

Abstract
The inability to rapidly restore the loss of function that results from severance (cutting or crushing) of PNS and CNS axons is a severe clinical problem. As a novel strategy to help alleviate this problem, we have developed in vitro procedures using Ca2+-free solutions of polyethylene glycol (PEG solutions), which within minutes induce functional and morphological continuity (PEG-induced fusion) between the cut or crushed ends of myelinated sciatic or spinal axons in rats. Using a PEG-based hydrogel that binds to connective tissue to provide mechanical strength at the lesion site and is nontoxic to nerve tissues in earthworms and mammals, we have also developed in vivo procedures that permanently maintain earthworm myelinated medial giant axons whose functional and morphological integrity has been restored by PEG-induced fusion after axonal severance. In all these in vitro or in vivo procedures, the success of PEG-induced fusion of sciatic or spinal axons and myelinated medial giant axons is measured by the restored conduction of action potentials through the lesion site, the presence of intact axonal profiles in electron micrographs taken at the lesion site, and/or the intra-axonal diffusion of fluorescent dyes across the lesion site. These and other data suggest that the application of polymeric fusiogens (such as our PEG solutions), possibly combined with a tissue adherent (such as our PEG hydrogels), could lead to in vivo treatments that rapidly and permanently repair cut or crushed axons in the PNS and CNS of adult mammals, including humans.
Keywords: axotomy, axonal regeneration, membrane fusion, neurotrauma, nerve repair, polyethylene glycol
Various strategies have been used to try to reestablish functional connections after PNS or CNS axons are severed. For PNS axons, strategies, such as nerve grafts (Jenq and Coggeshall, 1986), connective tissue matrices (Herbert et al., 1996), nerve growth guides (Aebischer et al., 1990), etc. (for review, see Das and Wallace, 1986; Lundborg, 1990), have been designed to enhance the number of severed axons that regenerate at 1–2 mm/d from surviving proximal stumps. However, even if they eventually reestablish some functions, these PNS outgrowths usually take from months to years to reach denervated target tissues in larger mammals, such as humans (Das and Wallace, 1986; Bittner, 1988, 1991). To try to reestablish functional connections after severance of CNS axons (whose proximal stumps normally do not regenerate), strategies have been designed to induce outgrowths from proximal stumps of surviving axons and/or to generate axonal outgrowths from donor tissues. Reestablishment of functional connections takes many months and has been difficult to convincingly demonstrate using such strategies, which include donor transplants of peripheral nerve sheaths (David and Aguayo, 1981) or embryonic tissues (Giovanini et al., 1997; Miya et al., 1997) and injections of antibodies to oligodendritic inhibitors of CNS axonal outgrowth (Schnell and Schwab, 1990).
As an alternate strategy to more rapidly reestablish function after severance of PNS or CNS axons in mammals in vivo, we suggest that solutions of fusiogens, such as polyethylene glycol (PEG), can rapidly (within minutes) and directly reestablish functional and morphological continuity between severed axonal ends. We also suggest that, if necessary, tissue adherents, such as PEG hydrogels, can be used to maintain the integrity of PEG-fused axons whose cut ends have been PEG-fused. To begin to test the efficacy of this strategy, we initially developed PEG solutions that could induce fusion between the severed ends of a myelinated medial giant axon (MGA) in the CNS of earthworms (Krause and Bittner, 1990; Krause et al., 1991) or unmyelinated MGAs in the CNS of crayfish in vitro or mammalian NG 108–15 cells in tissue culture (Bittner et al., 1986). We now report that modified PEG solutions rapidly fuse both cut and crushed ends of both myelinated mammalian sciatic axons (PNS axons) and spinal axons (CNS axons) in vitro according to the measures of functional and morphological continuity outlined above. We also report that PEG hydrogels that we have developed to adhere tightly to connective and other tissues in mammals and earthworms can be used to maintain the integrity of PEG-fused axons in vivo. For example, when our PEG-based hydrogel is applied to connective tissue sheaths of PEG-fused MGAs in vivo, MGAs PEG-fused in vivo remain functionally and morphologically intact for at least 20 d in nonanesthetized earthworms, and MGA-mediated behaviors are permanently reestablished.
Given these and other data, we suggest that PEG solutions could be combined with our PEG hydrogel in an efficacious strategy to directly, rapidly, and permanently repair severed axons in the PNS or CNS of adult mammals in vivo.
MATERIALS AND METHODS
In vitro preparations. Three centimeter lengths of sciatic nerves with their epineural sheaths intact were dissected from the hindlimbs of deeply anesthetized (60–80 mg/kg pentabarbitol) adult male or female rats (250–400 gm; n > 300). Some sciatic nerves (n > 20) were subsequently desheathed by removing the epineurium with a coarse-tipped and a fine-tipped Dumont model 5 forcep.
Three to five centimeter lengths of spinal cords were dissected free of the spinal column of deeply anesthetized adult rats (250–400 gm;n > 30) as described previously (Shi and Blight, 1996,1997). The spinal cord was divided in half at the midline, and the white matter of the dorsal columns was separated from the gray matter with a scalpel blade. Strips of white matter (spinal axons) 3- to 5-cm-long and several millimeters in diameter were maintained in oxygenated Krebs’ solution at room temperature (∼25°C) for at least 1 hr before use. Such spinal axons had no thick epineural sheaths.
Three to five centimeter lengths (n > 300) of ventral nerve cords (VNCs), the CNS of earthworms, were dissected from the middle body segments of adult animals anesthetized with 4% (w/v) Chlorotone (Eastman Kodak, Rochester, NY) (Krause and Bittner, 1990; Krause et al., 1991). As described previously (Günther, 1975, 1976; Roots and Lane, 1983; Okamura et al., 1985; Ballinger et al., 1997), the VNC contains two lateral giant axons (LGAs), each 50–100 μm in diameter, and a single MGA, 80–150 μm in diameter, including a 5- to 10-μm-wide sheath consisting of myelin whose biochemistry is slightly different from that of mammalian myelin and whose layered structure is somewhat looser than that of mammalian myelin. The MGA typically protrudes ∼50 μm from the cut end of each side of a severed VNC (Krause and Bittner, 1990; Krause et al., 1991,1994; Ballinger et al., 1997), making it particularly easy to appose the two ends. MGA and LGA action potentials (APs) can be uniquely identified in extracellular recordings of transmembrane currents from the VNC or from the entire animal (Bullock and Horridge, 1965;Günther, 1975, 1976; Krause and Bittner, 1990; Krause et al., 1991). That is, the MGA AP typically has the lowest threshold and fastest conduction velocity in the VNC and the second largest (extracellularly recorded) AP. The two LGAs are connected by gap junctions and typically exhibit coincident APs, which have the second fastest velocity and threshold but largest extracellularly recorded AP. All other (nongiant) axons in the VNC generate much (approximately threefold) slower APs with much (approximately fivefold) smaller extracellularly recorded APs than MGAs or LGAs. Once activatedin vivo by mechanical stimulation of sensory fibers at the rostral end of the earthworm, MGAs evoke a rapid contraction of the caudal end of the earthworm, an escape behavior that is critical for its survival in its normal environment (Bullock and Horridge, 1965).
In vivo preparations. VNCs were completely severed in earthworms (n > 150) anesthetized with 4% Chlorotone, and the cut ends of the MGAs were brought into close apposition with minuten pins as described below for in vitro preparations. Once the cut ends were closely apposed, in vitro andin vivo procedures were the same for application of various solutions or PEG hydrogels. APs were recorded from MGAs in vivo as described in Materials and Methods, Electrophysiological techniques. Operated earthworms were maintained in Petri dishes containing moistened filter paper for 3 d after surgery and thereafter maintained for up to 20 d in Petri dishes containing peat moss and potting soil.
Saline solutions. As given in detail in Table1 for each numbered solution listed below, sheathed or desheathed sciatic nerves or strips of spinal axons were bathed in various solutions: (1) a Ca2+-free hypotonic PBS; (2) a physiological “mammalian saline” at ∼25 or 37°C, as maintained by a Peltier unit; (3) a Ca2+-free, slightly hypotonic, mammalian saline formulated as listed in 2, except that CaCl2 was omitted and 0.5–2 mm EGTA was added; (4) a Ca2+-free hypotonic mammalian saline formulated as listed in 3, except that CaCl2 was omitted, NaCl was reduced by one-half, and 0.5–2 mm EGTA was added; (5) a physiological Krebs’ solution; and (6) a Ca2+-free (isotonic) Krebs’ solution formulated as in 5, except for equimolar replacement of CaCl2 with MgCl2 plus 0.5–2 mm EGTA. These and other solutions were usually equilibrated with a mixture of 95% O2–5% CO2. We detected no difference in results obtained with the mammalian saline versus Krebs’ solution or the Ca2+-free, slightly hypotonic, mammalian saline versus the Ca2+-free (isotonic) Krebs’ solution.
Table 1.
Composition of mammalian and earthworm salines
| Solution # | Designated name | Composition (in mm) |
|---|---|---|
| 1 | Ca2+-free hypotonic PBS | 77 NaCl, 2.6 KCl, 4.0 Na2HPO4, and 1.0 KH2PO4 |
| 2 | Physiological mammalian saline | 133 NaCl, 2.6 KCl, 2.0 CaCl2, and 10.0 HEPES, pH 7.4 |
| 3 | Ca2+-free slightly hypotonic mammalian saline | 133 NaCl, 2.6 KCl, 10.0 HEPES, and 0.5–2.0 EGTA, pH 7.4 |
| 4 | Ca2+-free hypotonic mammalian saline | 66.5 NaCl, 2.6 KCl, 10.0 HEPES, and 0.5–2.0 EGTA, pH 7.4 |
| 5 | Physiological Krebs’ solution | 124 NaCl, 2.0 KCl, 26 NaHCO3, 1.3 MgSO4, 1.2 KH2PO4, 1.2 CaCl2, 10.0 dextrose, and 6.0 Na ascorbate, pH 7.4 |
| 6 | Ca2+-free (isotonic) Krebs’ solution | 124 NaCl, 2.0 KCl, 26 NaHCO3, 2.5 MgSO4, 1.2 KH2PO4, 10.0 dextrose, 6.0 Na ascorbate, and 0.5–2.0 EGTA, pH 7.4 |
| 7 | Physiological earthworm saline | 40 NaCl, 10 Na2SO4, 0.5 K2SO4, 0.5 MgCl2, 3.0 CaCl2 1.3 TRIS, and 1.5 HEPES, pH 7.4 |
| 8 | Ca2+-free earthworm saline | 40 NaCl, 10 Na2SO4, 0.5 K2SO4, 0.5 MgCl2, 1.3 TRIS, 1.5 HEPES, and 0.5–1.0 EGTA, pH 7.4 |
VNCs containing the MGA were bathed at room temperature (∼25°C) in either (7) a physiological “earthworm saline” or (8) a Ca2+-free (slightly) hypotonic earthworm saline formulated as listed above, except that CaCl2 was omitted and 0.5–1.0 mm EGTA was added.
Electrophysiological techniques. To accomplish conventional extracellular stimulation and recording of either multiple simultaneously generated APs [“compound” APs (CAPs)] from bundles of sciatic axons or uniquely identifiable APs from a single MGA, sciatic nerves or VNCs were bathed in physiological salines in a single-chambered device similar to the one developed to record APs before and after PEG-induced fusion of MGAs (Krause and Bittner, 1990;Krause et al., 1991). CAPs or APs were led to an extracellular AC amplifier (P-15; Grass Instruments, West Warwick, RI) and displayed on an oscilloscope. APs from MGAs were occasionally extracellularly recorded in this apparatus from the entire animalin vivo by using damp Kimwipes (Kimberly-Clark, Roswell, GA) to immobilize the entire intact earthworm, positioned so that its ventral surface was pressed tightly against the stimulating and recording electrodes in our single-chambered device (Krause et al., 1991, their Fig. 1). Sciatic nerves were extracellularly stimulated with voltages (usually 5–15 V) that were supramaximal to elicit a CAP having the largest peak amplitude, i.e., voltages that elicited APs in the maximum number of sciatic axons.
For double sucrose gap recordings, sciatic nerves, spinal nerves, or VNCs were placed in the central chamber of a five-chambered recording device as described previously (Shi and Blight, 1996). CAPs or APs were stimulated or recorded in the end chambers, which were filled with 0.12m KCl. The larger end chambers were each separated from the larger central chamber by a smaller chamber through which sucrose was perfused. The central chamber was often continuously perfused (except during PEG application) with oxygenated 25°C Krebs’ solution by gravity flow. Solutions in the central chamber were continuously removed by vacuum suction. CAPs or APs were led to a DC intracellular amplifier (IE-251; Warner, Grand Haven, MI) to be conventionally displayed on an oscilloscope or a Dell 200 computer to be processed by a data analysis program (C-Scope; RC Electronics, Santa Barbara, CA) whose output was displayed on an x–yplotter.
APs from MGAs were also occasionally recorded intracellularly with glass microelectrodes filled with 3 m KCl. In such cases, the signals were displayed as described for MGA APs in double sucrose gap recordings.
Severance of sciatic axons, spinal axons, or MGAs. Sciatic nerves, strips of spinal axons, or VNCs were viewed with a dissecting microscope during all experimental procedures, including axonal severance (cutting or crushing) and subsequent PEG-induced fusion. In all “cut” preparations of sheathed or desheathed sciatic nerves, spinal axons (lacking sheaths), or sheathed VNCs, the nerve bundle was completely cut between the stimulating and recording electrodes with a shard of a microknife made from a Gillette (Boston, MA) Blue Blade so that the cut axonal ends always completely separated by 0.5–1.0 mm. Cut ends were tightly apposed by pushing on the sciatic nerve, spinal axons, or VNC with minuten pins or by light pressure applied with fine-tipped forceps. A nylon mesh stretched across a metal loop (which was not in contact with the nerve tissue) positioned by a micromanipulator was used to keep the tightly apposed ends in place or to make fine adjustments in the alignment of the cut ends by gently tugging on the surface axons and nylon mesh strands. In “crushed” preparations of sciatic axons, spinal axons, or MGAs, the sheathed sciatic nerve or VNC, respectively, was compressed with a dull microknife or fine-tipped forceps to produce a transparent zone (200- to 500-μm-wide) of axoplasm-free tissue, as viewed with the dissecting microscope. The epineurial sheath surrounding crushed sciatic axons or MGAs did not pull apart, i.e., the connective tissue sheathing the sciatic nerve or VNC was not severed. The completeness of cut or crush severance was always confirmed by electrically stimulating the sciatic nerve, strip of spinal axons, or VNC and observing that no CAPs from sciatic axons, spinal axons, or APs from MGAs were transmitted across the lesion site. Furthermore, most of the morphological and electrophysiological data presented in the Results were taken from cut (rather than crushed) preparations, thereby eliminating artifacts or alternate explanations associated with the presence of an incomplete lesion.
Application of PEG solutions. PEG of various molecular weights (in kilodaltons), as specified in the Results, was dissolved in distilled water, which often contained food coloring to enable us to visualize the location of the PEG solution. PEG was applied from a micropipette positioned using a micromanipulator on one side of the sciatic nerve, strip of spinal axons, or VNC so that the PEG-containing solution flowed in a narrow stream (∼500-μm-wide) over the closely apposed cut or crushed axons at the lesion site. The PEG was then removed by continuous suction applied to a glass micropipette positioned on the opposite side of the sciatic nerve, strip of spinal axons, or VNC as reported previously (Krause and Bittner, 1990; Krause et al., 1991).
Synthesis of PEG hydrogels. As described previously (Pathak et al., 1992; Sawhney et al., 1993), our PEG hydrogel was synthesized by photochemically inducing the polymerization of a precursor solution to form a cross-linked hydrogel directly on the tissue being treatedin situ. The precursor solution consisted of 23% 8 kDa PEG diacrylate (see below), 1 mm eosin Y (Sigma, St. Louis, MO), 100 mm triethanolamine (Sigma), and 1500 ppmN-vinylpyrrolidone (Aldrich, Milwaukee, WI) dissolved in PBS at a final pH of 7.4. The 8 kDa PEG diacrylate was formed from 8 kDa PEG, which has an alcohol at both ends, by reacting the terminal alcohols with acryloyl chloride to form the α,ω-diacrylate of PEG (Pathak et al., 1992). This precursor polymer is soluble in the aqueous precursor solution, but polymerization of the material (because it has an acrylate at both ends) forms a cross-linked polymer network that is highly swollen with water (∼90% of its mass is water). This polymerization was induced by exposing the precursor solution briefly (15–30 sec) to visible light at intensities and frequencies that are noncytotoxic, even for exposures lasting many hours.
In brief, the PEG-based hydrogel we developed has several valuable properties. (1) The hydrogel transforms within 30 sec from a liquid precursor into a solid gel that adheres strongly to the VNC and sciatic nerve tissues. PEG-based diacrylate precursors were polymerizedin situ from aqueous solution to form materials that adhered tightly to VNCs (n > 20) and sciatic nerves (n > 20) and remained there during degradation as reported for the peritoneal cavity (Hubbell et al., 1994; Chowdhury and Hubbell, 1996) and the carotid artery (West and Hubbell, 1996). The PEG diacrylate penetrates the biological macromolecular network in the extracellular matrix of the tissue. Conversion into a solid cross-linked hydrogel results in permanent interpenetration and thus adhesion. The PEG diacrylate is the precursor polymer that is further polymerized and cross-linked, the eosin Y and triethanolamine form a photoinitiation system, and the N-vinylpyrrolidone serves as a polymerization accelerator. (2) Our hydrogel transforms from a liquid precursor to a solid hydrogel on the surface of VNCs, sciatic nerves, or spinal cords without cytotoxicity as reported for other tissues (West and Hubbell, 1996). The PEG diacrylate precursor has essentially the same biocompatibility (lack of cytotoxicity) as native PEG of the same molecular weight, eosin Y is a common drug colorant, triethanolamine is commonly used in pharmaceutical formulations, andN-vinylpyrrolidone is acceptable as a contaminant in the medical polymer Povidone at much higher concentrations than used herein (Sawhney et al., 1993). (3) The hydrogel is biocompatible and noninflammatory in vivo. The transformed PEG hydrogel is highly biocompatible, in a large part because of the favorable interactions of PEG with proteins. PEG is very hydrophilic and nonionic, and it blocks interactions with cell-surface receptors, thus producing low levels of protein adsorption and inflammatory cell adhesion (West and Hubbell, 1996). (4) Our hydrogel can be designed to degrade over a few days to several months by incorporating ester (e.g., glycolic, lactic, caproic acid) linkages to provide sites for nonenzymatic hydrolysis (Sawhney et al., 1993; West and Hubbell, 1996). In the present case, biostable hydrogels, lacking such degradation sites, were usually selected for these studies. (5) Our hydrogel does not affect axonal structure or function (see Results).
Application of PEG hydrogels. PEG hydrogels were applied from microsyringe needles in vitro or in vivo to sciatic nerves of rats anesthetized with pentabarbitol as described above. After recovery from anesthesia, the animals were kept in individual cages. Wounds were closed with 7–0 or 8–0 sutures (Ethicon, Somerville, NJ). Topical anesthetics were applied to the wound site, which was examined daily for infection.
PEG hydrogels were applied to the VNC of earthworms maintained in 10-cm-diameter Petri dishes, sometimes containing filter paper or soil moistened with 0.1% Chlorotone to anesthetize the animal. The lesion site was sometimes closed with 7–0 or 8–0 sutures. MGA APs were occasionally recorded in vivo from the outer surface of earthworms as described in Materials and Methods, Electrophysiological techniques.
Histological–ultrastructural procedures. After recording CAPs, control (n = 2) or PEG-fused (n = 5) sciatic nerves were carefully fixed in place in the recording chamber (which was then discarded) with a solution of 3% paraformaldehyde, 3% glutaraldehyde, and 0.1% picric acid in 0.1m cacodylate buffer, pH 7.4. The tissue was post-fixed in 2% osmium tetroxide in 0.1 m cacodylate buffer, pH 7.4, dehydrated in a graded alcohol series, and embedded in Spurrs’s plastic as described previously (Sunio and Bittner, 1997). The sciatic nerve was sectioned longitudinally approximately halfway through the fusion site, taking both thick (0.5 μm) sections for imaging at lower magnifications to identify the original site of apposed cut ends and thin (silver or gold) sections for imaging at higher magnifications. To construct a better three-dimensional image of the fusion site, we sometimes remounted the remaining tissue and took thick and thin cross sections at the site of PEG-induced fusion (seeBallinger et al., 1997, for a description of this sectioning procedure).
Confocal or epifluorescence microscopy of dye-filled axons.The following fluorescent dyes taken up by sciatic axons were obtained from Molecular Probes (Eugene, Oregon): sulforhodamine 101 (548 nm excitation wavelength, 605 nm emission wavelength), sulforhodamine B-dextran (565 nm excitation wavelength, 586 nm emission wavelength), and Texas Red (584 nm excitation wavelength, 605 nm emission wavelength). Dye-labeled desheathed sciatic axons were viewed with confocal and differential interference contrast (DIC) microscopy using a Leica (Nussloch, Germany) TCS-4D fitted with two water immersion lenses [40×, 0.75 numerical aperture; 63×, 0.90 numerical aperture; both from Zeiss (Oberkochen, Germany)] and a 10×, 0.30 numerical aperture lens from Leica. (Sciatic axons were much more difficult to image confocally in sheathed sciatic nerves.) Digitized DIC and confocal images were stored and analyzed on a Macintosh 7200/90 equipped with NIH Image 1.6 and Adobe Systems (San Jose, CA) Photoshop 4.0 software. Dyes were extracellularly applied to sciatic or spinal nerve bundles by using a 26 gauge needle to inject ∼1 μl of a 15% solution of a fluorescent dye from a microsyringe into these nerve bundles at 3–6 mm from the lesion site. The injected sciatic nerves were maintained at 25°C for 6–14 hr to allow diffusion of the dyes through the entire length of control or PEG-fused sciatic axons. To ensure that the dye did not travel by an extracellular diffusion path, we continuously perfused oxygenated Krebs’ through the chamber containing the lesion site to remove any dye that may have leaked into the bath saline.
MGAs (n > 15) were iontophoretically injected (Krause and Bittner, 1990) on one side of the lesion site with micropipettes filled with Lucifer yellow CH (Molecular Probes). Sciatic axons, spinal axons, or MGAs filled with fluorescent dyes were also examined using an inverted Zeiss ICM-35 with the ability to image for epifluorescence and enhanced video microscopy of whole mounts cleared in methyl salicylate.
RESULTS
Development of our protocol for PEG-induced fusion of sciatic axonsin vitro
In an initial series of experiments (n > 250 sciatic nerves) to devise a protocol to fuse the cut or crushed ends of mammalian axons, we first extracellularly recorded control CAPs generated by 1 Hz maximal stimulation of intact sciatic axons in sheathed sciatic nerves (Fig.1A,B,i) bathed in mammalian saline (see Materials and Methods). Sciatic nerves were then bathed in a Ca2+-free saline (Materials and Methods; Tables 1,2) before completely cutting (n > 50) or crushing (n > 200) the sciatic nerves halfway between the stimulating and recording electrodes (see Materials and Methods). CAPs were usually slightly smaller in Ca2+-free salines in these initial (data not shown) and later (Fig. 1D,E) experiments. CAPs always completely disappeared after sciatic axons were completely transected by cutting or crushing (Fig.1A,B, t). The 1 Hz electrical stimulation was temporarily halted after the sciatic axons were severed, and a PEG solution was applied to the lesion site. After 0.2–2 min, the Ca2+-free saline was replaced with a physiological saline containing Ca2+. CAPs were counted as conducting through the lesion site (successful fusion) only if a CAP reappeared after PEG-induced fusion (Fig.1A,B, f) and then completely disappeared after the sciatic axons were retransected (Fig.1A,B, rt), i.e., transected so that the cut ends were completely separated by a gap of at least 0.5 mm. In brief, we applied the same electrophysiological criteria to CAPs to define a successful fusion event for mammalian sciatic axons (Fig. 1A,B; Table 2) that we applied in previous (Krause et al., 1991) or current (Fig.1F; Table 2) reports of successful fusion of earthworm MGAs; conduction of MGA APs across the stimulating and recording electrodes must be present before severance, completely eliminated by severance, restored by PEG, and completely eliminated by resevering.
Fig. 1.
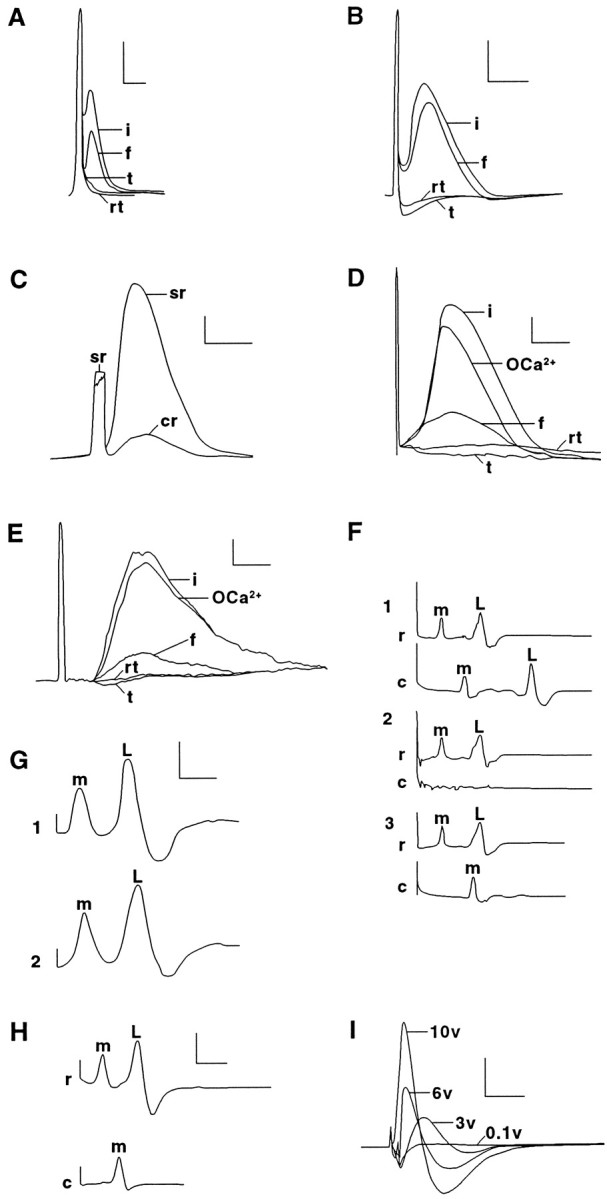
CAPs from sciatic (A–D,I) or spinal (E) axons or APs from MGAs (F–H) in intact and PEG-fused preparations extracellularly recorded using conventional (A–C, F–I) or double sucrose gap techniques (C–E). A–E, Individual CAPs recorded conventionally (A–C) or with double sucrose gap (C, E) from control rat sciatic or spinal axons (i traces) before replacing the Krebs’ (Table 1, solution 5) in the central chamber with Ca2+-free Krebs’ (Table 1, solution 6) containing 1 mm EGTA (OCa2+ traces) at ∼25°C. The sciatic or spinal axons were then transected to eliminate the CAP (t traces). PEG was applied to the apposed cut ends, and the central chamber was again perfused with Krebs’ (Table 1, solution 5). Within 10 min, the CAP again appeared in fused sciatic or spinal axons (f traces) and remained for ≥30 min. The CAP was again eliminated when the sciatic or spinal axons were retransected at the original lesion site (rt traces).C illustrates the increase in signal-to-noise ratios achieved using sucrose gap recordings (sr trace) compared with conventional recordings (cr trace) of an intact sciatic nerve. The sciatic nerve or strip of spinal axons was always extracellularly stimulated by a maximal voltage (e.g., ∼6 V inD and 10 V in E) that reliably produced a CAP with the greatest peak amplitude. F, Individual MGA and LGA APs recorded conventionally using the single-chamber device ofKrause et al. (1991). Control (intact) MGA (m) and LGA (L) APs (trace 1) placed in physiological earthworm saline (Table 1, solution 7) stimulated by a rostral electrode and recorded from rostrally (r) and caudally (c) placed electrodes before cutting the axons between the two recording electrodes to eliminate the APs from the caudal electrode (trace 2). The VNC was placed in Ca2+-free earthworm saline (Table 1, solution 8), and PEG was applied to the apposed cut ends of the MGA, but not the LGA, to induce PEG-fusion only of the MGA, and the central chamber was again perfused with earthworm saline (Table 1, solution 7). PEG hydrogel was then applied to the lesion site. Within 10 min, the MGA (but not LGA) AP was again recorded for 1–24 hr on the caudal side of the lesion site (trace 3). G, MGA and LGA APs recorded in vitro from a VNC before (trace 1) and after (trace 2) it was surrounded by our PEG-based hydrogel for 1 hr. H, MGA APs recorded from a VNC in which MGAs PEG fused in vivo by our PEG solution were surrounded by our PEG-based hydrogel and then maintained in vivo for 15 d. The VNC was then dissected from the animal. When the VNC was extracellularly stimulated rostral to the lesion site and extracellularly recorded rostrally and caudally to the site of PEG-induced fusion and hydrogel application, the rostral electrodes recorded MGA and LGA APs, and the caudal electrode recorded only MGA APs. That is, the MGA, but not the LGA, had been PEG-fused. The MGA was then injected with Lucifer yellow CH, which diffused through the site of PEG-induced fusion (Fig. 3D). I, CAPs conventionally recorded in vitro from a sciatic nerve exposed to PEG-based hydrogel for 10 d in vivo. The sciatic nerve was extracellularly stimulated with 0.1 (subthreshold), 3, 6, and 10 (maximal) V pulses. Calibration: A,B, F–I, 0.5 mV, 1 msec;C–E, 1 mV, 0.5 msec.
Table 2.
Percent of PEG-fused CAPs observed within 60 min after severing sciatic axons
| Lesion | |||||||
|---|---|---|---|---|---|---|---|
| P# | n | % Fused | Lesion | PEG | EGTA | Duration | Saline |
| Initial | |||||||
| 1 | 5 | 0 | crush | 50%, 1, 5 kDa | 0.5 | 30–60 | 1 |
| 2 | 9 | 0 | crush | 60%, 1/1.5 kDa | 1 | 30–60 | 1 |
| 3 | 24 | 0 | crush | 50%, 4 kDa | 1 | <15 | 4 |
| 4 | 29 | 0 | cut | 50%, 4 kDa | 1 | <15 | 3 |
| 5 | 15 | 0 | crush | 50%, 4 kDa | 1 | <15 | 4 |
| 6 | 19 | 0 | crush | 50%, 1.5 kDa | 0.5 | 30–60 | 3 |
| 7 | 8 | 0 | crush | 50%, 1.5 kDa | 0.5 | 30–60 | 3; 0. 1% DMSO |
| 8 | 4 | 0 | crush | 50%, 1.5 kDa | 0.5 | 30–60 | 3; 1% DMSO |
| 9 | 8 | 13 | crush | 50%, 4 kDa | 1 | 30–60 | 3 |
| 10 | 23 | 0 | crush | 1%, 2 kDa | 1 | 30–60 | 3 |
| 11 | 27 | 0 | crush | 10%, 2 kDa | 1 | 30–60 | 3 |
| Subsequent | |||||||
| 12 | 10 | 10 | cut | 50%, 2 kDa | 1 | 60 | 3 |
| 13 | 10 | 20 | cut | 50%, 2 kDa | 1 | 60, 2× | 3 |
| 14 | 22 | 23 | crush | 50%, 2 kDa | 1 | 60 | 3 |
| 15 | 22 | 36 | crush | 50%, 2 kDa | 1 | 60, 2× | 3 |
| 16 | 7 | 71 | crush | 50%, 2 kDa | 1 | 60 | 3 |
| 17 | 4 | 25 | cut | 50%, 2 kDa | 1 | 60 | 3 |
| 18 | 14 | 21 | crush | 50%, 2 kDa | 1 | 60, 2× | 3 |
| 19 | 6 | 17 | cut | 50%, 2 kDa | 1 | 60, 2× | 3 |
| 20 | 7 | 43 | crush | 50%, 2 kDa | 1 | 60 | 3; 100 μg/ml calpain |
| 21 | 5 | 0 | crush | 50%, 2 kDa | 1 | 60 | 3; 100 μg/μm leupeptin |
| 22 | 18 | 39 | cut | 50%, 2 kDa | 1 | 60–120 | 6 |
| 23 | 26 | 8 | cut | 50%, 2 kDa | 1 | 60–120 | 6, desheathed |
P#, Protocol reference number in Results. n, Total number of sciatic nerves tested. % Fused, Number of sciatic nerve preparations that exhibited action potentials conducting through the lesion site as assessed by a detectable CAP that disappeared after resevering the sciatic axons, divided by the total number of sciatic nerve preparations tested (n), times 100%. Lesion, Complete cut or crush as assessed by loss of CAP conduction through the lesion site. PEG, Percentage of PEG (w/w dissolved in distilled water) of molecular weight given in kilodaltons. EGTA, Millimolar EGTA added to the Ca2+-free saline in which the sciatic axons were severed just before PEG application. Duration, Duration (in seconds) of PEG application, applied once or twice (indicated by 2×). Ca2+-free saline, Solution
(from Table 1) in which sciatic axons were cut and PEG applied. All sciatic nerve preparations were sheathed, unless otherwise indicated.
We observed no instances of PEG-induced fusion (as measured by reappearance of a CAP) of cut or crushed sciatic axons with most of our initial attempts (Table 2) to modify the protocol that had been most successful in PEG-fusing earthworm giant axons: 4 kDa PEG at 50% w/w (in double-distilled water) applied for 30–60 sec to the lesion site, which was bathed in Ca2+-free salines of 70–90% tonicity with 1 mm EGTA (Krause and Bittner, 1990). For example, CAPs were not conducted across a crush site after application of 50% (w/w) PEG of 1.5 kDa or 60% PEG of 1–1.5 kDa (J. T. Baker Chemical Company, Phillipsburg, NJ) applied for 30–60 sec in Ca2+-free hypotonic PBS with 0.5 mm EGTA (Table 2, protocols 1, 2). No cases of successful fusion were noted when 50% 4 kDa PEG (J. T. Baker Chemical Company) was applied in Ca2+-free mammalian salines or Ca2+-free hypotonic mammalian salines for shorter times, e.g., <15 sec (Table 2, protocols 3–5). No cases of successful CAP conduction across a crush site were noted after 30–60 sec applications of 50% 1.5 kDa PEG in Ca2+-free, slightly hypotonic, mammalian saline when <1.0 mm EGTA was used, even if 0.1 or 1% DMSO was added (Table 2, protocols 6–8). Only one successful fusion of crushed sciatic axons was observed when PEG with a somewhat higher molecular weight was applied (Table 2, protocol 9). No cases of successful fusion of crushed sciatic axons were noted when lower concentrations (1–10%) of 2 kDa PEG (Aldrich) were applied in Ca2+-free, slightly hypotonic, mammalian saline containing 1.0 mm EGTA (Table 2, protocols 10, 11).
In contrast to these earlier trials, PEG-induced fusion was consistently observed in subsequent sets of trials using a 50% (w/w) solution of 2 kDa PEG (Aldrich) applied once or twice for 60–120 sec in Ca2+-free, slightly hypotonic, salines containing 1–2 mm EGTA. For example, cut or crushed sheathed sciatic axons exhibited fusion when 50% 2 kDa PEG was applied for 60 sec once or twice in a Ca2+-free, slightly hypotonic, mammalian saline containing 1 mm EGTA (Table 2, protocols 12–19). The percentage of sciatic nerve preparations exhibiting electrophysiological evidence of successful PEG-induced fusion ranged from 10 to 71% in different sets of trials (Table 2) performed by different personnel. In general, the rate of success increased with practice by each collaborator.
The addition of 100 μg/ml calpain to 50% 2 kDa PEG slightly increased the frequency of preparations exhibiting successful PEG-induced fusion of crushed sciatic axons, whereas the addition of 100 μg/ml leupeptin greatly decreased the frequency of successful PEG-induced fusion (Table 2, paradigms 20, 21, respectively). We also noted (Table 2, paradigms 22, 23) that sheathed sciatic axon preparations were much easier to fuse with PEG than desheathed preparations (used for confocal viewing because the sciatic nerve sheath distorts confocal imaging).
Given these data and our observations that (1) hypotonic salines were not necessary to fuse cut spinal axons and (2) Ca2+-free Krebs’ solutions containing 1–2 mm EGTA produced results similar to slightly hypotonic mammalian salines containing 1–2 mm EGTA, we performed almost all subsequent sucrose gap-recorded fusions of cut sciatic or spinal axons using a 50% solution of 2 kDa PEG (Aldrich) applied once or twice for 60–120 sec in an isotonic Ca2+-free Krebs’ solution (sciatic axons) containing 1–2 mmEGTA.
Sucrose gap assessment of sciatic axons and MGAs fused with PEG in vitro
To enhance the signal-to-noise ratio of CAPs recorded from PEG-fused sciatic (Fig. 1C,D) or spinal (Fig.1E) axons, we placed sheathed or desheathed sciatic nerves or strips of spinal axons from rats in a five-chambered device (Shi and Blight, 1996) for single or double sucrose gap recordings (see Materials and Methods). The ability of intact (control), severed, or fused sciatic or spinal axons to conduct CAPs through the central chamber was assessed in the following manner. Control CAPs generated by maximum stimulation of intact axons in one end chamber were conducted across the central chamber containing Krebs’ solution (Table 1, solution 5) and recorded in the opposite end chamber(Fig.1D,E, i). The axons in the central chamber were then immersed in the Ca2+-free Krebs’ solution containing 1 mm EGTA (Table 1, solution 6). Although CAP amplitude usually declined by 10–30%, this calcium-free solution did not eliminate the conduction of CAPs across the central chamber (n > 20 sciatic nerves) (Fig.1D,E, OCa2+). The sciatic or spinal axons were then cut completely at their midpoint in the central chamber, as assessed by visual inspection and by the complete elimination of CAP conduction across the lesion site in the central chamber (Fig. 1D,E,t).
To induce severed axonal ends to fuse, the cut surfaces of sciatic or spinal axons bathed in the Ca2+-free Krebs’ were first tightly apposed, and a continuous stream of 50% 2 kDa PEG was applied to the lesion site for 60–120 sec. The Ca2+-free solution in the central chamber bathing the PEG-exposed axons was replaced by oxygenated Krebs’ (Table 1, solution 5). CAPs often conducted across the lesion site within 2–15 min after PEG application and continued up to 120 min thereafter (Fig.1D,E, f), at which time recordings were discontinued. The peak amplitude of CAPs after PEG fusion ranged from 1 to 25% (usually 5–15%) of the amplitude of control CAPs recorded from the same sciatic or spinal axons before severance. The latency of the CAP often increased slightly, and the peak broadened after PEG-induced fusion. The peak amplitude of the CAP after PEG fusion was reduced, probably because PEG reestablished continuity between only a fraction of the total number of severed sciatic axons, as suggested by photomicrographs or electronmicrographs (Figs. 2,3A) or confocal images (Fig.4) of such lesion sites after the application of PEG solutions. The increased latency and/or broadening of the peak of the CAP after PEG-induced fusion might result from a slowed conduction velocity through the fusion site, possibly because of a decreased input resistance of PEG-fused membranes as reported for PEG-fused earthworm giant axons (Krause et al., 1991), or from a tendency for fusion to occur in smaller-diameter fibers in a bundle of PNS or CNS axons.
Fig. 2.
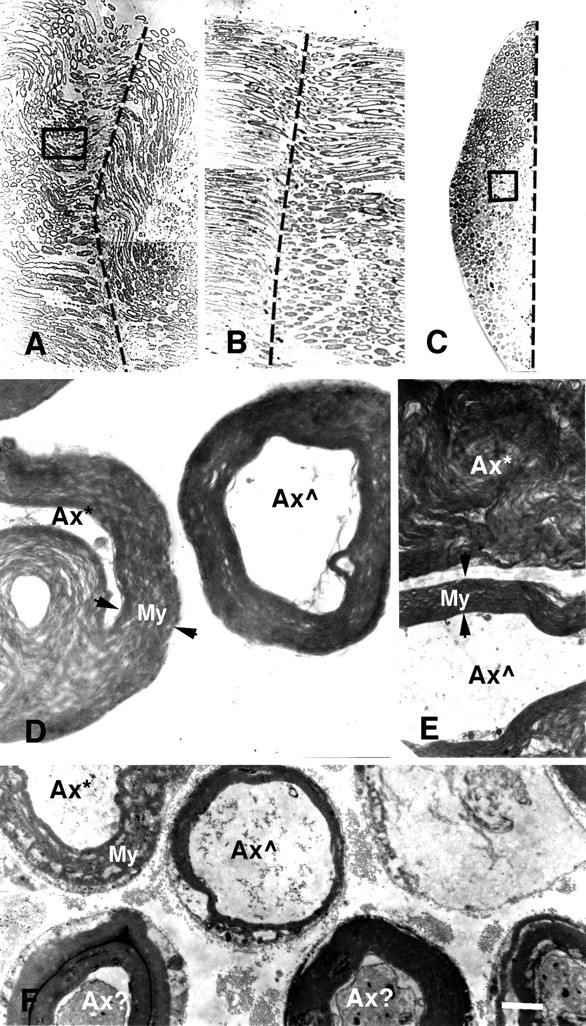
Photomicrographs (A–C) and electron micrographs (D–F) of longitudinal (A,B, D, E) and cross (C, F) sections of PEG-fused sciatic nerves after a complete cut (A,D, E) or crush (B,C, F). Vertical dashed lines in A–C show the site of severance and subsequent PEG application. Note that B andC are longitudinal and cross sections, respectively, taken through the site of PEG-induced fusion of a crushed sciatic nerve, of which a portion (box in C) is shown at higher magnification in F. As described in Materials and Methods and by Ballinger et al. (1997), the crushed sciatic axon was first sampled in longitudinal section (B) until a portion of the fusion site was detected (dashed line). The tissue was then remounted, and cross sections (C) were taken through another portion of the fusion site (dashed line).D and E are higher-magnification images of a portion of the cross section (box inA) of the entire sciatic nerve at the site of PEG-induced fusion. Axons exposed to PEG at the lesion site were in some disarray, and hence some individual axons were in transverse (D) or longitudinal (E) planes with respect to the long axis of an axon. Ax*, An axon with myelin or other membranes seriously delaminated, damaged, or dissolved by PEG; Ax , an axon with relatively intact axoplasm, myelin, and other membranes at the plane of PEG-induced fusion; Ax?, an axon with slightly damaged axoplasm, myelin, and other membranes at the plane of PEG-induced fusion;apposed arrowheads, the extent of myelin sheaths (My). For the cut axon shown in A,D, and E, the peak amplitude of the control CAP was 20 mV before the axons were transected. The CAP amplitude was 0 mV after the axons were cut in Ca2+-free hypotonic mammalian saline with 1.0 mm EGTA. A thin stream of 50% PEG 2000 kDa (Aldrich) was applied to the lesion site for 2 min. The preparation was then bathed in Krebs’ saline. Within 2 min after PEG application, the CAP was 0.5 mV, suggesting that PEG had induced fusion in a small fraction (2–5%) of crushed axonal ends. The CAP grew to 5 mV after the PEG-fused axons were maintained in Krebs’ for 1 hr, suggesting that as many as 20–30% of axons were now conducting through the lesion site. The sciatic nerve was then fixed as described in Materials and Methods. For the crushed sciatic nerve shown in B, C, and F, the peak amplitude of the control CAP was 14 mV before the axons were crushed. The CAP amplitude was 0 mV after the axons were crushed in Ca2+-free hypotonic mammalian saline with 1.0 mm EGTA. A thin stream of 50% PEG 2000 kDa (Aldrich) was applied to the lesion site for 1 min. The preparation was then bathed in the mammalian saline. Within 5 min after PEG application, the peak amplitude of the CAP was 1.0 mV, suggesting that PEG had induced fusion in a fraction (5–10%) of crushed axonal ends. The sciatic nerve was then fixed as described in Materials and Methods. Scale bar: A–C, 75 μm; D, 2 μm; E, F, 4 μm.
Fig. 3.
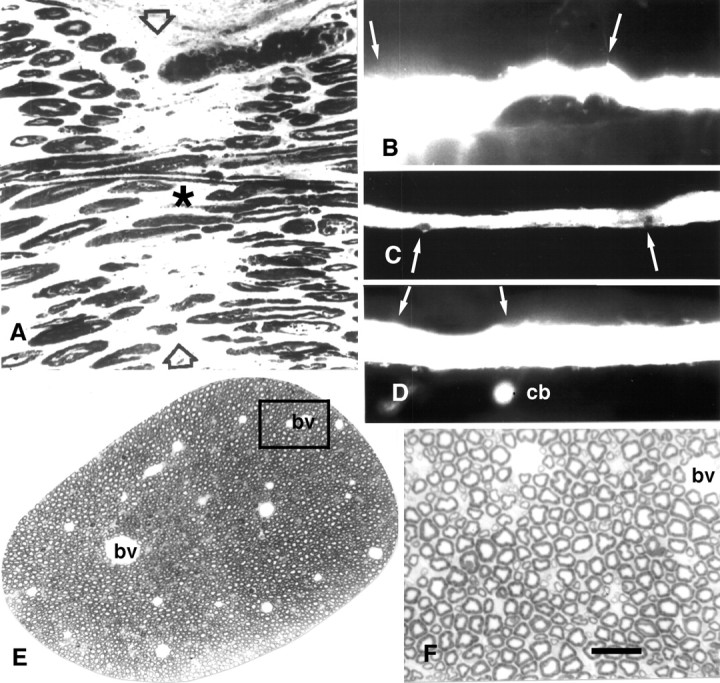
Photomicrographs of sciatic axons (A, E, F) and MGAs showing PEG fusion in vitro (A) orin vivo (C, D) and that the PEG hydrogel does no obvious morphological damage to MGAs (B–D) or sciatic axons (E,F). A, Photomicrograph of a longitudinal section taken through a cut sciatic axon that had been PEG-fused as described in Materials and Methods.Asterisk identifies a myelinated axon that could be traced through the lesion site. Arrowheads mark the site of PEG-induced fusion. B, Lucifer yellow fill of an MGA surrounded by PEG-based hydrogel for 15 d. The hydrogel was appled to the VNC in the region bounded by the two arrows.C, Lucifer yellow fill of an MGA fused with the PEG solution and then surrounded by the PEG-based hydrogel for 5 din vivo. Arrows show location of hydrogel. D, Lucifer yellow fill of a PEG-fused MGA surrounded by PEG-based hydrogel maintained for 15 d in vivo. Arrows show location of hydrogel.cb, The dye-filled cell body of the MGA. Data from same animal shown in Figure 1G. E, Photomicrograph of rat sciatic axon surrounded by PEG-based hydrogel for 24 hr. F, Enlargement of boxed area in E. bv, Blood vessel. Scale bar:A, F, 25 μm; B, 100 μm; C, D, 150 μm; E, 200 μm.
Fig. 4.
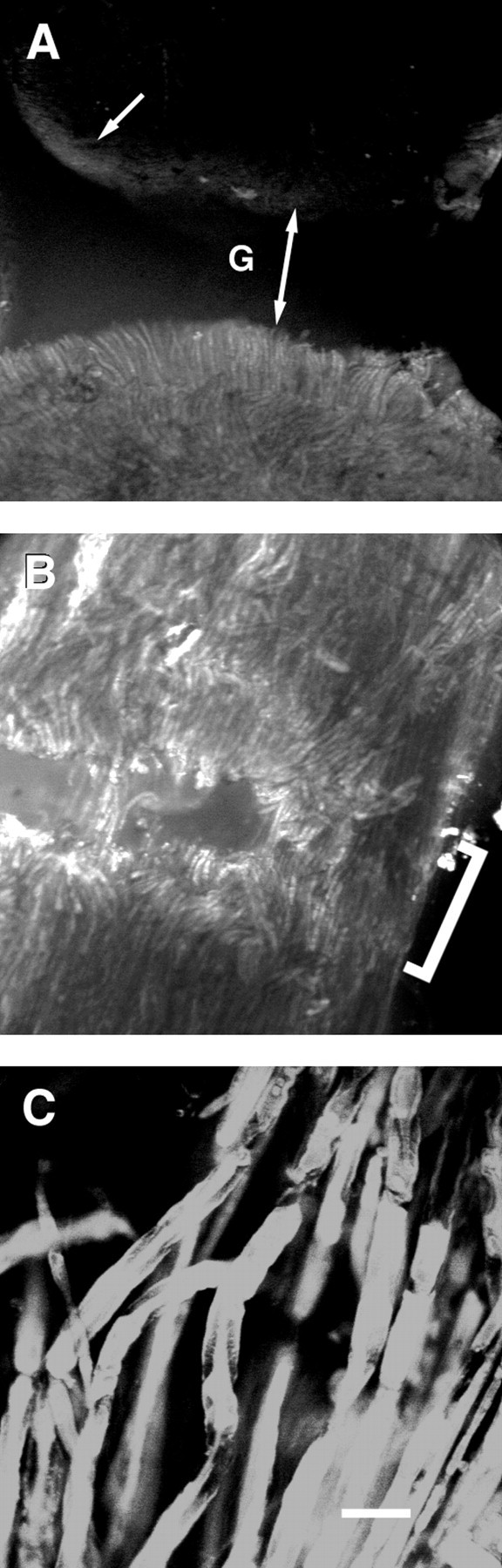
Confocal fluorescence images (photomicrographs) of control (A) and cut sciatic axons subsequently PEG-fused (B, C) and injected with sulforhodamine 101 24 hr before viewing. All figures are oriented so that the injected segment is at the bottom of each panel and the injection site is ∼3 mm from the lesion site.A, Lower-magnification image using a 10× Leica lens, showing control sciatic axon that was transected and closely apposed to the injected segment, but PEG was not applied to the lesion site (see Materials and Methods). The gap at lesion site is denoted by G and a double-headed arrow. Arrow points to the uptake of dye by connective tissue elements at the lesion site of the noninjected segment. B, Lower-magnification image using a 10× Leica lens of the lesion site (bracket) showing some dye-filled axons traversing the lesion site. C, Higher-magnification image using a 40× Zeiss lens showing dye-filled axons traversing the lesion site. For the cut sciatic nerve shown inB and C, the peak amplitude of the CAP was 6.2 mV before transection, 4.0 mV in Ca2+-free saline with 1.0 mm EGTA, 0 mV after transection, 1.9 mV within 10 min after PEG-induced fusion, and 0 mV when retransected. Scale bar: A, B, 110 μm;C, 25 μm.
We performed a series of control procedures using conventional or sucrose gap recordings to ensure that restored CAP conduction of PEG-fused sciatic and/or spinal axons represented a restoration of their axolemmal integrity at the lesion site rather than an artifactual conduction of electrical signals through the lesion site (Krause and Bittner, 1990; Krause et al., 1991). For example, CAPs did not conduct through the lesion site if the following: (1) PEG-fused sciatic or spinal axons were stimulated with subthreshold voltages (n > 40); (2) sciatic or spinal axons at the original fusion site were retransected (n > 50) (Fig.1D,E, rt); (3) Ca2+-free Krebs’ (or Ca2+-free mammalian saline), but not PEG, was applied to the closely abutted ends of cut (n > 20) or crushed sciatic (n> 10) or spinal (n > 10) axons; and (4) Ca2+-free Krebs’ (or Ca2+-free mammalian saline) and PEG were applied to loosely abutted ends of cut sciatic (n > 20) or spinal (n > 10) axons. As further controls for artifacts that might somehow be idiosyncratic to the five-chambered device or the sucrose gap technique, we showed that electrophysiological evidence that our PEG solutions fused the cut ends of MGAs could be obtained in three different ways: (1) double sucrose gap recordings using the five-chambered device; (2) conventional recordings using the five-chambered device in which all chambers were filled with earthworm saline; and (3) conventional recordings using the single-chambered device (Fig. 1F) from which APs from PEG-fused MGAs have already been published (Krause et al., 1991).
Ultrastructural assessments of sciatic axons fused with PEGin vitro
The anatomical continuity of sheathed sciatic axons was examined in electron micrographs of thin and thick sections of cut (n = 2) or crushed (n = 3) sciatic nerves that demonstrated PEG-induced fusion by restoration of CAP conduction using conventional or sucrose gap recording techniques (Fig.1A,B,D). Sciatic axons were often in disarray when viewed in longitudinal (Fig.2A,B,D,E) or cross (Fig. 2C,F) sections taken at many points through the lesion site. The myelin sheaths of most sciatic axons exposed to PEG showed extensive delamination and vesiculation in longitudinal (Fig.2D,E, Ax*) or cross (Fig. 2F, Ax*) sections. The myelin sheaths of other sciatic axons exposed to PEG exhibited intermediate amounts of delamination and vesiculation (Fig. 2F,Ax?). Only some sciatic axons had tightly packed laminar membranes in their myelin sheaths (Fig. 2D-F,Ax ), similar to those reported previously (Sunio and Bittner, 1997) for intact sciatic axons. Only those PEG-treated sciatic axons that did not have severe myelin delaminations or other damage typically exhibited a continuous axoplasm, sheath, and (possibly) axolemma at the lesion site when viewed in longitudinal (Fig. 2D,E, Ax ) or cross (Fig. 2F) sections. That is, PEG appeared to affect the myelin and other membranes of many sciatic axons at the fusion site but appeared to restore axonal and cytoplasmic continuity to only a relatively small fraction (usually 1–20%) of myelinated sciatic axons. (Individual unmyelinated sciatic axons were too small for us to detect in photomicrographs or to reliably examine the effects of PEG in low-power electronmicrographs.) The extent of repair of myelinated sciatic axons assessed ultrastructurally generally correlated with the extent of sciatic axon repair assessed electrophysiologically. For example, a greater percentage of myelinated sciatic axons appeared to be PEG-fused in the cut shown in Figure2A compared with the crush shown in B andC, a result consistent with the CAP data taken from these same preparations before fixation in which ∼25% of the peak amplitude of the control CAP was restored for the cut sciatic axons and ∼7% of the peak amplitude was restored for the crushed sciatic axons (Fig. 2). In longitudinal sections taken through a lesion site, myelinated axons could occasionally be traced through a site of PEG fusion (Fig. 3A, asterisk).
Confocal and epifluorescence assessments of sciatic axons fused with PEG in vitro
In another protocol to examine anatomical continuity of PEG-fused sciatic axons, desheathed sciatic nerves that conducted CAPs across the lesion site were evaluated by noting the diffusion across the lesion site of hydrophilic fluorescent dyes contained within individual sciatic axons. The lesion site was readily visible as a distinct gap in low-power fluorescence images of control preparations in which the two cut ends of a bundle of sciatic axons (n = 5) were closely apposed but PEG was not applied to the lesion site (Fig.4A). In these control preparations, a dye injected into a segment on one side of the lesion did not transfer to axons in the segment on the opposite side of the lesion site (Fig.4A), although small amounts of dye did label connective tissue elements at the cut edges (Fig. 4A,arrow). The lesion site was identifiable as a series of gaps in low-power fluorescence images of PEG-fused preparations (Fig.4B, bracket). In contrast to control preparations, a small fraction of dye-filled sciatic axons did traverse the lesion site (Fig. 4B,C) in preparations (n = 5) of cut sciatic axons that also showed electrophysiological evidence of PEG-induced fusion as assessed by the reappearance of a CAP after PEG application.
Functional and morphological measures in vitro andin vivo for nontoxicity of PEG hydrogels in control axons
We first examined how different formulations of PEG hydrogels affected the function (e.g., AP peak amplitude, threshold, and/or conduction velocity or CAP peak amplitude) or morphology (e.g., light microscopic or ultrastructural appearance) of MGAs or sciatic axonsin vitro and in vivo. As one example of an initial series of in vitro functional tests on over 200 MGAs, MGA (n > 20) APs were hyperexcitable (fired repetitively to a standard stimulus pulse of 0.5 msec in duration) when exposed to a 23% concentration of the PEG hydrogel with one eosin Y initiator but were indistinguishable from control APs with a second eosin initiator compared with control MGA APs (n > 20). Conversely, MGA (n > 20) APs were hypoexcitable and difficult to generate with a 28% concentration of the PEG hydrogel with either eosin initiator. Based on the results of such tests, we used the following formulation of the PEG hydrogel (see Materials and Methods) for all of the protocols described below: 23% w/v of 8 kDa PEG diacrylate dissolved in PBS, pH 7.4, containing 1 mmeosin Y, 100 mm triethanolamine, and 1500 ppmN-vinylpyrrolidone.
In a subsequent series of in vitro tests, we determined that this formulation of a PEG hydrogel was nontoxic to control MGAs and sciatic axons. For example, when this PEG hydrogel was applied to VNCs (n = 10) for 1–24 hr in vitro, we were able to conventionally record extracellular APs from MGAs in 90% of all animals tested (Fig. 1H). (The MGA is occasionally damaged in dissecting the VNC from an earthworm, and MGA APs cannot be recorded from 5–10% of dissected VNCs in control preparations.) Furthermore, this ability to extracellularly record an MGA AP and the peak amplitude of intracellularly recorded APs (67 ± 4 mV) or their conduction velocity (13 ± 3 m/sec) were not significantly different (p > 0.1%) compared with MGAs (n = 10) taken from paired intact (control) regions of the VNC from the same animal (90%; 65 ± 3 mV; 14 ± 3 m/sec, respectively).
Similar data were obtained from VNCs dissected from earthworms to which PEG hydrogel was applied in vivo for 1–30 d and sampled at the postoperative times given in Table 3. For example, an ability to elicit MGA APs and their conduction velocity were not significantly different (p > 0.1%) from data obtained from control MGAs (see above) or from sham-operated worms (Table 3). In this protocol, hydrogel-treated and sham-operated animals were anesthetized for 1 d at 16°C on Chlorotone-soaked filter paper and then placed in anesthetic-free soil. After recovery from anesthesia, the earthworms exhibited behaviors normally elicited by MGA activation, including a rapid contraction of the caudal end of the earthworm when the rostral end was mechanically stimulated.
Table 3.
Conduction of MGA APs in intact or PEG-fused MGAs versus postoperative time
| PO | n | % MGAs | CV(n) ± SD | Conditions |
|---|---|---|---|---|
| Intact MGAs, sham-operated,in vivo | ||||
| 1 d | 16 | 88 | 14 (7) ± 3 | |
| 5 d | 15 | 87 | 11 (6) ± 4 | |
| 15 d | 16 | 94 | 15 (6) ± 3 | |
| 30 d | 20 | 95 | 13 (5) ± 4 | |
| Intact MGAs, PEG hydrogel, in vivo | ||||
| 1 d | 24 | 92 | 11 (7) ± 4 | |
| 5 d | 24 | 96 | 12 (7) ± 5 | |
| 15 d | 21 | 81 | 13 (6) ± 3 | |
| 30 d | 15 | 87 | 14 (5) ± 4 | |
| PEG-fused MGAs, in vitro | ||||
| 0.5–24 hr | 31 | 16 | ||
| 0.5–24 hr | 17 | 47 | PEG hydrogel | |
| PEG-fused MGAs, in vivo | ||||
| 1 d | 99 | 27 | Anesthetic | |
| 1 d | 12 | 25 | PEG hydrogel | |
| 5–15 d | 11 | 27 | PEG hydrogel | |
| 20 d | 2 | 50 | PEG hydrogel | |
PO, Postoperative time. n, Total number of MGAs tested. % MGAs, Number of MGAs tested that conducted an AP through the lesion site, site of sham operation, and/or site of hydrogel application, divided by the total number of MGA preparations tested (n), times 100%. CV(n) ± SD, Conduction velocity (m/sec) and SD of number (n) of MGA APs traveling in a rostral-to-caudal direction through the lesion site (APs are normally conducted rostrally-to-caudally in MGAs in the intact animal). PEG hydrogels were not applied, and earthworms were not anesthetized, unless explicitly stated. p > 0.10 (Student’st test) for any paired set of CVs from sham-operated versus hydrogel-treated MGAs.
Morphological examinations of VNCs (Fig. 3B–D) confirmed data from these functional tests showing that the hydrogel “did no harm” to intact MGAs in vivo. For example, the diameter of MGAs or LGAs filled with Lucifer yellow CH were unaffected, or only slightly decreased, by application of the PEG hydrogel for 5 or 15 d (Fig. 3B). Video-enhanced microscopic or ultrastructural examination showed only minor exfoliation at the site of hydrogel application (data not shown). The portion of VNC exposed to the PEG hydrogel often showed an accumulation of connective tissue that adhered to the VNC. Sham-operated worms showed similar accumulations of connective tissue, although these structures were usually less extensive and less frequent compared with PEG hydrogel-treated VNCs maintained in vivo for weeks.
This formulation of a PEG hydrogel was also nontoxic to rat sciatic axons in vitro and in vivo according to various functional and structural tests. For example, when this PEG hydrogel was applied to rat sciatic nerves (n = 20) in vitro, the amplitude and conduction velocity of CAPs did not decrease compared with intact (control) sciatic axons taken from the same animal. When PEG hydrogel was applied to rat sciatic nerves (n > 20) in vivo, sciatic axons continued to conduct normal CAPs for days to weeks after application when the sciatic axons were removed from the animal and recorded in vitro (Fig. 1I). The sciatic functional index, an in vivo measure of rat sciatic function (de Medinaceli et al., 1982), was not obviously affected by application of PEG hydrogel to sciatic nerves for 1–30 d compared with control or sham-operated rats (A. Lore, K. Loftin, and G. Bittner, unpublished observations). Finally, rat sciatic axons surrounded by this PEG hydrogel for weeks in vivo exhibited normal morphology (Fig.3E,F) compared with control sciatic axons (Sunio and Bittner, 1997). In brief, our PEG hydrogels adhered tightly (see Materials and Methods) and were nontoxic to intact MGAs and rat sciatic axons tested in vitro andin vivo.
Use of the PEG hydrogel to maintain PEG-fused MGAs in vitro and in vivo
We first applied this PEG hydrogel to PEG-fused MGAs in vitro and in vivo to ensure that the PEG hydrogel was nontoxic to PEG-fused MGAs. For example, the application of PEG hydrogels to VNCs containing an MGA fused with our PEG solutionin vitro did not decrease our probability of observing PEG-fused MGAs when the VNC was examined for conduction of MGA APs through the lesion site from 0.5 to 24 hr (Table 3, PEG-fused MGAsin vitro). In fact, the PEG hydrogel may even have enhanced our ability to observe PEG-fused MGAs, which varied from 8 to 71%, in different in vitro trials that did not use the PEG hydrogel (Tables 2, 3).
If our PEG hydrogel was not applied to PEG-fused MGAs in vivo, those MGAs remained PEG-fused for 24 hr (Table 3, PEG-fused MGAs in vivo) only if the earthworm was immobilized by maintaining it on filter paper or soil moistened with an anesthetic (0.05–0.1% Chlorotone). That is, once the earthworm was no longer immobilized by the anesthetic, the VNC of PEG-fused MGAs quickly pulled apart at the site of fusion induced by application of the PEG solution. By applying our PEG hydrogel to the site where cut ends of an MGA had been induced to fuse in vivo, APs in PEG-fused MGAs could be maintained in nonanesthetized animals (Fig. 1H) sampled at 1, 5–15, or 20 d after transection (Table 3, PEG-fused MGAs in vivo), i.e., postfusion times that almost certainly equate to a permanent reestablishment of structural and functional continuity for earthworm axons. Other data were consistent with this hypothesis. For example, tactile stimulation of the rostral ends of these animals with PEG-fused MGAs elicited a rapid contraction of the caudal end of the earthworm. Furthermore, before sampling for microscopy, MGA APs traversed the lesion site in whole animal recordings (see Materials and Methods). When the VNCs were removed from these earthworms (n = 14) and recorded in vitro, MGA APs conducted across the site of PEG-induced fusion (Fig. 1H). When Lucifer yellow CH was injected into some (n = 4) of these PEG-fused MGAs ∼1 mm caudal to the lesion site at the time of sampling (5–15 d after transection) (Fig. 3C,D), the dye remained within the injected MGA and diffused across the lesion site within minutes. All of these data suggest that PEG solutions combined with PEG hydrogels can reestablish and permanently maintain morphological and functional continuity in vivo between the previously severed halves of PEG-fused earthworm MGAs, a myelinated invertebrate CNS axon that mediates behaviors important to the survival of the animal. Because there has been a rather conservative evolution of the structural and functional properties of invertebrate and vertebrate (including mammalian) axons, these data from rats and earthworms also suggest that mammalian axons might be repaired in vivo by PEG solutions and, if need be, that the integrity of PEG-fused axons could be maintained in vivo by PEG hydrogels.
DISCUSSION
Our in vitro data show that PEG solutions can rapidly (within minutes) establish functional (Fig. 1A–E; Table 2) and morphological (Figs. 2A–F,3A,E,F,4A–C) continuity between the cut or crushed ends of myelinated mammalian PNS and/or CNS axons. Our in vitro andin vivo data also show that PEG hydrogels adhere tightly (see Materials and Methods) and are nontoxic to intact earthworm MGAs (Figs. 1G, 3B; Table 3) and rat sciatic axons (Figs. 1I,3E,F). Finally, our in vivo data show that PEG solutions combined with PEG hydrogels can rapidly and permanently repair severed earthworm myelinated axons (MGAs) according to functional (Fig. 1H; Table 3) and morphological (Fig. 3C,D) criteria. These data suggest that PEG solutions, possibly combined with a PEG hydrogel, might also permanently reestablish functional and morphological continuity in vivo between the cut or crushed (severed) ends of myelinated mammalian PNS and CNS axons.
Possible mechanisms by which PEG solutions induce axonal fusion
We propose the following model to explain how PEG-induced fusion restores functional and morphological continuity between the ends of cut or crushed axons. PEG is a polymeric fusiogen that removes water from hydrophilic groups on the plasmalemmal surface, thereby inducing closely apposed cell membranes to fuse (Sowers, 1987; Lee and Lentz, 1997). Ca2+-free salines containing EGTA prevent the cut ends of unmyelinated (Eddleman et al., 1997, 1998; Godell et al., 1997) and myelinated (Krause and Bittner, 1990; Krause et al., 1991,1994; Ballinger et al., 1997) axons from sealing by preventing the formation of Ca2+-induced vesicles, which normally seal a somewhat constricted cut end. That is, a much greater area can be more closely apposed between the interface of two open axonal ends than between the interface of two constricted ends filled with a mass of vesicles. [Myelin membranes may also fuse with the axolemma. Slightly hypotonic salines produce axoplasmic swellings that open cut and crushed axonal ends and may facilitate PEG-induced fusion by allowing closer apposition of crushed axonal ends within unsevered perineural or epineural sheaths (Krause and Bittner, 1990; Krause et al., 1991).] However, even open ends of closely apposed axons are incompletely fused by PEG, and those PEG-fused axons initially have many plasmalemmal discontinuities. When such axons are subsequently placed in a Ca2+-containing physiological saline, Ca2+ inflow at the plasmalemmal discontinuities induces the formation of endocytotic vesicles and other membranous structures, which, in turn, seal those discontinuities (Krause et al., 1994; Ballinger et al., 1997; Eddleman et al., 1997, 1998). This repair of plasmalemmal lesions takes from several seconds to an hour, depending on the size and type of membrane lesion (Krause et al., 1994;Steinhardt et al., 1994; Ballinger et al., 1997; Eddleman et al., 1997,1998). Calpain enhances the ability of cut axonal ends to constrict and seal (Xie and Barrett, 1991; Eddleman et al., 1997; Godell et al., 1997), thereby inhibiting PEG-fusion between severed ends. In contrast, leupeptin (an inhibitor of calpain) inhibits constriction and sealing of severed axonal ends (Eddleman et al., 1997; Godell et al., 1997), thereby enhancing PEG-induced fusion.
Agents other than PEG might also be used to induce the fusion of severed axonal ends in mammals. For example, closely apposed ends of severed earthworm MGAs have been fused by laser beams (Yogev et al., 1991) and electric fields (Todorov et al., 1992). Similarly, agents other than our PEG-based hydrogel might be used to add mechanical strength to the sheaths of axons repaired by PEG solutions.
Possible clinical applicability of PEG solutions and PEG hydrogels
We have considered four problems that need to be resolved for successful implementation of our strategy to use PEG solutions to rapidly and permanently reestablish the function of severed PNS or CNS axons in mammals in vivo.
First, the restoration of all original functions requires that, in a bundle of PNS or CNS axons, the cut nerve ends be perfectly aligned and all axons successfully PEG-fused. This problem is observed clinically in accidents that cut PNS axons, after which the amount and specificity of naturally occurring axonal regeneration is not high unless the ends of severed fascicles are carefully aligned by microsurgery (Lundborg, 1990; Seckel, 1990). [This problem is somewhat less severe for crush-type lesions to CNS or PNS axons because crushed ends often remain aligned (Blight, 1989; Lundborg, 1990; Seckel, 1990).]
Second, PEG solutions do not induce axonal fusion unless the severed axonal ends are tightly apposed. Axonal ends usually separate somewhat after cut lesions, and an obvious gap often occurs between crushed axonal ends after many CNS (especially spinal) and some PNS contusion injuries (Blight, 1989).
Third, the mechanical strength at the lesion site is low when severed PNS or CNS axons are induced to fuse by PEG solutions applied in vitro or in vivo. PEG solutions do not reconnect severed connective tissue elements (e.g., collagen) that provide mechanical strength to nerve bundles.
Fourth, the distal stumps of severed mammalian axons usually degenerate within hours to a few days after severance (Ramon y Cajal, 1928; Das and Wallace, 1986; Bittner, 1991). Without intervention to lengthen the survival times of severed distal axonal processes, the time window for PEG-induced fusion would be limited to several hours after injury.
We believe that each of these four problems can be at least partially resolved.
First, if the goal is partial restoration of function rather than complete recovery, then the survival or regeneration of only 10% of CNS axons in mammals produces significant behavioral recovery (Eidelberg et al., 1977; Das and Wallace, 1986). Hence, the successful PEG-induced fusion of only 10% of axons in a CNS bundle whose ends are carefully (but not perfectly) aligned could produce rapid return of some functions that otherwise would never be restored. Furthermore, successful PEG-induced fusion of carefully aligned PNS axons would restore function much more rapidly (within minutes) compared with regeneration by growth cone outgrowth at 1–2 mm/d. (We have observed that mammalian sciatic axons or earthworm MGAs that are not successfully PEG-fused are not prevented from regenerating at 1–2 mm/day.)
Second, cut CNS or PNS nerve ends can be brought into close apposition by various surgical procedures (Das and Wallace, 1986). Furthermore, our current in vitro data from crushed rat sciatic axons and our current and published (Krause and Bittner, 1990; Krause et al., 1991) in vitro data from crushed earthworm MGAs show that the success rate for PEG-induced fusion of crushed myelinated axons is rather high when their axonal ends are opened and brought into close apposition in Ca2+-free, slightly hypotonic, salines containing 1–2 mm EGTA.
Third, our PEG hydrogel provides high mechanical strength at the lesion site and allows MGAs fused by PEG solutions to transmit APs across the lesion site for many posttransection days in nonanesthetized animals. Because this hydrogel exhibits similar properties on many invertebrate and mammalian soft tissues, it should provide sufficient mechanical strength to nerve sheaths in vivo to allow PEG-fused PNS and CNS axons in mammals to remain fused once the animal recovers from anesthesia.
Fourth, the rapid Wallerian degeneration of mammalian myelinated axons can now be greatly retarded via procedures that are not technically demanding. For example, we have recently shown that in vivocooling to 13°C (Sea et al., 1995) or injection of cyclosporin A (Sunio and Bittner, 1997) allows the distal stumps of many myelinated axons to survive for at least 6–10 d in rats, as does injection of antibodies to complement three receptors (Lunn et al., 1989).
In summary, we expect that our in vitro protocols to PEG fuse mammalian myelinated axons might be combined with our PEG hydrogel techniques to greatly decrease the time in vivo required for return of function after cut or crush lesions to PNS or CNS axons in mammals (including humans). These PEG-induced fusion techniques might also be combined with microsurgery, nerve growth guides (Jenq and Coggeshall, 1986; Aebischer et al., 1990; Herbert et al., 1996), or other strategies to increase the speed, amount, or specificity of regeneration of severed PNS axons in mammals. Our PEG-fusion techniques might also be combined with transplants of peripheral nerve sheaths (David and Aguayo, 1981), embryonic tissues (Giovanini et al., 1997;Miya et al., 1997), or injections of antibodies to oligodendritic inhibitors of CNS axonal outgrowth (Schnell and Schwab, 1990) to restore function to severed mammalian CNS axons.
Footnotes
These studies were funded by National Institutes of Health Grants NS31256 and HD31484, a Texas Advanced Technology grant to G.D.B., personal funds of G.D.B., and National Science Foundation Grant BES-9696020 to J.A.H. We thank Dr. Riyi Shi (Center for Paralysis Research, Purdue University, West Lafayette, IN) for demonstrating the use of a three-chambered device for double sucrose gap recordings. We also thank Dr. Jennifer L. West for demonstrating how to apply PEG-based hydrogel adhesives.
Correspondence should be addressed to Dr. George D. Bittner, Department of Zoology, University of Texas, Austin, TX 78712-1064.
REFERENCES
- 1.Aebischer P, Guenard V, Valentini RF. The morphology of regenerating peripheral nerves is modulated by the surface microgeometry of polymeric guidance channels. Brain Res. 1990;531:211–218. doi: 10.1016/0006-8993(90)90776-8. [DOI] [PubMed] [Google Scholar]
- 2.Ballinger ML, Blanchette AR, Krause TL, Smyers MS, Fishman HM, Bittner GD. Delaminating myelin membranes help seal the cut ends of severed earthworm giant axons. J Neurobiol. 1997;33:945–960. doi: 10.1002/(sici)1097-4695(199712)33:7<945::aid-neu6>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 3.Bittner GD. Long term survival of severed distal axonal stumps in vertebrates and invertebrates. Am Zool. 1988;28:1165–1179. [Google Scholar]
- 4.Bittner GD. Long term survival of anucleate axons and its implications for nerve regeneration. Trends Neurosci. 1991;14:188–193. doi: 10.1016/0166-2236(91)90104-3. [DOI] [PubMed] [Google Scholar]
- 5.Bittner GD, Ballinger ML, Raymond MA. Reconnection of severed nerve axons with polyethylene glycol. Brain Res. 1986;367:351–365. doi: 10.1016/0006-8993(86)91617-3. [DOI] [PubMed] [Google Scholar]
- 6.Blight AR. Effect of 4-aminopyridine on axonal conduction-block in chronic spinal cord injury. Brain Res Bull. 1989;22:47–52. doi: 10.1016/0361-9230(89)90126-3. [DOI] [PubMed] [Google Scholar]
- 7.Bullock TH, Horridge GA. Structure and function of the nervous system of invertebrates, Vols 1 and 2. Freeman; San Francisco: 1965. [Google Scholar]
- 8.Chowdhury SM, Hubbell JA. Adhesion prevention with ancrod released via a tissue adherent hydrogel. J Surg Res. 1996;61:58–64. doi: 10.1006/jsre.1996.0081. [DOI] [PubMed] [Google Scholar]
- 9.Das GD, Wallace RB. Neural transplantation and regeneration. Springer; New York: 1986. [Google Scholar]
- 10.David S, Aguayo AJ. Axonal elongation into peripheral nervous system “bridges” after central nervous system injury in adult rats. Science. 1981;241:931–933. doi: 10.1126/science.6171034. [DOI] [PubMed] [Google Scholar]
- 11.de Medinaceli L, Freed WJ, Wyatt RJ. An index of the functional condition of rat sciatic nerve based on measurements made from walking tracks. Exp Neurol. 1982;77:634–643. doi: 10.1016/0014-4886(82)90234-5. [DOI] [PubMed] [Google Scholar]
- 12.Eddleman CS, Ballinger ML, Smyers MS, Godell CM, Fishman HM, Bittner GD. Repair of plasmalemmal lesions by vesicles. Proc Natl Acad Sci USA. 1997;94:4745–4750. doi: 10.1073/pnas.94.9.4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eddleman CS, Ballinger ML, Smyers MS, Fishman HM, Bittner GD. Endocytotic formation of vesicles and other membranous structures induced by Ca2+ and axoplasmic injury. J Neurosci. 1998;18:4029–4041. doi: 10.1523/JNEUROSCI.18-11-04029.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eidelberg E, Straehley D, Erspamer R, Watkins CJ. Relationship between residual hindlimb-assisted locomotion and surviving axons after incomplete spinal cord injuries. Exp Neurol. 1977;56:312–322. doi: 10.1016/0014-4886(77)90350-8. [DOI] [PubMed] [Google Scholar]
- 15.Giovanini MA, Reier PJ, Eskin TA, Wirth E, Anderson DK. Characteristics of human fetal spinal cord grafts in the adult rat spinal cord: influences of lesion and grafting conditions. Exp Neurol. 1997;148:523–543. doi: 10.1006/exnr.1997.6703. [DOI] [PubMed] [Google Scholar]
- 16.Godell CM, Smyers MS, Eddleman CS, Ballinger ML, Fishman HM, Bittner GD. Calpain promotes the sealing of severed giant axons. Proc Natl Acad Sci USA. 1997;94:4751–4756. doi: 10.1073/pnas.94.9.4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Günther J. Neuronal syncytia in the giant fibers of earthworms. J Neuroctyol. 1975;4:55–62. doi: 10.1007/BF01099095. [DOI] [PubMed] [Google Scholar]
- 18.Günther J. Impulse conduction in the myelinated giant fibers of the earthworm: structure and function of the dorsal nodes in the median giant fiber. J Comp Neurol. 1976;168:505–532. doi: 10.1002/cne.901680405. [DOI] [PubMed] [Google Scholar]
- 19.Herbert CB, Bittner GD, Hubbell JA. Effects of fibrinolysis on neurite growth from dorsal root ganglia cultured in two- and three-dimensional fibrin gels. J Comp Neurol. 1996;365:380–391. doi: 10.1002/(SICI)1096-9861(19960212)365:3<380::AID-CNE4>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 20.Hubbell JA, Palsson BO, Papoutsakis ET. Tissue engineering and cell therapies. Preface. Biotechnol Bioeng. 1994;43:541. doi: 10.1002/bit.260430702. [DOI] [PubMed] [Google Scholar]
- 21.Jenq C-B, Coggeshall RE. The effects of an autologous transplant on patterns of regeneration in rat sciatic nerve. Brain Res. 1986;364:45–56. doi: 10.1016/0006-8993(86)90986-8. [DOI] [PubMed] [Google Scholar]
- 22.Krause TL, Bittner GD. Rapid morphological fusion of severed myelinated axons by polyethylene glycol. Proc Natl Acad Sci USA. 1990;87:1471–1475. doi: 10.1073/pnas.87.4.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krause TL, Marquis RM, Lyckman AW, Ballinger ML, Bittner GD. Rapid artificial restoration of electrical continuity across a crush lesion of a giant axon. Brain Res. 1991;561:350–353. doi: 10.1016/0006-8993(91)91615-8. [DOI] [PubMed] [Google Scholar]
- 24.Krause TL, Fishman HM, Ballinger ML, Bittner GD. Extent and mechanism of sealing in transected giant axons of squid and earthworms. J Neurosci. 1994;14:6638–6651. doi: 10.1523/JNEUROSCI.14-11-06638.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee J, Lentz BR. Evolution of lipidic structures during model membrane fusion and the relation of this process to cell membrane fusion. Biochemistry. 1997;36:6251–6259. doi: 10.1021/bi970404c. [DOI] [PubMed] [Google Scholar]
- 26.Lundborg G. Nerve regeneration problems in a clinical perspective. Restor Neurol Neurosci. 1990;1:297–302. doi: 10.3233/RNN-1990-13418. [DOI] [PubMed] [Google Scholar]
- 27.Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of wallerian degeneration does not hinder regeneration in peripheral nerve. Eur J Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- 28.Miya D, Giszter S, Mori F, Adipudi V, Tessler A, Murray M. Fetal transplants alter the development of function after spinal cord transection in newborn rats. J Neurosci. 1997;17:4856–4872. doi: 10.1523/JNEUROSCI.17-12-04856.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Okamura N, Stoskopf M, Yamaguchi H, Kishimoto Y. Lipid composition of the neuron system of earthworms (Lumbricus terrestris). J Neurochem. 1985;45:1875–1979. doi: 10.1111/j.1471-4159.1985.tb10546.x. [DOI] [PubMed] [Google Scholar]
- 30.Pathak CP, Sawhney AS, Hubbell JA. Rapid photopolymerization of immunoprotective gels in contact with cells and tissue. J Am Chem Soc. 1992;114:8311–8312. [Google Scholar]
- 31.Ramon y Cajal S. Degeneration and regeneration of the nervous system (May RM, translator). Oxford UP; London: 1928. [Google Scholar]
- 32.Roots BI, Lane NJ. Myelinating glia of earthworm giant axons: thermally induced intramembranous changes. Tissue Cell. 1983;15:695–709. doi: 10.1016/0040-8166(83)90044-7. [DOI] [PubMed] [Google Scholar]
- 33.Sawhney AS, Pathak CP, Hubbell JA. Bioerodible hydrogels based on photopolymerized poly(ethylene glycol)-co-Poly(α-hydroxy acid)-diacrylate macromeres. Macromolecules. 1993;26:581–587. [Google Scholar]
- 34.Schnell L, Schwab ME. Axonal regeneration in the rat spinal cord produced by an antibody against myelin-associated neurite growth inhibitors. Nature. 1990;343:269–272. doi: 10.1038/343269a0. [DOI] [PubMed] [Google Scholar]
- 35.Sea T, Ballinger ML, Bittner GD. Cooling of peripheral myelinated axons retards wallerian degeneration. Exp Neurol. 1995;133:85–95. doi: 10.1006/exnr.1995.1010. [DOI] [PubMed] [Google Scholar]
- 36.Seckel B. Enhancement of peripheral nerve regeneration. Muscle Nerve. 1990;13:785–800. doi: 10.1002/mus.880130904. [DOI] [PubMed] [Google Scholar]
- 37.Shi R, Blight AR. Compression injury of mammalian spinal cord in vitro and the dynamics of action potential conduction failure. J Neurophysiol. 1996;76:1572–1580. doi: 10.1152/jn.1996.76.3.1572. [DOI] [PubMed] [Google Scholar]
- 38.Shi R, Blight AR. Differential effects of low and high concentrations of 4-aminopyridine on axonal conduction in normal and injured spinal cord. Neuroscience. 1997;77:553–562. doi: 10.1016/s0306-4522(96)00477-0. [DOI] [PubMed] [Google Scholar]
- 39.Sowers AE. Cell fusion. Plenum; New York: 1987. [Google Scholar]
- 40.Steinhardt RA, Bi G, Alderton JM. Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science. 1994;263:390–393. doi: 10.1126/science.7904084. [DOI] [PubMed] [Google Scholar]
- 41.Sunio A, Bittner GD. Cyclosporin A retards the wallerian degeneration of peripheral mammalian axons. Exp Neurol. 1997;146:46–56. doi: 10.1006/exnr.1997.6484. [DOI] [PubMed] [Google Scholar]
- 42.Todorov AT, Yogev D, Qi P, Fendler JH, Rodziewicz GS. Electric-field-induced reconnection of severed axons. Brain Res. 1992;582:329–334. doi: 10.1016/0006-8993(92)90151-x. [DOI] [PubMed] [Google Scholar]
- 43.West JL, Hubbell JA. Separation of the arterial wall from blood contact using hydrogel barriers reduces intimal thickening after balloon injury in the rat: the roles of medial and luminal factors in arterial healing. Proc Nat Acad Sci USA. 1996;93:13188–13193. doi: 10.1073/pnas.93.23.13188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xie XY, Barrett JN. Membrane resealing in cultured rat septal neurons after neurite transection: evidence for enhancement by Ca2+-triggered protease activity and cytoskeletal disassembly. J Neurosci. 1991;11:3257–3267. doi: 10.1523/JNEUROSCI.11-10-03257.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yogev D, Todorov AT, Qi P, Fendler JH, Rodziewicz GS. Laser induced reconnection of severed axons. Biochem Biophys Res Commun. 1991;180:874–880. doi: 10.1016/s0006-291x(05)81146-5. [DOI] [PubMed] [Google Scholar]