal cell loss, VM, and PHH could be prevented to a significant extent by LPA1 removal or blockade and, to a lesser extent, by LPA3 removal (Figs. 4 and 5).
These data are among the first to model hydrocephalus in mice at an age that neurobiologically corresponds to the ages of premature human infants at risk for PHH, namely, GA 22 to 34 weeks (1, 17). The postnatal PHH model reported here, which responded to LPA exposure at a developmental stage after neurogenesis has ceased within the cerebral cortex and the subjacent ganglionic eminence, implicates ependymal cell–intrinsic effects. This mechanism is fundamentally distinct from a fetal model of LPA-induced hydrocephalus, wherein LPA exposure affected migration and fate of mitotic and early postmitotic LPA1-expressing neural progenitor cells (NPCs) and ultimately led to some ependymal effects (9, 28). Both models produce hydrocephalus with 100% penetrance, but the fetal model manifests alterations in NPC fate that reduce ependymal cell generation and induce aqueductal stenosis, whereas the new postnatal model directly affected the ependymal cells, resulting in their dysfunction, death, and removal in the absence of physical CSF blockages. This is a key difference, as the neonatal model may shed new light on communicating or idiopathic cases of hydrocephalus.
The importance of ependymal degeneration in the initiation of LPA-induced PHH corroborates previous hydrocephalus literature (12–14). Several studies have demonstrated that genetic mutations that disrupt ciliary function or perturb ependymal development often induce spontaneous hydrocephalus. For instance, deletion of SNX27 was shown to disrupt ependymal development and ciliogenesis (12); a mutation in Hydin causes structural axoneme defects that reduce ciliary bending (14), and a mutation of ependymal ciliary protein CCDC39 induces structural abnormalities and reduced cilia-driven CSF flow (13). All three models have high rates of spontaneous hydrocephalus. Here, we identify LPA signaling as a key mediator of PHH that affects ciliary and ependymal integrity, consistent with these other models.
To more fully characterize hydrocephalus in murine models, we developed an innovative method to measure ICP. Elevated ICP is an important clinical hallmark of PHH that is difficult to measure in small animals. As a result, VM is commonly used as a proxy for hydrocephalus. This circumstance leaves open the question of whether animal models actually produce increased ICP or whether these models induce cellular losses that can cause VM in the absence of high-pressure hydrocephalus. We monitored ICP in early postnatal mice using a slim fiber-optic pressure cable, emulating clinical approaches. To our knowledge, this new postnatal model is the first to demonstrate increased ICP in mice beyond the more common end point of VM alone. The use of this model and ICP-measuring technique should allow more accurate assessment of PHH-related variables, including effects of candidate therapeutics.
The complete penetrance of intraventricular LPA exposure in producing postnatal hydrocephalus is notable. The CNS damage and severity of hydrocephalus followed a general dose response, whereby higher concentrations of LPA produced more pronounced PHH sequelae, particularly the degree of VM (fig. S5). Higher concentrations of LPA would be expected to access more LPARs and activate more LPAR subtypes. This may, in part, explain the incomplete rescue of LPA1-null and/or LPA3-null mutants, the rare rescue of other receptor subtype null mutants, and the variable pharmacological inhibition by Ki16425 or AM095. Further modification of this model to use reduced LPA dosing in combination with physiologically relevant carrier molecules, such as albumin, may allow more robust identification of new medical interventions. In addition to LPAR subtypes, pharmacological interventions could target downstream signaling pathways such as GPCR kinase 2, which inhibits LPA1 activation in neural cells (29); Rho/ROCK, which may influence ependymal adhesion or induce cytoskeletal changes affecting ependymal cilia (30); or apoptotic pathways suggested by cleaved Cas3 activity (31), to further elucidate mechanistic and therapeutic insights for neonatal PHH.
The dysfunction observed in our PHH model was progressive in nature, ultimately leading to the death and removal of ciliated ependymal cells. Examination at multiple time points delineated temporal stages of ependymal damage, raising questions about which stages might be therapeutically tractable. The effects of LPA exposure on ciliary motility were detected rapidly, as demonstrated in living slice cultures, and manifested as reductions in ciliary movement speed. Within hours in vivo, we observed significant ciliary changes that included membrane and axoneme disruption, followed by cell death and removal (Figs. 2 and 3).
It is possible that some aspects of ciliary dysfunction might be reversible. In this regard, a notable feature of this new postnatal model was the increase in innate immune cells observed through several methods. SEM showed cells possibly engulfing cilia within 3 hours following LPA injection; Iba1 immunofluorescence demonstrated recruitment to the ependymal surface 6 hours after LPA exposure, and the number of innate immune cells in the brain was quantifiably increased 24 hours following LPA injection, as determined by flow cytometry, supporting the possibility of macrophage and microglial involvement in PHH (Fig. 3). It is conceivable that ependymal cell death and ciliary loss might be prevented or reversed, in part, by the reduction of innate immune cell infiltration. However, clinical anti-inflammatory therapeutic interventions have shown equivocal results (2, 32), so further study is necessary to determine the role of this observed immune component in PHH progression.
LPA1 and LPA3 signaling pathways are promising targets for pharmacological intervention, since these receptors are members of a family of transmembrane GPCRs that are already candidates for many human medicines (33). In addition, multiple LPAR antagonists are under development for medical indications including fibrosis and cancer (34). The superior efficacy of the LPA1 inhibitor AM095 in preventing LPA-induced VM supports further development of LPAR antagonists with improved pharmacokinetic profiles for use in model systems and future clinical trials. Considering that IVH is observed in 15 to 20% of premature infants (2, 3), a case may be made for a prevention trial in the most susceptible infants at increased risk during the earliest GAs. Further characterization of our neonatal PHH model could reveal novel medical interventions to reduce PHH sequelae and circumvent the need for neurosurgical interventions.
MATERIALS AND METHODS
Experimental design
The objective of this study was to develop a PHH model targeting neonatal ages. We hypothesized that LPA would induce hydrocephalus at different ages, dependent on the LPAR-expressing cellular populations. For example, LPA might generate occlusions by stimulating residual neuroprogenitors or increasing CSF production through the choroid plexus. However, after beginning the study, we realized that the ventricular ependyma expresses high levels of LPARs at early postnatal ages and modified our hypotheses to determine how LPA affects this specific cellular population at several time points after injection.
This study used animals from C57BL/6J and Balb/c backgrounds along with several knockout variants. All procedures were conducted in accordance with Institutional Animal Care and Use Committee guidelines of The Scripps Research Institute and the Sanford Burnham Prebys Medical Discovery Institute. Animals were treated with vehicle or LPA injected directly into the lateral ventricle, monitored before euthanasia, and brains were then harvested at several time points for histological and immunohistochemical analysis. Quantitative end points included VM by ventricular volume quantification, CSF accumulation by ICP measurements, and ependymal cell retention by quantification of the area of S100 fluorescent cell bodies.
Mice were randomly assigned treatments within each litter, and each experiment had littermate controls. Treatment and genotype identity were blinded from quantitative procedures. For VM quantification, images were acquired, and treatment identity was removed before processing. For ICP measurements, mice were assigned a number by tail-marking with no identifying treatment information, and genotypes were processed after data acquisition and animal euthanasia. S100 quantifications were run through an ImageJ macro that treated all samples identically.
As this study induced hydrocephalus with 100% incidence in LPA-treated animals, experiments could be conducted with high confidence and low error expectations. Therefore, sample size was selected to equal or exceed five individual animals per experiment. Multiple litters were used to obtain the required sample size for each experimental finding; therefore, each experiment was replicated at least once. The ex vivo brain slice experiment used three individual littermate brains for analysis. All findings were reproducible as reported, with PHH occurring in 100% of injected WT animals. Results did not change on the basis of time performed, litter variations, use of foster mothers, mouse strain, individual’s sex, a particular LPA or vehicle stock, or any other assessed parameters.
Data collection end points included tissue harvest at several time points after injection. These time points were selected on the basis of morphological and immunohistochemical changes at each stage, starting 7 days after injection when hydrocephalus is highly apparent. Brains were sectioned to sample the entire lateral ventricle system of the brain at 100-μm intervals for VM quantification, without exclusions. For immunohistochemical applications, brains were sectioned every 200 μm to capture the lateral ventricles on a single slide. For pressure measurements, at least 10 data points were acquired from each individual mouse for analysis. Additional data collection details and statistical analyses can be found in the methods below. No data points or outliers were excluded from quantifications.
Animal models
P8 littermates of C57BL/6J and Balb/c background were used for this study. No differences in PHH development were observed between strains (fig. S5). Lpar1-5 knockout mice (35–39) were crossed onto C57BL/6J (LPA2, LPA4, and LPA5) or Balb/c (LPA1, LPA2, and LPA3) backgrounds. Pups were fostered with WT mothers when the birth mothers demonstrated aggression or knockout mothers could not leave the breeding colony.
LPA preparation and concentration
In anticipation of an expected 97% loss between LPA reconstitution and injection, resulting from lipid interactions with pipettes, tubes, and syringe surfaces in the absence of lipid carrier proteins, the following protocol was developed to achieve a final injection concentration of LPA equivalent to 112 μM (fig. S1). Powdered 18:1 LPA (857130P, Avanti Polar Lipids) was dissolved in methanol, then aliquoted into low-retention tubes, vacuum-dried, and stored at −20°C. LPA was reconstituted by sonicating in Hanks’ balanced salt solution (HBSS; 14175-095, Gibco) for a 5 mM stock. Five microliters of this stock was administered intracranially. Following calculated losses and on the basis of an estimated CSF volume of 24.4 μl, this 112 μM injection concentration resulted in a working concentration of 18 μM in CSF or 1.8 μM in the whole brain. Control animals were injected with 5 μl of vehicle. P8 CSF and brain volumes were estimated on the basis of mathematical extrapolation of data presented by Chuang et al. (40).
For the final inhibitor experiment, powdered LPA was reconstituted in 0.1% fatty acid–free bovine serum albumin (BSA) in phosphate-buffered saline (PBS), aliquoted, and diluted to a 500 μM LPA stock in 0.01% BSA before injection. This resulted in an approximate concentration of 80 μM in CSF or 8 μM in whole brain. No losses were included in these calculations, as BSA significantly enhances LPA availability in aqueous solution.
Surgery
P8 pups were immobilized on a stereotactic frame using a custom head rest and sedated with either intraperitoneally administered Nembutal (40 mg/kg) or gaseous isoflurane. The skin atop the head was opened 0.5 cm from the bregma, and the needle was inserted through the skull at approximately 3.2 mm caudal by 0.7 mm lateral by 1.2 mm deep. Mice were injected into the right lateral ventricle with 5 μl of vehicle (HBSS) or 18:1 LPA using a 35- to 36-gauge needle and a glass 10-μl NanoFil syringe (World Precision Instruments). Postoperative flunixin meglumine (1 mg/kg; 0061-0851-03, Merck) was administered subcutaneously for 2 days for pain management, and mice were monitored daily until tissue harvest.
Pharmacology
To study the efficacy of pharmacological LPAR antagonists AM095 and Ki16425, we diluted the concentration of LPA to 10% (500 μM LPA in the stock tube) and added a carrier protein, 0.01% BSA, to the LPA and drug solutions, as BSA increases the solubility and bioavailability of LPA, Ki16425, and AM095 in solution (6). This LPA formulation was used for all LPAR inhibitor experiments, including Fig. 4F and fig. S7. Mice were pretreated with an intracranial injection of a 2.5-μl solution containing AM095 (1 mg/kg) (4 mM; HY-16029, MedChem), Ki16425 (1 mg/kg) (4 mM; 355025-24-0, Cayman Chemical), or vehicle (PBS with 0.01% BSA and 5% dimethyl sulfoxide) 15 min before injection at the same location with 5 μl of 18:1 LPA in PBS with 0.01% BSA.
Histology and microscopy
Whole brains were harvested from experimental animals at the time points indicated. For paraffin-embedded sections, brains were fixed in formalin acid alcohol (4% formaldehyde and 5% glacial acetic acid in 70% ethanol), paraffinized in a Tissue-Tek VIP, and embedded with a Tissue-Tek TEC. For frozen sections, tissues were fixed in 4% paraformaldehyde in PBS, cryoprotected with 10 and 20% sucrose in PBS, and embedded in optimal cutting temperature compound (4583, Tissue-Tek) or Neg-50 (6502, Thermo Fisher Scientific). Sections were cut to a 10-μm thickness on a Leica microtome or cryostat.
A ZEISS Imager.M2 was used to obtain all images. For bright-field imaging, brain sections were deparaffinized and rehydrated through xylene and ethanol washes, stained with H&E, dehydrated in ethanol and xylenes, and then coverslipped and preserved with dibutylphthalate polystyrene xylene mounting media. Electron microscopy was performed at The Scripps Research Institute Core Microscopy Facility with a Philips CM100 TEM and Hitachi S4800 SEM, with tissue processing, sectioning, and imaging performed with the core staff.
For immunohistochemical analyses, deparaffinized or frozen sections were washed in tris-buffered saline (TBS) (pH 7.4), blocked in TBST (0.1% Triton X-100 in TBS) with 5% normal goat serum (NGS), and incubated with primary antibody in TBST with 1% NGS overnight at the dilutions listed below. After washing in TBST, sections were incubated with secondary antibody diluted 1:1000 in TBST for 1 hour and then washed sequentially in TBST, TBST with 1:10,000 4′,6-diamidino-2-phenylindole nuclear counterstain, and TBS. Slides were coverslipped with VECTASHIELD (H-1000, Vector Laboratories) and kept at 4°C until imaging. Antibodies used were S100A/B (1:300; NB200-538, Novus Biologicals) on paraffinized tissue, AcTub (1:250; T6793, Sigma-Aldrich) on paraffinized tissue, cleaved Cas3 (1:200; #9664, Cell Signaling Technology) on frozen tissue, and Iba1 (1:250; 019-19741, Wako) on frozen tissue. ISEL+ was performed on paraffinized tissue with the 2.5 brown detection kit (322310, Advanced Cell Diagnostics) and horseradish peroxidase labeling (Vector ImmPRESS, MP-7401) as per the manufacturer’s instructions.
VM quantification
Lateral ventricle volume was measured in coronal sections collected every 100 μm from the most anterior to posterior sections containing lateral ventricle. The ventricular area of each section was quantified in Adobe Photoshop CS6. The wand, quick selection, and lasso tools were used to individually select the left and right ventricular openings. In instances where ventricles were joined, a <5-pixel line was drawn down the midline of the brain to bisect the ventricles. Choroid plexus was included during quantification, as the free-floating nature of this organ causes it to occupy inconsistent locations in the ventricle. All ventricular areas were then summed and multiplied by 100 μm to create a volume approximation.
ICP recording
A BIOPAC Systems fiber-optic pressure cable was inserted through the barrel of a 21-gauge needle with a filed-down bevel. The sensor was wetted and calibrated in ultrapure water before each use. Pressure recordings were taken by retracting the sensor into the needle barrel, puncturing the skull of a heavily sedated mouse, and extending the pressure probe into the lateral ventricle. The probe was allowed to equilibrate for 5 s before recording every fifth measurement from the Opsens control panel. A total of at least 10 measurements were obtained and averaged together for each individual. To prevent fluid escape and pressure release during recording, the edge of the needle was sealed around the skull with putty or wax. After recording, the probe was carefully withdrawn, the mouse was euthanized, and brain tissue was collected.
Brain slice culture for live imaging
Brains were collected from two to three P8 animals, kept in iced HBSS with bubbled oxygen, and cut into 250-μm sections with a Leica VT1000 S vibratome. Sections containing lateral ventricle were incubated in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) at 37°C for 5 min with membrane-permeable dyes: Hoechst nuclear stain (5 μg/ml) (H3570, Invitrogen), Lectin DyLight 488 (5 μg/ml) (DL-1174, Vector Laboratories) for ependymal and vascular staining, and Vybrant CM-DiI (5 μg/ml) (V22888, Invitrogen). CM-DiI was used to nonspecifically label cell membranes; however, ependymal cilia had higher staining intensities than other cellular membranes, likely because of their surface area and direct exposure to solution. Following two 3-min washes in DMEM, brains were transferred to a glass-bottomed 12-well plate (P12G-1.5-14-F, MatTek) containing 1 ml of high-glucose DMEM and weighed down with harp slice grids (HSG-5BD MEA, ALA Scientific). Sections were incubated for 1 hour to induce adhesion before imaging on a Nikon N-SIM with an A1R confocal scanner and equipped with a plate-heating chamber. This microscope has a resonance scanner that acquires simultaneous images at 405-, 488-, and 568-nm wavelengths. By performing a band scan, we were able to capture 10-s time-lapse images at 113.2 fps, which allows for visualization of ciliary beating. These videos were analyzed with Imaris to track the movement of CM-DiI–stained cilia. Points were excluded for colocalization with Hoechst or Lectin 488, restricted by frame-to-frame distance to track the back-and-forth movement of cilia, and filtered by tracking duration to prevent quantification of cilia beating out of frame. Tracking analysis video was exported at 23 fps (1/5 speed).
Brain immune cell isolation and quantification
Twenty-four hours after injection with vehicle or LPA into P8 mouse ventricles (P8+1d), animals were perfused with PBS, and brains were harvested. The Miltenyi gentleMACS Octo dissociator was used to create single-cell suspensions from whole brains. Immune cells were then isolated by a discontinuous Percoll (17-0891-02, GE Healthcare) gradient before fixation in 0.1% paraformaldehyde. Cells were incubated with anti-CD16/32 (Fc receptor block; 14-0161, eBioscience) before staining in Brilliant Stain Buffer Plus [566385, BD Biosciences (BD)] with antibody cocktail to identify macrophages and activated microglia (CD45+CD11b+/hiF4/80+Ly6G−). Antibodies were purchased from either BD or eBioscience and were as follows: CD45-BV711 (30-F11, BD), CD11b-BV510 (M1/70, BD), F4/80-AF488 (BM8, eBioscience), and Ly6G-PE-Cy7 (1A8, eBioscience). Liquid counting beads (BD) were added immediately before data were obtained on a BD FACSAria Fusion flow cytometer and analyzed using FlowJo v10.4 software (Tree Star Inc.).
Statistical analysis
Statistical analyses were performed in GraphPad Prism 7.0. By setting the upper and lower limits of the 95% confidence interval based on the vehicle values, equivalence testing determined that uninjected and vehicle-injected cohorts were experimentally equivalent; therefore, comparisons to control groups were reported as P values determined between the means of experimental and vehicle-treated control cohorts. Tests between a single experimental group and a control were performed by unpaired two-tailed t test. Unless noted otherwise in the figure legend, one-way analysis of variance (ANOVA) followed by Bonferroni’s multiple comparison test was used to determine differences between the means of multiple experimental groups. Dot plots report means (center bar) and SEM (error bars). Dotted lines indicate 2 SDs above the vehicle or control mean. The F test was used to determine whether sample variances were equal. Survival comparisons were made using the Wilcoxon test and reported as a Kaplan-Meier graph.
Supplementary Material
Acknowledgments
We thank T. Shimizu for the gift of the LPA4-null mice; D. Jones, L. Wolszon, R. Rivera, and G. Kaeser for editing; R. Rivera for maintaining the mouse colony; and J. Nhan, M. Merete, and A. Cerda for quantification assistance. Funding: This work was supported through NIH NS084398 (J.C.), NIH MH051699 (J.C.), DOD W81XWH-17-1-0455 (J.C.), NIH T32GM007752 (N.C.L. and A.J.F.), AHA 16SDG27020014 (V.A.B.), NIH HL141880 (V.A.B.), and NIH NS103940 (Y.K.). Author contributions: J.C. conceived the project, oversaw experiments, and wrote the paper. N.C.L. planned and performed experiments, analyzed data, and wrote the paper. P.S.-P., A.J.F., Y.K., and V.A.B. performed experiments and analyzed data. G.K. performed histological work for the project. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. All knockout mouse strains require material transfer agreement approval to obtain.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaax2011/DC1
Fig. S1. LPA losses during sample preparation.
Fig. S2. Live ciliary tracking.
Fig. S3. SEM cilia coverage of the ventricular surface.
Fig. S4. LPAR RNA expression in the ventricular region.
Fig. S5. Dependence of LPA-induced PHH on age, sex, strain, and injection concentration.
Fig. S6. Data from Fig. 5E analyzed by genotype subgroup and PHH distribution for LPA1-null pressure measurements.
Fig. S7. LPAR antagonist efficacy for PHH prevention.
Movie S1. Real-time video of live ciliary beating.
Movie S2. Example of ciliary tracking analysis.
REFERENCES AND NOTES
- 1.Kahle K. T., Kulkarni A. V., Limbrick D. D. Jr., Warf B. C., Hydrocephalus in children. Lancet 387, 788–799 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Robinson S., Neonatal posthemorrhagic hydrocephalus from prematurity: Pathophysiology and current treatment concepts. J. Neurosurg. Pediatr. 9, 242–258 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stoll B. J., Hansen N. I., Bell E. F., Walsh M. C., Carlo W. A., Shankaran S., Laptook A. R., Sanchez P. J., Van Meurs K. P., Wyckoff M., Das A., Hale E. C., Ball M. B., Newman N. S., Schibler K., Poindexter B. B., Kennedy K. A., Cotten C. M., Watterberg K. L., D'Angio C. T., DeMauro S. B., Truog W. E., Devaskar U., Higgins R. D.; Eunice Kennedy Shriver National Institute of Child Health and Human Development Neonatal Research Network , Trends in care practices, morbidity, and mortality of extremely preterm neonates, 1993-2012. JAMA 314, 1039–1051 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dewan M. C., Rattani A., Mekary R., Glancz L. J., Yunusa I., Baticulon R. E., Fieggen G., Wellons J. C. III, Park K. B., Warf B. C., Global hydrocephalus epidemiology and incidence: Systematic review and meta-analysis. J. Neurosurg. 1, 1–15 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Yung Y. C., Stoddard N. C., Chun J., LPA receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 55, 1192–1214 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aoki J., Taira A., Takanezawa Y., Kishi Y., Hama K., Kishimoto T., Mizuno K., Saku K., Taguchi R., Arai H., Serum lysophosphatidic acid is produced through diverse phospholipase pathways. J. Biol. Chem. 277, 48737–48744 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Yung Y. C., Stoddard N. C., Mirendil H., Chun J., Lysophosphatidic acid signaling in the nervous system. Neuron 85, 669–682 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hecht J. H., Weiner J. A., Post S. R., Chun J., Ventricular zone gene-1 (vzg-1) encodes a lysophosphatidic acid receptor expressed in neurogenic regions of the developing cerebral cortex. J. Cell Biol. 135, 1071–1083 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yung Y. C., Mutoh T., Lin M.-E., Noguchi K., Rivera R. R., Choi J. W., Kingsbury M. A., Chun J., Lysophosphatidic acid signaling may initiate fetal hydrocephalus. Sci. Transl. Med. 3, 99ra87 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faubel R., Westendorf C., Bodenschatz E., Eichele G., Cilia-based flow network in the brain ventricles. Science 353, 176–178 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Lun M. P., Monuki E. S., Lehtinen M. K., Development and functions of the choroid plexus-cerebrospinal fluid system. Nat. Rev. Neurosci. 16, 445–457 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X., Zhou Y., Wang J., Tseng I.-C., Huang T., Zhao Y., Zheng Q., Gao Y., Luo H., Zhang X., Bu G., Hong W., Xu H., SNX27 deletion causes hydrocephalus by impairing ependymal cell differentiation and ciliogenesis. J. Neurosci. 36, 12586–12597 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abdelhamed Z., Vuong S. M., Hill L., Shula C., Timms A., Beier D., Campbell K., Mangano F. T., Stottmann R. W., Goto J., A mutation in Ccdc39 causes neonatal hydrocephalus with abnormal motile cilia development in mice. Development 145, dev154500 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lechtreck K.-F., Delmotte P., Robinson M. L., Sanderson M. J., Witman G. B., Mutations in Hydin impair ciliary motility in mice. J. Cell Biol. 180, 633–643 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fukumizu M., Takashima S., Becker L. E., Neonatal posthemorrhagic hydrocephalus: Neuropathologic and immunohistochemical studies. Pediatr. Neurol. 13, 230–234 (1995). [DOI] [PubMed] [Google Scholar]
- 16.McAllister J. P., Guerra M. M., Ruiz L. C., Jimenez A. J., Dominguez-Pinos D., Sival D., den Dunnen W., Morales D. M., Schmidt R. E., Rodriguez E. M., Limbrick D. D., Ventricular zone disruption in human neonates with intraventricular hemorrhage. J. Neuropathol. Exp. Neurol. 76, 358–375 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clancy B., Darlington R. B., Finlay B. L., Translating developmental time across mammalian species. Neuroscience 105, 7–17 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Workman A. D., Charvet C. J., Clancy B., Darlington R. B., Finlay B. L., Modeling transformations of neurodevelopmental sequences across mammalian species. J. Neurosci. 33, 7368–7383 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Avasthi P., Marshall W. F., Stages of ciliogenesis and regulation of ciliary length. Differentiation 83, S30–S42 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Devitt A., Marshall L. J., The innate immune system and the clearance of apoptotic cells. J. Leukoc. Biol. 90, 447–457 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Yung Y. C., Kennedy G., Chun J., Identification of neural programmed cell death through the detection of DNA fragmentation in situ and by PCR. Curr. Protoc. Neurosci. Chapter 3, Unit 3.8 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Funke M., Zhao Z., Xu Y., Chun J., Tager A. M., The lysophosphatidic acid receptor LPA1 promotes epithelial cell apoptosis after lung injury. Am. J. Respir. Cell Mol. Biol. 46, 355–364 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holtsberg F. W., Steiner M. R., Keller J. N., Mark R. J., Mattson M. P., Steiner S. M., Lysophosphatidic acid induces necrosis and apoptosis in hippocampal neurons. J. Neurochem. 70, 66–76 (1998). [DOI] [PubMed] [Google Scholar]
- 24.Ohuchi H., Hamada A., Matsuda H., Takagi A., Tanaka M., Aoki J., Arai H., Noji S., Expression patterns of the lysophospholipid receptor genes during mouse early development. Dev. Dyn. 237, 3280–3294 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Allen Institute for Brain Science, Allen Mouse Brain Atlas (Allen Institute for Brain Science, 2015). [Google Scholar]
- 26.Swaney J. S., Chapman C., Correa L. D., Stebbins K. J., Broadhead A. R., Bain G., Santini A. M., Darlington J., King C. D., Baccei C. S., Lee C., Parr T. A., Roppe J. R., Seiders T. J., Ziff J., Prasit P., Hutchinson J. H., Evans J. F., Lorrain D. S., Pharmacokinetic and pharmacodynamic characterization of an oral lysophosphatidic acid type 1 receptor-selective antagonist. J. Pharmacol. Exp. Ther. 336, 693–700 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Ohta H., Sato K., Murata N., Damirin A., Malchinkhuu E., Kon J., Kimura T., Tobo M., Yamazaki Y., Watanabe T., Yagi M., Sato M., Suzuki R., Murooka H., Sakai T., Nishitoba T., Im D.-S., Nochi H., Tamoto K., Tomura H., Okajima F., Ki16425, a subtype-selective antagonist for EDG-family lysophosphatidic acid receptors. Mol. Pharmacol. 64, 994–1005 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Park R., Moon U. Y., Park J. Y., Hughes L. J., Johnson R. L., Cho S.-H. H., Kim S., Yap is required for ependymal integrity and is suppressed in LPA-induced hydrocephalus. Nat. Commun. 7, 10329 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herr K. J., Herr D. R., Lee C. W., Noguchi K., Chun J., Stereotyped fetal brain disorganization is induced by hypoxia and requires lysophosphatidic acid receptor 1 (LPA1) signaling. Proc. Natl. Acad. Sci. U.S.A. 108, 15444–15449 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi J., Wu X., Surma M., Vemula S., Zhang L., Yang Y., Kapur R., Wei L., Distinct roles for ROCK1 and ROCK2 in the regulation of cell detachment. Cell Death Dis. 4, e483 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Revankar C. M., Vines C. M., Cimino D. F., Prossnitz E. R., Arrestins block G protein-coupled receptor-mediated apoptosis. J. Biol. Chem. 279, 24578–24584 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Gilard V., Chadie A., Ferracci F.-X., Brasseur-Daudruy M., Proust F., Marret S., Curey S., Post hemorrhagic hydrocephalus and neurodevelopmental outcomes in a context of neonatal intraventricular hemorrhage: An institutional experience in 122 preterm children. BMC Pediatr. 18, 288 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vassilatis D. K., Hohmann J. G., Zeng H., Li F., Ranchalis J. E., Mortrud M. T., Brown A., Rodriguez S. S., Weller J. R., Wright A. C., Bergmann J. E., Gaitanaris G. A., The G protein-coupled receptor repertoires of human and mouse. Proc. Natl. Acad. Sci. U.S.A. 100, 4903–4908 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stoddard N. C., Chun J., Promising pharmacological directions in the world of lysophosphatidic acid signaling. Biomol. Ther. 23, 1–11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Contos J. J., Fukushima N., Weiner J. A., Kaushal D., Chun J., Requirement for the lpA1 lysophosphatidic acid receptor gene in normal suckling behavior. Proc. Natl. Acad. Sci. U.S.A. 97, 13384–13389 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Contos J. J., Ishii I., Fukushima N., Kingsbury M. A., Ye X., Kawamura S., Brown J. H., Chun J., Characterization of lpa2 (Edg4) and lpa1/lpa2 (Edg2/Edg4) lysophosphatidic acid receptor knockout mice: Signaling deficits without obvious phenotypic abnormality attributable to lpa2. Mol. Cell. Biol. 22, 6921–6929 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ye X., Hama K., Contos J. J. A., Anliker B., Inoue A., Skinner M. K., Suzuki H., Amano T., Kennedy G., Arai H., Aoki J., Chun J., LPA3-mediated lysophosphatidic acid signalling in embryo implantation and spacing. Nature 435, 104–108 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sumida H., Noguchi K., Kihara Y., Abe M., Yanagida K., Hamano F., Sato S., Tamaki K., Morishita Y., Kano M., Iwata C., Miyazono K., Sakimura K., Shimizu T., Ishii S., LPA4 regulates blood and lymphatic vessel formation during mouse embryogenesis. Blood 116, 5060–5070 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Lin M.-E., Rivera R. R., Chun J., Targeted deletion of LPA5 identifies novel roles for lysophosphatidic acid signaling in development of neuropathic pain. J. Biol. Chem. 287, 17608–17617 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chuang N., Mori S., Yamamoto A., Jiang H., Ye X., Xu X., Richards L. J., Nathans J., Miller M. I., Toga A. W., Sidman R. L., Zhang J., An MRI-based atlas and database of the developing mouse brain. Neuroimage 54, 80–89 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laurence K. M., Coates S., The natural history of hydrocephalus: Detailed analysis of 182 unoperated cases. Arch. Dis. Child. 37, 345–362 (1962). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Al-Shroof M., Karnik A. M., Karnik A. A., Longshore J., Sliman N. A., Khan F. A., Ciliary dyskinesia associated with hydrocephalus and mental retardation in a Jordanian family. Mayo Clin. Proc. 76, 1219–1224 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Cinalli G., Spennato P., Nastro A., Aliberti F., Trischitta V., Ruggiero C., Mirone G., Cianciulli E., Hydrocephalus in aqueductal stenosis. Childs Nerv. Syst. 27, 1621–1642 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Blegvad C., Skjolding A. D., Broholm H., Laursen H., Juhler M., Pathophysiology of shunt dysfunction in shunt treated hydrocephalus. Acta Neurochir. 155, 1763–1772 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Wessell A. P., Kole M. J., Cannarsa G., Oliver J., Jindal G., Miller T., Gandhi D., Parikh G., Badjatia N., Aldrich E. F., Simard J. M., A sustained systemic inflammatory response syndrome is associated with shunt-dependent hydrocephalus after aneurysmal subarachnoid hemorrhage. J. Neurosurg. 1, 1–8 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/10/eaax2011/DC1
Fig. S1. LPA losses during sample preparation.
Fig. S2. Live ciliary tracking.
Fig. S3. SEM cilia coverage of the ventricular surface.
Fig. S4. LPAR RNA expression in the ventricular region.
Fig. S5. Dependence of LPA-induced PHH on age, sex, strain, and injection concentration.
Fig. S6. Data from Fig. 5E analyzed by genotype subgroup and PHH distribution for LPA1-null pressure measurements.
Fig. S7. LPAR antagonist efficacy for PHH prevention.
Movie S1. Real-time video of live ciliary beating.
Movie S2. Example of ciliary tracking analysis.